

This study joins our previous efforts to elucidate how NS5A transits between hypo- and hyperphosphorylated states via phosphorylation on a series of highly conserved serine residues. Of the serine residues, serine 229 is the most interesting since phosphorylation-mimicking and phosphorylation-ablating mutations at this serine residue are both lethal. With a new high-quality antibody specific to serine 229 phosphorylation, we concluded that serine 229 must remain wild type so that it can dynamically cycle through phosphorylation and dephosphorylation that govern NS5A between hypo- and hyperphosphorylated states. Both are required for the HCV life cycle. When phosphorylated, serine 229 signals phosphorylation on serine 232 and 235 in a sequential manner, leading NS5A to the hyperphosphorylated state. As serine 235 phosphorylation is reached, serine 229 is dephosphorylated, stopping signal for hyperphosphorylation. This balances NS5A between two phosphorylation states and in intracellular structures that warrant a productive HCV life cycle.
KEYWORDS: hepatitis C virus, protein kinases, protein phosphorylation, proteomics, viral replication
ABSTRACT
The nonstructural protein NS5A of hepatitis C virus (HCV) is a phosphorylated protein that is indispensable for viral replication and assembly. We previously showed that NS5A undergoes sequential serine S232/S235/S238 phosphorylation resulting in NS5A transition from a hypo- to a hyperphosphorylated state. Here, we studied functions of S229 with a newly generated antibody specific to S229 phosphorylation. In contrast to S232, S235, or S238 phosphorylation detected only in the hyperphosphorylated NS5A, S229 phosphorylation was found in both hypo- and hyperphosphorylated NS5A, suggesting that S229 phosphorylation initiates NS5A sequential phosphorylation. Immunoblotting showed an inverse relationship between S229 phosphorylation and S235 phosphorylation. When S235 was phosphorylated as in the wild-type NS5A, the S229 phosphorylation level was low; when S235 could not be phosphorylated as in the S235A mutant NS5A, the S229 phosphorylation level was high. These results suggest an intrinsic feedback regulation between S229 phosphorylation and S235 phosphorylation. It has been known that NS5A distributes in large static and small dynamic intracellular structures and that both structures are required for the HCV life cycle. We found that S229A or S229D mutation was lethal to the virus and that both increased NS5A in large intracellular structures. Similarly, the lethal S235A mutation also increased NS5A in large structures. Likewise, the replication-compromised S235D mutation also increased NS5A in large structures, albeit to a lesser extent. Our data suggest that S229 probably cycles through phosphorylation and dephosphorylation to maintain a delicate balance of NS5A between hypo- and hyperphosphorylated states and the intracellular distribution necessary for the HCV life cycle.
IMPORTANCE This study joins our previous efforts to elucidate how NS5A transits between hypo- and hyperphosphorylated states via phosphorylation on a series of highly conserved serine residues. Of the serine residues, serine 229 is the most interesting since phosphorylation-mimicking and phosphorylation-ablating mutations at this serine residue are both lethal. With a new high-quality antibody specific to serine 229 phosphorylation, we concluded that serine 229 must remain wild type so that it can dynamically cycle through phosphorylation and dephosphorylation that govern NS5A between hypo- and hyperphosphorylated states. Both are required for the HCV life cycle. When phosphorylated, serine 229 signals phosphorylation on serine 232 and 235 in a sequential manner, leading NS5A to the hyperphosphorylated state. As serine 235 phosphorylation is reached, serine 229 is dephosphorylated, stopping signal for hyperphosphorylation. This balances NS5A between two phosphorylation states and in intracellular structures that warrant a productive HCV life cycle.
INTRODUCTION
Hepatitis C virus (HCV) is an RNA virus that affects 71 million people worldwide (1). Its RNA genome encodes a long polyprotein consisting of three structural proteins (core, E1, and E2) and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (2, 3) that are cleaved by host peptidases and virus-encoded proteases to release individually functional proteins (4–6). Although the structural proteins make up the viral particles, each nonstructural protein performs distinct functions necessary for the viral life cycle (7).
The NS5A protein does not have known enzymatic activities (8–10). It has three structural domains (I, II, and III) connected by two low-complexity sequence (LCS) regions (8). Except for domain I, whose crystal structure has been solved (11, 12), the rest of the protein is thought to be highly disordered. NS5A forms a dimer via interaction between two domain I regions (13). Domain I and the LCS I region constitute the minimal protein portion in the NS5A dimer involved in RNA binding (9, 14, 15). In addition, NS5A interacts with a plethora of host and viral proteins with versatile functions in host cell responses, viral genome replication, and viral particle assembly (8–10).
When analyzed by SDS-PAGE and immunoblotting, NS5A appears as two bands at 56 and 58 kDa, representing, respectively, hypo- and hyperphosphorylated states (16). Many studies have pinpointed a cluster of highly conserved serine residues in particular S225, S229, S232, and S235 in the LCS I region responsible for NS5A hyperphosphorylation and viral genome replication (17–24). Recently, a threonine-rich cluster downstream to the LCS I region was also shown to contribute to NS5A hyperphosphorylation (25). Thus, the so-called hyperphosphorylated NS5A consists of multiple phosphorylated NS5A variants. In genotypes 1, 3, 4, and 5, alanine mutations in the above-mentioned serine residues reduced NS5A hyperphosphorylation and enhanced viral genome replication (20, 26). Harak et al. demonstrated that the ability of the above mutants to replicate did not correlate with NS5A hyperphosphorylation but rather correlated with reduced activation of the lipid kinase phosphatidylinositol 4-kinase IIIα (PI4KIIIα), whose overactivation was shown to be detrimental to viral replication (26). On the contrary, numerous studies showed that the same mutations in genotype 2 reduced NS5A hyperphosphorylation and reduced viral replication (17–19, 22–24, 27). Why the same alanine mutations reduced genotype 2 replication was unclear.
