

The PM-specific localization of HIV-1 Gag is a crucial early step in infectious progeny production. The interaction between the MA highly basic region (MA-HBR) of Gag and the PM-specific lipid PI(4,5)P2 is critical for Gag localization to the PM. Additionally, in vitro evidence has indicated that MA-RNA binding prevents nonspecific binding of Gag to non-PI(4,5)P2-containing membranes. However, cell-based evidence supporting a role for HIV-1 MA-RNA binding in PM-specific subcellular localization has been scarce; thus, it remained possible that in cells, just the high basic charge or the PI(4,5)P2 binding ability is sufficient for MA to direct Gag specifically to the PM. The present study reveals for the first time an excellent correlation between RNA binding of the MA-HBR and inhibition of promiscuous Gag localization, both within the cells, and thereby provides cell-based evidence supporting a mechanism in which HIV-1 MA binding to RNA ensures the specific localization of Gag to the PM.
KEYWORDS: HIV-1 Gag; PI(4,5)P2; RNA; acidic lipids; highly basic region; membrane binding; plasma membrane; retrovirus assembly; subcellular localization
ABSTRACT
The HIV-1 Gag matrix (MA) domain mediates the localization of Gag to the plasma membrane (PM), the site for infectious virion assembly. The MA highly basic region (MA-HBR) interacts with phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2], a PM-specific acidic lipid. The MA-HBR also binds RNAs. To test whether acidic lipids alone determine PM-specific localization of Gag or whether MA-RNA binding also plays a role, we compared a panel of MA-HBR mutants that contain two types of substitutions at MA residues 25 and 26 or residues 29 and 31: Lys→Arg (KR) (25/26KR and 29/31KR) and Lys→Thr (KT) (25/26KT and 29/31KT). Consistent with the importance of the HBR charge in RNA binding, both KT mutants failed to bind RNA via MA efficiently, unlike the corresponding KR mutants. Both 25/26KT Gag-yellow fluorescent protein (YFP) and 29/31KT Gag-YFP bound nonspecifically to the PM and intracellular membranes, presumably via the myristoyl moiety and remaining MA basic residues. In contrast, 25/26KR Gag-YFP bound specifically to the PM, suggesting a role for the total positive charge and/or MA-bound RNA in navigating Gag to the PM. Unlike 29/31KT Gag-YFP, 29/31KR Gag-YFP was predominantly cytosolic and showed little intracellular membrane binding despite having a higher HBR charge. Therefore, it is likely that MA-RNA binding blocks promiscuous Gag membrane binding in cells. Notably, the introduction of a heterologous multimerization domain restored PI(4,5)P2-dependent PM-specific localization for 29/31KR Gag-YFP, suggesting that the blocking of PM binding is more readily reversed than that of intracellular membrane binding. Altogether, these cell-based data support a model in which MA-RNA binding ensures PM-specific localization of Gag via suppression of nonspecific membrane binding.
IMPORTANCE The PM-specific localization of HIV-1 Gag is a crucial early step in infectious progeny production. The interaction between the MA highly basic region (MA-HBR) of Gag and the PM-specific lipid PI(4,5)P2 is critical for Gag localization to the PM. Additionally, in vitro evidence has indicated that MA-RNA binding prevents nonspecific binding of Gag to non-PI(4,5)P2-containing membranes. However, cell-based evidence supporting a role for HIV-1 MA-RNA binding in PM-specific subcellular localization has been scarce; thus, it remained possible that in cells, just the high basic charge or the PI(4,5)P2 binding ability is sufficient for MA to direct Gag specifically to the PM. The present study reveals for the first time an excellent correlation between RNA binding of the MA-HBR and inhibition of promiscuous Gag localization, both within the cells, and thereby provides cell-based evidence supporting a mechanism in which HIV-1 MA binding to RNA ensures the specific localization of Gag to the PM.
INTRODUCTION
HIV-1 progeny virions exit most cells at the plasma membrane (PM) (1, 2). The matrix (MA) domain of the HIV-1 structural polyprotein Gag mediates the targeting and binding of Gag to the PM, which is a crucial stage of virus particle production (3–9). The two essential features of the MA domain required for the PM-specific localization and binding of Gag are an N-terminal myristate moiety and a highly basic region (MA-HBR) (10–16). The myristate moiety allows Gag to form hydrophobic interactions with membranes when it is not sequestered in the MA globular domain (17–24). The MA-HBR comprises a highly conserved cluster of basic residues spanning residues 14 to 31 (13, 14, 23, 25). This basic patch drives electrostatic and head group-specific interactions of Gag with phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2], an acidic phospholipid found primarily in the inner leaflet of the PM (15, 20, 26–34). In addition to acidic phospholipids, RNA, which has been shown to bind the MA domain (35–43), may play an important role in the regulation of Gag-membrane binding (28, 32, 44–48). Among the HBR basic residues, a nuclear magnetic resonance (NMR)-based study showed that residues 29 and 31 are particularly important for PI(4,5)P2 interaction in the absence of RNA (49).
Wild-type (WT) Gag that is translated in vitro using rabbit reticulocyte lysates binds liposomes consisting of a neutral lipid, phosphatidylcholine (PC), and an acidic lipid, phosphatidylserine (PS) (PC+PS liposomes) poorly but shows enhanced membrane binding either when Gag is treated with RNase or when PI(4,5)P2 is included in the liposomes (32, 34, 50). In cells, besides NC, MA-HBR mediates significant RNA binding to WT Gag (46, 47). Notably, regardless of the presence of NC, Gag present in the cytosol binds to PC+PS liposomes only upon RNase treatment (46), suggesting a role for MA-bound RNA in cells. In good agreement with these studies, RNase treatment of cell homogenates derived from HIV-1-expressing cells resulted in a significant shift of Gag from the cytosolic to the membrane fraction (47). These observations suggest that WT Gag is susceptible to negative regulation of membrane binding by MA-bound RNA and that Gag-membrane binding occurs only when this RNA is removed by RNase or counteracted by PI(4,5)P2. Sequencing of RNAs cross-linked to MA revealed that the major RNA species bound to MA in cells is tRNA and that MA-tRNA binding is reduced with membrane-bound Gag compared to cytosolic Gag (47). Consistent with the role for MA-tRNA binding, tRNA-mediated inhibition of Gag-liposome binding has been observed in vitro (46, 48, 51–53).
Based on these studies, our working model is that RNA bound to MA-HBR prevents Gag from electrostatically binding to acidic phospholipids such as PS, which are present ubiquitously in the cell (54). In this model, PI(4,5)P2 helps Gag overcome RNA-mediated negative regulation, thereby promoting Gag binding to the PM, while RNA prevents Gag from binding to other acidic lipids present in non-PM membranes (32, 44). The hypothesis that MA-RNA binding prevents the promiscuous localization of Gag has not been directly investigated in the context of HIV-1 Gag expressed in cells. Our previous study of Gag chimeras containing various retroviral MA domains showed a correlation between the size of basic patches, RNA sensitivity in an in vitro liposome binding assay, and PM-specific Gag localization in cells (29). However, MA-RNA binding in cells was not measured in that study. Moreover, confounding effects of structural variations of the various retroviral MA domains, other than the size of the basic patches, could not be excluded. In addition, although unlikely, it remains possible that Gag chimeras with different retroviral MA domains may be differentially endocytosed after nascent virion assembly at the PM, resulting in apparent differences in Gag localization.
In the present study, to examine the correlation between MA-RNA binding and subcellular Gag localization while addressing the limitations in previous studies, we compared the effects of two types of amino acid substitutions in the HIV-1 MA-HBR on the ability of MA to bind RNA in cells and on the specificity of the subcellular localization of HIV-1 Gag. The first type is Lys-to-Thr (KT) changes. We previously showed that MA-HBR mutants with KT changes at MA residues 25 and 26 (25/26KT) or MA residues 29 and 31 (29/31KT) display promiscuous subcellular localization to both the PM and intracellular membranes (14, 32). MA-RNA binding is reduced upon substitutions of Lys in the MA-HBR to neutral amino acids such as Thr in previous in vitro studies (35, 36). In cells, such substitutions caused a reduction in tRNA populations bound to Gag relative to total RNA populations bound to Gag (47). However, the magnitude of the effect of the HBR mutations on the RNA binding ability of MA in cells remained to be determined. The second type of amino acid substitution in MA-HBR that we introduced is Lys-to-Arg (KR) changes. These changes, which do not affect the overall basic charge of the MA-HBR, unlike the KT changes, are expected to preserve the RNA binding ability, since liposome binding of a mutant Gag in which all basic residues of the MA-HBR were switched (K→R and R→K) is still sensitive to an RNA-mediated block of PC+PS liposome binding (55). However, this prediction had not been tested either in cells or for specific HBR residues. The comparison of KT and KR substitutions in the MA-HBR in this study revealed that basic-to-neutral changes in two Lys residues are sufficient to diminish MA-RNA binding in cells to the background level and that there is a strong correlation between MA-RNA binding and the subcellular localization of Gag. Altogether, the findings in this study support our working model that MA-RNA binding inhibits the promiscuous localization of Gag, thereby ensuring Gag localization to the PM in the presence of PI(4,5)P2.
(This article was submitted to an online preprint archive [56].)
RESULTS
Gag derivatives with Lys-to-Arg changes in the MA-HBR bind RNA more efficiently in cells than those with Lys-to-Thr changes.
To compare the RNA binding capacities of MA, we introduced three modifications into Gag constructs and examined RNA binding to WT MA and MA-HBR mutants in cells using photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP). The first modification is an MA amino acid substitution (1GA) that blocks N-terminal myristoylation and thereby prevents Gag from binding membranes. A previous cell-based PAR-CLIP study (47) was conducted using Gag constructs that can bind membranes, which would cause dissociation of RNA from MA according to the model described above (see the introduction). Therefore, we eliminated membrane binding so as to allow us to determine the total RNA binding capacity of Gag. Second, to focus on the RNA binding ability of the MA domain, we deleted most of the NC domain (delNC), which is the major RNA binding domain of Gag. Finally, we fused yellow fluorescent protein (YFP) to the Gag C terminus (Gag-YFP) to facilitate microscopy analysis of the constructs analyzed for MA-RNA binding.
