

Patients afflicted by triple-negative breast cancer have decreased survival and limited therapeutic options. Reovirus infection results in cell death of a variety of cancers, but it is unknown if different reovirus types lead to triple-negative breast cancer cell death. In this study, we generated two novel reoviruses that more efficiently infect and kill triple-negative breast cancer cells. We show that infection in the presence of DNA-damaging agents enhances infection and triple-negative breast cancer cell killing by reovirus. These data suggest that a combination of a genetically engineered oncolytic reovirus and topoisomerase inhibitors may provide a potent therapeutic option for patients afflicted with triple-negative breast cancer.
KEYWORDS: DNA damage, anticancer therapy, breast cancer, cancer, high-throughput screening, innate immunity, oncolytic viruses, oncolytics, reovirus, topoisomerase inhibitors
ABSTRACT
Breast cancer is the second leading cause of cancer-related deaths in women in the United States. Triple-negative breast cancer constitutes a subset of breast cancer that is associated with higher rates of relapse, decreased survival, and limited therapeutic options for patients afflicted with this type of breast cancer. Mammalian orthoreovirus (reovirus) selectively infects and kills transformed cells, and a serotype 3 reovirus is in clinical trials to assess its efficacy as an oncolytic agent against several cancers. It is unclear if reovirus serotypes differentially infect and kill triple-negative breast cancer cells and if reovirus-induced cytotoxicity of breast cancer cells can be enhanced by modulating the activity of host molecules and pathways. Here, we generated reassortant reoviruses by forward genetics with enhanced infective and cytotoxic properties in triple-negative breast cancer cells. From a high-throughput screen of small-molecule inhibitors, we identified topoisomerase inhibitors as a class of drugs that enhance reovirus infectivity and cytotoxicity of triple-negative breast cancer cells. Treatment of triple-negative breast cancer cells with topoisomerase inhibitors activates DNA damage response pathways, and reovirus infection induces robust production of type III, but not type I, interferon (IFN). Although type I and type III IFNs can activate STAT1 and STAT2, triple-negative breast cancer cellular proliferation is only negatively affected by type I IFN. Together, these data show that reassortant viruses with a novel genetic composition generated by forward genetics in combination with topoisomerase inhibitors more efficiently infect and kill triple-negative breast cancer cells.
IMPORTANCE Patients afflicted by triple-negative breast cancer have decreased survival and limited therapeutic options. Reovirus infection results in cell death of a variety of cancers, but it is unknown if different reovirus types lead to triple-negative breast cancer cell death. In this study, we generated two novel reoviruses that more efficiently infect and kill triple-negative breast cancer cells. We show that infection in the presence of DNA-damaging agents enhances infection and triple-negative breast cancer cell killing by reovirus. These data suggest that a combination of a genetically engineered oncolytic reovirus and topoisomerase inhibitors may provide a potent therapeutic option for patients afflicted with triple-negative breast cancer.
INTRODUCTION
Breast cancer is the leading cause of cancer and the second leading cause of deaths by cancer in women in the United States (https://seer.cancer.gov/). Triple-negative breast cancer (TNBC) constitutes approximately 15% of breast cancers and has a higher rate of relapse and shorter overall survival after metastasis than other subtypes of breast cancer (1). In addition, compared to other forms of breast cancer, TNBC more frequently affects the young and is more prevalent in African American women, and tumors are larger and biologically more aggressive (2). TNBC is characterized by the lack of expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2/neu) and can be classified into seven subtypes based on its genetic signature (2). Although targeted therapies against hormone receptor-positive and HER2-positive breast cancer have been efficacious, the absence of these molecules on TNBC cells has limited treatment to cytotoxic chemotherapy, radiotherapy, and surgery (3, 4). This raises a need for targeted therapeutics against this type of cancer.
The concept that viruses can promote tumor regression is nearly as old as the discovery of viruses (5). The deregulated expression of viral receptors, endocytic uptake molecules, and proteases; altered metabolic states; and impaired innate immunity make cancer cells ideally suited for virus infection and replication (6–8). In addition to directly impacting cancer cell biology, oncolytic viruses can elicit antitumor immune responses and serve as adjuvants for other cancer therapies (9–11). Several viruses are under study to assess their oncolytic properties against several cancers (6, 7). Nonfusogenic mammalian orthoreovirus (reovirus) is a nonenveloped double-stranded RNA (dsRNA) virus in the Reoviridae family. A serotype 3 reovirus (Reolysin) is in phase I and II clinical trials (ClinicalTrials.gov identifiers NCT01622543 and NCT01656538) to assess its efficacy against a variety of cancers (https://clinicaltrials.gov). Reovirus can be delivered to patients via intratumoral and intravenous administration and can be effective in combination therapy (12). Reovirus has an inherent preference to replicate in tumor cells, making it ideally suited for use in oncolytic virotherapies (13, 14). However, the cellular and viral factors that promote preferential reovirus infection of cancer cells are not fully elucidated.
Reovirus has a segmented genome with three large (L), three medium (M), and four small (S) dsRNA gene segments (15). There are three different reovirus serotypes (types 1, 2, and 3) based on the neutralization ability of antibodies raised against the σ1 attachment protein that is encoded by the S1 gene segment (16, 17). Reoviruses infect most mammals, and although humans are infected during childhood, infection seldom results in disease (16, 18–20). Reovirus induces programmed cell death in vitro and in vivo (21–28). Although both type 1 and type 3 reoviruses can induce apoptosis, type 3 reoviruses induce apoptosis and necroptosis more efficiently in most cells (16, 21, 22). Serotype-dependent differences in apoptosis induction segregate with the S1 and M2 gene segments (29–31). However, there is a limited understanding of the viral factors that determine preferential replication and killing of cancer cells.
