

Flaviviruses are a diverse group of insect-borne viruses responsible for numerous significant public health threats. Previously, we used a computational biology approach to define molecular signatures of antiviral DC responses following activation of innate immune signaling or infection with West Nile virus (WNV). In this work, we identify STAT5 as a regulator of DC activation and antiviral immune responses downstream of innate immune signaling that was not activated during either WNV or Zika virus (ZIKV) infection. WNV and ZIKV actively blocked STAT5 phosphorylation downstream of RIG-I, IFN-β, and IL-4, but not GM-CSF, signaling. However, other related flaviviruses, dengue virus serotypes 1 to 4 and the yellow fever 17D vaccine strain, did not antagonize STAT5 phosphorylation. Mechanistically, WNV and ZIKV showed differential inhibition of Jak kinases upstream of STAT5, suggesting divergent countermeasures to inhibit STAT5 activation. Combined, these findings identify STAT5 as a target of antagonism by specific pathogenic flaviviruses to subvert antiviral immune responses in human DCs.
KEYWORDS: interferon, RIG-I-like receptors, West Nile virus, dendritic cells, mRNA-seq
ABSTRACT
Flaviviruses are a diverse group of arthropod-borne viruses responsible for numerous significant public health threats; therefore, understanding the interactions between these viruses and the human immune response remains vital. West Nile virus (WNV) and Zika virus (ZIKV) infect human dendritic cells (DCs) and can block antiviral immune responses in DCs. Previously, we used mRNA sequencing and weighted gene coexpression network analysis (WGCNA) to define molecular signatures of antiviral DC responses following activation of innate immune signaling (RIG-I, MDA5, or type I interferon [IFN] signaling) or infection with WNV. Using this approach, we found that several genes involved in T cell cosignaling and antigen processing were not enriched in DCs during WNV infection. Using cis-regulatory sequence analysis, STAT5 was identified as a regulator of DC activation and immune responses downstream of innate immune signaling that was not activated during either WNV or ZIKV infection. Mechanistically, WNV and ZIKV actively blocked STAT5 phosphorylation downstream of RIG-I, IFN-β, and interleukin-4 (IL-4), but not granulocyte-macrophage colony-stimulating factor (GM-CSF), signaling. Unexpectedly, dengue virus serotypes 1 to 4 (DENV1 to DENV4) and the yellow fever 17D vaccine strain (YFV-17D) did not antagonize STAT5 phosphorylation. In contrast to WNV, ZIKV inhibited JAK1 and TYK2 phosphorylation following type I IFN treatment, suggesting divergent mechanisms used by these viruses to inhibit STAT5 activation. Combined, these findings identify STAT5 as a target of antagonism by specific pathogenic flaviviruses to subvert the immune response in infected DCs.
IMPORTANCE Flaviviruses are a diverse group of insect-borne viruses responsible for numerous significant public health threats. Previously, we used a computational biology approach to define molecular signatures of antiviral DC responses following activation of innate immune signaling or infection with West Nile virus (WNV). In this work, we identify STAT5 as a regulator of DC activation and antiviral immune responses downstream of innate immune signaling that was not activated during either WNV or Zika virus (ZIKV) infection. WNV and ZIKV actively blocked STAT5 phosphorylation downstream of RIG-I, IFN-β, and IL-4, but not GM-CSF, signaling. However, other related flaviviruses, dengue virus serotypes 1 to 4 and the yellow fever 17D vaccine strain, did not antagonize STAT5 phosphorylation. Mechanistically, WNV and ZIKV showed differential inhibition of Jak kinases upstream of STAT5, suggesting divergent countermeasures to inhibit STAT5 activation. Combined, these findings identify STAT5 as a target of antagonism by specific pathogenic flaviviruses to subvert antiviral immune responses in human DCs.
INTRODUCTION
West Nile virus (WNV) is a neurotropic flavivirus, and it has been the leading cause of arboviral neuroinvasive disease since its introduction to the United States in 1999, accounting for >95% of reported cases (1). After inoculation by an infected mosquito, 20 to 25% of WNV-infected individuals develop symptoms ranging from mild flu-like symptoms to severe neurologic disease, including meningitis, encephalitis, and acute flaccid paralysis (2). Longer-lasting sequelae of WNV-induced fever and neuroinvasive disease include ocular abnormalities, arthralgias, psychological impairment, and permanent memory loss (3). Recently, WNV has also been shown in mice to translocate across the placenta and infect fetal neuronal tissue during pregnancy, prompting fetal demise (4). The lack of FDA-approved antiviral therapeutics or vaccines for human WNV infection reinforces the need to further understand innate immune signaling during WNV pathogenesis.
Dendritic cells (DCs) are vital components of both the innate and adaptive immune responses during flavivirus infection. DCs are professional antigen-presenting cells that, upon infection, can process and present antigens on major histocompatibility complex (MHC) molecules, express costimulatory markers, and produce cytokines necessary for activation of the adaptive immune response (5). Mouse models of WNV pathogenesis have demonstrated that DCs are initial targets of infection by WNV and that innate immune signaling in DCs is essential for clearance of neuroinvasive disease (6–9). In the absence of type I interferon (IFN) signaling in murine DCs, WNV shows increased replication in myeloid cells and enhanced tissue tropism, resulting in increased mortality in mice (7). Adoptive transfer studies in mice have also demonstrated that immunocompetent DCs are essential for proper priming of WNV-specific T cell responses and viral clearance from the central nervous system (8). Recent studies have also established that WNV and a related neurotropic flavivirus, Zika virus (ZIKV), productively infect human monocyte-derived dendritic cells (moDCs) and suppress expression of costimulatory markers on infected DCs (10). WNV-infected DCs also display a reduced capacity to induce allogeneic CD4+ and CD8+ T cell proliferation (11). However, the exact mechanism of viral antagonism of human DC activation during WNV infection remains unknown.
During infection in DCs, intracellular WNV RNA is detected by the RIG-I like receptors (RLRs) RIG-I and MDA5, which interact with the adaptor protein mitochondrial antiviral signaling (MAVS) to induce transcription of proinflammatory cytokines, antiviral effectors, and type I IFN (12, 13). Upon release of type I IFN (IFN-α/β), this signal is further potentiated through the type I IFN receptor (IFNAR), causing phosphorylation of bound Janus kinases (JAKs) and subsequent signal transducer and activator of transcription (STAT) proteins (14). Once phosphorylated, STAT proteins will dimerize and translocate to the nucleus, bind to IFN-stimulated response elements (ISREs) on DNA, and initiate robust induction of interferon-stimulated genes (ISGs) to restrict viral replication. Canonically, IFNAR signals through STAT1 and STAT2 heterodimers, both of which are targets of flavivirus antagonism to inhibit antiviral responses (10, 15–18).
In addition, IFNAR can signal through STAT5, a pleiotropic STAT protein activated downstream of numerous cytokines and growth factors. STAT5 exists as two homologs, STAT5a and STAT5b, which share 96% homology at the protein level and homodimerize upon activation and bind gamma-activated sequence (GAS) elements on DNA (19). STAT5 signaling plays a variety of roles involving DC development and activation. Generation of conventional DCs requires STAT5 signaling by inhibition of plasmacytoid DC development through induction of interferon regulatory factor 8 (IRF8) (20). Conditional knockout mouse studies have also established that STAT5 signaling is essential for thymic stromal lymphopoietin (TSLP)-induced activation of DCs and allergic Th2 responses in the lungs (21). Inhibition of STAT5 signaling in monocyte-derived dendritic cells (moDCs) also inhibits lipopolysaccharide-induced DC maturation, costimulatory marker expression, and stimulation of Th1 immune responses (22). Despite the role of STAT5 in DC development and maturation, the importance of STAT5 signaling in DCs during viral infection is currently unknown.