The 1GA/delNC/Gag-YFP constructs containing KR or KT mutations in MA residues 25 and 26 (25/26KR and 25/26KT) or MA residues 29 and 31 (29/31KR and 29/31KT) (Fig. 1) were compared with isogenic constructs with the WT HBR sequence or a mutant HBR sequence in which all MA-HBR basic residues were switched to neutral residues (6A2T) (Fig. 1). To determine the amount of RNA bound to MA in cells, we employed a PAR-CLIP assay (47, 57), with modifications (Fig. 2A). HeLa cells were transfected with one of the constructs described above or a non-Gag control, pUC19; cultured in medium containing 4-thiouridine (4-SU), a photoactivatable ribonucleoside analogue; and subsequently exposed to UV light, which cross-links RNA binding proteins and 4-SU-containing RNA bound to the proteins. Following cell lysis, Gag constructs were immunoprecipitated using HIV immunoglobulin (HIV-Ig), and the RNA bound to the constructs was end labeled with 32P prior to SDS-PAGE and electrotransfer to a polyvinylidene difluoride (PVDF) membrane. The RNA binding efficiency of the constructs was determined by comparison of the signal intensity of RNA by phosphorimager analysis versus the signal intensity of Gag constructs detected by immunoblotting on the same membrane.
FIG 1.

Amino acid substitutions introduced into the MA-HBR. (A) Schematic illustration of HIV-1 Gag with the structure of HIV-1 MA (PDB accession number 2HMX) showing the basic residues of the HBR in blue, with residues mutated in this study circled. (B) Sequences of the HIV-1 Gag MA-HBR analyzed in this study. Lys-to-Arg (KR) or Lys-to-Thr (KT) changes were introduced at MA residues 25 and 26 or residues 29 and 31. In the 6A2T mutant, all MA-HBR basic residues were switched to neutral Ala or Thr.
FIG 2.
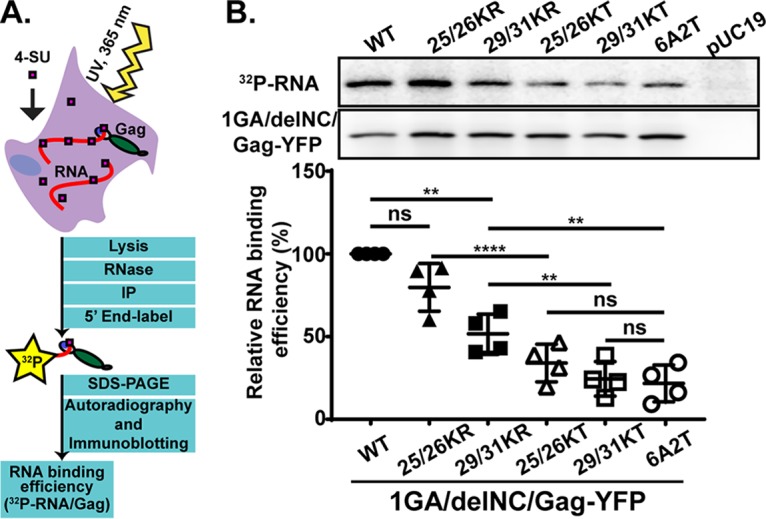
Gag derivatives with KR mutations in the MA-HBR bind RNA efficiently compared to those with KT mutations. (A) Schematic illustration of the PAR-CLIP assay. A cytomegalovirus (CMV) promoter-driven Gag-YFP construct lacking the myristoylation site (1GA) and most of the NC domain (delNC) was used as the backbone for the Gag derivatives analyzed in this assay. A mutant Gag with all MA-HBR basic residues switched to neutral Ala or Thr, 6A2T, was used as a negative control. To identify bands representing Gag, a non-Gag plasmid, pUC19, was used as an additional control. The 1GA/delNC/Gag-YFP constructs were expressed in HeLa cells and cross-linked to 4-SU-containing RNA in the cells. Gag proteins were recovered by immunoprecipitation (IP), Gag-bound RNA was end labeled with 32P, and signals for Gag proteins and Gag-bound RNA were detected by SDS-PAGE followed by immunoblotting and autoradiography, respectively. (B, top) Representative results for 32P-labeled Gag-bound RNA detected by autoradiography and total Gag detected by immunoblotting and chemiluminescence. (Bottom) Relative RNA binding efficiency (percent) of MA-HBR mutants determined by quantifying the intensity of RNA signals normalized by the Gag band intensity on the same PVDF membrane. Results from 4 independent experiments are shown as means ± standard deviations. P values were determined using Student’s t test, using raw data. ns, not significant; **, P ≤ 0.01; ****, P ≤ 0.0001.
Consistent with previous results (46), the Gag construct bearing 6A2T MA did not bind RNA efficiently, showing an ∼4-fold reduction in the amount of RNA bound relative to the WT (Fig. 2B). Both the constructs bearing 25/26KT and 29/31KT MA bound significantly less RNA than the WT construct, similar to what was observed for 6A2T, indicating that both Lys25 and -26 and Lys29 and -31 contribute to MA-RNA binding to similar extents in cells. On the other hand, there was no statistically significant difference in the RNA binding efficiencies between the 25/26KR MA and WT MA. The Gag construct with the other KR changes, 29/31KR, also bound significantly more RNA than its KT counterpart albeit less efficiently than the WT. These results indicate that the RNA binding efficiency of MA is dependent on the overall positive charge of the MA-HBR basic patch. Additionally, the identity of basic amino acids at residues 29 and/or 31 also plays a role in WT-level RNA binding. Having observed differential effects on MA-RNA binding between KR and KT substitutions, we investigated the correlation (or lack thereof) between MA-RNA binding and the subcellular localization of Gag, as described below, focusing first on the effects of the substitutions at MA residues 25 and 26 and next on those at residues 29 and 31.
Gag derivatives containing 25/26KR substitutions exhibit PM-specific localization, unlike those with 25/26KT changes.
To compare subcellular distributions of Lys25/26 mutants, we expressed myristoylated and YFP-tagged Gag constructs bearing these changes, in both the full-length and delNC contexts, in HeLa cells and observed the YFP localization in cells (Fig. 3). Based on the YFP distribution patterns, we identified 4 different phenotypes (Fig. 3A and B): (i) localization predominantly at the PM (green bars), (ii) localization to both the PM and intracellular compartments (blue bars), (iii) localization to only intracellular compartments (orange bars), and (iv) only a hazy cytosolic localization, with no punctate intensities (pink bars). These 4 distribution patterns were also validated by comparison of the YFP signal with that of Alexa Fluor 594-conjugated concanavalin A (ConA-AF594), which serves as the PM marker (Fig. 3C).
FIG 3.

Gag derivatives containing KR substitutions in the MA-HBR do not show promiscuous localization in cells, unlike those with KT changes. (A and B) HeLa cells were transfected with full-length Gag-YFP (A) and delNC/Gag-YFP (B), which contain the WT MA sequence or 25/26KR, 25/26KT, 29/31KR, or 29/31KT. At 14 h posttransfection, cells were stained with Alexa Fluor 594-conjugated concanavalin A (ConA-AF594), fixed with 4% paraformaldehyde in PBS, and analyzed using a fluorescence microscope. Note that subcellular distributions of delNC/Gag-YFP constructs mirror those of the corresponding full-length Gag-YFP constructs for both WT and MA-HBR mutants. Forty-two to 85 cells were analyzed under each condition across 3 independent experiments. The localization patterns determined by epifluorescence microscopy were confirmed by confocal microscopy. (Top) Representative confocal images. The blue arrowhead indicates the Gag-YFP signal in the intracellular compartments (IC). (Bottom) Bar graphs representing percentages of cells showing the indicated patterns of subcellular distribution for each Gag-YFP construct. ConA-AF594 staining was used as a plasma membrane (PM) marker (not shown). (C, top) Images showing an example of Gag-YFP showing PM-specific localization and ConA-AF594 staining of the PM. Green arrowheads indicate overlap of the two signals at the cell surface in a merged image. (Bottom) Images showing an example of Gag-YFP exhibiting PM and IC localization. The blue arrowhead indicates the Gag-YFP signal in the intracellular compartments. (D) HeLa cells were transfected with full-length WT Gag-YFP or 29/31KA Gag-YFP. At 16 h posttransfection, cells were stained with ConA-AF594, fixed with 4% paraformaldehyde in PBS, and analyzed using a fluorescence microscope. A total of 45 to 62 cells in 3 independent experiments were analyzed. The localization patterns determined by epifluorescence microscopy were confirmed by confocal microscopy. (Top) Representative confocal images. The blue arrowhead indicates the Gag-YFP signal in the intracellular compartments. (Bottom) Bar graphs representing percentages of cell populations as described above.
As described in the introduction, the 25/26KT mutant bound both the PM and intracellular membranes. In contrast, the 25/26KR mutant presented a PM-specific localization similar to that of the WT. Importantly for correlating the localization data with MA-RNA binding results (Fig. 2), the subcellular localization patterns of delNC/Gag-YFP were similar to those of the corresponding full-length Gag-YFP for both the WT and the MA-HBR mutants. When we examined the virus-like particle (VLP) release efficiency of the full-length Gag-YFP construct, we found that despite the difference in subcellular localization, both the Lys25/26 mutants displayed 2-fold increases in VLP release efficiency compared to the WT (Fig. 4). A previous study showed that 25/26KT Gag produced in vitro using rabbit reticulocyte lysates binds neutral PC-only liposomes efficiently, unlike WT Gag. Gag binding to neutral lipid membranes is via hydrophobic, and not electrostatic, interactions. Therefore, the efficient binding of 25/26KT Gag to PC-only liposomes reflects enhanced myristate-driven hydrophobic interactions, most likely due to increased myristate exposure (32). Using the same liposome binding assay, we found that the 25/26KR mutant also exhibits increased hydrophobic interactions (Fig. 5A and B). Of note, myristoylation-deficient full-length Gag-YFP (1GA/Gag-YFP) containing the 25/26KR or 25/26KT MA sequence was unable to bind any cellular membranes (Fig. 5C), eliminating the involvement of an alternative, myristate-independent mechanism for membrane binding of Lys25/26 mutants. Although it is formally possible that an unknown cellular function promotes the release of myristoylated Lys25/26 mutants but not the WT, these in vitro and cell-based data collectively suggest that the enhanced VLP release efficiency of Gag constructs with the Lys25/26 substitutions is likely a result of increased hydrophobic interactions dependent on the myristoyl moiety.