For years, NS5A was thought to transit from a hypo- to a hyperphosphorylated state via phosphorylation of the serine residues in the LCS I region (8, 28). These serine residues are spaced two or three amino acids apart and correspond to the phosphorylation preference of casein kinase Iα (CKIα) or glycogen synthase kinase 3β (GSK3β), which phosphorylate a serine/threonine residue at position 0 when, respectively, an upstream (−3) or a downstream (+4) serine/threonine residue is phosphorylated or primed first (29, 30). Because both kinases have been implicated in NS5A phosphorylation (19, 31, 32), it can be anticipated that a sequential phosphorylation cascade from the NH2 terminus to the COOH terminus or in the reverse direction would enable NS5A transition from hypo- to hyperphosphorylated state. This sequential phosphorylation cascade could result in a number of differentially phosphorylated NS5A variants that recruit different proteins for diverse functions (9).
Using three antibodies specific to S232, S235, and S238 phosphorylation, we recently showed that S235 phosphorylated NS5A is the primary hyperphosphorylated NS5A variant (24) and that NS5A undergoes sequential S232/S235/S238 phosphorylation by CKIα, leading to the hyperphosphorylated state (27). Extending this phosphorylation cascade further upstream, S229 phosphorylation should prime sequential S232/S235/S238 phosphorylation, as indicated in an earlier study where S229 phosphorylation was shown to prime S232 phosphorylation in in vitro CKIα assay (33). However, NS5A hyperphosphorylation remains even when S229 is mutated to alanine (17, 18). Moreover, both a phosphorylation-ablating alanine mutation and a phosphorylation-mimicking aspartate mutation at S229 blunt HCV replication (17, 18), leaving the functions of S229 phosphorylation mysterious.
In the present study, we made an NS5A antibody specific to S229 phosphorylation and used it to show that S229 likely cycles between dephosphorylated and phosphorylated states, thereby maintaining a delicate balance of NS5A between hypo- and hyperphosphorylated states via sequential phosphorylation, which is critical to the life cycle of genotype 2a HCV.
RESULTS AND DISCUSSION
S229 phosphorylation was detected in hypo- and hyperphosphorylated NS5A in HCV-infected Huh7.5.1 cells.
As a continuing effort to study sequential S232/S235/S238 NS5A phosphorylation cascade (Fig. 1A) (27), we made an antibody specific to S229 phosphorylation. The antibody was generated by immunizing rabbits with an S229 phosphorylated long peptide (Fig. 1B). On the dot blot (Fig. 1B), the antibody detected this long S229 phosphorylated peptide in a dose-dependent manner and not the same length peptide without S229 phosphorylation. The antibody also detected a shorter S229 phosphorylated peptide in a dose-dependent manner, indicating high specificity. Indeed, the S229 phosphorylation-specific antibody did not cross-react with other peptides with phosphorylation at S222, S232, S235, or S238 discovered with phosphoproteomics (19). In HCV (J6/JFH1, genotype 2a)-infected Huh7.5.1 cells, the level of S229 phosphorylation was very low and increasing the scanning light intensity was necessary to show the weak S229 phosphorylation signal (Fig. 1C). Immunoprecipitation with the 9E10 NS5A antibody (34), followed by immunoblotting for S229 phosphorylation, confirmed the weak S229 phosphorylation signal and the appearance of S229 phosphorylation in both hypo- and hyperphosphorylated NS5A (Fig. 1D). At 4, 12, and 24 h postinfection, when the total NS5A was barely detected by the 9E10 antibody (Fig. 1C), S229 phosphorylation appeared to be in the hypophosphorylated NS5A (p56). However, due to the lack of definitive NS5A signals, which could be due to antibody sensitivity issues, S229 phosphorylation at these time points should be considered with caution. Starting at 48 h postinfection, S229 phosphorylation began to show predominantly in the hyperphosphorylated NS5A and yet with a visible appearance in the hypophosphorylated NS5A. Quantitation shows a higher level of S229 phosphorylation in the hyperphosphorylated NS5A versus hypophosphorylated NS5A throughout the rest of the experimental period (Fig. 1C, bottom panel). The unit S229 phosphorylation level per NS5A species was the highest at 48 h postinfection and thereafter sharply declined with time. It is important to note that the experimental period exceeds the HCV life cycle, and therefore the above observations should not be overinterpreted.
FIG 1.

Characterization of the S229 phosphorylation-specific antibody. (A) Schematic of NS5A structure. Serine residues in the low-complexity sequence (LCS) I region are numbered. (B) Dot blot analysis for the NS5A S229 phosphorylation-specific antibody (anti-pS229). Synthetic S229 phosphorylated and nonphosphorylated peptides were serially diluted and dotted on a nitrocellulose membrane before detection with the antibody. Sequences of the peptides are shown with the phosphorylated-serine residues colored red. Arrow denotes the pS229 peptide used for antibody generation. (C) Immunoblot showing time course S229 phosphorylation in J6/JFH1-infected Huh7.5.1 cells. The bottom panel shows pS229 signal intensity in relation to NS5A (total and hypo- or hyperphosphorylated NS5A). Due to the lack of apparent NS5A bands at 0, 4, 12, and 24 h, values at these time points were set as zero. (D) Immunoblotting (IB) for S229 phosphorylation in NS5A immunoprecipitated (IP) with the NS5A-specific 9E10 antibody. The sample was Huh7.5.1 cells infected with J6/JFH1 HCV for 3 days. In, input; Un, unbound; W, wash; E, eluate.