Here, we employed a coexpression network-based analysis combined with promoter scanning analysis, and we identified STAT5, a critical transcription factor for regulating antiviral responses and activation within human DCs, downstream of RLR and type I IFN signaling. WNV and ZIKV infection induced minimal STAT5 signaling, corresponding with a failure to upregulate innate immune mediators and molecules involved in DC activation. WNV and ZIKV antagonism of STAT5 in moDCs was also receptor specific, inhibiting type I IFN and interleukin-4 (IL-4), but not granulocyte-macrophage colony-stimulating factor GM-CSF, and inducing STAT5 phosphorylation downstream of JAK kinase signaling. Surprisingly, WNV and ZIKV, but not dengue virus serotypes 1 to 4 (DENV1 to DENV4) or yellow fever virus vaccine strain 17D (YFV-17D), blocked STAT5 signaling in Vero cells with differential upstream blockade of JAK kinase signaling, suggesting that targeting STAT5 may be a virus-specific strategy of WNV and ZIKV to subvert antiviral responses.
(This article was submitted to an online preprint archive [23].)
RESULTS
STAT5 is a regulatory node of antiviral DC responses.
Previous work from our lab utilized a systems biology approach to assess the global antiviral response during WNV infection within primary human monocyte-derived DCs. Using moDCs from 5 different donors, we performed mRNA sequencing (mRNA-seq) following treatment with innate immune agonists targeting the RIG-I, MDA5, and IFNAR signaling pathways as well as infection with WNV at 12 and 24 hpi, signifying log phase viral growth (11). Using weighted gene coexpression network analysis (WGCNA) and Metacore pathway analysis, we determined that innate immune agonist treatments and WNV infection at 24 hpi induced notable gene expression within module 5 (M5), a subset of differentially expressed genes (DEGs) highly enriched for pathways involved in type I IFN signaling, pattern recognition receptor (PRR) signaling, and innate antiviral responses. WGCNA clusters DEGs into modules based on coexpression, suggesting the presence of common transcriptional regulators driving gene expression within a module. To define the transcriptional regulatory network of the M5 antiviral module, we performed cis-regulatory sequence analysis to computationally predict regulatory nodes using iRegulon, which identifies enrichment of transcription factor binding motifs within the top highly connected genes comprising M5 (24). Consistent with pathway enrichment for antiviral pathways, our analysis identified the ISGF3 transcription complex, IRF1, and NF-κB within the top predicted transcriptional regulators of M5 following RIG-I stimulation. Unexpectedly, we also found notable enrichment for STAT5, a transcriptional regulator with a previously described role in promoting DC activation (21, 22) (Fig. 1A).
FIG 1.

Systems biology reveals STAT5 as a regulatory node of antiviral DC responses. (A) Transcription factor regulatory network of module 5 gene expression as predicted by iRegulon (left panel). The top predicted transcriptional regulators (yellow nodes) are shown with a connecting line to predicted target genes (orange nodes). The consensus sequence for promoter regions targeted by STAT5 as generated by iRegulon and annotated as transfac_public-M00459 (top right panel) is shown. The number of predicted target genes and the normalized enrichment score (NES) for a given regulator are shown below (bottom right panel). (B) The STAT5 regulatory node (center) is shown with the predicted target genes indicated (small nodes), as determined by iRegulon. (C) Heat map of predicted STAT5 target genes after stimulation with innate immune agonists showing the log2 normalized fold change relative to the level in uninfected, untreated cells is shown (>2-fold change; significance, P < 0.01). Genes that did not reach the significance threshold are depicted in black. Each column within a treatment condition is marked by a unique color and represents a different donor (n = 5 donors). (D) moDCs were treated with RIG-I agonist (10 or 100 ng/1e6 cells) for 90 min or 18 h. (E) moDCs were treated with RIG-I or MDA5 agonist for 90 min (10, 100, 1,000, and 10,000 ng/1e6 cells). (F) moDCs were treated with IFN-β (1,000 IU/ml) for 30 min. For panels D to F, Western blot analysis was performed for the indicated proteins. Western blots are representative of data obtained from 3 to 8 donors.
STAT5 regulates expression of genes associated with innate immunity and DC activation.
Given that a role for STAT5 has not been previously implicated during flavivirus infection, we next evaluated the expression levels of predicted STAT5 target genes. Predicted STAT5 target genes included multiple genes associated with innate immunity (e.g., IRF1, Toll-like receptor 7 [TLR7], and TRIM25) and DC activation (e.g., CD80, CXCL11, and CCL2) (Fig. 1B). RIG-I agonist treatment induced upregulation of predicted STAT5 target genes. Treatment with transfected poly(I·C) and type I IFN also activated transcription of several STAT5 target genes although to a lesser extent than RIG-I agonist treatment. Given recent work implicating STAT5 signaling upstream of DC activation, in part through binding to the promoter regions of CD80 and CD83, we hypothesized that STAT5 might be an important regulator of DC activation downstream of RLR signaling (21, 22). Indeed, multiple predicted STAT5 target genes were involved in processes related to DC activation, including molecules involved in T cell cosignaling (e.g., CD80, IDO1, and SLAMF1) and cytokine signaling (e.g., CCL2, CCL13, CXCL11, IL2RA, JAK2, SOCS, and CISH) (Fig. 1C). Treatment with RIG-I or MDA5 agonist induced significant upregulation of STAT5 targets involved in DC activation, corresponding with dose-dependent phosphorylation of STAT5 at tyrosine residue 694, a critical event for STAT5 dimerization and DNA binding (25). Notably, STAT5 phosphorylation coincided with the kinetics and magnitude of IRF3 phosphorylation (Fig. 1D and E). IFN-β signaling also promoted STAT5 phosphorylation, confirming previous work describing the activation of STAT5 by type I IFN signaling in other cell types (Fig. 1F) (26–28). Collectively, cis-regulatory analysis revealed STAT5 as a transcriptional regulator of numerous antiviral genes within human moDCs and implicates STAT5 as a regulator of DC responses downstream of the RLR and type I IFN signaling axes.
WNV and ZIKV actively antagonize STAT5 activation.
In contrast to RLR and type I IFN stimulation, STAT5 signaling was substantially less enriched during WNV infection, where almost 80% of predicted STAT5 target genes, including those involved in DC activation, were not significantly expressed over levels in mock-infected cells (Fig. 2A). The minimal induction of STAT5 target genes corresponded to a lack of STAT5 phosphorylation following WNV infection during (24 hpi; 53.3% E protein-positive [protein+] cells) and after (48 hpi) log phase viral growth, despite increased amounts of STAT5 total protein by 48 hpi (Fig. 2B to D). Given that WNV infection induces type I IFN secretion at 48 hpi (11) and the selective lack of STAT5 phosphorylation during WNV infection of moDCs, we hypothesized that WNV antagonizes type I IFN-mediated STAT5 signaling. Indeed, WNV infection potently blocked STAT5 phosphorylation following both RIG-I stimulation and IFN-β treatment at 24 and 48 hpi (Fig. 2C and D). Infection with UV-inactivated WNV (UV-WNV) failed to block RIG-I-induced STAT5 phosphorylation, suggesting that viral replication is required for inhibition of STAT5 signaling. In notable contrast to STAT5, both STAT1 and STAT2 were phosphorylated during WNV infection, suggesting that STAT5 may be differentially modulated by WNV. ZIKV, a closely related neurotropic flavivirus that productively infects human moDCs (10), also antagonized STAT5 phosphorylation downstream of RIG-I and type I IFN signaling despite relatively low levels of infection (14.7% E protein+ cells) (Fig. 2B and E). Combined, our data strongly suggest that STAT5 is a target of antagonism by both WNV and ZIKV in human moDCs.