FIG 4.
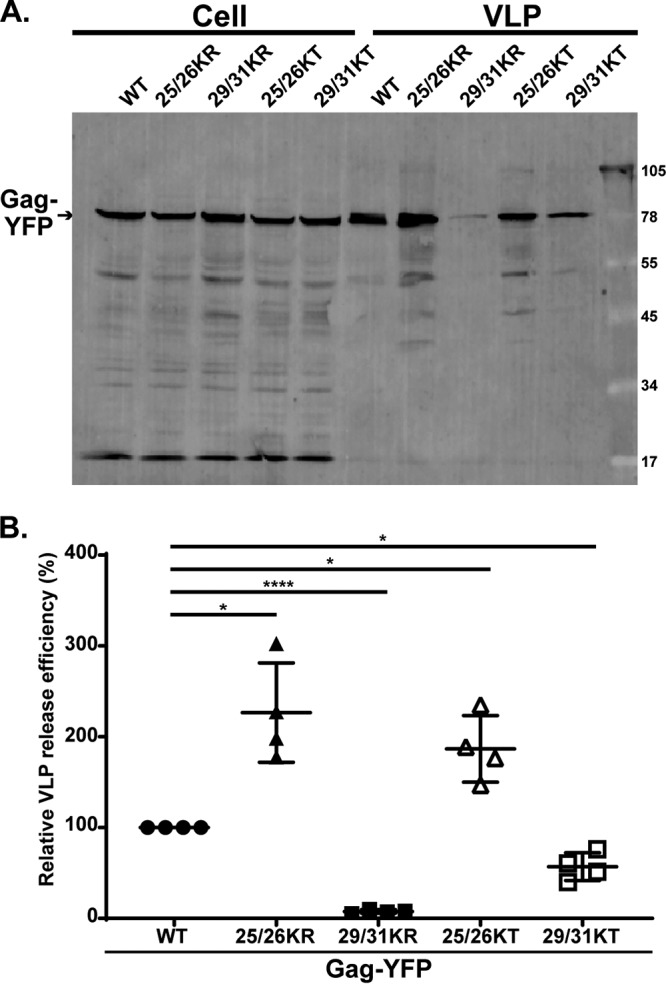
MA-HBR mutations alter VLP release efficiency. (A) HeLa cells were transfected with WT Gag-YFP or Gag-YFP containing 25/26KR, 25/26KT, 29/31KR, or 29/31KT changes. At 14 h posttransfection, cell and virus-like particle (VLP) lysates were collected and subjected to SDS-PAGE, and Gag proteins were detected by immunoblotting using HIV immunoglobulin. (B) Relative VLP release efficiency representing the amount of VLP-associated Gag as a fraction of the total Gag present in VLP and cell lysates and normalized to the VLP release efficiency of WT Gag. Results from 4 independent experiments are shown as means ± standard deviations. The average VLP release efficiency of WT Gag was 8.3%. P values were determined from raw data using Student’s t test. *, P ≤ 0.05; ****, P ≤ 0.0001.
FIG 5.
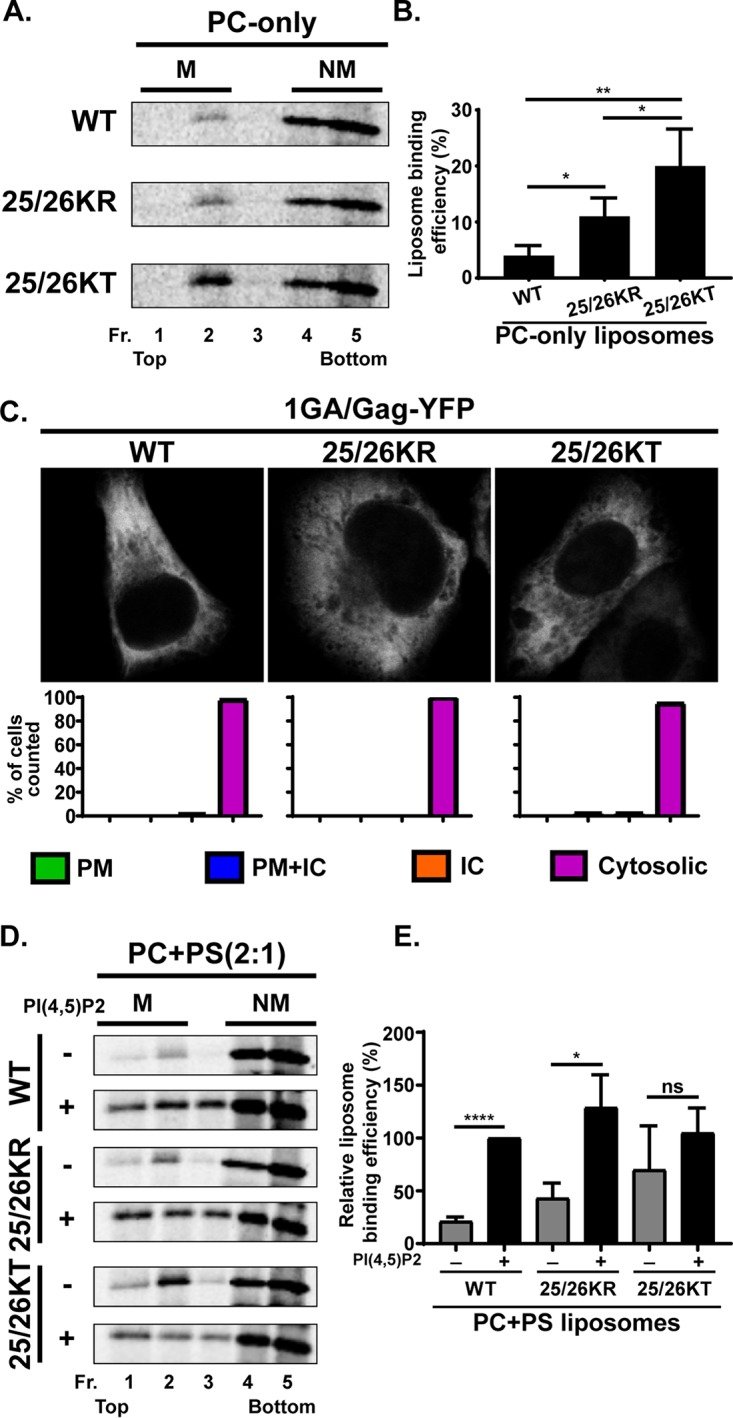
Membrane binding of Gag derivatives containing Lys25/26 substitutions. (A) 35S-labeled HIV-1 WT Gag, 25/26KR Gag, and 25/26KT Gag were synthesized in vitro using rabbit reticulocyte lysates and incubated with PC-only liposomes. The reaction mixtures were subjected to membrane flotation centrifugation, and a total of five 1-ml fractions were collected from each sample. M, membrane-bound Gag; NM, non-membrane-bound Gag. (B) Liposome binding efficiency calculated as the percentage of membrane-bound Gag compared to the total Gag synthesized in the reaction. Results from four independent experiments are shown as means ± standard deviations. P values were determined by Student’s t test. *, P ≤ 0.05; **, P ≤ 0.01. (C) HeLa cells were transfected with nonmyristoylated Gag-YFP (1GA/Gag-YFP), which contains the WT MA sequence or 25/26KR or 25/26KT substitutions. At 16 h posttransfection, cells were stained with ConA-AF594, fixed with 4% paraformaldehyde in PBS, and analyzed using a fluorescence microscope. Forty to 52 cells were analyzed under each condition across 3 independent experiments. The localization patterns determined by epifluorescence microscopy were confirmed by confocal microscopy. Representative confocal images are shown. Bar graphs represent percentages of cells showing the indicated patterns of subcellular distribution for each Gag-YFP construct. ConA-AF594 staining was used as a PM marker as described in the legend of Fig. 3 (not shown). (D) 35S-labeled HIV-1 WT Gag, 25/26KR Gag, and 25/26KT Gag were synthesized in vitro using rabbit reticulocyte lysates and incubated with PC+PS (2:1) liposomes (−) or liposomes containing 7.25 mol% PI(4,5)P2 (+). The reaction mixtures were analyzed as described above for panel A. (E) Relative liposome binding efficiency calculated as the percentage of membrane-bound Gag to the total Gag synthesized in the reaction and normalized to the efficiency of WT Gag binding to PI(4,5)P2-containing liposomes. The average efficiency of WT Gag binding to PI(4,5)P2-containing liposomes was 42.5%. Results from three independent experiments are presented as means ± standard deviations. P values were determined by Student’s t test. ns, not significant; *, P ≤ 0.05; ****, P ≤ 0.0001.
25/26KR Gag is dependent on PI(4,5)P2 for efficient membrane binding, unlike 25/26KT Gag.
While 25/26KR MA shows WT-level RNA binding and mediates the PM-specific localization of Gag-YFP, unlike WT MA, it engaged in increased hydrophobic interactions with membranes, similar to its corresponding KT mutant MA (32) (Fig. 5A and B). A previous study demonstrated that the 25/26KT mutant exhibits increased PI(4,5)P2-independent liposome binding (32). Therefore, we examined whether 25/26KR MA still shows PI(4,5)P2-dependent liposome binding. We found that the presence of PI(4,5)P2 significantly increases the liposome binding efficiency of the 25/26KR mutant but does not affect the 25/26KT mutant (Fig. 5D and E). These results indicate that the 25/26KR mutant is dependent on PI(4,5)P2 for efficient membrane binding, unlike the 25/26KT mutant, even though both mutants showed increased hydrophobic interactions relative to the WT.
Upon depletion of cellular PI(4,5)P2, 25/26KR Gag-YFP exhibits a promiscuous subcellular localization.