In this study, we show that coinfection and serial passaging of parental reoviruses in TNBC cells yield reassortant viruses with enhanced oncolytic capacities compared to parental reoviruses. Reassortant reoviruses have a predominant type 1 genetic composition, with some type 3 gene segments as well as synonymous and nonsynonymous point mutations. We show that reassortant reoviruses have enhanced infective and cytotoxic capacities in TNBC cells compared to parental viruses. To further enhance the oncolytic properties of these reassortant viruses, we used a high-throughput screen of small-molecule inhibitors and identified DNA-damaging topoisomerase inhibitors as a class of drugs that reduces TNBC cell viability while enhancing reovirus infectivity. Infection of TNBC cells in the presence of topoisomerase inhibitors results in induction of DNA damage, increased levels of type III but not type I interferon (IFN), and enhanced cell killing. Although type I and type III IFNs can activate STAT1 and STAT2, triple-negative breast cancer cellular proliferation is only negatively affected by type I IFN. Together, our results show that reassortant reoviruses with a novel genetic composition have enhanced oncolytic properties and that pairing of topoisomerase inhibitors with reovirus potentiates TNBC cell killing.
(This article was submitted to an online preprint archive [32].)
RESULTS
Generation of reassortant viruses in triple-negative breast cancer cells by forward genetics.
Reovirus serotypes have distinct infective, replicative, and cell-killing properties, and the segmented nature of the reovirus genome allows the generation of viruses with novel properties through gene reassortment following coinfection (33, 34). To generate reoviruses with enhanced replicative properties in TNBC cells, MDA-MB-231 cells were coinfected with the prototype laboratory strains type 1 Lang (T1L), type 2 Jones (T2J), and type 3 Dearing (T3D) and serially passaged in these cells 10 or 20 times (Fig. 1A). Following serial passage, individual viral clones were isolated by a plaque assay, and the gene segment identity for each clone (44 clones following 10 passages and 45 clones following 20 passages) was determined by SDS-gel electrophoresis (Fig. 1B). Of the 44 isolates analyzed following 10 serial passages, 8 distinct electropherotypes were identified, with 23 isolates (52%) having the same electropherotype (r2Reovirus) (Fig. 1C). Following 20 serial passages, 6 distinct isolates were identified, including two (r9 and r10) that were not observed after passage 10 (Fig. 1D). The most predominant electropherotypes following 20 serial passages were r1Reovirus and r2Reovirus, constituting 33% and 27%, respectively, of all isolates. Illumina next-generation sequencing (NGS) revealed that r1Reovirus is composed of seven gene segments from T1L and three from T3D (L2, M2, and S2), while r2Reovirus is composed of nine gene segments from T1L and one from T3D (M2) (Fig. 2). In addition, both viruses have previously unidentified nonsynonymous point mutations that result in an Ala-to-Thr substitution at amino acid 160 in L3, an Ile-to-Val substitution at amino acid 250 in S3, and a Val-to-Ile substitution at amino acid 49 in S4. A Pro-to-Thr substitution at amino acid 161 is also found in r1Reovirus. In addition, r1Reovirus and r2Reovirus have several synonymous point mutations (see Table S1 in the supplemental material). Interestingly, the r1Reovirus S2 gene segment, but no other gene segment, has single-residue variations that range from 35% to 65%. Sanger sequencing of the S2 gene segment from 10 r1Reovirus plaque isolates showed a wide array of mutations distinct from the initial virus isolate (data not shown). These data suggest that the S2 gene segment of r1Reovirus is genetically unstable. We did not detect single-residue variations in gene segments from either parental T1L, T2J, or T3D or r2Reovirus, suggesting that this is not an intrinsic property of the S2 gene segment carried from parental viruses. Together, these data indicate that coinfection and serial passaging of reoviruses in MDA-MB-231 cells lead to the generation of reassortant reoviruses with novel genetic compositions.
FIG 1.

Generation of reoviruses by forward genetics in MDA-MB-231 cells. (A) Triple-negative breast cancer MDA-MB-231 cells were coinfected with T1L, T2J, and T3D and serially passaged 10 or 20 times. Virus isolates were obtained following a plaque assay on L929 cells and sequenced by Illumina next-generation sequencing. (B) Polyacrylamide gel electrophoresis of reovirus parental strains T1L, T2J, and T3D; r1Reovirus (r1); and r2Reovirus (r2). Strains are differentiated by migration patterns of three large (L), three medium (M), and four small (S) gene segments. (C) Percentage of viral isolates with a specific electropherotype following 10 serial passages in MDA-MB-231 cells (n = 44). r1Reovirus accounts for 11% of isolates, while r2Reovirus accounts for 52%. (D) Percentage of viral isolates with a specific electropherotype following 20 serial passages in MDA-MB-231 cells (n = 45). r1Reovirus accounts for 33% of isolates, while r2Reovirus accounts for 27%.
FIG 2.

Genetic composition of r1Reovirus and r2Reovirus. The genetic composition of parental and reassortant r1Reovirus and r2Reovirus was determined by Illumina next-generation sequencing. r1Reovirus has seven gene segments from T1L and three from T3D (L2, M2, and S2) and four nonsynonymous point mutations (L3, A160T; S3, P161T I250V; S4, V49I). r2Reovirus has nine gene segments from T1L and one from T3D (M2) and three nonsynonymous point mutations (L3, A160T; S3, I250V; S4, V49I). Both r1Reovirus and r2Reovirus have several synonymous point mutations.
Reassortant reoviruses infect MDA-MB-231 cells more efficiently than parental reoviruses.
Reovirus attaches to cells via a strength-adhesion mechanism whereby the viral attachment fiber σ1 binds to cell surface carbohydrate and the proteinaceous receptor JAM-A or NgR1 (35–39). To determine the attachment efficiencies of r1Reovirus and r2Reovirus in comparison to parental reoviruses, MDA-MB-231 cells were adsorbed with a vehicle (mock) or Alexa Fluor 633 (A633)-labeled T1L, T3D, type 3 Cashdollar (T3C$) (the reovirus strain currently in clinical trials), or reassortant reoviruses at a multiplicity of infection (MOI) of 5 × 104 particles/cell and assessed for cell surface reovirus by flow cytometry (Fig. 3A). Reassortant reoviruses attach to cells with an efficiency similar to that of T1L but less efficiently than the type 3 reoviruses T3D and T3C$. As reassortant reoviruses contain a T1L S1 gene segment, it is not surprising that they attach to cells at levels similar to those of parental T1L. These data also indicate that other genetic changes found in r1Reovirus and r2Reovirus do not impact the ability of these viruses to attach to cells.