FIG 2.

WNV actively blocks STAT5 phosphorylation and expression of STAT5 target genes. (A) Heat map of predicted STAT5 target genes after stimulation with RIG-I agonist or infection with WNV (12 and 24 hpi) showing the log2 normalized fold change relative to levels in uninfected, untreated cells is shown (>2-fold change; significance, P < 0.01). Genes that did not reach the significance threshold are depicted in black. Each column within a treatment condition is marked by a unique color and represents a different donor (n = 5 donors). (B) Representative flow cytometry plot of viral E protein+ cells after infection in moDCs after WNV or ZIKV infection (24 or 48 h; MOI of 10, based on Vero cell titer). SSC-H, side scatter height. (C) moDCs were treated with RIG-I agonist (100 ng/1e6 cells) for 90 min following no infection (Mock), infection with UV-inactivated WNV (MOI of 10; UV), or infection with replication-competent WNV (MOI of 10; WNV). (D and E) Human moDCs were left uninfected (Mock) or infected with WNV or ZIKV (24 or 48 h; MOI of 10, based on Vero cell titer). Cells were left untreated or pulse treated with IFN-β (1,000 IU/ml) for 30 min. Data in panel B are representative of results obtained from two independent experiments. Data shown in panels C and D are representative of results obtained from 3 to 8 donors.
WNV and ZIKV block STAT5 phosphorylation in a pathway-specific manner.
Next, we asked if WNV and ZIKV blocked STAT5 activation downstream of additional cytokine signaling pathways. Common gamma-chain family cytokines, such as IL-4, as well as multiple growth factors, including GM-CSF, signal through their respective receptors to promote STAT5 phosphorylation (29, 30) (Fig. 3A). Similar to our findings with type I IFN signaling, WNV infection dampened IL-4-induced STAT5 phosphorylation in moDCs (Fig. 3B). In contrast, WNV failed to antagonize GM-CSF signaling, whereby the increased STAT5 protein induced during infection led to increased STAT5 phosphorylation. ZIKV also potently inhibited IL-4-mediated, but not GM-CSF-mediated, STAT5 phosphorylation in human DCs (Fig. 3C). Together, our findings suggest that WNV blocks STAT5 phosphorylation to antagonize STAT5-dependent gene induction in human moDCs in a pathway-specific manner.
FIG 3.
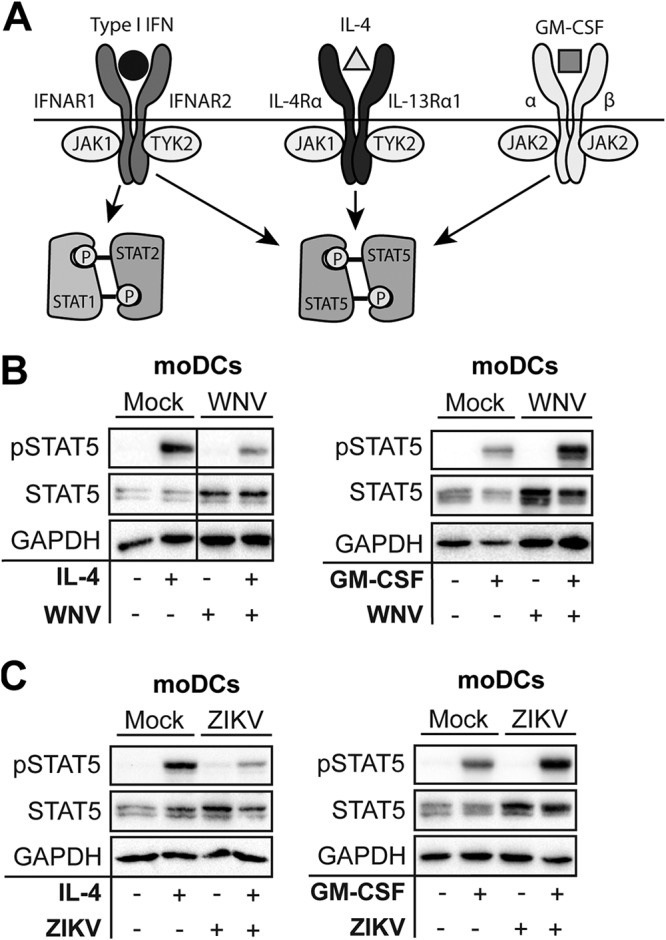
WNV and ZIKV inhibit STAT5 phosphorylation (P) in a pathway-dependent manner. (A) Schematic of STAT signaling downstream of type I IFN, IL-4, and GM-CSF signaling. (B and C) moDCs were infected with WNV or ZIKV (MOI of 10, based on Vero cell titer) for 48 h and treated with IL-4 (10 ng/ml) for 30 min or with GM-CSF (10 ng/ml) for 30 min. For panels B and C, Western blot analysis was performed for the indicated proteins. Data are representative of results obtained from 3 to 8 donors.
Blockade of STAT5 phosphorylation is flavivirus specific.
Secretion of type I IFN by infected moDCs may initiate negative regulators that downregulate type I IFN signaling independent of viral antagonism of STAT proteins. To remove confounding interpretations inherent with IFN-competent cells, we employed a Vero cell model of WNV infection, which allows for synchronous infection of cells that lack endogenous type I IFN signaling. Infection of Vero cells with WNV did not induce phosphorylation of STAT1, STAT2, or STAT5, consistent with the lack of an endogenous type I IFN response. Similar to our studies in human moDCs, pulse treatment of WNV-infected Vero cells (MOI of 0.01; 61.5% viral E protein+ cells) with IFN-β revealed a substantial blockade of STAT5 phosphorylation (Fig. 4A and B). STAT1 and STAT2 phosphorylations were also blocked, but in contrast to that of STAT5, the blockade of STAT1 and STAT2 was less pronounced. Blockade of STAT5 phosphorylation paralleled the large increase seen in WNV RNA and infectious virus release between 18 and 24 hpi during log phase viral growth (11). Together, these data strongly suggest that antagonism of STAT5 is an active immune evasion mechanism in moDCs and not a bystander effect of endogenous type I IFN signaling during WNV infection. We next asked whether STAT5 antagonism was unique to neurotropic flaviviruses (WNV and ZIKV) or if this mechanism is conserved among other flaviviruses. We infected Vero cells with ZIKV PR-2015 at an MOI that achieved levels of infection comparable to those in our studies with WNV (Fig. 4B). ZIKV antagonized STAT5 phosphorylation in a dose-dependent manner at higher MOIs (MOI of 1; 85% viral E protein+ cells) rather than at lower MOIs (MOI of 0.25; 65% viral E protein+ cells) downstream of type I IFN signaling in Vero cells. ZIKV infection at the higher MOI of 1 showed levels of STAT5 antagonism similar to those observed in human moDCs (Fig. 4C and D). Consistent with recent work on STAT antagonism by ZIKV, we also observed diminished STAT1 phosphorylation, as well as decreased STAT2 phosphorylation that corresponded with degradation of STAT2 total protein (10, 16). In contrast to WNV at similar infection levels (percentage of viral E protein+ cells: DENV1, 53.9%; DENV2, 64%; DENV3, 60.5%; DENV4, 55.7%; YFV-17D, 51.7%), STAT5 phosphorylation was not blocked in Vero cells infected with DENV1 to DENV4 and YFV-17D following IFN-β pulse treatment (Fig. 5A to F). Consistent with previous studies of DENV2 antagonism of type I IFN signaling (17), we observed STAT2 degradation in Vero cells infected with DENV1, -2, and -4, while STAT1 phosphorylation was detected after IFN-β pulse treatment. Markedly, DENV3 did not show degradation of STAT2 but showed a slight reduction in STAT2 phosphorylation, suggesting that antagonism of STAT2 by DENV may have a serotype-specific effect in Vero cells. Altogether, these findings demonstrate that antagonism of STAT5 is a virus-specific strategy used by WNV and ZIKV to potentially subvert the antiviral landscape in human DCs during infection.