The PM localization of WT Gag is dependent on PI(4,5)P2 (15, 26, 27). To investigate whether the PM localization of 25/26KR Gag is also dependent on PI(4,5)P2, we next examined the effect of the expression of 5-phosphatase IV (5ptaseIV), which depletes cellular PI(4,5)P2, on the subcellular distribution of Gag-YFP derivatives. We coexpressed WT Gag-YFP or Gag-YFP with Lys25/26 substitutions with full-length 5ptaseIV (FL) or its catalytically inactive derivative (Δ1) (Fig. 6). As previously reported (26), WT Gag-YFP lost PM-specific localization and displayed a predominantly hazy cytosolic but sometimes intracellular punctate distribution when coexpressed with FL 5ptaseIV. The 25/26KT mutant showed a predominantly promiscuous localization regardless of the presence of FL or Δ1 5ptaseIV, demonstrating that this MA substitution mutant is not dependent on PI(4,5)P2 for cellular membrane binding. In contrast, the localization of the 25/26KR mutant changed from a predominantly PM-specific localization to a more promiscuous localization when it was coexpressed with FL but not Δ1 5ptaseIV. These results indicate that the PM-specific localization of 25/26KR Gag is dependent on the presence of PI(4,5)P2 in the cell.
FIG 6.
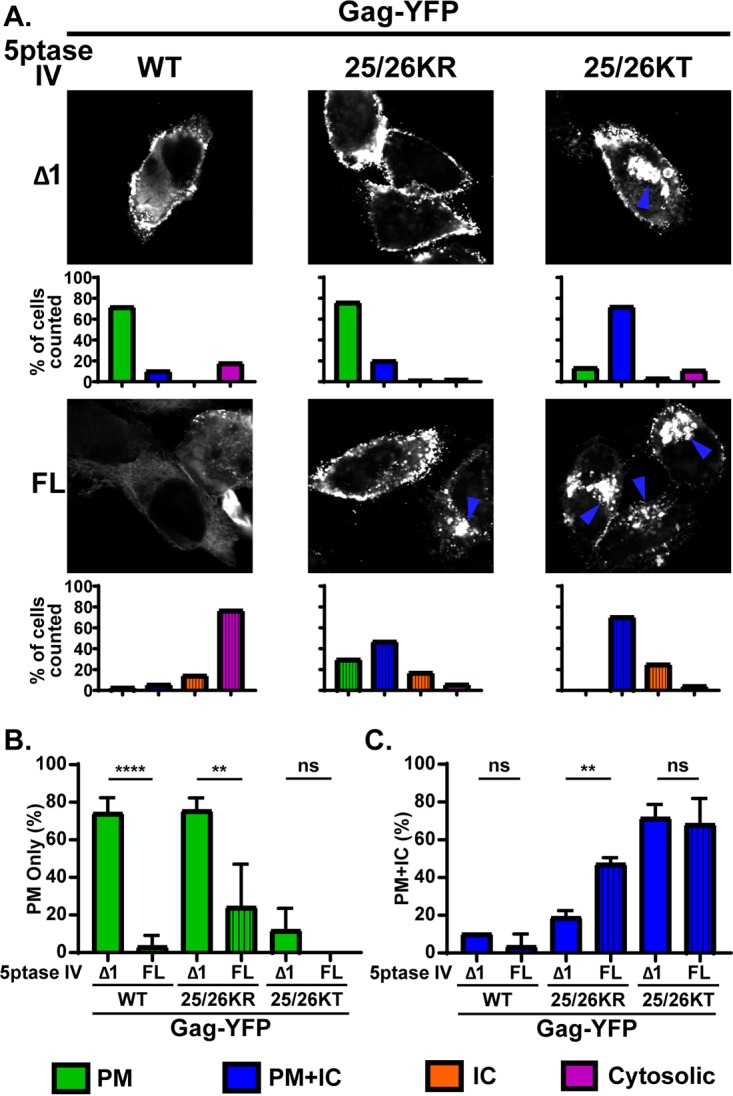
Depletion of cellular PI(4,5)P2 leads to promiscuous subcellular localization of 25/26KR Gag-YFP. (A) HeLa cells were transfected with WT Gag-YFP, 25/26KR Gag-YFP, or 25/26KT Gag-YFP along with myc-tagged FL 5ptaseIV or the catalytically inactive Δ1 derivative. At 14 h posttransfection, cells were stained with ConA-AF594 (not shown), fixed with 4% paraformaldehyde in PBS, permeabilized, immunostained with mouse monoclonal anti-myc antibody and anti-mouse IgG conjugated with Alexa Fluor 405 (not shown), and analyzed using a fluorescence microscope. Only cells positive for both myc-tagged 5ptaseIV and Gag-YFP were included in the analysis. Sixty-nine to 134 cells were analyzed under each condition across 3 independent experiments. Representative confocal images of Gag-YFP are shown. The blue arrowhead indicates the Gag-YFP signal in the intracellular compartments. The localization patterns were examined as described in the legend of in Fig. 3, and the percentages of total cells showing the indicated subcellular distribution patterns under each condition are shown in bar graphs. (B and C) Percentages of cells showing a PM-only distribution of Gag-YFP (B) and PM and intracellular compartment distribution of Gag-YFP (C) compared between cells expressing FL and Δ1 5ptaseIV across 3 independent experiments. Data are presented as means ± standard deviations. P values were determined by Student’s t test. ns, not significant; **, P ≤ 0.01; ****, P ≤ 0.0001.
Overall, our results thus far suggest that in the case of the Lys25/26 mutants, Gag that binds RNA efficiently maintains a PI(4,5)P2-dependent PM-specific localization, while Gag that binds RNA poorly shows a PI(4,5)P2-independent promiscuous localization. Thus, these results indicate that for Gag derivatives containing Lys25/26 substitutions, there is a correlation between RNA binding and PI(4,5)P2-dependent PM-specific localization, as previously observed with Gag chimeras containing heterologous retroviral MA domains (29).
Gag derivatives containing 29/31KR substitutions show a predominantly cytosolic distribution.
Since 29/31KR MA exhibits significantly higher RNA binding than 29/31KT MA (Fig. 2), we next sought to test whether the RNA binding efficiency correlates with subcellular localization in the case of Gag constructs containing changes at MA residues 29 and 31, as was observed for Gag mutants with changes at residues 25 and 26. To this end, we first examined the distribution of myristoylated full-length Gag-YFP and delNC/Gag-YFP derivatives (Fig. 3). Consistent with previous reports, Gag-YFP with the 29/31KT changes displayed a promiscuous localization, with its signals being present at both the PM and intracellular membranes. Interestingly, the 29/31KR mutant displayed a predominantly cytosolic distribution of Gag-YFP, indicating a loss of efficient membrane binding. It is important to note that unlike 29/31KT Gag-YFP, 29/31KR Gag-YFP showed negligible intracellular membrane binding despite having a higher positive charge than 29/31KT MA, which could mediate electrostatic interactions with acidic lipids more efficiently. We found that both delNC and full-length Gag-YFP showed analogous subcellular patterns for each Lys29/31 mutant. Of note, substitution of Lys29 and -31 to not only Thr but also another neutral amino acid, Ala (Fig. 3D), or an acidic amino acid, Glu (14), led to a promiscuous Gag localization. Therefore, it is unlikely that the promiscuous localization of 29/31KT Gag-YFP is due to the inadvertent introduction of a motif targeting Gag to intracellular compartments. Rather, these observations suggest that any changes in Lys29 and -31, except for the Lys-to-Arg changes, which maintain the total basic charge of the MA-HBR, would result in a promiscuous Gag localization. These results, together with the results of the comparison between two Lys25/26 mutants, strongly suggest that MA-RNA binding prevents Gag binding to intracellular membranes regardless of whether Gag can bind the PM. In congruence with the microscopy results, we found that the VLP release efficiency determined in the context of full-length Gag-YFP was significantly impaired for the 29/31KT mutant and almost abolished for the 29/31KR mutant (Fig. 4).
Replacing the NC domain with a heterologous dimerization motif enables 29/31KR Gag-YFP to localize specifically to the PM in a PI(4,5)P2-dependent fashion.
Even though the lack of binding to intracellular membranes is a shared phenotype between 25/26KR Gag-YFP and 29/31KR Gag-YFP, 29/31KR Gag-YFP showed a cytosolic distribution in most cells, unlike 25/26KR Gag-YFP. We hypothesized that 29/31KR Gag, unlike WT and 25/26KR Gag, is unable to counteract the negative regulation of membrane binding by RNA that is bound to its MA-HBR. A previous study suggests that while the overall positive charge of the MA-HBR is sufficient for RNA-mediated suppression of Gag-liposome binding irrespective of the identity of the residues, the identity of at least one of the basic residues is important for MA-PI(4,5)P2 interaction (55). In addition, a recent NMR study found that Lys29 and -31 are important for binding of MA to PI(4,5)P2, even in an experimental system in which RNA is absent (49).
Based on these observations, we reasoned that due to attenuated MA-PI(4,5)P2 binding, 29/31KR Gag is unable to counteract RNA-mediated suppression of membrane binding. It is then conceivable, according to our model, that if membrane binding of this mutant Gag can be enhanced enough to offset the negative regulation imposed by MA-RNA interactions, this mutant would localize specifically to the PM but not the intracellular compartments because MA-bound RNA would still prevent promiscuous membrane binding. Multimerization increases the binding of HIV-1 MA to the PM as well as to liposomes of different compositions (48). Thus, we sought to test the hypothesis mentioned above by attempting to augment the multimerization of 29/31KR Gag. Previous studies reported that a leucine zipper dimerization motif (LZ) promotes Gag multimerization more efficiently than the NC domain (58, 59). Therefore, in order to improve Gag multimerization, we replaced the NC domain of Gag with LZ (38, 60–63). We then expressed the WT and the Lys29/31 substitution mutants in the context of Gag-YFP and GagLZ-YFP in cells and examined their subcellular localization patterns (Fig. 7).
FIG 7.