FIG 3.
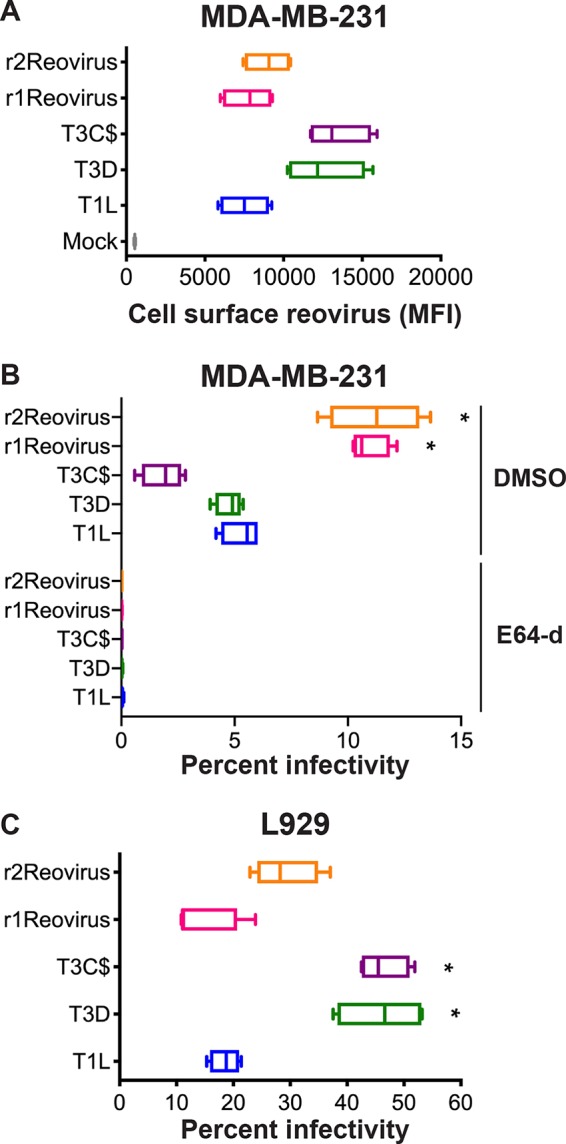
Attachment to and infection of MDA-MB-231 cells by reassortant reoviruses. (A) MDA-MB-231 cells were adsorbed with A633-labeled T1L, T3D, T3C$, or reassortant reoviruses at an MOI of 5 × 104 particles/cell and assessed for cell surface reovirus by flow cytometry. Results are expressed as box-and-whisker plots of cell surface reovirus mean fluorescence intensities (MFI) for quadruplicate independent experiments. (B) MDA-MB-231 cells were treated with DMSO or 4 μM E64-d; adsorbed with T1L, T3D, T3C$, or reassortant reoviruses at an MOI of 100 PFU/cell; and assessed for infectivity after 18 h by indirect immunofluorescence using reovirus-specific antiserum. (C) L929 cells were adsorbed with T1L, T3D, T3C$, or reassortant reoviruses at an MOI of 5 PFU/cell and assessed for infectivity after 18 h by indirect immunofluorescence using reovirus-specific antiserum. Results are expressed as box-and-whisker plots of percent infectivity for quadruplicate independent experiments. *, P < 0.0005 (in comparison to T1L, as determined by two-way ANOVA with Tukey’s multiple-comparison test).
To determine how genetic changes in r1Reovirus and r2Reovirus affect reovirus infection of TNBC cells, MDA-MB-231 cells were pretreated with dimethyl sulfoxide (DMSO) or the cysteine protease inhibitor E64-d, which blocks reovirus cell entry by preventing proteolysis during endocytic uptake (40); adsorbed with mock, T1L, T3D, T3C$, or reassortant reoviruses at an MOI of 100 PFU/cell; and assessed for infectivity after 18 h by indirect immunofluorescence using reovirus-specific antiserum (Fig. 3B). In contrast to attachment, r1Reovirus and r2Reovirus infect MDA-MB-231 cells more efficiently than parental reoviruses or T3C$, with both reassortant viruses infecting cells over 2-fold more efficiently. Infection with all viruses tested was impaired by E64-d, indicating a similar requirement for proteolytic processing during entry. These data indicate that reassortant reoviruses establish infection more efficiently in MDA-MB-231 cells than parental reoviruses and that infection of these cells requires proteasomal processing of the virion during cell entry.
To determine if the increased infectivity of the reassortant viruses is limited to MDA-MB-231 cells, the infectivity of parental and reassortant reoviruses was assessed on murine L929 fibroblasts, which are highly susceptible to reovirus infection and are used to propagate the virus (Fig. 3C). L929 cells were adsorbed with mock, T1L, T3D, T3C$, or reassortant reoviruses at an MOI of 5 PFU/cell and assessed for infectivity after 18 h by indirect immunofluorescence using reovirus-specific antiserum. In contrast to what was observed in MDA-MB-231 cells, reassortant reoviruses infect L929 cells at levels similar to those of parental T1L but less efficiently than both T3D and T3C$. These data indicate that r1Reovirus and r2Reovirus more efficiently infect TNBC cells but not L929 cells. This suggests that the genetic changes found in the reassortant viruses confer enhanced infection in the TNBC cells used for serial passage at a step after attachment.
Replication kinetics of reassortant reoviruses are similar to those of T1L but faster than those of T3D.
To determine the replication efficiency of parental and reassortant reoviruses, MDA-MB-231 cells were adsorbed with mock, T1L, T3D, T3C$, or reassortant reoviruses at an MOI of 10 PFU/cell and assessed for viral replication over a 3-day course of infection (Fig. 4). Despite the differences observed in infectivity, all viruses except T3D replicated with similar kinetics, with T3C$ having faster replication kinetics by day 1 postinfection (Fig. 4B) and reaching higher peak titers than all other viruses tested. T1L, T3C$, r1Reovirus, and r2Reovirus had similar replication kinetics at days 2 and 3 postinfection. T3D replication kinetics were slower, with lower viral yields, than those of all other viruses tested. Interestingly, although T3C$ differs from T3D by only 22 amino acids, its replication kinetics are more similar to those of T1L and the reassortant reoviruses than to those of T3D. These data indicate that although reassortant reoviruses establish infection in MDA-MB-231 cells more efficiently than parental reoviruses, replication kinetics are similar to those of T1L but significantly enhanced compared to those of T3D.