The appearance of S229 phosphorylation in both hypo- and hyperphosphorylated NS5A suggests that S229 phosphorylation could serve as an initiating signal that fires sequential S232/S235/S238 phosphorylation by CKIα (29). In line with this, our previous study showed that alanine mutation at S229 (S229A) reduced (see Fig. 3 in reference 27), whereas aspartate mutation at S229 (S229D) restored NS5A phosphorylation at S232, S235, and S238 (see Fig. 6 in reference 27). The facts that S229A mutation significantly reduced but did not completely abolish NS5A phosphorylation at S232, S235, or S238 (see Fig. 5 in reference 27) suggest that (i) S229 phosphorylation, although not absolutely required, plays a major part in sequential S232/S235/S238 phosphorylation and that (ii) kinases other than CKIα, such as DNA-dependent kinase (DNA-PK; see below) (35), could phosphorylate S232 and lead to hyperphosphorylation.
S229 phosphorylation was elevated in S235A mutant NS5A.
To test whether S229 phosphorylation is affected by phosphorylation on other serine residues, we detected S229 phosphorylation in HEK293T cells transfected with NS3-NS5A-expressing constructs: wild type (WT) or with an alanine mutation at various serine residues. Previously, we showed that NS5A phosphorylation occurs similarly in the HEK293T and the T7-Huh7 cells (24, 27, 36). As seen in the immunoblots (Fig. 2A), the S229 phosphorylation level was low in wild-type NS3-NS5A-expressing HEK293T cells, similar to what was observed in HCV-infected Huh7.5.1 cells (Fig. 1C). In fact, S229 phosphorylation was low in almost all except S235A mutant NS3-NS5A-transfected HEK293T cells. Figure 2B summarizes results from three independent experiments. As seen, the S229 phosphorylation level was higher in the S235A mutant than in wild-type NS5A. This surprisingly high level of S229 phosphorylation was also observed in the T7-Huh7 cells transfected with S235A mutant NS3-NS5A (Fig. 2C and D) or S235A mutant NS3-NS5B (Fig. 2E and F) construct. Previously in S235A mutant NS5A (see Fig. 3A in reference 27), we noticed a new p57 band that was phosphorylated at S232. In the present study, we found that the S229 phosphorylated NS5A band of the S235A mutant was slightly above the p56 band and below the S232 phosphorylated p57 band (Fig. 2A), suggesting sequential S229/S232/S235 phosphorylation that gradually elevates the hypophosphorylated p56 band toward the hyperphosphorylated p58 band. We omitted discussion on S238 phosphorylation because S238A mutation does not affect NS5A hyperphosphorylation or reporter virus activity (19).
FIG 2.

S229 phosphorylation level was elevated in S235A mutant NS5A. (A and C) Immunoblotting for S229 phosphorylation in HEK293T cells or T7-Huh cells transfected with wild-type (WT) or various serine-to-alanine mutant NS3-NS5A constructs. (B and D) Summaries of the immunoblotting results in panels A and C, respectively. (E and G) Immunoblotting for S229 phosphorylation in T7-Huh7 cells transfected with WT or various serine-to-alanine or serine-to-aspartate mutant NS3-NS5B constructs. (F and H) Summary of the immunoblotting results in panels E and G, respectively. (I and J) Immunoblots and summary for S229 phosphorylation in T7-Huh7 cells transfected with WT, S235A, and S235D mutant NS3-NS5B construct. Values are means ± the standard errors (SE) from three independent experiments for S229 phosphorylation normalized for loading (the amount of total NS5A) and standardized against that of the S235A or S235D mutant NS5A. The values were not standardized conventionally with that of the WT NS5A because S229 signals for WT NS5A were not detected in several cases, and the values became negative after correction for background in the mock control. ND, not detected; asterisk, statistical significance compared to WT (*, P < 0.05; **, P < 0.01; ***, P < 0.005 [Student t test]).
NS5A hyperphosphorylation was intrinsically regulated.
The low S229 phosphorylation level in HCV-infected Huh7.5.1 cells and in wild-type NS3-NS5B-transfected HEK293T and T7-Huh cells suggests that S229 is probably dephosphorylated when sequential NS5A phosphorylation reaches S235 phosphorylation, the primary NS5A hyperphosphorylation event required for HCV replication (24). We thus hypothesized that one of the functions of S229 phosphorylation by an unknown kinase is to prime sequential S232/S235 phosphorylation by CKIα (19, 27). When S235 is phosphorylated, an unknown phosphatase dephosphorylates S229. In line with this, the S229 phosphorylation level was lower in S235D (Fig. 2G and H) than in S235A (Fig. 2E and F) NS3-NS5B-transfected T7-Huh7 cells. Analysis of wild-type, S235A, and S235D NS5A for S229 phosphorylation on a single blot confirmed the results (Fig. 2I and J). The fact that S229 in the S235D mutant was not dephosphorylated to the same level as wild-type NS5A (Fig. 2G and H) is likely because aspartate mutation at S235 does not fully represent phosphorylation at S235 that leads to S229 dephosphorylation. When S235 is not phosphorylated as in the S235A mutant NS5A (Fig. 2E and F), S229 phosphorylation was elevated, attempting to reinforce the signal for sequential S232/S235 phosphorylation.