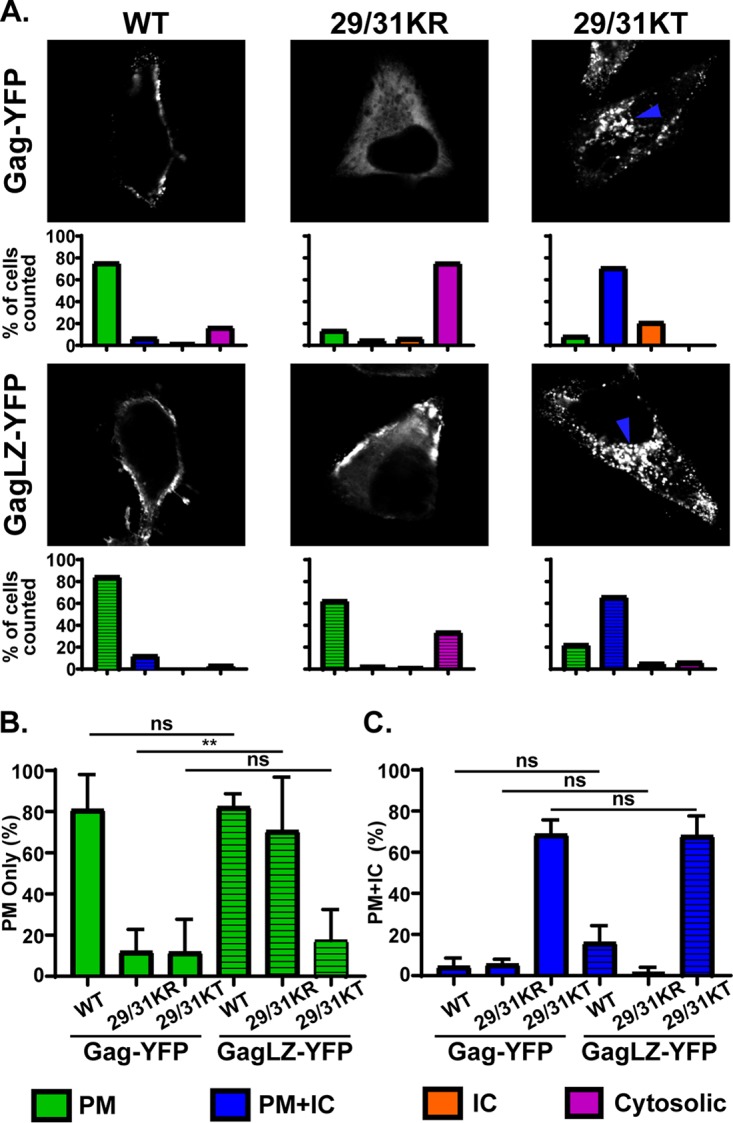
29/31KR Gag exhibits increased PM-specific localization when the NC domain is exchanged for the leucine zipper dimerization motif (LZ). (A) HeLa cells were transfected with WT Gag-YFP, WT GagLZ-YFP, or their derivatives with 29/31K substitutions. At 14 h posttransfection, cells were stained with ConA-AF594 (not shown), fixed with 4% paraformaldehyde in PBS, and analyzed using a fluorescence microscope. Sixty-one to 125 cells were analyzed under each condition across 3 independent experiments. (Top) Representative confocal images of Gag-YFP. The blue arrowhead indicates the Gag-YFP signal in the intracellular compartments. (Bottom) Localization patterns were examined as described in the legend of Fig. 3, and the percentages of total cells showing the indicated subcellular distribution patterns are shown in bar graphs. (B and C) Percentages of cells showing PM-only (B) and PM and intracellular compartment (C) distributions compared between Gag-YFP and GagLZ-YFP across 3 independent experiments. Data are shown as means ± standard deviations. P values were determined by Student’s t test. ns, not significant; **, P ≤ 0.01.
We found that WT GagLZ-YFP predominantly localized at the PM, as observed for WT Gag-YFP. 29/31KT GagLZ-YFP showed a promiscuous localization, like 29/31KT Gag-YFP, indicating that merely replacing the NC domain with LZ did not lead to a PM-specific localization. Consistent with our prediction, 29/31KR GagLZ-YFP demonstrated a significantly enhanced PM localization compared to 29/31KR Gag-YFP. Importantly, this construct showed very little promiscuous localization, unlike 29/31KT GagLZ-YFP, suggesting that MA with 29/31KR changes retains the ability to ensure PM-specific localization. Unexpectedly, 29/31KR GagLZ-YFP still failed to release VLPs efficiently (Fig. 8).
FIG 8.

29/31KR GagLZ-YFP fails to release VLPs. (A) HeLa cells were transfected with Gag-YFP or GagLZ-YFP containing WT MA or 29/31KR MA sequences. At 16 h posttransfection, cell and VLP lysates were collected and analyzed as described in the legend of Fig. 4. (B) Relative VLP release efficiency representing the amount of VLP-associated Gag as a fraction of total Gag present in VLP and cell lysates and normalized to the VLP release efficiency of WT Gag. Results from 3 independent experiments are shown as means ± standard deviations. The average VLP release efficiency of WT Gag was 9.6%. ns, not significant; ***, P ≤ 0.001.
We then wanted to test directly in cells whether LZ-driven PM localization is still PI(4,5)P2 dependent. To this end, we coexpressed GagLZ-YFP constructs with the FL and Δ1 5ptaseIV enzymes (Fig. 9). We found that both WT GagLZ-YFP and 29/31KR GagLZ-YFP lost the PM-specific localization when the FL 5ptaseIV enzyme was coexpressed. These results suggest that WT GagLZ-YFP and 29/31KR GagLZ-YFP bind the PM in a PI(4,5)P2-dependent manner. Interestingly, in the presence of FL 5ptaseIV, WT GagLZ-YFP showed a promiscuous localization to intracellular compartments and the PM, whereas 29/31KR GagLZ-YFP showed a predominantly cytosolic localization, as observed for WT Gag-YFP (Fig. 6).
FIG 9.

29/31KR GagLZ-YFP localizes to the PM in a PI(4,5)P2-dependent manner. HeLa cells were transfected with WT GagLZ-YFP or 29/31KR GagLZ-YFP along with myc-tagged FL 5ptaseIV or the catalytically inactive Δ1 derivative. At 14 to 16 h posttransfection, cells were stained with ConA-AF594 (not shown), fixed with 4% paraformaldehyde in PBS, immunostained with mouse monoclonal anti-myc antibody and anti-mouse IgG conjugated with Alexa Fluor 405 (not shown), and analyzed as described in the legend of Fig. 6. Thirty-seven to 78 cells in 3 independent experiments were analyzed under each condition. (Top) Representative confocal images of Gag-YFP. The blue arrowhead indicates the Gag-YFP signal in the intracellular compartments. (Bottom) The localization patterns were examined as described in the legend of Fig. 3, and the percentages of cells showing the indicated subcellular distribution patterns are shown in bar graphs.
Overall, the presented data suggest that when the MA-HBR in Gag binds RNA, Gag localizes specifically to the PM if it can counteract the RNA-mediated block of membrane binding either via interaction with PI(4,5)P2, which occurs natively in the case of WT and 25/26KR Gag, or when enhanced by improved multimerization, as observed for 29/31KR GagLZ. In contrast, when Gag is deficient in MA-RNA binding, as observed for KT mutants, it fails to localize specifically to the PM and instead binds promiscuously to different cellular membrane compartments. Altogether, the analyses of Lys25/26 and Lys29/31 substitutions in the MA-HBR revealed a strong correlation between MA-RNA binding and suppression of nonspecific Gag localization to intracellular membranes (summarized in Fig. 10).
FIG 10.

Relationships between MA-RNA binding and subcellular localization of Gag. Note that the full-length Gag constructs and the corresponding delNC Gag constructs show similar subcellular localization patterns for both WT and MA-HBR mutants.
DISCUSSION
In the present study, we examined the effects of amino acid substitutions in the MA-HBR on the relative amount of RNA bound to HIV-1 Gag in cells and on the subcellular localization of Gag. We found that MA-HBR mutants that were incapable of binding RNA efficiently displayed a promiscuous subcellular localization (i.e., 25/26KT Gag and 29/31KT Gag), consistent with previous observations obtained with Gag chimeras containing heterologous retroviral MA sequences (29). In contrast, MA-HBR mutants that bound RNA efficiently did not show a promiscuous localization; they either localized specifically to the PM (25/26KR Gag) or remained mainly in the cytosol (29/31KR Gag). The latter presented a PM-specific and not a promiscuous localization when its membrane binding ability was enhanced via LZ-mediated multimerization. The correlation established between MA-RNA binding and the lack of Gag localization to intracellular membranes in this study further provides support for our current working model in which RNA-mediated inhibition of Gag membrane binding ensures the PM-specific localization of Gag.
The liposome binding experiments using neutral liposomes indicate that both 25/26KT and 25/26KR Gag engage in greater hydrophobic interactions with membranes than WT Gag, likely due to increased myristate exposure relative to WT Gag (32; this study). Despite the enhanced hydrophobic interactions with membranes, 25/26KR Gag maintains a PM-specific localization, like WT Gag. However, the outcomes of PI(4,5)P2 depletion differ between WT and 25/26KR Gag. Upon FL 5ptaseIV expression, WT Gag loses membrane binding and exhibits a predominantly cytosolic distribution, whereas 25/26KR Gag retains membrane binding albeit localizing promiscuously in the majority of the Gag-YFP-expressing cells. The two different outcomes of PI(4,5)P2 depletion highlight the two functions of PI(4,5)P2, enhancing membrane binding of Gag and supporting PM-specific localization.
We found that 25/26KR Gag exhibits an increased VLP release efficiency compared to WT Gag, likely due to enhanced membrane binding as discussed above. Therefore, the Lys-to-Arg mutation at residues 25 and 26 should confer a fitness advantage to HIV-1. However, Lys25 and -26 are highly conserved across various clades of HIV-1 (64). We speculate that Arg, instead of Lys, at positions 25 and 26 may negatively affect other aspects of the HIV-1 life cycle. Similar to 25/26KR Gag, 25/26KT Gag also shows a superior VLP release efficiency, even though 25/26KT Gag does not localize specifically to the PM, unlike its KR counterpart. The high VLP release efficiency of 25/26KT Gag may be due to even more robust hydrophobic interactions with membranes than for 25/26KR Gag (Fig. 5), which may make up for the Gag mislocalization. The increased hydrophobic interactions of 25/26KT Gag, which does not bind RNA, relative to 25/26KR Gag, which binds RNA at the WT level, are consistent with the observation that the removal of RNA increases myristate-dependent hydrophobic interactions (32). However, 25/26KR Gag still showed enhanced hydrophobic membrane binding relative to WT Gag. Therefore, further investigation is required to determine the mechanism(s) by which Lys25 and -26 modulates myristate exposure.