FIG 4.

Reassortant viruses replicate with kinetics similar to those of T1L and T3C$, but faster than those of T3D, in MDA-MB-231 cells. T1L, T3D, T3C$, r1Reovirus, and r2Reovirus were adsorbed at an MOI of 10 PFU/cell, and viral titers (A) and viral yields (B) were determined by a plaque assay on L929 cells at 0 to 3 days postinfection. The results are presented as mean viral titers (± standard errors of the means [SEM]) (A) or mean viral yields (±SEM) (B) compared to values at day 0 postinfection.
r1Reovirus and r2Reovirus impact TNBC cell viability with faster kinetics than parental reoviruses.
Type 3 reoviruses induce cell death more efficiently than type 1 reoviruses in vitro and in vivo, and T3C$ is currently in clinical trials to test its efficacy as an oncolytic against a variety of cancers (31, 41). To determine the efficacy of virally induced cytotoxicity in TNBC cells, MDA-MB-231 cells were adsorbed with mock, T1L, T3D, T3C$, r1Reovirus, or r2Reovirus at an MOI of 500 PFU/cell, or treated with staurosporine as a positive control, and assessed for cell viability for 7 days (Fig. 5A). Compared to mock-infected cells, all reoviruses tested impaired cell viability, with reassortant reoviruses impairing cell viability with the fastest kinetics. In reassortant reovirus-infected cells, cell viability peaked at day 2 postinfection, reaching levels similar to those of cells treated with staurosporine by day 5 postinfection. Cell viability peaked at day 3 postinfection in T1L-, T3D-, and T3C$-infected cells, reaching staurosporine treatment levels by day 5 with T1L and day 6 with T3C$. At day 3 postinfection, cell viability was significantly impaired in reassortant reovirus-infected cells but not in cells infected with T1L, T3D, or T3C$ (Fig. 5A). Overall, the impact of reassortant viruses on cell viability was 1 day ahead of those of T1L and T3C$ and 2 to 3 days ahead of that of T3D. To determine if similar effects on cell viability could be observed in another TNBC cell line, MDA-MB-436 cells were infected with mock, T1L, T3D, T3C$, or r2Reovirus and assessed for cell viability over 6 days (Fig. 5B). Similar to what was observed in MDA-MB-231 cells, r2Reovirus induced cell death with significantly faster kinetics than either parental T1L or T3D or T3C$. At day 4 postinfection, r2Reovirus was the only virus tested to significantly impair MDA-MB-436 cell viability (Fig. 5B). These data show that reassortant viruses negatively affect cell viability of TNBC cells more efficiently than parental reoviruses and the oncolytic T3C$ strain. These data also suggest that T3D is not efficient at inducing cell death in at least a subset of TNBC cells.
FIG 5.

Impact of reovirus infection on viability of TNBC and L929 cells. MDA-MB-231 (A), MDA-MB-436 (B), and L929 (C) cells were adsorbed with T1L, T3D, T3C$, r1Reovirus, or r2Reovirus at an MOI of 500 PFU/cell or treated with 1 μM staurosporine, and cell viability was assessed at the times shown. Results are presented as MFI and SEM for four independent experiments. The bottom panels show cell viability for all cell lines in panels A to C for days 3, 4, and 1 postinfection. Error bars represent SEM. *, P < 0.01; **, P ≤ 0.001; ***, P ≤ 0.0001 (in comparison to T1L, as determined by two-way ANOVA with Tukey’s multiple-comparison test).
To determine if r1Reovirus and r2Reovirus differ from parental reoviruses in their ability to impair the viability of non-TNBC cells, L929 cells were adsorbed with mock, T1L, T3D, T3C$, r1Reovirus, or r2Reovirus at an MOI of 500 PFU/cell and assessed for cell viability over a 3-day time course (Fig. 5C). In contrast to what was observed in MDA-MB-231 cells, all reoviruses tested impaired cell viability with relatively similar kinetics, except for T3C$, which impaired L929 cell viability with significantly faster kinetics. These data indicate that reassortant viruses induce cell death with faster kinetics than parental reoviruses in TNBC cells and, to a lesser extent, in L929 cells. Given that r2Reovirus had enhanced infectivity and cytotoxicity in MDA-MB-231 cells compared to parental viruses and r1Reovirus has a genomically unstable S2 gene segment, experiments in the rest of this study were performed with r2Reovirus.
Identification of small molecules that impact reovirus infectivity of MDA-MB-231 cells.
The efficacy of reovirus as a mono-oncolytic therapeutic has been limited. Combinatorial therapeutics can enhance efficacy by targeting different pathways that lead to enhanced cancer cell death (42). To identify small-molecule inhibitors that enhance the oncolytic potential of reovirus, a high-throughput screen to assess the effect of small molecules from NIH Clinical Collections I and II (NCC) on reovirus infectivity was performed. The NCC is composed of compounds that have been through phase I to III clinical trials. To test the effects of compounds in the NCC on reovirus infectivity, MDA-MB-231 cells were pretreated with the vehicle (DMSO), 4 μM E64-d, or 10 μM NCC compounds for 1 h. r2Reovirus was added to cells at an MOI of 20 PFU/cell; incubated for 20 h postinfection in the presence of DMSO, 2 μM E64-d, or 5 μM NCC compounds; and scored for infectivity by indirect immunofluorescence using reovirus-specific antiserum (Fig. 6A and Table S2). Of the 700 compounds in the NCC, 20 increased reovirus infectivity, whereas 17 decreased infectivity (Fig. 6B). Six microtubule-inhibiting compounds impaired reovirus infectivity, corroborating a need for microtubule function in reovirus cell entry (43). The sodium ATPase pump inhibitor digoxin and two serotonin antagonists also impaired reovirus infection, corroborating a role for the sodium ATPase pump and serotonin receptors in reovirus infection (44, 45). Four topoisomerase inhibitors, doxorubicin, epirubicin, etoposide (topoisomerase II inhibitors), and topotecan (topoisomerase I inhibitor), significantly enhanced reovirus infectivity. Topoisomerase inhibitors can sensitize TNBC cells to cell death, but it is unknown how they impact reovirus-mediated cell death (46).