We recently found that CKII preferentially binds hyperphosphorylated NS5A (i.e., with S232 and S235 phosphorylation) in a dimethyl-label based quantitative proteomics study (35). CKII phosphorylates serine or threonine (i.e., S229) when the +3 position is a negatively charged aspartate or glutamate residue (37), which is often used to mimic phosphorylated serine (i.e., phosphorylated S232) or threonine (17–20, 38). These facts make CKII an ideal kinase candidate that binds S232 phosphorylated NS5A and phosphorylates S229. In fact, CKII has been implicated in NS5A phosphorylation (39, 40). The theory presented above also explains several observations. For example, why is S229 not phosphorylated in the S232A mutant NS5A (Fig. 2E) that abolishes S235 phosphorylation (see Fig. 5 in reference 27)? This is because S232A abolishes S232 phosphorylation, a prerequisite for CKII-mediated S229 phosphorylation. Similarly, because S225A also abolishes S232 phosphorylation (see Fig. 5A in reference 27), there was no CKII-mediated S229 phosphorylation in S225A mutant NS5A (Fig. 2E). In addition, if CKII is to phosphorylate S229 when the +3 amino acid is an aspartate as in the S232D mutant, why was S229 not phosphorylated in the S232D mutant NS5A (Fig. 2G and H)? This is because S232D permits S235 phosphorylation (see Fig. 6C in reference 27), which in turn results in S229 dephosphorylation (Fig. 2G). Similarly, S229 phosphorylation was not observed in the S238D mutant NS5A (Fig. 2G) that permitted S235 phosphorylation (see Fig. 6C in reference 27).
Despite an ideal theory, our attempts to test CKII’s role in S229 phosphorylation were complicated with cytotoxicity upon CKII inhibition with inhibitors or small hairpin RNA-mediated knockdown. Two CKII inhibitors were used for the test: TMCB and TBB (see Materials and Methods). As seen in Fig. 3A and B, both TMCB and TBB dose dependently reduced S229 phosphorylation in S235A mutant NS3-NS5B-transfected T7-Huh7 cells; however, these drugs worked at very high concentrations close to cytotoxic doses (Fig. 3I and J) that also reduced the total NS5A amounts (Fig. 3A and B). As a result, the proportional S229 phosphorylation level per unit of NS5A did not change upon CKII inhibition (Fig. 3E and F). Thus, we could not be certain whether TMCB or TBB reduces S229 phosphorylation, thereby reducing NS5A stability, or whether they indiscriminately reduce NS5A in general. On the contrary, S232 phosphorylation was not affected by CKII inhibition even at high doses when the total NS5A amounts were reduced (Fig. 3C and D). This mathematically resulted in an increase in S232 phosphorylation per unit NS5A (Fig. 3E and F). Because of the apparent cytotoxic effects and the characteristics of the CKII phosphorylation preference, we compared S229 phosphorylation per unit of S232 phosphorylation (Fig. 3G and H). As seen, the S229 over S232 phosphorylation ratio decreased in a dose-dependent manner upon CKII inhibition by TCMB or TBB. Because of the cytotoxic effects of the inhibitors, small hairpin RNA-mediated CKII knockdown was used; however, CKII knockdown resulted in severe cell death before the experiments could be performed. Thus, the CKII inhibitors exhibit apparent cytotoxic effects, which are associated with reduced total NS5A, reduced S229 phosphorylation, and no change in S232 phosphorylation on immunoblots. It is of interest to check whether other kinases, e.g., PI4KIIIα, involved in the synthesis of hypo- and hyperphosphorylated NS5A participate in S229 phosphorylation (41, 42).
FIG 3.

Effects of CKII inhibition on cell viability, NS5A, and NS5A phosphorylation. (A and B) Effects of TMCB and TBB on NS5A S229 phosphorylation in NS3-NS5B transfected T7-Huh7 cells. (C and D) Effects of TMCB and TBB on NS5A S232 phosphorylation in the same cells. (E and F) Summary of the effects of TMCB and TBB on NS5A S229 and S232 phosphorylation. (G and H) Summary of the effects of TMCB and TBB on S229 over S232 phosphorylation ratio. (I and J) Cell viability assay for TMCB and TBB. The S235A mutant NS3-NS5B-transfected T7-Huh7 cells were exposed to TMCB for 1 day or TBB for 6 h prior to immunoblotting and viability assay. An asterisk (*) indicates statistical significance compared to 0 μM (P < 0.05 [Student t test]).
S225D did not induce S229 phosphorylation (Fig. 2G), and yet sequential S232/S235 phosphorylation was still observed in the S225D mutant (see Fig. 6 in reference 27), suggesting that S225 phosphorylation probably does not fall within the sequential S229/S232/S235 phosphorylation cascade. However, the fact that S225A and S225D respectively abolish (see Fig. 5 in reference 27) and rescue (see Fig. 6 in reference 27) S232/S235 phosphorylation suggests that S225 participates in S232/S235 sequential phosphorylation independent of S229 phosphorylation.
A complete HCV life cycle requires a wild-type S229.
It has been known that S235 phosphorylation is the major hyperphosphorylation event of NS5A (24) required for genotype 2a HCV replication (17–19). Our reporter HCV activity assay confirmed the above observations (Fig. 4A). S235A mutation abolished whereas S235D mutation rescued the reporter HCV activity (Fig. 4B). It has been known that S229A or S229D (or S229E) mutation blunted the reporter HCV activity and genome replication (17, 18), indicating that S229 is a key residue in HCV replication (43). Our reporter HCV activity assay also confirmed this (Fig. 4B). Based on these observations, we would like to propose that S229 must remain a wild-type serine residue that cycles between phosphorylation and dephosphorylation states to fire signals for NS5A sequential phosphorylation for the purpose of reaching S235 phosphorylation. When S229 cannot be phosphorylated as in the case of S229A mutation, S229A does not fire signal for sequential NS5A phosphorylation. As a result, an unknown kinase such as DNA-PK (35) kicks in to phosphorylate S232 that primes S235 phosphorylation in the S229A mutant NS5A (see Fig. 5A and C in reference 27); however, the phosphorylation levels were too low to sustain the reporter virus activity (Fig. 4B). When S229 is mutated to an aspartate that cannot be dephosphorylated after S235 is phosphorylated (see Fig. 6C in reference 27), the reporter virus activity is also low (Fig. 4B), suggesting that S229 and S235 cannot be phosphorylated at the same time or simultaneously phosphorylated for too long for a productive HCV life cycle. To test this, the reporter virus activity was measured in the Huh7.5.1 cells transfected with a doubly mutated virus construct (S229A/S235A, S229A/S235D, S229D/S235A, or S229D/S235D). As seen in Fig. 4C, as long as S235 is mutated to alanine that cannot be phosphorylated, the reporter virus activity was low regardless S229A or S229D. As expected, the reporter activities of S229D/S235D and S229A/S235D were both low because either S229A or S229D blocked S229 to cycle between dephosphorylated and phosphorylated state. Our explanation for the observations presented above seems rather plausible, although there could be alternative explanations. For example, wild-type S229 perhaps performs functions independent of its phosphorylation, given its low phosphorylation level.