We found that RNA binding of 29/31KR Gag was not as efficient as that of WT Gag, although it was better than that of 29/31KT Gag. While we observed previously that the RNA sensitivity of Gag-liposome binding is maintained when all Lys and Arg residues in the MA-HBR are switched with each other (55), the presence of Lys, but not Arg, at residues 29 and 31 seems to be important for WT-level RNA binding of Gag. Although both Arg and Lys are positively charged basic amino acids, Arg has a terminal guanidium group, while Lys has a single terminal amino group. These groups also differ in their geometry (i.e., planar versus tetrahedral). Therefore, it is possible that Lys and Arg participate in a different number and/or orientation of electrostatic interactions with RNA or lipids (65–68).
Even though 29/31KR Gag showed reduced RNA binding, this level of MA-RNA binding was sufficient for suppressing binding to intracellular membranes. It is noteworthy that 29/31KR Gag failed to bind even the PM in most cells. Strongly cationic model proteins have been shown to associate with the PM (69). However, in the case of HIV-1 Gag, the high positive charge retained in 29/31KR MA is insufficient for the PM localization, likely due to the RNA-mediated block. As mentioned above, Lys29 and -31 are important for PI(4,5)P2 interaction even in the absence of RNA (49). Based on this knowledge, we speculate that due to an inefficient interaction with PI(4,5)P2, 29/31KR Gag is unable to overcome the negative regulation by RNA. Therefore, the balance between MA-RNA and MA-PI(4,5)P2 binding, which is carefully orchestrated for WT Gag, is tipped in the favor of MA-RNA binding in the case of 29/31KR Gag. In an attempt to find a mutant that retains a partial ability to interact with PI(4,5)P2, which could result in a partial shift of the balance back to MA-PI(4,5)P2 binding, we created the single mutants 29KR Gag and 31KR Gag and examined their subcellular localization in HeLa cells. However, we found that 31KR Gag fully recapitulated the phenotype of 29/31KR by exhibiting a predominantly cytosolic distribution, while 29KR Gag showed mainly a WT-like PM-specific localization (data not shown). Thus, Lys but not Arg is required at MA residue 31, whereas residue 29 can be either Lys or Arg for WT-like membrane binding and PM-specific localization of Gag.
The PM-specific localization of 29/31KR GagLZ is likely a manifestation of the enhanced multimerization of 29/31KR Gag restoring the balance toward MA-PI(4,5)P2 binding. Probable mechanisms through which LZ may promote membrane binding may be an avidity effect due to increased multimerization, myristate exposure induced by multimerization (22), a reduction in the amount of RNA bound to MA associated with myristate exposure (53), or some combination of these factors. We cannot rule out the possibility that the 29/31KR mutation affects MA-MA interactions in the context of LZ-mediated Gag multimerization and thereby affects membrane binding of Gag; however, MA-HBR residues Lys29 and -31 are not among the residues identified as being important for MA trimerization (70).
Previous studies have shown that NC deletion abrogates Gag localization to specific PM domains, such as uropods in polarized T cells or the virus-containing compartments (VCCs) in macrophages, whereas insertion of LZ restores such localization (71, 72). In contrast, we do not see a difference in the subcellular localization of Gag between full-length Gag and delNC Gag for any of the MA mutants examined (Fig. 3). Taken together, these data suggest that the presence of NC and, hence, NC-dependent multimerization do not dictate overall Gag localization to the PM but are important for the subsequent movement of Gag to specific domains within the PM.
We observed no commensurate increase in VLP release for 29/31KR GagLZ-YFP compared to 29/31KR despite the shift in Gag localization from the cytosol to the PM. Analogous to our observation, a recent study found that when the MA domain of HIV-1 Gag was replaced with the N-terminal PI(4,5)P2 binding region of avian sarcoma virus Gag, the chimeric Gag localized to the PM but did not release VLPs efficiently (73). The order of Gag-PM binding and multimerization into Gag puncta varies between different retroviruses (74, 75), suggesting that membrane binding and multimerization need to be coordinated for optimal particle assembly. Thus, it is conceivable that the release defect of 29/31KR GagLZ-YFP may be due in part to a change in Gag multimerization caused by the absence of the native NC domain or the presence of the LZ motif or YFP tag that becomes apparent in the context of the 29/31KR mutation.
Overall, this paper provides a strong correlation between the ability of HIV-1 Gag to bind RNA via MA and the specificity of Gag localization to the PM. In the absence of PI(4,5)P2, WT Gag and 29/31KR GagLZ remain in the cytosol (Fig. 6 and 9). Therefore, RNA binding is likely sufficient for preventing nonspecific binding to other membranes for these Gag proteins. On the other hand, 25/26KR Gag and WT GagLZ, which are likely to have higher affinities for lipid bilayers due to increased myristate exposure or avidity, show a promiscuous localization upon PI(4,5)P2 depletion (Fig. 6 and 9). In these cases, both RNA and PI(4,5)P2 likely work in conjunction to prevent promiscuous localization. Altogether, the comparisons between the MA-HBR mutants indicate the role of MA-bound RNA as an important player in targeting Gag specifically to the PM.
MATERIALS AND METHODS
Plasmids.
pCMVNLGagPolRRE Gag-YFP, which expresses HIV-1 GagNL4-3 fused to YFP in an HIV-1 Rev-dependent manner, was constructed as described previously for pCMVNLGagPolRRE Gag-mRFP (76), using standard molecular cloning techniques. pCMVNLGagPolRRE delNC/Gag-YFP was generated by replacing the SpeI-XmaI region of pCMVNLGagPolRRE (77) with the corresponding region of pNL4-3/delNC/Gag-YFP, described previously (78). pCMVNL GagLZ-YFP (previously named pCMVNLGag Venus LZ) was described in previous reports (29, 55). pGEMNLNR, an expression vector used for in vitro transcription and translation of Gag, was described previously (26). To create pCMVNLGagPolRRE 25/26KR Gag-YFP, pCMVNLGagPolRRE 29/31KR Gag-YFP, pCMVNLGagPolRRE 29KR Gag-YFP, pCMVNLGagPolRRE 31KR Gag-YFP, and pCMVNLGagPolRRE 29/31KA Gag-YFP, the corresponding MA-HBR point mutations were introduced into pCMVNLGagPolRRE Gag-YFP using PCR mutagenesis. pCMVNLGagPolRRE 25/26KT Gag-YFP and pCMVNLGagPolRRE 29/31KT Gag-YFP were constructed by replacing the SpeI-BssHII region of pCMVNLGagPolRRE Gag-YFP with the corresponding regions of pGEMNLNR 25/26KT Gag and pGEMNLNR 29/31KT Gag (26, 32). pCMVNLGagPolRRE delNC/Gag-YFP constructs containing the MA-HBR mutations, i.e., 25/26KR, 25/26KT, 29/31KR, and 29/31KT, were constructed using standard molecular cloning techniques. pCMVNLGagPolRRE 6A2T delNC/Gag-YFP was created by replacing the SpeI-BssHII region of pCMVNLGagPolRRE delNC/Gag-YFP with the corresponding region of pGEMNLNR 6A2T Gag (26, 32). pGEMNLNR plasmids containing the MA-HBR mutations, i.e., 25/26KR and 29/31KR, were constructed using standard molecular biology techniques. The 1GA mutation was introduced into the pCMVNLGagPolRRE Gag-YFP and pCMVNLGagPolRRE delNC/Gag-YFP constructs by PCR mutagenesis as described previously (75). pCMVNL GagLZ-YFP containing the MA-HBR mutations, i.e., 29/31KR and 29/31KT, and pGEMNLNR containing the MA-HBR mutations, i.e., 25/26KR and 29/31KR, were constructed using standard molecular cloning techniques.
pCMV-Rev was described previously (76) (kindly provided by S. Venkatesan, National Institutes of Health). Mammalian expression plasmids encoding myc-tagged 5-phosphatase IV (FL 5ptaseIV), pcDNA4TO/Myc5ptaseIV, and its catalytically inactive Δ1 derivative (Δ1 5ptaseIV) were previously described (26, 79). pCMV-Vphu, which harbors the codon-optimized version of the HIV-1 Vpu gene, was previously described (80) (a kind gift from K. Strebel).
Cells and transfection.
HeLa cells were cultured as described previously (32). For microscopy, 30,000 HeLa cells were plated into each well of eight-well chamber slides (Lab-Tek; Nalgene Nunc International), cultured for 24 h, and transfected with plasmids encoding the indicated Gag-YFP derivatives using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For VLP release and PAR-CLIP experiments, 4 × 105 cells were plated into each well of six-well plates (Corning), cultured for 24 h, and transfected as described above.
VLP release assay and immunoblotting.
VLP release assays were carried out as described previously (32). HIV-1 Gag in cell and VLP lysates was detected by immunoblotting using HIV immunoglobulin (HIV-Ig; AIDS Research and Reference Reagent Program) as a primary antibody. Alexa Fluor 488-conjugated goat anti-human IgG (Invitrogen) was used for the detection of primary antibodies for HIV-1 Gag. Fluorescence signals were detected and quantified using a Typhoon Trio imager (GE Healthcare).
Immunostaining and fluorescence microscopy.
HeLa cells transfected with plasmids expressing Gag-YFP were incubated for 1 min at room temperature with Alexa Fluor 594-conjugated concanavalin A (ConA-AF594; Invitrogen) for visualization of the plasma membrane cells at 14 to 16 h posttransfection. Cells were then fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). The expression of 5ptaseIV (both full-length [FL] and Δ1 enzymes) in cells was detected by immunostaining with mouse anti-myc antibody (9E10; Santa Cruz Biotechnologies) after permeabilization of the cells. For quantitative analysis of Gag localization phenotypes, images of about 20 fields were recorded using an Olympus IX70 inverted fluorescence microscope at a ×60 magnification, and a range of 37 to 134 cells were evaluated for Gag localization patterns. For 5ptaseIV coexpression studies, only cells that were positive for both Gag and 5ptaseIV (either FL or Δ1) were chosen for evaluation. Confocal microscopy using a Leica SP5 inverted confocal microscope was carried out to further verify localization patterns determined by epifluorescence microscopy.