FIG 6.

Screening of NIH Clinical Collection small molecules for reovirus infectivity. MDA-MB-231 cells were treated with the vehicle (DMSO), 4 μM E64-d, or 10 μM compounds from the NIH Clinical Collection for 1 h and infected with r2Reovirus at an MOI of 20 PFU/cell in the presence of DMSO, 2 μM E64-d, or 5 μM compounds from the NIH Clinical Collection for 20 h. Cells were scored for infectivity by indirect immunofluorescence using reovirus-specific antisera. (A) Infectivity from average Z scores for compounds in the NIH Clinical Collection for duplicate experiments. (B) Compounds from the NIH Clinical Collection that increase (green) (top) or decrease (red) (bottom) infectivity by a Z-score of 2 or more. Data are shown as Z-scores for two independent experiments (run) and the average from both experiments.
Topoisomerase inhibitors enhance reovirus infection of MDA-MB-231 cells without altering viral replication.
To determine if topoisomerase inhibitors affect reovirus infection of TNBC cells, MDA-MB-231 cells were treated with increasing concentrations of doxorubicin, epirubicin, and topotecan for 1 h at 37°C; infected with mock or r2Reovirus at an MOI of 100 PFU/cell; and scored for infectivity by indirect immunofluorescence using reovirus-specific antiserum (Fig. 7). Reovirus infectivity increased slightly when cells were treated with 0.1 μM and more significantly when cells were treated with 1.0 μM all three drugs. Treatment of cells with 10 μM doxorubicin or epirubicin decreased infectivity compared to treatment with 1.0 μM, likely due to cellular cytotoxicity. In contrast, treatment of cells with 10 μM topotecan enhanced reovirus infectivity more than any other concentration tested. To determine if topoisomerase inhibitors affect reovirus replication in TNBC cells, MDA-MB-231 cells were treated with the vehicle (DMSO) or 1 μM doxorubicin, epirubicin, or topotecan; adsorbed with mock or r2Reovirus at an MOI of 10 PFU/cell; and assessed for replication over a 3-day time course (Fig. 8). Treatment of cells with doxorubicin or epirubicin slightly decreased viral titers by day 3 postinfection compared to DMSO. Treatment of cells with topotecan slightly affected viral titers at day 0, but replication kinetics were similar to those under all other conditions at days 1 to 3, with slightly higher viral yields at day 3. These data indicate that topoisomerase inhibitors augment reovirus infectivity in a concentration-dependent manner while not significantly altering the ability of reovirus to replicate in these cells.
FIG 7.

Topoisomerase inhibitors enhance reovirus infection of TNBC cells. MDA-MB-231 cells were treated for 1 h with the vehicle (DMSO), 8 μM E64-d, or increasing concentrations doxorubicin, epirubicin, or topotecan and infected with r2Reovirus at an MOI of 100 PFU/cell for 20 h. Cells were assessed for infectivity by indirect immunofluorescence using reovirus-specific antisera. Data are shown as percent infectivity for quadruplicate independent experiments. **, P ≤ 0.01; ***, P < 0.001; ****, P < 0.0001 (in comparison to DMSO, as determined by one-way ANOVA with Dunnett’s multiple-comparison test).
FIG 8.

Topoisomerase inhibitor drugs do not impair r2Reovirus replication in MDA-MB-231 cells. MDA-MB-231 cells were treated with the vehicle (DMSO) or 1 μM topoisomerase inhibitors, adsorbed with r2Reovirus at an MOI of 10 PFU/cell, and assessed for viral replication by a plaque assay on L929 cells at days 0 to 3 postinfection. Results are presented as mean viral titers (±SEM) (A) and mean viral yields (±SEM) (B) from day 0.
Topoisomerase inhibitors enhance reovirus-mediated cell killing of MDA-MB-231 cells.
To determine if topoisomerase inhibitors confer additive or synergistic effects on reovirus-mediated cytotoxicity, MDA-MB-231 cells were treated with the vehicle (DMSO) or increasing concentrations of doxorubicin, epirubicin, or topotecan for 1 h at 37°C; infected with r2Reovirus at an MOI of 200 PFU/cell; and assessed for cell viability over 3 days (Fig. 9). Treatment with 0.1 μM all three drugs did not significantly impact cell viability in the presence or absence of r2Reovirus. In the absence of virus, 1.0 μM doxorubicin and epirubicin impaired cell viability to levels similar to those with virus alone. The addition of reovirus moderately enhanced cytotoxicity compared to that with either agent alone. These effects can be especially observed at day 3 postinfection (Fig. 9B). Treatment with 10 μM doxorubicin or epirubicin had significant cytotoxic properties in the absence of reovirus. In contrast, 1.0 μM topotecan led to significantly diminished cell viability in the absence of reovirus, and the addition of reovirus conferred an additive effect on the cytotoxic effects of both topotecan and reovirus. A synergistic cytotoxic effect was observed when reovirus was combined with 10 μM topotecan compared to either agent alone. Together, these data indicate that the combination of topoisomerase inhibitors with reovirus, especially topotecan, enhances the cytopathic properties of drugs and virus in a TNBC cell line.
FIG 9.

Cell viability of MDA-MB-231 cells is impaired by reovirus and topoisomerase inhibitors. (A) MDA-MB-231 cells were treated with the vehicle (DMSO) or increasing concentrations of doxorubicin, epirubicin, or topotecan for 1 h; infected with r2Reovirus at an MOI of 200 PFU/cell; and assessed for cell viability at days 0 to 3 postinfection. Data are shown as MFIs for quadruplicate independent experiments. (B) Cell viability under all the conditions described above for panel A for day 3 postinfection. Error bars represent SEM. **, P < 0.01; ***, P < 0.001 (by one-way ANOVA with Tukey’s multiple-comparison test).