FIG 4.

The complete HCV life cycle requires a wild-type S229. (A) Schematic of the full-length HCV construct with Renilla luciferase reporter. (B and C) Luciferase activity assay of the reporter virus in the Huh7.5.1 cells transfected with in vitro transcript of wild-type (WT) or mutant with the indicated serine-to-alanine or serine-to-aspartate mutation at S229 or S235. (C) Values are means ± the SE from three independent experiments, standardized against that observed at 4 h after transfection. An asterisk (*) indicates statistical significance compared to the 4-h posttransfection time point (P < 0.05 [Student t test]).
S229 phosphorylation regulates intracellular NS5A distributions.
Using confocal immunofluorescence microscopy, we found that NS5A was present in large and small structures that only partially colocalized with S235 phosphorylated NS5A, which represents the major hyperphosphorylated NS5A variant in NS3-NS5B-transfected T7-Huh7 cells (Fig. 5). Likewise, the S235 hyperphosphorylated NS5A only partially colocalized with NS3 representing the replication complex (Fig. 6). Using live-cell imaging, Wolk et al. previously showed two distinct structures where NS5A resides: a large structure representing membranous web with restricted motility and a small structure representing replication complex with fast and long-distance motility (44). Both are required for the HCV life cycle. In addition to this model, we would like to propose that S229, perhaps by cycling between phosphorylation and dephosphorylation, harnesses sequential S232/S235 phosphorylation to maintain a delicate balance of NS5A between hypo- and hyperphosphorylated states and the intracellular localization crucial to the HCV life cycle. Consistent with this, when S229 was mutated to alanine or aspartate, S235 hyperphosphorylated NS5A was restricted to large structures (Fig. 5). Quantification of the immunofluorescence micrographs showed that about 18% of the WT NS5A resided in large structures (Fig. 7A, the fourth quartile with a particle volume of >1.02 μm3). In contrast, significantly more NS5A resided in large structures when S229 was mutated to alanine or aspartate (Fig. 7A). On average, about 36 and 26% of the NS5A were found in large structures for S229A and S229D, respectively (Fig. 7A). S229A or S229D mutation also increased S235 hyperphosphorylated NS5A to colocalize with NS3 in large structures (Fig. 6). On average (Fig. 7B), about 17% NS3 in the WT NS5A transfected cells was found in large structures. In contrast, significantly more NS3 was found in large structures in S229A (35%) or S229D (27%) NS5A-transfected cells (Fig. 7B). Since both S229A and S229D mutations are lethal (Fig. 4), these results indicate that it is necessary for NS5A to maintain a balanced intracellular distribution in both large and small structures for the HCV life cycle. In line with this, S235A mutation that increased NS5A in large structures (Fig. 7C, as well as Fig. 8) blunted viral replication (Fig. 4). In contrast, S235D that permitted NS5A distribution in large and small structures (Fig. 7C, as well as Fig. 8) was capable of replication (Fig. 4), albeit compromised.
FIG 5.

S229A and S229D mutations altered intracellular distribution of NS5A. Confocal immunofluorescence micrographs of NS5A (red) costained with S235 phosphorylated NS5A (pS235, green) in the T7-Huh7 cells transfected with wild-type (WT), S229A, or S229D mutant NS3-NS5B mutant constructs. The nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole; blue). Boxes indicate areas enlarged.
FIG 6.

S229A and S229D mutations altered intracellular distribution of NS5A with regard to NS3. Confocal immunofluorescence micrographs of NS3 (red) costained with S235 phosphorylated NS5A (pS235, green) in the T7-Huh7 cells transfected with wild-type (WT), S229A, or S229D mutant NS3-NS5B mutant constructs. The nuclei were stained with DAPI (blue). Boxes indicate areas enlarged.
FIG 7.

Distribution of intracellular NS5A or NS3 particles based on their volume sizes. (A) Intracellular NS5A particles in WT, S229A, or S229D NS3-NS5B- transfected T7-Huh7 cells. (B) Intracellular NS3 particles in WT, S229A, or S229D NS3-NS5B-transfected T7-Huh7 cells. (C) Intracellular NS5A particles in WT, S235A, or S235D NS3-NS5B-transfected T7-Huh7 cells. Asterisk indicates statistical significance (***, P < 0.005 [χ2 test]).
FIG 8.

S235A altered NS5A intracellular distribution. Confocal immunofluorescence micrographs of NS5A (red) costained with S229 phosphorylation (pS229, green) in the T7-Huh7 cells transfected with wild-type (WT), S235A, or S235D mutant NS3-NS5B mutant constructs. The nuclei were stained with DAPI (blue). Boxes indicate areas enlarged.
In summary, our results are compatible with a role of S229 phosphorylation in initiating NS5A sequential phosphorylation (Fig. 9). Perhaps by cycling through phosphorylation and dephosphorylation, S229 maintains a delicate balance of NS5A between hypo- and hyperphosphorylated states and the intracellular distribution necessary for the HCV life cycle.
FIG 9.