FIG 4.

WNV and ZIKV antagonize STAT5 phosphorylation in the absence of IFN signaling. (A) Vero cells were infected with WNV-TX (MOI of 0.01, based on Vero cell titer) or ZIKV-PRVABC59 (MOI of 0.25 or 1, based on Vero cell titer), and the percentage of E protein+ cells was determined by flow cytometry at 24 hpi. (B) Vero cells were left uninfected (Mock) or infected with WNV-TX (MOI of 0.01, based on Vero cell titer) at 24 hpi. (C and D) Vero cells were left uninfected (Mock) or infected with ZIKV (MOI of 0.25 or 1, based on Vero cell titer) at 24 hpi. Data are representative of results obtained from three independent experiments.
FIG 5.

DENV1 to DENV4 and YFV-17D do not antagonize STAT5 phosphorylation at infection levels similar to those of WNV. (A) Vero cells were infected with DENV1 to DENV4 or YFV-17D (MOI of 1, based on Vero cell titer), and the percentages of E protein+ cells were determined by flow cytometry at 24 hpi. Vero cells were left uninfected (Mock) or infected with YFV-17D (MOI of 0.1 or 1, based on Vero cell titer) (B) or with DENV1 to DENV4 (MOI of 0.1 or 1, based on Vero cell titer) for 24 h (C to F). Cells were then left untreated or pulse treated with IFN-β (1,000 IU/ml) for 30 min. For data shown in panels B to F, Western blot analysis was performed for the indicated proteins. Data are representative of results obtained from two independent experiments.
ZIKV, but not WNV, blocks activation of TYK2 and JAK1.
The pathway-specific inhibition of STAT5 through type I IFN and IL-4 signaling, but not GM-CSF signaling, within moDCs provides insight into the host target of viral antagonism. The type I IFN receptor and the type II IL-4 receptor associate with JAK1 and TYK2 to mediate tyrosine phosphorylation of STAT1 and STAT2, while TYK2 constitutively associates with STAT5 and mediates its tyrosine phosphorylation (28, 29). GM-CSF signaling through the GM-CSF receptor predominately activates JAK2, but not JAK1 or TYK2, suggesting that TYK2 and JAK1 may be targeted to block STAT5 phosphorylation (30, 31). To assess JAK inhibition, we infected Vero cells at an MOI of 0.1 with WNV for 12, 18, and 24 h. Consistent with the lack of endogenous type I IFN production, WNV infection alone did not induce TYK2 phosphorylation. Although we did observe JAK1 phosphorylation, this is likely explained by the production of other cytokines that can activate JAK1 (32). In contrast to blockade of STAT protein phosphorylation, we observed no blockade of TYK2 and enhanced JAK1 phosphorylation following IFN-β pulse treatment of infected cells (Fig. 6A). Despite its similarities to WNV in inhibiting STAT5 phosphorylation, ZIKV efficiently blocked phosphorylation of both JAK1 and TYK2 downstream of type I IFN signaling (Fig. 6B). Previous work has shown that flaviviruses are capable of downregulating IFNAR1 from the cell surface (33, 34), so, we assessed whether ZIKV infection decreased expression of IFNAR1 or IFNAR2 from the Vero cell surface (Fig. 6C). Although at 24 hpi we observed blockade of JAK1 and TYK2 phosphorylation in ZIKV-infected Vero cells, we also observed an increase in both IFNAR1 and IFNAR2 expression compared to that in mock-infected cells. Combined, these findings suggest that WNV and ZIKV antagonize STAT5 signaling through two separate mechanisms: ZIKV at the level of JAK kinase phosphorylation and WNV downstream of JAK kinases, potentially through more direct inhibition of STAT5 phosphorylation.
FIG 6.

ZIKV, but not WNV, blocks TYK2 and JAK1 activation to compromise STAT5 phosphorylation. (A and B) Vero cells were left uninfected (Mock) or infected with WNV (MOI of 0.1, based on Vero cell titer) or ZIKV-PRVABC59 (MOI of 1, based on Vero cell titer) for 24 h. Cells were then either left untreated or pulse treated with IFN-β (1,000 IU/ml) for 30 min. (C) Vero cells were left uninfected (Mock) or infected with WNV (MOI of 1, based on Vero cell titer) for 24 h, and levels of surface IFNAR1 and IFNAR2 expression were measured by flow cytometry. For panels A and B, Western blot analysis was performed for the indicated proteins. Data in A and B are representative of results obtained from two independent experiments.
DISCUSSION
In this study, we combined traditional virologic and immunologic measures with transcriptomic and computational approaches to define the global antiviral response during WNV infection in human primary cells. Using cis-regulatory sequence analysis, STAT5, a transcription factor previously described as a regulator of DC activation, was identified as an important regulatory node of antiviral DC responses downstream of innate immune signaling. In contrast, STAT5 signaling was minimally activated during WNV infection in human moDCs, corresponding with minimal expression of genes of inflammatory responses or molecules involved in T cell priming. Mechanistically, WNV and ZIKV blocked STAT5 phosphorylation downstream of RIG-I, IFN-β, and IL-4, but not GM-CSF, signaling, suggesting pathway-specific antagonism of STAT5 activation in moDCs. Notably, neither the related flaviviruses DENV1 to DENV4 nor the vaccine strain YFV-17D inhibited STAT5 phosphorylation. Mechanistically, WNV and ZIKV displayed differential inhibition of JAK1 and TYK2 phosphorylation, indicating two distinct mechanisms to antagonize STAT5 signaling. Combined, our data identify antagonism of STAT5 phosphorylation as a conserved immune countermeasure used by certain pathogenic flaviviruses.
cis-Regulatory sequence analysis revealed STAT5 as a regulatory node of multiple components of DC activation downstream of RLR and type I IFN signaling. Indeed, we observed significant enrichment for multiple STAT5 target genes involved in DC activation, which corresponded with increased gene expression and secretion of proinflammatory cytokines and upregulation of proteins involved in T cell activation (11). While this confirms previous studies that have implicated STAT5 upstream of DC activation, our work reveals a new facet of STAT5 activation during flavivirus infection through engagement of innate immune signaling (21, 22). The rapid kinetics of STAT5 phosphorylation following RIG-I or MDA5 stimulation suggest that RLR signaling may directly induce phosphorylation of STAT5 through activation of a tyrosine kinase, such as Src or Lyn, both of which are induced by RLR signaling (35–39). Alternatively, rapid production of type I IFN, which also promotes STAT5 activation, may mediate STAT5 phosphorylation following RLR signaling. Combined, our data suggest that STAT5 is an important regulatory node downstream of the RLR and type I IFN signaling axis.