Liposome binding assay.
Preparation of liposomes, in vitro Gag translation, and sucrose gradient flotation centrifugation were performed as described previously (32). In brief, full-length myristoylated Gag was translated in vitro using Promega’s TnT Sp6 coupled reticulocyte lysate system and incubated with sonicated liposomes composed of a neutral lipid, phosphatidylcholine, and an acidic lipid, phosphatidylserine, in a 2:1 molar ratio (PC+PS [2:1] liposomes), with or without the addition of 7.25 mol% PI(4,5)P2. Following sucrose flotation centrifugation, five fractions were collected, with the top two fractions representing the membrane-bound, floating Gag. Following SDS-PAGE, Gag bands were detected by phosphorimager analysis and quantified using ImageQuant 1D TL v8.1 image analysis software. The liposome binding efficiency (percent) was calculated as the percentage of membrane-bound Gag versus the total Gag in all 5 fractions.
PAR-CLIP assay.
To determine the amount of cellular RNA bound to transfected Gag, a PAR-CLIP assay was performed as described previously (47, 57), with some modifications. Briefly, in each well of a 6-well plate, 4 × 105 HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum in the presence of the antibiotics penicillin and streptomycin. The cells were then transfected with either a plasmid encoding 1GA/delNC/Gag-YFP containing the indicated MA sequence or pUC19. The cells were cultured for 24 h posttransfection before the addition of fresh medium containing 100 μM 4-thiouridine (4-SU; Sigma), a photo-cross-linkable ribonucleoside analogue. After 16 h, the cells were exposed to 365-nm UV light at 300 mJ/cm2 to cross-link RNA and RNA binding proteins. Cells were lysed with NP-40 lysis buffer (50 mM HEPES buffer [pH 7.5] containing 150 mM KCl, 2 mM EDTA, 0.5% [vol/vol] NP-40 substitute, and 0.5 mM dithiothreitol [DTT] and supplemented with a protease inhibitor cocktail), and the lysate was treated with 20 U/ml RNase A (Promega) and 60 U/ml RQ1 DNase (Promega). Gag was immunoprecipitated by incubating the cell lysates with HIV-Ig bound to Dynabeads protein G (Invitrogen) for 1 h on a rotating shaker at 4°C. The RNA bound to Gag was first dephosphorylated at the 5′ end with calf intestinal alkaline phosphatase (New England BioLabs [NEB]), followed by T4 polynucleotide kinase-catalyzed (NEB) rephosphorylation of the RNA at the 5′ end with 32P ([γ-32P]ATP; PerkinElmer). The Gag-RNA complex was eluted from Dynabeads by heating in PAR-CLIP elution buffer (50 mM Tris HCl [pH 6.8] containing 2 mM EDTA-NaOH, 10% [vol/vol] glycerol, 2% [vol/vol] SDS, 100 mM DTT, and 0.1% [wt/vol] bromophenol blue) for 5 min at 95°C with shaking at 1,400 rpm. After resolving the bands by SDS-PAGE and subsequent electrotransfer to a PVDF membrane, the membrane was probed with HIV-Ig as the primary antibody and the corresponding secondary antibody conjugated to horseradish peroxidase (goat anti-human IgG; Invitrogen). Gag-specific bands were detected after incubation with SuperSignal West Pico chemiluminescent substrate (Pierce) on a Syngene Pxi multiapplication imager, and the intensities of the bands were measured using GeneTools analysis software. The 32P-end-labeled RNA bands were also detected on the same membrane using autoradiography on a Typhoon FLA 9500 laser scanner and quantified using ImageQuant 1D TL v8.1 image analysis software. The relative RNA binding efficiency (percent) was calculated as the percentage of the total RNA band intensity versus the total Gag band intensity for each construct and compared to the normalized value for WT 1GA/delNC/Gag-YFP.
Statistical analysis.
Student’s t tests were performed using GraphPad Prism. P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We thank members of our laboratory for helpful discussions and critical reviews of the manuscript. We thank S. Kutluay for her helpful comments in the optimization of the PAR-CLIP technique. The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: HIV-Ig (from the NABI and NHLBI). The microscopy data presented in this study were collected at the Biomedical Research Core Facilities Microscopy core, University of Michigan.
This work was supported by NIH grant R37 AI071727 (to A.O.).
REFERENCES
- 1.Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol 4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finzi A, Orthwein A, Mercier J, Cohen EA. 2007. Productive human immunodeficiency virus type 1 assembly takes place at the plasma membrane. J Virol 81:7476–7490. doi: 10.1128/JVI.00308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adamson CS, Freed EO. 2007. Human immunodeficiency virus type 1 assembly, release, and maturation. Adv Pharmacol 55:347–387. doi: 10.1016/S1054-3589(07)55010-6. [DOI] [PubMed] [Google Scholar]
- 4.Bieniasz PD. 2009. The cell biology of HIV-1 virion genesis. Cell Host Microbe 5:550–558. doi: 10.1016/j.chom.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freed EO. 2015. HIV-1 assembly, release and maturation. Nat Rev Microbiol 13:484–496. doi: 10.1038/nrmicro3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swanstrom R, Wills J. 1997. Synthesis, assembly, and processing of viral proteins, p 263–334. In Coffin JM, Hughes SH, Varmus HE (ed), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- 7.Sundquist WI, Kräusslich HG. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2:a006924. doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garnier L, Bowzard JB, Wills JW. 1998. Recent advances and remaining problems in HIV assembly. AIDS 12(Suppl A):S5–S16. [PubMed] [Google Scholar]
- 9.Balasubramaniam M, Freed EO. 2011. New insights into HIV assembly and trafficking. Physiology (Bethesda) 26:236–251. doi: 10.1152/physiol.00051.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Göttlinger HG, Sodroski JG, Haseltine WA. 1989. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A 86:5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bryant M, Ratner L. 1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A 87:523–527. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou W, Parent LJ, Wills JW, Resh MD. 1994. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J Virol 68:2556–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan X, Yu X, Lee TH, Essex M. 1993. Mutations in the N-terminal region of human immunodeficiency virus type 1 matrix protein block intracellular transport of the Gag precursor. J Virol 67:6387–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ono A, Orenstein JM, Freed EO. 2000. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J Virol 74:2855–2866. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol(4,5)bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A 101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freed EO, Orenstein JM, Buckler-White AJ, Martin MA. 1994. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J Virol 68:5311–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ono A, Freed EO. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol 73:4136–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paillart JC, Göttlinger HG. 1999. Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of gag membrane targeting. J Virol 73:2604–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spearman P, Horton R, Ratner L, Kuli-Zade I. 1997. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J Virol 71:6582–6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A 103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saad JS, Loeliger E, Luncsford P, Liriano M, Tai J, Kim A, Miller J, Joshi A, Freed EO, Summers MF. 2007. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J Mol Biol 366:574–585. doi: 10.1016/j.jmb.2006.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. 2004. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A 101:517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou W, Resh MD. 1996. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J Virol 70:8540–8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermida-Matsumoto L, Resh MD. 1999. Human immunodeficiency virus type 1 protease triggers a myristoyl switch that modulates membrane binding of Pr55(gag) and p17MA. J Virol 73:1902–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. 1996. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci U S A 93:3099–3104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. 2008. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient Gag membrane binding. J Virol 82:2405–2417. doi: 10.1128/JVI.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan R, Uchil PD, Jin J, Shui G, Ott DE, Mothes W, Wenk MR. 2008. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J Virol 82:11228–11238. doi: 10.1128/JVI.00981-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inlora J, Chukkapalli V, Derse D, Ono A. 2011. Gag localization and virus-like particle release mediated by the matrix domain of human T-lymphotropic virus type 1 Gag are less dependent on phosphatidylinositol-(4,5)-bisphosphate than those mediated by the matrix domain of HIV-1 Gag. J Virol 85:3802–3810. doi: 10.1128/JVI.02383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inlora J, Collins DR, Trubin ME, Chung JY, Ono A. 2014. Membrane binding and subcellular localization of retroviral Gag proteins are differentially regulated by MA interactions with phosphatidylinositol-(4,5)-bisphosphate and RNA. mBio 5:e02202-14. doi: 10.1128/mBio.02202-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alfadhli A, Barklis RL, Barklis E. 2009. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 387:466–472. doi: 10.1016/j.virol.2009.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anraku K, Fukuda R, Takamune N, Misumi S, Okamoto Y, Otsuka M, Fujita M. 2010. Highly sensitive analysis of the interaction between HIV-1 Gag and phosphoinositide derivatives based on surface plasmon resonance. Biochemistry 49:5109–5116. doi: 10.1021/bi9019274. [DOI] [PubMed] [Google Scholar]
- 32.Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci U S A 107:1600–1605. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shkriabai N, Datta SA, Zhao Z, Hess S, Rein A, Kvaratskhelia M. 2006. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry 45:4077–4083. doi: 10.1021/bi052308e. [DOI] [PubMed] [Google Scholar]
- 34.Chukkapalli V, Ono A. 2011. Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J Mol Biol 410:512–524. doi: 10.1016/j.jmb.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purohit P, Dupont S, Stevenson M, Green MR. 2001. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 7:576–584. doi: 10.1017/s1355838201002023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hearps AC, Wagstaff KM, Piller SC, Jans DA. 2008. The N-terminal basic domain of the HIV-1 matrix protein does not contain a conventional nuclear localization sequence but is required for DNA binding and protein self-association. Biochemistry 47:2199–2210. doi: 10.1021/bi701360j. [DOI] [PubMed] [Google Scholar]