Activation of DNA damage repair and innate immune signaling pathways following reovirus infection with topoisomerase inhibitors.
Reovirus infection activates innate immune signaling that results in the production of interferon (IFN) (47, 48). Topoisomerase inhibitors, but not reovirus, induce DNA damage repair pathways and can induce innate immune signaling (49). To determine if reovirus infection of TNBC cells impacts DNA damage repair and innate immune pathways, MDA-MB-231 cells were treated with DMSO, doxorubicin, epirubicin, or topotecan for 1 h at 37°C and infected with mock or r2Reovirus. Whole-cell lysates were then collected at 0, 1, and 2 days postinfection (dpi) and immunoblotted for phosphorylated and total STAT1, STAT2, STAT3, ATM, and p53 (Fig. 10A). Reovirus infection in the presence of topotecan resulted in increased levels of phosphorylated STAT1 and STAT2 at day 1 postinfection (Fig. 10B). Total levels of STAT1 and STAT2 were slightly elevated in cells treated with doxorubicin, epirubicin, and topotecan compared to those in cells treated DMSO. STAT3 is constitutively activated in 40% of breast cancers and is associated with the epithelial-to-mesenchymal transition (50, 51). Phosphorylated STAT3 was detected in the absence of reovirus regardless of the presence of topoisomerase inhibitors. Infection resulted in decreased levels of phosphorylated STAT3 at 1 and 2 dpi, also independent of doxorubicin, epirubicin, or topotecan. These data indicate that reovirus infection of MDA-MB-231 cells promotes the activation of innate immune pathways and that infection in the presence of topotecan, but not doxorubicin or epirubicin, enhances the activation of both STAT1 and STAT2. Reovirus infection also dampens the activation of STAT3 independent of topoisomerase inhibitors.
FIG 10.

Reovirus activates STAT1 and STAT2 signaling, and topoisomerase inhibitors activate DNA damage response pathways. (A) MDA-MB-231 cells were treated with the vehicle (DMSO) or 2 μM doxorubicin, epirubicin, or topotecan for 1 h; infected with reovirus at an MOI of 100 PFU/cell; and incubated with DMSO or 1 μM topoisomerase inhibitors for 0 to 2 days postinfection. Whole-cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies specific for phosphorylated and total STAT1, STAT2, STAT3, ATM, p53, and GAPDH and reovirus. Residues recognized by phosphorylation-specific antibodies are shown in parentheses. (B and C) Quantitative densitometry of immunoblots from three independent experiments. All data are normalized to values for GAPDH and DMSO (mock) for each corresponding day. Error bars indicate SEM.
Reovirus infection in the absence of topoisomerase inhibitors slightly affected levels of phosphorylated and total ATM and p53, with phosphorylated ATM levels trending upward over the times tested (Fig. 10C). Treatment of cells with topoisomerase inhibitors in the absence of reovirus increased levels of phosphorylated ATM and p53 compared to those in DMSO-treated cells at all time points tested. The activation of ATM and p53 by topoisomerase inhibitors was not affected by the presence of reovirus. These data suggest that reovirus does not affect the activation of DNA damage signaling by topoisomerase inhibitors.
Reovirus infection of TNBC cells results in increased levels of type III interferon.
To assess if the increased levels of phosphorylated STAT1 and STAT2 correlate with IFN production during reovirus infection, MDA-MB-231 cells were treated with DMSO, doxorubicin, epirubicin, or topotecan for 1 h at 37°C and infected with r2Reovirus at an MOI of 100 PFU/cell, and RNA and supernatants were collected at 0, 8, 12, 24, and 48 h postinfection (Fig. 11). Reovirus mRNA levels were largely unaffected by the presence or absence of topoisomerase inhibitors up to 12 h postinfection and slightly increased with doxorubicin and epirubicin at 24 and 48 h postinfection compared to those with DMSO and topotecan (Fig. 11A), confirming that topoisomerase inhibitors do not significantly affect reovirus replication. Despite robust infection, negligible levels of IFNB1 mRNA were observed in the presence or absence topoisomerase inhibitors (Fig. 11B). In the gut epithelium, IFN-λ plays an essential role in the regulation of enteric reovirus infection (52, 53). However, little is known on the effects of IFN-λ in breast cancer proliferation or its role during reovirus infection of breast epithelial cells. In contrast to what was observed with IFNB1, significant levels of IFNL1 mRNA were observed starting at 8 h postinfection and up to 48 h postinfection in infected cells (Fig. 11C). Also, in infected cells, IFNL1 mRNA levels were higher in DMSO- and topotecan-treated cells at 8 and 12 h postinfection than in doxorubicin- and epirubicin-treated cells, with the latter peaking at 24 h postinfection. Interestingly, robust levels of IFNL1 mRNA were observed at 24 h and 48 h in uninfected cells treated with doxorubicin and epirubicin. To determine if increasing levels of IFNL1 mRNA result in increasing levels of protein, IFN-λ levels were assessed by an enzyme-linked immunosorbent assay (ELISA) (Fig. 11D). Secreted IFN-λ was detected only in infected cells, except for low levels at 48 h in uninfected cells. IFN-λ was first observed at 12 h postinfection only in epirubicin-treated cells. By 24 h postinfection, IFN-λ was observed at similar levels in cells treated with DMSO, doxorubicin, and topotecan but not epirubicin. At 48 h postinfection, high levels of IFN-λ were observed under all infection conditions, with the highest levels observed in topotecan-treated cells. These data further support that topoisomerase inhibitors do not affect overall reovirus replication kinetics and that reovirus infection of MDA-MB-231 cells results in increased levels of type III, but not type I, IFN mRNA and protein. Although topoisomerase inhibitors had a modest effect on the induction of IFNL1 mRNA following reovirus infection, the presence of topotecan had the largest effect on the levels of secreted IFN-λ.
FIG 11.