In contrast to RLR and IFN-β signaling, WNV infection did not upregulate most predicted STAT5 target genes, and STAT5 was not phosphorylated during infection despite secretion of IFN-β and IFN-α. Corresponding with minimal STAT5 enrichment, WNV infection failed to promote upregulation of inflammatory mediators and molecules involved in antigen presentation and T cell cosignaling. These findings are similar to those of previous work, in which WNV infection also failed to induce inflammatory cytokine secretion (40). Infection of moDCs with a nonpathogenic WNV isolate, WNV Kunjin, also induced minimal production of IL-12, despite notable upregulation of both CD86 and CD40 (41). This suggests that an inability to induce inflammatory cytokine responses may be shared among WNV strains, while pathogenic strains have evolved unique mechanisms to subvert antigen presentation and T cell activation. The lack of activation of WNV-infected human moDCs is also similar to our recent work with ZIKV (10). In contrast to WNV and ZIKV, infection of moDCs with the YFV vaccine strain (YFV-17D) upregulates multiple inflammatory mediators and surface expression of CD80 and CD86 (42). The ability of YFV-17D to induce strong DC activation may reflect the loss of viral antagonism during the attenuation process, similar to the ability of WNV Kunjin to induce upregulation of CD86 and CD40 (41). Alternatively, the ability of YFV-17D to induce DC activation may be an inherent property of certain flaviviruses. Indeed, DENV has also been found to activate inflammatory responses and upregulate costimulatory molecules following infection despite its ability to degrade STAT2 in infected cells (17, 43, 44). We demonstrate here that, unlike WNV and ZIKV, YFV-17D and DENV1 to DENV4 do not inhibit STAT5 phosphorylation in Vero cells. This raises the possibility that the inability to block STAT5 may explain the activation induced during YFV-17D and DENV infection. Altogether, we demonstrate that WNV, similar to ZIKV, induces minimal DC activation during productive infection, contrasting with both DENV and YFV-17D.
Blockade of STAT5 signaling by WNV was found to be multifrontal, as multiple STAT5-signaling cytokines induced downstream of RIG-I signaling (GM-CSF, IL-4, and IL-15) are not produced during infection with WNV (11). The lack of GM-CSF secretion may also overcome the need for WNV to block GM-CSF-induced STAT5 phosphorylation. Similar to WNV, ZIKV has also been found to induce minimal secretion of cytokines, including IL-4, GM-CSF, IL-12, IL-15, and RANTES, during infection of human moDCs (10). In contrast, DENV and YFV-17D infection in human moDCs has been reported to induce proinflammatory cytokine expression and secretion, including IL-12 and RANTES (45–48). This robust cytokine response could potentially reflect the inability of DENV and YFV-17D to inhibit STAT5-signaling cytokines. Combined, these findings suggest that the ability to antagonize STAT5 signaling may be an important feature of WNV and ZIKV pathogenesis.
Surprisingly, viral blockade of STAT5 phosphorylation during WNV infection, but not ZIKV infection, did not correspond with impaired activation of TYK2 and JAK1, members of the Janus-associated kinase (JAK) family that phosphorylate STAT5 downstream of type I IFN and IL-4, but not GM-CSF, signaling. Numerous flaviviruses, including the closely related Japanese encephalitis virus (JEV) and Langat virus, have been shown to inhibit TYK2 activation through the NS5 protein to subvert JAK/STAT signaling (18, 49). Nevertheless, our work suggests that inhibition of TYK2 or JAK1 activation is likely not the mechanism used by WNV to block STAT5 phosphorylation. Indeed, STAT1 and STAT2, which are also phosphorylated by TYK2 and JAK1, were not blocked as strongly as STAT5, suggesting that viral inhibition may be occurring at a step unique to STAT5 signaling. STAT5 itself is likely not targeted directly, given that GM-CSF-induced STAT5 signaling remains intact during WNV infection. One possibility is that WNV may specifically disrupt the interaction between STAT5 and TYK2 or JAK1. WNV infection may also directly induce negative regulators of STAT5 signaling, such as facilitating interactions between STAT5 and the protein tyrosine phosphatase, SHP-2, a well-known regulator of cytosolic STAT5 signaling (50). Another possibility includes induction of the suppressor of cytokine signaling family, members of which broadly regulate JAK/STAT signaling and can be modulated during flavivirus infection (51). Importantly, UV-inactivated virus, which undergoes cellular binding and entry but is replication incompetent, was unable to inhibit STAT5. This suggests that viral replication is required for STAT5 antagonism, potentially through a secreted viral protein that would affect both infected and uninfected cells. The observation that DENV1 to DENV4 and YFV-17D do not block STAT5 phosphorylation provides a valuable tool to further define the viral and host factors within subsets of flaviviruses that mediate STAT5 blockade.
In summary, our systems biology approach identified STAT5 as a regulator of DC activation that is blocked by WNV as a mechanism to subvert DC activation and T cell priming. ZIKV, but not YFV-17D or DENV1 to DENV4, also blocked STAT5 signaling, suggesting that viral antagonism of STAT5 may be a common strategy of particular subsets of pathogenic flaviviruses to evade the pressures of host immunity. Our study advances our understanding of how pathogenic flaviviruses subvert antiviral immunity during human infection.
MATERIALS AND METHODS
Ethics statement.
Human peripheral blood mononuclear cells (PBMCs) were obtained from deidentified healthy adult blood donors and processed immediately. All individuals who participated in this study provided informed consent in writing in accordance to the protocol approved by the Institutional Review Board of Emory University (IRB no. 00045821), entitled “Phlebotomy of healthy adults for the purpose of evaluation and validation of immune response assays.”
Cell lines.
Vero cells (WHO Reference Cell Banks) were maintained in complete Dulbecco’s modified Eagle’s medium (DMEM). Complete DMEM was prepared as follows: DMEM (Corning) supplemented with 10% fetal bovine serum (Optima, Atlanta Biologics), 2 mM l-glutamine (Corning), 1 mM HEPES (Corning), 1 mM sodium pyruvate (Corning), 1× minimal essential medium (MEM) nonessential amino acids (Corning), and 1× antibiotics/antimycotics (Corning). Complete RPMI (cRPMI) medium was prepared as follows: RPMI 1640 medium (Corning) supplemented with 10% fetal bovine serum (Optima, Atlanta Biologics), 2 mM l-glutamine (Corning), 1 mM sodium pyruvate (Corning), 1× MEM nonessential amino acids (Corning), and 1× antibiotics/antimycotics (Corning).
Generation of monocyte-derived dendritic cells.
To generate human moDCs, CD14+ monocytes were differentiated in cRPMI medium supplemented with 100 ng/ml GM-CSF and IL-4 for 5 to 6 days, as previously described (10). In brief, freshly isolated PBMCs obtained from healthy donor peripheral blood (lymphocyte separation medium; StemCell Technologies) were subjected to CD14+ magnetic bead positive selection using a MojoSort human CD14 selection kit (BioLegend). Purified CD14+ monocytes were cultured in complete RPMI medium supplemented with 100 ng/ml each of recombinant human IL-4 and GM-CSF (PeproTech) at a cell density of 2e6 cells/ml. After 24 h of culture, medium and nonadherent cells were removed and replaced with fresh medium and cytokines. Suspension cells (moDCs) were harvested after 5 to 6 days of culture and were consistently CD14− CD11c+ HLA-DR+ DC-SIGN+ CD1a+ by flow cytometry. For experimentation, moDCs were maintained in complete RPMI medium without GM-CSF or IL-4. For experiments measuring STAT5 phosphorylation, moDCs were rested in cRPMI medium without GM-CSF or IL-4 for 24 h prior to experimentation.
Viruses.
WNV stocks were generated from an infectious clone, WNV isolate TX 2002-HC, and passaged once in Vero cells, as previously described (52). ZIKV strain PRVABC59 was obtained from the Centers for Disease Control and Prevention as previously described (53). YFV-17D was subpassaged from YF-VAX (Aventis Pasteur) in SW-480 cells, followed by passaging in Vero cells as previously described (42). The DENV1, DENV3, and DENV4 strains were derived from low-passage-number clinical isolates and generated from infectious clones (IC) as previously described (54–56). The DENV2 IC was generated in the laboratory of R. S. Baric in a similar manner to the DENV1, DENV3, and DENV4 IC. The generation and characterization of the DENV2 S16803 IC will be described in a future publication. WNV and ZIKV were titrated by plaque assay on Vero cells with a 1% agarose overlay and crystal violet counterstain, as previously described (52). DENV was titrated by focus-forming assay as previously described (56). moDCs were infected with WNV or ZIKV at an MOI of 10 for 1 h at 37°C in cRPMI medium (without GM-CSF or IL-4). After 1 h, virus was washed off, and cells were resuspended in fresh cRPMI medium and incubated at 37°C for 3 to 72 h.