- 37.Burniston MT, Cimarelli A, Colgan J, Curtis SP, Luban J. 1999. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J Virol 73:8527–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang CY, Chang YF, Wang SM, Tseng YT, Huang KJ, Wang CT. 2008. HIV-1 matrix protein repositioning in nucleocapsid region fails to confer virus-like particle assembly. Virology 378:97–104. doi: 10.1016/j.virol.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cimarelli A, Luban J. 1999. Translation elongation factor 1-alpha interacts specifically with the human immunodeficiency virus type 1 Gag polyprotein. J Virol 73:5388–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ott DE, Coren LV, Gagliardi TD. 2005. Redundant roles for nucleocapsid and matrix RNA-binding sequences in human immunodeficiency virus type 1 assembly. J Virol 79:13839–13847. doi: 10.1128/JVI.79.22.13839-13847.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Webb JA, Jones CP, Parent LJ, Rouzina I, Musier-Forsyth K. 2013. Distinct binding interactions of HIV-1 Gag to Psi and non-Psi RNAs: implications for viral genomic RNA packaging. RNA 19:1078–1088. doi: 10.1261/rna.038869.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramalingam D, Duclair S, Datta SA, Ellington A, Rein A, Prasad VR. 2011. RNA aptamers directed to human immunodeficiency virus type 1 Gag polyprotein bind to the matrix and nucleocapsid domains and inhibit virus production. J Virol 85:305–314. doi: 10.1128/JVI.02626-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lochrie MA, Waugh S, Pratt DG, Clever J, Parslow TG, Polisky B. 1997. In vitro selection of RNAs that bind to the human immunodeficiency virus type-1 gag polyprotein. Nucleic Acids Res 25:2902–2910. doi: 10.1093/nar/25.14.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alfadhli A, Still A, Barklis E. 2009. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J Virol 83:12196–12203. doi: 10.1128/JVI.01197-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alfadhli A, McNett H, Tsagli S, Bächinger HP, Peyton DH, Barklis E. 2011. HIV-1 matrix protein binding to RNA. J Mol Biol 410:653–666. doi: 10.1016/j.jmb.2011.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chukkapalli V, Inlora J, Todd GC, Ono A. 2013. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J Virol 87:7155–7159. doi: 10.1128/JVI.00075-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kutluay SB, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, Bieniasz PD. 2014. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 159:1096–1109. doi: 10.1016/j.cell.2014.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dick RA, Kamynina E, Vogt VM. 2013. Effect of multimerization on membrane association of Rous sarcoma virus and HIV-1 matrix domain proteins. J Virol 87:13598–13608. doi: 10.1128/JVI.01659-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mercredi PY, Bucca N, Loeliger B, Gaines CR, Mehta M, Bhargava P, Tedbury PR, Charlier L, Floquet N, Muriaux D, Favard C, Sanders CR, Freed EO, Marchant J, Summers MF. 2016. Structural and molecular determinants of membrane binding by the HIV-1 matrix protein. J Mol Biol 428:1637–1655. doi: 10.1016/j.jmb.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dick RA, Vogt VM. 2014. Membrane interaction of retroviral Gag proteins. Front Microbiol 5:187. doi: 10.3389/fmicb.2014.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Todd GC, Duchon A, Inlora J, Olson ED, Musier-Forsyth K, Ono A. 2017. Inhibition of HIV-1 Gag-membrane interactions by specific RNAs. RNA 23:395–405. doi: 10.1261/rna.058453.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlson LA, Bai Y, Keane SC, Doudna JA, Hurley JH. 2016. Reconstitution of selective HIV-1 RNA packaging in vitro by membrane-bound Gag assemblies. Elife 5:e14663. doi: 10.7554/eLife.14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaines CR, Tkacik E, Rivera-Oven A, Somani P, Achimovich A, Alabi T, Zhu A, Getachew N, Yang AL, McDonough M, Hawkins T, Spadaro Z, Summers MF. 2018. HIV-1 matrix protein interactions with tRNA: implications for membrane targeting. J Mol Biol 430:2113–2127. doi: 10.1016/j.jmb.2018.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leventis PA, Grinstein S. 2010. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys 39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 55.Llewellyn GN, Grover JR, Olety B, Ono A. 2013. HIV-1 Gag associates with specific uropod-directed microdomains in a manner dependent on its MA highly basic region. J Virol 87:6441–6454. doi: 10.1128/JVI.00040-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thornhill D, Olety B, Ono A. 2019. Relationships between MA-RNA binding in cells and suppression of HIV-1 Gag mislocalization to intracellular membranes. bioRxiv doi: 10.1101/629998. [DOI] [PMC free article] [PubMed]
- 57.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. 2010. PAR-CliP—a method to identify transcriptome-wide the binding sites of RNA binding proteins. J Vis Exp 2010:2034. doi: 10.3791/2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klein KC, Reed JC, Tanaka M, Nguyen VT, Giri S, Lingappa JR. 2011. HIV Gag-leucine zipper chimeras form ABCE1-containing intermediates and RNase-resistant immature capsids similar to those formed by wild-type HIV-1 Gag. J Virol 85:7419–7435. doi: 10.1128/JVI.00288-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Dou J, Ding L, Spearman P. 2007. Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J Virol 81:12899–12910. doi: 10.1128/JVI.01280-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Accola MA, Strack B, Göttlinger HG. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J Virol 74:5395–5402. doi: 10.1128/JVI.74.12.5395-5402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crist RM, Datta SA, Stephen AG, Soheilian F, Mirro J, Fisher RJ, Nagashima K, Rein A. 2009. Assembly properties of human immunodeficiency virus type 1 Gag-leucine zipper chimeras: implications for retrovirus assembly. J Virol 83:2216–2225. doi: 10.1128/JVI.02031-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Qian H, Love Z, Barklis E. 1998. Analysis of the assembly function of the human immunodeficiency virus type 1 Gag protein nucleocapsid domain. J Virol 72:1782–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stauffer S, Rahman SA, de Marco A, Carlson LA, Glass B, Oberwinkler H, Herold N, Briggs JAG, Müller B, Grünewald K, Kräusslich HG. 2014. The nucleocapsid domain of Gag is dispensable for actin incorporation into HIV-1 and for association of viral budding sites with cortical F-actin. J Virol 88:7893–7903. doi: 10.1128/JVI.00428-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Foley B, Leitner T, Apetrei C, Hahn B, Mizrachi I, Mullins J, Rambaut A, Wolinsky S, Korber B (ed). 2018. HIV Sequence Compendium 2018. LA-UR 18-25673 Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM. [Google Scholar]
- 65.Calnan BJ, Tidor B, Biancalana S, Hudson D, Frankel AD. 1991. Arginine-mediated RNA recognition: the arginine fork. Science 252:1167–1171. doi: 10.1126/science.252.5009.1167. [DOI] [PubMed] [Google Scholar]
- 66.Li L, Vorobyov I, Allen TW. 2013. The different interactions of lysine and arginine side chains with lipid membranes. J Phys Chem B 117:11906–11920. doi: 10.1021/jp405418y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sokalingam S, Raghunathan G, Soundrarajan N, Lee SG. 2012. A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein. PLoS One 7:e40410. doi: 10.1371/journal.pone.0040410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu Z, Cui Q, Yethiraj A. 2013. Why do arginine and lysine organize lipids differently? Insights from coarse-grained and atomistic simulations. J Phys Chem B 117:12145–12156. doi: 10.1021/jp4068729. [DOI] [PubMed] [Google Scholar]
- 69.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. 2008. Membrane phosphatidylserine regulates surface charge and protein localization. Science 319:210–213. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]
- 70.Tedbury PR, Novikova M, Ablan SD, Freed EO. 2016. Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc Natl Acad Sci U S A 113:E182–E190. doi: 10.1073/pnas.1516618113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Llewellyn GN, Hogue IB, Grover JR, Ono A. 2010. Nucleocapsid promotes localization of HIV-1 gag to uropods that participate in virological synapses between T cells. PLoS Pathog 6:e1001167. doi: 10.1371/journal.ppat.1001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Inlora J, Chukkapalli V, Bedi S, Ono A. 2016. Molecular determinants directing HIV-1 Gag assembly to virus-containing compartments in primary macrophages. J Virol 90:8509–8519. doi: 10.1128/JVI.01004-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watanabe SM, Medina GN, Eastep GN, Ghanam RH, Vlach J, Saad JS, Carter CA. 2018. The matrix domain of the Gag protein from avian sarcoma virus contains a PI(4,5)P. J Biol Chem 293:18841–18853. doi: 10.1074/jbc.RA118.003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eichorst JP, Chen Y, Mueller JD, Mansky LM. 2018. Distinct pathway of human T-cell leukemia virus type 1 Gag punctum biogenesis provides new insights into enveloped virus assembly. mBio 9:e00758-18. doi: 10.1128/mBio.00758-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ivanchenko S, Godinez WJ, Lampe M, Kräusslich HG, Eils R, Rohr K, Bräuchle C, Müller B, Lamb DC. 2009. Dynamics of HIV-1 assembly and release. PLoS Pathog 5:e1000652. doi: 10.1371/journal.ppat.1000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hogue IB, Grover JR, Soheilian F, Nagashima K, Ono A. 2011. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J Virol 85:9749–9766. doi: 10.1128/JVI.00743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ono A, Freed EO. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci U S A 98:13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hogue IB, Hoppe A, Ono A. 2009. Quantitative fluorescence resonance energy transfer microscopy analysis of the human immunodeficiency virus type 1 Gag-Gag interaction: relative contributions of the CA and NC domains and membrane binding. J Virol 83:7322–7336. doi: 10.1128/JVI.02545-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kisseleva MV, Wilson MP, Majerus PW. 2000. The isolation and characterization of a cDNA encoding phospholipid-specific inositol polyphosphate 5-phosphatase. J Biol Chem 275:20110–20116. doi: 10.1074/jbc.M910119199. [DOI] [PubMed] [Google Scholar]
- 80.Nguyen KL, Llano M, Akari H, Miyagi E, Poeschla EM, Strebel K, Bour S. 2004. Codon optimization of the HIV-1 vpu and vif genes stabilizes their mRNA and allows for highly efficient Rev-independent expression. Virology 319:163–175. doi: 10.1016/j.virol.2003.11.021. [DOI] [PubMed] [Google Scholar]