Topoisomerase inhibitors and r2Reovirus induce higher levels of IFNL1 over time than either agent alone. MDA-MB-231 cells were treated with the vehicle (DMSO) or 2 μM doxorubicin, epirubicin, or topotecan and infected with mock or r2Reovirus at an MOI of 100 PFU/cell. (A to C) RNA was isolated from cells at times shown, and qPCR was performed to assess mRNA levels of IFNB1 (A), IFNL1 (B), and reovirus S1 (C). The dashed line in panel C represents background baseline levels observed for mock. Data are shown as fold changes normalized to the value for a housekeeping gene for duplicate independent experiments. Error bars indicate SEM. (D) Levels of IFN-λ in cell supernatants, determined by an ELISA. Data are shown as picograms per milliliter of IFN-λ for duplicate independent experiments.
Type III IFNs do not affect cell viability of TNBC cells.
Infection of MDA-MB-231 cells results in the production of type III IFN. To determine if type I or type III IFNs impact cell viability of TNBC cells, MDA-MB-231 cells were treated with DMSO, increasing amounts of recombinant human IFN-λ or IFN-β, or 1 μM doxorubicin or infected with r2Reovirus at an MOI of 100 PFU/cell and assessed for cell viability over 3 days (Fig. 12A). Treatment of cells with IFN-λ did not affect cell viability. In contrast, treatment of cells with IFN-β decreased cell viability in a dose-dependent manner, with cell viability levels reaching those seen during reovirus infection with the highest dose tested. To determine if MDA-MB-231 cells can sense type I and type III IFNs, cells were left untreated or treated with increasing amounts of IFN-λ or IFN-β and assessed for the activation status of STAT1 and STAT2 after 1 h (Fig. 12B). Compared to untreated cells, phosphorylated STAT1 and STAT2 were observed following treatment with both IFN-λ and IFN-β, suggesting that MDA-MB-231 cells can respond to type I and type III IFNs. These data suggest that while infection of MDA-MB-231 cells results in robust production of type III IFN, the cytotoxic effects of reovirus infection are not directly due to antiproliferative effects of the IFN-λ produced by these TNBC cells.
FIG 12.

IFN-λ does not impact MDA-MB-231 cellular proliferation but activates STATs. (A) MDA-MB-231 cells were treated with DMSO, increasing amounts of recombinant human IFN-λ or recombinant human IFN-β, or 1 μM doxorubicin or infected with r2Reovirus at an MOI of 100 PFU/cell for 1 h and assessed for cell viability at the times shown. Data are shown as average MFIs for quadruplicate independent experiments. Error bars indicate SEM. (B) MDA-MB-31 cells were left untreated or treated with increasing amounts of recombinant human IFN-λ or IFN-β for 1h. Whole-cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies specific for phosphorylated and total STAT1, STAT2, and GAPDH. Residues recognized by phosphorylation-specific antibodies are shown in parentheses.
DISCUSSION
Reovirus has an inherent preference to replicate in tumor cells, making it ideally suited for use in oncolytic therapy (13, 14). Reovirus can be delivered to patients via intratumoral and intravenous administration and can be effective in combination therapy (12). A type 3 reovirus (T3C$) is currently in phase I and II clinical trials against a variety of cancers in combination with several drugs (ClinicalTrials.gov identifiers NCT01622543 and NCT01656538). In this study, we generated novel reassortant reoviruses with enhanced replicative properties in TNBC cells by coinfection of a TNBC cell line with prototype strains T1L, T2J, and T3D and serial passage. Reassortant reoviruses attach to cells with an efficiency similar to that of T1L, whereas type 3 reoviruses attach with enhanced efficacy. T1L uses GM2 glycans to attach to cells, whereas T3D interacts with α2,3-linked sialic acid (38, 54). A high expression level of α2,3-sialic acid in breast cancer is associated with greater metastatic potential (55), suggesting that the slight enhancement in attachment observed with type 3 reoviruses could be due to high levels of α2,3-sialic acid present on the surface of MDA-MB-231 cells.
Reassortant viruses did not have mutations in σ1, and the most predominant viruses following serial passaging all had a type 1 σ1. These data suggest that carbohydrate binding did not drive the selection of the reassortant viruses. JAM-A is expressed in normal mammary epithelial cells, and high JAM-A expression in breast cancer patients correlates with decreased survival and increased recurrence (56, 57). MDA-MB-231 cells express JAM-A (57), although relatively low JAM-A levels may be responsible for the low infectivity observed for all reoviruses tested in comparison to infection in L929 cells. These data suggest that receptor engagement is not responsible for the enhanced infectivity observed with the reassortant viruses.
During cell entry, reovirus traverses to endosomes, where cathepsin proteases cleave the outer capsid protein σ3, forming an infectious subvirion particle (ISVP) (40, 58). Both reassortants have a nonsynonymous mutation in the σ3-encoding S4 gene segment that results in a V49I substitution. This mutation has not been identified to impact reovirus disassembly kinetics, but it is possible that it could expedite viral cell entry kinetics. However, reassortant viruses were equally as sensitive to E64-d treatment as parental viruses. Although reassortant viruses infected MDA-MB-231 cells more efficiently than T1L, T3D, and T3C$, replication kinetics of the reassortant viruses were similar to those of T1L and T3C$ and faster than those of T3D. These data indicate that type 1 reoviruses replicate with enhanced kinetics compared to those of T3D but that genetic differences between T3D and T3C$ are sufficient to allow T3C$ to replicate as efficiently as type 1 viruses. These data also suggest that the enhanced cytotoxic properties of the reassortant viruses over parental viruses are not due to enhanced replication kinetics in MDA-MB-231 cells.
The reovirus L3, S2, and S3 gene segments have distinct roles in reovirus replication. The L3-encoded λ1 protein is a major inner capsid protein that has phosphohydrolase activity and participates in viral transcription (59, 60). The S2-encoded σ2 protein is essential for the assembly of viral cores (61). The S3-encoded nonstructural protein σNS is required for viral factory formation (62). The similarity in replication efficiency observed between T1L and the reassortant viruses suggests that the A160T mutation in L3, the I250V mutation in S3 (found in both reassortants), and the P161T mutation in S3 (in r1Reovirus only) do not impact overall replication efficiency. However, it is possible that point mutations in these gene segments in the reassortant viruses impact the activity of the viral proteins that result in enhanced infectivity or cytotoxicity in the context of TNBC cells. Further characterization of the point mutations found in the reassortant viruses will help elucidate their impact on viral fitness.