Quantitation of infectious virus.
Infectious virus was quantitated using a plaque assay on Vero cells with a 1% agarose overlay and crystal violet counterstain, as previously described (52).
Innate immune agonists.
To stimulate RIG-I signaling, 100 ng of RIG-I agonist derived from the 3′ untranslated region (UTR) of hepatitis C virus (57) was transfected per 1e6 cells using a TransIT-mRNA transfection kit (Mirus). For stimulation of MDA5 signaling, 100 ng of high-molecular-weight poly(I·C) was transfected per 1e6 cells using LyoVec transfection reagent (Invivogen). To stimulate type I IFN signaling, cells were incubated with 100 IU/ml of human recombinant IFN-β. In select experiments, different doses of agonists were used, and this is indicated within the respective figure legends.
RNA sequencing and bioinformatics.
moDCs were generated from 5 donors and either treated with innate immune agonists for 12 h (RIG-I, MDA5, or IFN-β) or infected with WNV (12 hpi and 24 hpi). Total RNA was purified (Quick-RNA MiniPrep kit; Zymo Research), and mRNA sequencing libraries were prepared for RNA sequencing (TruSeq chemistry; Illumina). RNA sequencing was performed on an Illumina HiSeq 2500 System (100-bp single-end reads). Sequencing reads were mapped to the human reference genome 38. Weighted gene coexpression module analysis was performed on DESeq2 normalized mapped reads (TIBCO Spotfire with Integromics, version 7.0) from RIG-I agonist-, MDA5 agonist-, IFN-β-, and mock-treated samples. First, the data sets were reduced to focus the network analysis on the 5,446 most variable genes (as determined by variation value greater than 1) using the variance function in R. We constructed a signed weighted correlation network by generating a matrix pairwise correlation between all annotated gene pairs. The resulting biweight midcorrelation matrix was transformed into an adjacency matrix using the soft thresholding power (β1) of 12. The adjacency matrix was used to define the topological overlap matrix (TOM) based on a dissimilarity measurement of 1 − TOM. Genes were hierarchically clustered using average linkage, and modules were assigned using the dynamic tree-cutting algorithm (module eigengenes were merged if the pairwise calculation was larger than 0.75). This resulted in the construction of six modules. Transcriptional regulators within the M5 module were computationally predicted with iRegulon (24), using the top most connected M5 genes using an eigengene-based connectivity cutoff of 0.4. Differentially expressed genes within the M5 module were identified as having a >2-fold change (significance at a P value of <0.01) relative to levels in uninfected and untreated cells. Pathway analysis was performed on M5 genes using MetaCore pathway map analysis (version 6.29; Thomson Reuters).
Flow cytometry.
Cells were prepared for analysis as previously described (10). For intracellular staining of WNV E protein, human moDCs and Vero cells were fixed and permeabilized (Transcription Factor Staining Buffer kit; Tonbo Biosciences). The cells were reblocked for 5 min on ice with 0.25 μl/sample human TruStain FcX and 0.25 μl/sample normal mouse serum (ThermoFisher Scientific) in Perm Buffer and E protein for 20 min on ice using 0.5 μl/sample of allophycocyanin-labeled E16 (E16-APC) (for WNV) (58) or the pan-flavivirus 4G2-APC antibody (for ZIKV, DENV1 to DENV4, and YFV-17D) in Perm Buffer. Unconjugated monoclonal 4G2 antibody was conjugated to APC using a Novus Lighting-Link kit per the manufacturer’s instructions. Flow cytometry data were analyzed using FlowJo, version 10, software. Primary antibodies are listed in Table 1.
TABLE 1.
Primary antibodies used in this study
| Primary antibodya | Source or reference | Catalog no. |
|---|---|---|
| Rabbit anti-human phospho-STAT1 (Tyr701, clone D4A7) | Cell Signaling Technologies | 7649 |
| Rabbit anti-human STAT1 (clone 9172) | Cell Signaling Technologies | 9172 |
| Rabbit anti-human phospho-STAT2 (Tyr689, Upstate) | EMD Millipore | 07-224 |
| Rabbit anti-human STAT2 (clone D9J7L) | Cell Signaling Technologies | 72604 |
| Rabbit anti-human phospho-STAT5 (Tyr694, clone D47E7) | Cell Signaling Technologies | 4322 |
| Rabbit anti-human STAT5 (clone 9363) | Cell Signaling Technologies | 9363 |
| Rabbit anti-human phospho-IRF3 (Ser396, clone 4D4G) | Cell Signaling Technologies | 4947 |
| Rabbit anti-human total IRF3 (clone D83B9) | Cell Signaling Technologies | 4302 |
| Rabbit anti-human total Tyk2 (clone D4I5T) | Cell Signaling Technologies | 14193 |
| Rabbit anti-human phospho-Tyk2 (Tyr1054/1055, clone D7T8A) | Cell Signaling Technologies | 68790 |
| Rabbit anti-human total JAK1 (clone 6G4) | Cell Signaling Technologies | 3344 |
| Rabbit anti-human phospho-JAK1 (Tyr1034/1035, clone D7N4Z) | Cell Signaling Technologies | 74129 |
| Rabbit anti-human GAPDH (clone D16H11) | Cell Signaling Technologies | 5174 |
| Mouse anti-human CD11c (BV710, clone B-Ly6) | BD Biosciences | 563130 |
| Mouse anti-human HLA-DR (PE-CF594, clone G46-6) | BD Biosciences | 562304 |
| Mouse anti-human CD1a (BV421, clone HI149) | BioLegend | 300128 |
| Mouse anti-human CD14 (PE, clone M5E2) | BioLegend | 301806 |
| Unconjugated monoclonal humanized E16 antibody | 58 | |
| Unconjugated monoclonal mouse 4G2 antibody | 53 |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PE, phycoerythrin.
Western blotting.
Whole-cell lysates were collected in modified radioimmunoprecipitation assay buffer (10 mM Tris [pH 7.5], 150 mM NaCl, 1% sodium deoxycholate, and 1% Triton X-100 supplemented with Halt protease inhibitor cocktail [ThermoFisher] and Halt phosphatase inhibitor cocktail [ThermoFisher]). Protein lysates were separated by SDS-PAGE, and Western blotting was performed using a ChemiDoc XRS+ imaging system (Bio-Rad). Western blots were analyzed using Image Lab, version 5.2.1, software (Bio-Rad) and prepared for publication using Adobe Illustrator. Primary antibodies are listed in Table 1.
Statistics.
All statistical analysis was performed using GraphPad Prism, version 8, software. The number of donors varied by experiment and is indicated within the figure legends. Statistical significance was determined at a P value of <0.05 using a Kruskal-Wallis test (when more than two groups lacking paired measurements were compared) and a Wilcoxon test (when two groups with paired measurements were compared). All comparisons were made between treatment or infection conditions with a time point-matched, uninfected and untreated control.
Data availability.
The raw data of all RNA sequencing in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) under GEO accession number GSE136342.
ACKNOWLEDGMENTS
We thank Children’s Healthcare of Atlanta and the Emory University Pediatric Flow Cytometry Core for providing access to flow cytometry and the Yerkes Genomics Core for performing RNA sequencing.