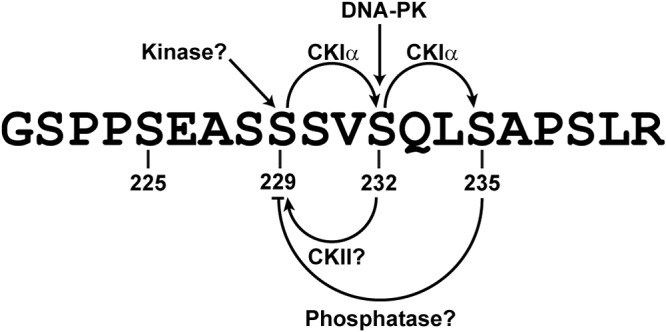
Schematic of NS5A sequential phosphorylation cascade.
MATERIALS AND METHODS
Cell culture.
Three types of cell lines: human kidney cells (HEK293T), human hepatocellular carcinoma cells (Huh7.5.1), and T7 polymerase harboring human hepatocellular carcinoma cells (T7-Huh7) were used in this study. These cells were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (Biological Industries) in a 5% CO2 incubator (Astec, Inc.) at 37°C.
Reagents.
Commercialized anti-NS5A domain I antibody (BioFront Technologies, Inc., catalog no. 7B5), and β-actin antibody (Sigma-Aldrich, catalog no. A5316) were used for immunoblotting. Serine 232, 235, and 238 phosphorylation-specific antibodies were generated and characterized previously (19, 24, 27). A rabbit serine 229 phosphorylation-specific polyclonal antibody was generated against a synthetic peptide (SEASpSSVSQLSAPSLRATC) by GeneTex, Inc., Taiwan. The antibody was purified using two affinity chromatography methods, first with immunizing phosphopeptide and followed by a nonphosphopeptide with the same sequence. IRDye680-labeled anti-mouse IgG (P/N 926-68070) and IRDye800-labeled anti-rabbit IgG (P/N 926-32213) secondary antibodies were purchased from Li-Core (Lincoln, NE). The CKII inhibitors TMCB (2-[4,5,6,7-tetrabromo-2-(dimethylamino)-1H-benzo[d]imidazol-1-yl]acetic acid) and TBB (4,5,6,7-tetrabromobenzotriazole) were purchased from Abcam (catalog no. ab120289 and catalog no. ab120998).
The expression constructs pcDNA3.1 NS3-NS5A (24), pTM NS3-NS5A (27), and pTM NS3-NS5B (45) were transfected into cells with the Lipofectamine 2000 reagent (catalog no. 11668-027, Invitrogen). Alanine mutations at S222, S225, S228, S229, S230, S232, S235, and S238 in the NS3-NS5A and NS3-NS5B constructs were made by using site-directed mutagenesis (24, 27).
HCV infection.
Culture-derived infectious HCV particles (HCVcc; J6/JFH1, genotype 2a) were prepared as described previously (36). Huh7.5.1 cells were seeded in 3.5-cm dishes and infected with HCVcc (J6/JFH1) at a multiplicity of infection of 0.001.
Immunoprecipitation.
Lysate of HCV-infected Huh7.5.1 cell (200 μg) was incubated with the 9E10 NS5A antibody (34) in 200 μl of phosphate-buffered saline (PBS) at 4°C overnight. This mixture was incubated with prewashed protein G-coated magnetic beads (80 μl, catalog no. 28-9440-08; GE Healthcare Life Sciences) at 4°C for 4 h. The unbound fraction was collected, and the beads were washed twice with 200 μl of PBS. The beads were heated (15 min) in 40 μl of 2% SDS solution to elute bound proteins before immunoblotting analysis.
Immunoblotting.
Immunoblotting was conducted as described previously (19). HCV-infected or expression construct-transfected cells were washed with PBS and lysed in a lysis buffer (50 mM HEPES, 150 mM NaCl, 5 mM EDTA, and 0.2% NP-40). The lysate was centrifuged briefly before the supernatant was collected and quantified for protein concentration. The proteins were then separated with SDS-PAGE and transferred to a nitrocellulose membrane for blocking and detection with antibodies. The proteins were visualized and quantified using a Li-Cor Odyssey scanner and software.
Luciferase assay.
The full-length Renilla report HCV construct (C19Rluc2AUbi) was obtained from Charles Rice’s group (46). It was linearized with XbaI digestion (catalog no. R0145; New England BioLabs) and purified using phenol-chloroform extraction. Purified DNA was transcribed into RNA by using a MEGAscript T7 transcription kit (catalog no. AM1334; Invitrogen). The RNA was transfected into the Huh7.5.1 cells using DMRIE-C transfection reagent (catalog no. 10459014; Thermo) according to the manufacturer’s recommended protocol. The transfected cells were lysed, and the luciferase activity was measured using a dual-luciferase reporter assay system (catalog no. E1910; Promega).
Cell viability assay.
T7-Huh7 cells (4 × 104) were seeded onto a 96-well plate for 1 day before being exposed to inhibitors (TMCB for 1 day or TBB for 6 h). After several washes with PBS, 50 μl of culture medium containing 10% MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] reagent was added to the wells, followed by incubation at 37°C for 45 min. The medium was then replaced with 100 μl of dimethyl sulfoxide and incubated with rotation at 145 rpm at room temperature for 20 min. The light absorbance was measured at 540 nm. Each experiment was performed in triplicate wells.
Confocal immunofluorescence microscopy.
Detailed procedures were described previously (19). Briefly, construct-transfected T7-Huh7 cells were fixed and probed with anti-NS5A, anti-NS3, S229 phosphorylation-specific, or S235 phosphorylation-specific antibodies. Immunofluorescence images were captured by the Zeiss LSM 880 laser scanning microscope with Airyscan and analyzed using ZEN software.
Quantification of immunofluorescence images.
Immunofluorescence micrographs were quantified with the IMARIS software (Bitplane) according to a similar algorithm (47). For each micrograph, about 30 cells were identified, and the sizes in μm3 of the detected particles corresponding to NS5A or NS3 immunostaining were measured. Within one set of experiments (WT, S235A, or S235D transfected cells; 30 cells each), about 2,500 particles were measured for sizes. These particles were sorted small to large and divided into four quartiles: the 0-to-25th percentile, the 25th-to-50th percentile, the 50th-to-75th percentile, and the 75th-to-100th percentile. The particles that fall within each quartile were identified for WT, S235A, and S235D, and the percentages of their particles in each quartile were calculated. A χ2 test was used to test whether there is a significant difference in the distribution of particles in the quartiles among WT, S235A, or S235D NS5A.
ACKNOWLEDGMENTS
We thank the First Core Facility at the College of Medicine, National Taiwan University, for making the NS3-NS5A and NS3-NS5B constructs in the pcDNA3.1 and the pTM plasmid.
This study was supported by the National Health Research Institutes, Taiwan (NHRI-EX108-10620BI to M.-J.Y.).
REFERENCES
- 1.Gotte M, Feld JJ. 2016. Direct-acting antiviral agents for hepatitis C: structural and mechanistic insights. Nat Rev Gastroenterol Hepatol 13:338–351. doi: 10.1038/nrgastro.2016.60. [DOI] [PubMed] [Google Scholar]
- 2.Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. 2004. Structural biology of hepatitis C virus. Hepatology 39:5–19. doi: 10.1002/hep.20032. [DOI] [PubMed] [Google Scholar]
- 3.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 4.McLauchlan J, Lemberg MK, Hope G, Martoglio B. 2002. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J 21:3980–3988. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hijikata M, Mizushima H, Akagi T, Mori S, Kakiuchi N, Kato N, Tanaka T, Kimura K, Shimotohno K. 1993. Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J Virol 67:4665–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reed KE, Grakoui A, Rice CM. 1995. Hepatitis C virus-encoded NS2-3 protease: cleavage-site mutagenesis and requirements for bimolecular cleavage. J Virol 69:4127–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moradpour D, Penin F. 2013. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol 369:113–142. doi: 10.1007/978-3-642-27340-7_5. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Staschke K, De Francesco R, Tan SL. 2007. Phosphorylation of hepatitis C virus NS5A nonstructural protein: a new paradigm for phosphorylation-dependent viral RNA replication? Virology 364:1–9. doi: 10.1016/j.virol.2007.01.042. [DOI] [PubMed] [Google Scholar]
- 9.Ross-Thriepland D, Harris M. 2015. Hepatitis C virus NS5A: enigmatic but still promiscuous 10 years on! J Gen Virol 96:727–738. doi: 10.1099/jgv.0.000009. [DOI] [PubMed] [Google Scholar]
- 10.Horner SM, Gale M Jr. 2013. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med 19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. 2009. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J Virol 83:4395–4403. doi: 10.1128/JVI.02352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tellinghuisen TL, Marcotrigiano J, Rice CM. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435:374–379. doi: 10.1038/nature03580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim PJ, Chatterji U, Cordek D, Sharma SD, Garcia-Rivera JA, Cameron CE, Lin K, Targett-Adams P, Gallay PA. 2012. Correlation between NS5A dimerization and hepatitis C virus replication. J Biol Chem 287:30861–30873. doi: 10.1074/jbc.M112.376822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang J, Huang L, Cordek DG, Vaughan R, Reynolds SL, Kihara G, Raney KD, Kao CC, Cameron CE. 2010. Hepatitis C virus nonstructural protein 5A: biochemical characterization of a novel structural class of RNA-binding proteins. J Virol 84:12480–12491. doi: 10.1128/JVI.01319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaneko T, Tanji Y, Satoh S, Hijikata M, Asabe S, Kimura K, Shimotohno K. 1994. Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem Biophys Res Commun 205:320–326. doi: 10.1006/bbrc.1994.2667. [DOI] [PubMed] [Google Scholar]
- 17.Masaki T, Matsunaga S, Takahashi H, Nakashima K, Kimura Y, Ito M, Matsuda M, Murayama A, Kato T, Hirano H, Endo Y, Lemon SM, Wakita T, Sawasaki T, Suzuki T. 2014. Involvement of hepatitis C virus NS5A hyperphosphorylation mediated by casein kinase Iα in infectious virus production. J Virol 88:7541–7555. doi: 10.1128/JVI.03170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fridell RA, Valera L, Qiu D, Kirk MJ, Wang C, Gao M. 2013. Intragenic complementation of hepatitis C virus NS5A RNA replication-defective alleles. J Virol 87:2320–2329. doi: 10.1128/JVI.02861-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chong WM, Hsu SC, Kao WT, Lo CW, Lee KY, Shao JS, Chen YH, Chang J, Chen SS, Yu MJ. 2016. Phosphoproteomics identified an NS5A phosphorylation site involved in hepatitis C virus replication. J Biol Chem 291:3918–3931. doi: 10.1074/jbc.M115.675413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol 79:3187–3194. doi: 10.1128/JVI.79.5.3187-3194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eyre NS, Hampton-Smith RJ, Aloia AL, Eddes JS, Simpson KJ, Hoffmann P, Beard MR. 2016. Phosphorylation of NS5A serine-235 is essential to hepatitis C virus RNA replication and normal replication compartment formation. Virology 491:27–44. doi: 10.1016/j.virol.