This work was funded in part by National Institutes of Health grants U19AI083019 (M.S.S.), R56AI110516 (M.S.S.), R21AI113485 (M.S.S.), 2U19AI090023 (B.P.), 5R37DK057665 (B.P.), 5R37AI048638 (B.P.), 2U19AI057266 (B.P.), and ORIP/OD P51OD011132 (M.S.S. and B.P.), the Emory University Department of Pediatrics Junior Faculty Focused Award (M.S.S.), Children’s Healthcare of Atlanta, the Emory Vaccine Center, and the Georgia Research Alliance (M.S.S.).
The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Burakoff A, Lehman J, Fischer M, Staples JE, Lindsey NP. 2018. West Nile virus and other nationally notifiable arboviral diseases—United States, 2016. MMWR Morb Mortal Wkly Rep 67:13–17. doi: 10.15585/mmwr.mm6701a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chancey C, Grinev A, Volkova E, Rios M. 2015. The global ecology and epidemiology of West Nile virus. Biomed Res Int 2015:376230. doi: 10.1155/2015/376230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel H, Sander B, Nelder MP. 2015. Long-term sequelae of West Nile virus-related illness: a systematic review. Lancet Infect Dis 15:951–959. doi: 10.1016/S1473-3099(15)00134-6. [DOI] [PubMed] [Google Scholar]
- 4.Platt DJ, Smith AM, Arora N, Diamond MS, Coyne CB, Miner JJ. 2018. Zika virus-related neurotropic flaviviruses infect human placental explants and cause fetal demise in mice. Sci Transl Med 10:eaao7090. doi: 10.1126/scitranslmed.aao7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pulendran B. 2015. The varieties of immunological experience; of pathogens, stress, and dendritic cells. Annu Rev Immunol 33 33:563–606. doi: 10.1146/annurev-immunol-020711-075049. [DOI] [PubMed] [Google Scholar]
- 6.Suthar MS, Diamond MS, Gale M Jr. 2013. West Nile virus infection and immunity. Nat Rev Microbiol 11:115–128. doi: 10.1038/nrmicro2950. [DOI] [PubMed] [Google Scholar]
- 7.Pinto AK, Ramos HJ, Wu X, Aggarwal S, Shrestha B, Gorman M, Kim KY, Suthar MS, Atkinson JP, Gale M Jr, Diamond MS. 2014. Deficient IFN signaling by myeloid cells leads to MAVS-dependent virus-induced sepsis. PLoS Pathog 10:e1004086. doi: 10.1371/journal.ppat.1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durrant DM, Robinette ML, Klein RS. 2013. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J Exp Med 210:503–516. doi: 10.1084/jem.20121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, Diamond MS, Gale M Jr. 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog 6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowen JR, Quicke KM, Maddur MS, O’Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, Suthar MS. 2017. Zika virus antagonizes type I interferon responses during infection of human dendritic cells. PLoS Pathog 13:e1006164. doi: 10.1371/journal.ppat.1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmerman MG, Bowen JR, McDonald CE, Pulendran B, Suthar MS. 2019. West Nile virus infection blocks inflammatory response and T cell costimulatory capacity of human monocyte-derived dendritic cells. J Virol 93:e00664-19. doi: 10.1128/JVI.00664-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M Jr. 2013. The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol 87:11416–11425. doi: 10.1128/JVI.01488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loo YM, Gale M Jr. 2011. Immune signaling by RIG-I-like receptors. Immunity 34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, Gale M Jr. 2006. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J Virol 80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sánchez-Seco MP, Evans MJ, Best SM, García-Sastre A. 2016. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison J, Laurent-Rolle M, Maestre AM, Rajsbaum R, Pisanelli G, Simon V, Mulder LCF, Fernandez-Sesma A, García-Sastre A. 2013. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog 9:e1003265. doi: 10.1371/journal.ppat.1003265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, Park GS, Boer E, Wolfinbarger JB, Bloom ME. 2005. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol 79:12828–12839. doi: 10.1128/JVI.79.20.12828-12839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hennighausen L, Robinson GW. 2008. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev 22:711–721. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esashi E, Wang YH, Perng O, Qin XF, Liu YJ, Watowich SS. 2008. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity 28:509–520. doi: 10.1016/j.immuni.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell BD, Kitajima M, Larson RP, Stoklasek TA, Dang K, Sakamoto K, Wagner KU, Kaplan DH, Reizis B, Hennighausen L, Ziegler SF. 2013. The transcription factor STAT5 is critical in dendritic cells for the development of TH2 but not TH1 responses. Nat Immunol 14:364–371. doi: 10.1038/ni.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toniolo PA, Liu S, Yeh JE, Moraes-Vieira PM, Walker SR, Vafaizadeh V, Barbuto JA, Frank DA. 2015. Inhibiting STAT5 by the BET bromodomain inhibitor JQ1 disrupts human dendritic cell maturation. J Immunol 194:3180–3190. doi: 10.4049/jimmunol.1401635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimmerman MG, Bowen JR, McDonald CE, Young E, Baric RS, Pulendran B, Suthar MS. 2019. STAT5: a target of antagonism by neurotropic flaviviruses. bioRxiv 10.1101/606608. [DOI] [PMC free article] [PubMed]
- 24.Janky R, Verfaillie A, Imrichova H, Van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D, Svetlichnyy D, Kalender Atak Z, Fiers M, Marine JC, Aerts S. 2014. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol 10:e1003731. doi: 10.1371/journal.pcbi.1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gouilleux F, Wakao H, Mundt M, Groner B. 1994. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J 13:4361–4369. doi: 10.1002/j.1460-2075.1994.tb06756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanabe Y, Nishibori T, Su L, Arduini RM, Baker DP, David M. 2005. Role of STAT1, STAT3, and STAT5 in IFN-ɑβ responses in T lymphocytes. J Immunol 174:609–613. doi: 10.4049/jimmunol.174.2.609. [DOI] [PubMed] [Google Scholar]
- 27.Meinke A, Barahmand-Pour F, Wohrl S, Stoiber D, Decker T. 1996. Activation of different Stat5 isoforms contributes to cell-type-restricted signaling in response to interferons. Mol Cell Biol 16:6937–6944. doi: 10.1128/mcb.16.12.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fish EN, Uddin S, Korkmaz M, Majchrzak B, Druker BJ, Platanias LC. 1999. Activation of a CrkL-stat5 signaling complex by type I interferons. J Biol Chem 274:571–573. doi: 10.1074/jbc.274.2.571. [DOI] [PubMed] [Google Scholar]
- 29.Lutz MB, Schnare M, Menges M, Rossner S, Rollinghoff M, Schuler G, Gessner A. 2002. Differential functions of IL-4 receptor types I and II for dendritic cell maturation and IL-12 production and their dependency on GM-CSF. J Immunol 169:3574–3580. doi: 10.4049/jimmunol.169.7.3574. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe S, Itoh T, Arai K. 1996. JAK2 is essential for activation of c-fos and c-myc promoters and cell proliferation through the human granulocyte-macrophage colony-stimulating factor receptor in BA/F3 cells. J Biol Chem 271:12681–12686. doi: 10.1074/jbc.271.21.12681. [DOI] [PubMed] [Google Scholar]
- 31.Al-Shami A, Mahanna W, Naccache PH. 1998. Granulocyte-macrophage colony-stimulating factor-activated signaling pathways in human neutrophils. Selective activation of Jak2, Stat3, and Stat5b. J Biol Chem 273:1058–1063. doi: 10.1074/jbc.273.2.1058. [DOI] [PubMed] [Google Scholar]
- 32.Hammaren HM, Virtanen AT, Raivola J, Silvennoinen O. 2019. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 118:48–63. doi: 10.1016/j.cyto.2018.03.041. [DOI] [PubMed] [Google Scholar]
- 33.Evans JD, Crown RA, Sohn JA, Seeger C. 2011. West Nile virus infection induces depletion of IFNAR1 protein levels. Viral Immunol 24:253–263. doi: 10.1089/vim.2010.0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lubick KJ, Robertson SJ, McNally KL, Freedman BA, Rasmussen AL, Taylor RT, Walts AD, Tsuruda S, Sakai M, Ishizuka M, Boer EF, Foster EC, Chiramel AI, Addison CB, Green R, Kastner DL, Katze MG, Holland SM, Forlino A, Freeman AF, Boehm M, Yoshii K, Best SM. 2015. Flavivirus antagonism of type I interferon signaling reveals prolidase as a regulator of IFNAR1 surface expression. Cell Host Microbe 18:61–74. doi: 10.1016/j.chom.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnsen IB, Nguyen TT, Bergstroem B, Fitzgerald KA, Anthonsen MW. 2009. The tyrosine kinase c-Src enhances RIG-I (retinoic acid-inducible gene I)-elicited antiviral signaling. J Biol Chem 284:19122–19131. doi: 10.1074/jbc.M808233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim YJ, Koo JE, Hong EH, Park ZY, Lim KM, Bae ON, Lee JY. 2015. A Src-family-tyrosine kinase, Lyn, is required for efficient IFN-beta expression in pattern recognition receptor, RIG-I, signal pathway by interacting with IPS-1. Cytokine 72:63–70. doi: 10.1016/j.cyto.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Chin H, Arai A, Wakao H, Kamiyama R, Miyasaka N, Miura O. 1998. Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood 91:3734–3745. [PubMed] [Google Scholar]
- 38.Xiao W, Ando T, Wang HY, Kawakami Y, Kawakami T. 2010. Lyn- and PLC-beta3-dependent regulation of SHP-1 phosphorylation controls Stat5 activity and myelomonocytic leukemia-like disease. Blood 116:6003–6013. doi: 10.1182/blood-2010-05-283937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okutani Y, Kitanaka A, Tanaka T, Kamano H, Ohnishi H, Kubota Y, Ishida T, Takahara J. 2001. Src directly tyrosine-phosphorylates STAT5 on its activation site and is involved in erythropoietin-induced signaling pathway. Oncogene 20:6643–6650. doi: 10.1038/sj.onc.1204807. [DOI] [PubMed] [Google Scholar]
- 40.Silva MC, Guerrero-Plata A, Gilfoy FD, Garofalo RP, Mason PW. 2007. Differential activation of human monocyte-derived and plasmacytoid dendritic cells by West Nile virus generated in different host cells. J Virol 81:13640–13648. doi: 10.1128/JVI.00857-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kovats S, Turner S, Simmons A, Powe T, Chakravarty E, Alberola-Ila J. 2016. West Nile virus-infected human dendritic cells fail to fully activate invariant natural killer T cells. Clin Exp Immunol 186:214–226. doi: 10.1111/cei.12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B. 2006. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med 203:413–424. doi: 10.1084/jem.20051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez-Madoz JR, Bernal-Rubio D, Kaminski D, Boyd K, Fernandez-Sesma A. 2010. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. J Virol 84:4845–4850. doi: 10.1128/JVI.02514-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olagnier D, Peri S, Steel C, van Montfoort N, Chiang C, Beljanski V, Slifker M, He Z, Nichols CN, Lin R, Balachandran S, Hiscott J. 2014. Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS Pathog 10:e1004566. doi: 10.1371/journal.ppat.1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gandini M, Reis SRNI, Torrentes-Carvalho A, Azeredo EL, da Silva Freire M, Galler R, Kubelka CF. 2011. Dengue-2 and yellow fever 17DD viruses infect human dendritic cells, resulting in an induction of activation markers, cytokines and chemokines and secretion of different TNF-alpha and IFN-alpha profiles. Mem Inst Oswaldo Cruz 106:594–605. doi: 10.1590/S0074-02762011000500012. [DOI] [PubMed] [Google Scholar]
- 46.Lim DS, Yawata N, Selva KJ, Li N, Tsai CY, Yeong LH, Liong KH, Ooi EE, Chong MK, Ng ML, Leo YS, Yawata M, Wong SB. 2014. The combination of type I IFN, TNF-alpha, and cell surface receptor engagement with dendritic cells enables NK cells to overcome immune evasion by dengue virus. J Immunol 193:5065–5075. doi: 10.4049/jimmunol.1302240. [DOI] [PubMed] [Google Scholar]
- 47.Costa VV, Ye W, Chen Q, Teixeira MM, Preiser P, Ooi EE, Chen J. 2017. Dengue virus-infected dendritic cells, but not monocytes, activate natural killer cells through a contact-dependent mechanism involving adhesion molecules. mBio 8:e00741-17. doi: 10.1128/mBio.00741-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho LJ, Wang JJ, Shaio MF, Kao CL, Chang DM, Han SW, Lai JH. 2001. Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. J Immunol 166:1499–1506. doi: 10.4049/jimmunol.166.3.1499. [DOI] [PubMed] [Google Scholar]
- 49.Lin RJ, Chang BL, Yu HP, Liao CL, Lin YL. 2006. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J Virol 80:5908–5918. doi: 10.1128/JVI.02714-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu CL, Jin YJ, Burakoff SJ. 2000. Cytosolic tyrosine dephosphorylation of STAT5. Potential role of SHP-2 in STAT5 regulation. J Biol Chem 275:599–604. doi: 10.1074/jbc.275.1.599. [DOI] [PubMed] [Google Scholar]
- 51.Sharma N, Kumawat KL, Rastogi M, Basu A, Singh SK. 2016. Japanese encephalitis virus exploits the microRNA-432 to regulate the expression of suppressor of cytokine signaling (SOCS) 5. Sci Rep 6:27685. doi: 10.1038/srep27685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suthar MS, Brassil MM, Blahnik G, Gale M Jr. 2012. Infectious clones of novel lineage 1 and lineage 2 West Nile virus strains WNV-TX02 and WNV-Madagascar. J Virol 86:7704–7709. doi: 10.1128/JVI.00401-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O’Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, Schinazi RF, Chakraborty R, Suthar MS. 2016. Zika virus infects human placental macrophages. Cell Host Microbe doi: 10.1016/j.chom.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gallichotte EN, Widman DG, Yount BL, Wahala WM, Durbin A, Whitehead S, Sariol CA, Crowe JE Jr, de Silva AM, Baric RS. 2015. A new quaternary structure epitope on dengue virus serotype 2 is the target of durable type-specific neutralizing antibodies. mBio 6:e01461-15. doi: 10.1128/mBio.01461-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Messer WB, Yount B, Hacker KE, Donaldson EF, Huynh JP, de Silva AM, Baric RS. 2012. Development and characterization of a reverse genetic system for studying dengue virus serotype 3 strain variation and neutralization. PLoS Negl Trop Dis 6:e1486. doi: 10.1371/journal.pntd.0001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Widman DG, Young E, Nivarthi U, Swanstrom JA, Royal SR, Yount BL, Debbink K, Begley M, Marcet S, Durbin A, de Silva AM, Messer WB, Baric RS. 2017. Transplantation of a quaternary structure neutralizing antibody epitope from dengue virus serotype 3 into serotype 4. Sci Rep 7:17169. doi: 10.1038/s41598-017-17355-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M Jr. 2008. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, Mehlhop E, Marri A, Chung KM, Ebel GD, Kramer LD, Fremont DH, Diamond MS. 2005. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med 11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data of all RNA sequencing in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) under GEO accession number GSE136342.