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 22.Ross-Thriepland D, Mankouri J, Harris M. 2015. Serine phosphorylation of the hepatitis C virus NS5A protein controls the establishment of replication complexes. J Virol 89:3123–3135. doi: 10.1128/JVI.02995-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goonawardane N, Gebhardt A, Bartlett C, Pichlmair A, Harris M. 2017. Phosphorylation of serine 225 in hepatitis C virus NS5A regulates protein-protein interactions. J Virol 91:e00805-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu SC, Lo CW, Pan TC, Lee KY, Yu MJ. 2017. Serine 235 is the primary NS5A hyperphosphorylation site responsible for hepatitis C virus replication. J Virol 91:e00194-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schenk C, Meyrath M, Warnken U, Schnolzer M, Mier W, Harak C, Lohmann V. 2018. Characterization of a threonine-rich cluster in hepatitis C virus nonstructural protein 5A and its contribution to hyperphosphorylation. J Virol 92:e00737-18. doi: 10.1128/JVI.00737-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harak C, Meyrath M, Romero-Brey I, Schenk C, Gondeau C, Schult P, Esser-Nobis K, Saeed M, Neddermann P, Schnitzler P, Gotthardt D, Perez-Del-Pulgar S, Neumann-Haefelin C, Thimme R, Meuleman P, Vondran FW, De Francesco R, Rice CM, Bartenschlager R, Lohmann V. 2016. Tuning a cellular lipid kinase activity adapts hepatitis C virus to replication in cell culture. Nat Microbiol 2:16247. doi: 10.1038/nmicrobiol.2016.247. [DOI] [PubMed] [Google Scholar]
- 27.Hsu SC, Tsai CN, Lee KY, Pan TC, Lo CW, Yu MJ. 2018. Sequential S232/S235/S238 phosphorylation of the hepatitis C virus nonstructural protein 5A. J Virol 92:e01295-18. doi: 10.1128/JVI.01295-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neddermann P. 2009. NS5A phosphorylation and hyperphosphorylation. Methods Mol Biol 510:95–110. doi: 10.1007/978-1-59745-394-3_8. [DOI] [PubMed] [Google Scholar]
- 29.Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. 1990. Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem 265:14264–14269. [PubMed] [Google Scholar]
- 30.Fiol CJ, Mahrenholz AM, Wang Y, Roeske RW, Roach PJ. 1987. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J Biol Chem 262:14042–14048. [PubMed] [Google Scholar]
- 31.Reed KE, Xu J, Rice CM. 1997. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J Virol 71:7187–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quintavalle M, Sambucini S, Di Pietro C, De Francesco R, Neddermann P. 2006. The alpha isoform of protein kinase CKI is responsible for hepatitis C virus NS5A hyperphosphorylation. J Virol 80:11305–11312. doi: 10.1128/JVI.01465-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quintavalle M, Sambucini S, Summa V, Orsatti L, Talamo F, De Francesco R, Neddermann P. 2007. Hepatitis C virus NS5A is a direct substrate of casein kinase Iα, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J Biol Chem 282:5536–5544. doi: 10.1074/jbc.M610486200. [DOI] [PubMed] [Google Scholar]
- 34.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 35.Pan TC, Lo CW, Chong WM, Tsai CN, Lee KY, Chen PY, Liao JC, Yu MJ. 2019. Differential proteomics reveals discrete functions of proteins interacting with hypo- versus hyperphosphorylated NS5A of the hepatitis C virus. J Proteome Res 18:2813–2825. doi: 10.1021/acs.jproteome.9b00130. [DOI] [PubMed] [Google Scholar]
- 36.Lee KY, Chen YH, Hsu SC, Yu MJ. 2016. Phosphorylation of serine 235 of the hepatitis C virus nonstructural protein NS5A by multiple kinases. PLoS One 11:e0166763. doi: 10.1371/journal.pone.0166763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller ML, Jensen LJ, Diella F, Jorgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten T, Olhovsky M, Pasculescu A, Alexander J, Knapp S, Blom N, Bork P, Li S, Cesareni G, Pawson T, Turk BE, Yaffe MB, Brunak S, Linding R. 2008. Linear motif atlas for phosphorylation-dependent signaling. Sci Signal 1:ra2. doi: 10.1126/scisignal.1159433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coito C, Diamond DL, Neddermann P, Korth MJ, Katze MG. 2004. High-throughput screening of the yeast kinome: identification of human serine/threonine protein kinases that phosphorylate the hepatitis C virus NS5A protein. J Virol 78:3502–3513. doi: 10.1128/JVI.78.7.3502-3513.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Lee D, Choe J. 1999. Hepatitis C virus NS5A protein is phosphorylated by casein kinase II. Biochem Biophys Res Commun 257:777–781. doi: 10.1006/bbrc.1999.0460. [DOI] [PubMed] [Google Scholar]
- 41.Reiss S, Harak C, Romero-Brey I, Radujkovic D, Klein R, Ruggieri A, Rebhan I, Bartenschlager R, Lohmann V. 2013. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog 9:e1003359. doi: 10.1371/journal.ppat.1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ross-Thriepland D, Harris M. 2014. Insights into the complexity and functionality of hepatitis C virus NS5A phosphorylation. J Virol 88:1421–1432. doi: 10.1128/JVI.03017-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolk B, Buchele B, Moradpour D, Rice CM. 2008. A dynamic view of hepatitis C virus replication complexes. J Virol 82:10519–10531. doi: 10.1128/JVI.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Backes P, Quinkert D, Reiss S, Binder M, Zayas M, Rescher U, Gerke V, Bartenschlager R, Lohmann V. 2010. Role of annexin A2 in the production of infectious hepatitis C virus particles. J Virol 84:5775–5789. doi: 10.1128/JVI.02343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol 80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caster AH, Kahn RA. 2012. Computational method for calculating fluorescence intensities within three-dimensional structures in cells. Cell Logist 2:176–188. doi: 10.4161/cl.23150. [DOI] [PMC free article] [PubMed] [Google Scholar]