Of all the viruses tested in MDA-MB-231 cells, r1Reovirus and r2Reovirus impaired cell viability with the fastest kinetics, and only T3D was severely deficient in killing these cells. The poor induction of cell death by T3D may be related to its dampened replication in these cells. Differences in the induction of apoptosis by reovirus strains segregate with the M2 and S1 gene segments (31). Apoptosis is activated by fragments of the M2-encoded μ1 protein generated during reovirus cell entry (26, 30, 31, 63, 64). The μ1 protein impacts reovirus infectivity by enhancing reovirus attachment to cells (65). S1 is genetically linked to reovirus induction of apoptosis through the activities of both σ1 and σ1s, although it is unclear if the effects of σ1s on the induction of cell death are independent of its ability to regulate viral protein synthesis and induce cell cycle arrest (66, 67). We did not observe significant levels of cell cycle arrest in MDA-MB-231 cells infected with reassortant reoviruses (data not shown). It is unclear if the enhanced cytopathic properties of reassortant viruses in the context of TNBC cells map to the T3D M2 gene segment, the various nonsynonymous changes, or a combination of both.
Screening of small molecules from the NIH Clinical Collection identified 20 molecules that increase infectivity and 17 molecules that decrease infectivity in MDA-MB-231 cells. Six microtubule-inhibiting drugs, digoxin, and two serotonin antagonists affected reovirus infectivity, corroborating the role of microtubules, the sodium-potassium ATPase pump, and serotonin receptors in reovirus infection (43–45). Of the 17 molecules that enhanced infectivity, 4 are topoisomerase I (topotecan) or topoisomerase II (doxorubicin, epirubicin, and etoposide) inhibitors. Treatment of cells with topoisomerase inhibitors resulted in increased infectivity, with no effect on virus attachment (data not shown) or viral replication, except for slight increases in viral RNA at 24 and 48 h postinfection. Topoisomerase inhibitors promote DNA double-strand breaks leading to cell death (68–71). Reovirus infection does not induce DNA double-strand breaks and promotes cell death through the induction of extrinsic and intrinsic apoptosis or necroptosis (21, 22, 26, 31, 72, 73). It is possible that topoisomerase inhibitors positively affect the uptake of viral particles during cell entry that results in enhanced infectivity and that doxorubicin and epirubicin further impact a step late in the viral life cycle that results in enhanced transcription of viral RNA. It is also possible that the additive cytotoxicity observed in MDA-MB-231 cells when both reovirus and topoisomerase inhibitors are present is mediated through the activation of complementary cell death pathways.
Reovirus infection does not impair the DNA double-strand break response activated by treatment with topoisomerase inhibitors. Late during infection, in the presence of topoisomerase inhibitors, levels of phosphorylated and total p53 were lower than those in uninfected cells. It remains to be determined if the effects of reovirus infection on p53 are at the transcriptional, translational, or posttranslational level. Reovirus infection can induce higher levels of activated MDM2, which leads to p53 degradation (74). In the context of reovirus infection, it is possible that topoisomerase inhibitors promote p53 stabilization through impairing the activation of MDM2 by the virus. It is also possible that the effects on total p53 at late times postinfection are due to virus-dependent host translational shutoff. In support of this, total levels of STAT1, STAT2, STAT3, and ATM were also lower at late times of infection.
Reovirus infection of MDA-MB-231 cells resulted in robust expression of type III, but not type I, IFN mRNA and protein. Infection in the presence of topoisomerase inhibitors did not significantly affect the levels of IFNL1 mRNA. Interestingly, doxorubicin and epirubicin treatment in the absence of infection results in the induction of IFNL1 mRNA starting at 24 h, reaching levels similar to those detected in virus-infected cells by 48 h. Induction of DNA double-strand breaks by topoisomerase inhibitors can result in p53-dependent regulation of type I IFN through a STING-dependent but cGAS-independent pathway (49). MDA-MB-231 cells express STING (data not shown), suggesting that topoisomerase inhibitors could be inducing the transcription of type III IFN downstream of the induction of the DNA damage response through a similar mechanism. However, topoisomerase inhibitors did not induce type I IFN transcription in the presence or absence of reovirus.
Levels of IFN-λ were first observed at 12 to 24 h postinfection in the presence or absence of topoisomerase inhibitors, with the highest levels of IFN-λ detected at 48 h postinfection in the presence of topotecan. IFN-λ1, IFN-λ2, and IFN-λ3 are expressed in breast cancer cells, although their role in mediating innate immunity in these cells is not well characterized (75). Type I and type III IFNs are transcriptionally regulated by the transcription factor IFN regulatory factor 3 (IRF3) (76, 77). Reovirus can antagonize IFN production by sequestering IRF3 to viral inclusions (78), and infection of gut epithelial cells in vitro and in vivo results in upregulated levels of IFN-λ mRNA (53, 78, 79). It is possible that in MDA-MB-231 cells, reovirus is unable to sequester IRF3 to viral inclusions, resulting in robust production of type III IFN. Reovirus infection of TNBC cells resulted in high levels of secreted IFN-λ, with over 200 pg/ml being detected at 48 h postinfection in the presence or absence of topoisomerase inhibitors. Levels of IFN-λ in the presence of topotecan at 48 h postinfection reached over 800 pg/ml, levels which were higher than those observed in dendritic cells that had been exposed to a RIG-I agonist (80). It is unclear why topotecan, but not doxorubicin or epirubicin, results in significantly higher IFN-λ levels, especially considering that IFNL1 mRNA levels were not different in infected cells in the presence of the different topoisomerase inhibitors. Under all conditions, IFNL1 mRNA was detected 4 to 16 h prior to secreted IFN-λ. This delay is likely responsible for the lack of detectable secreted IFN-λ when cells were treated with doxorubicin or epirubicin in the absence of infection. Interestingly, in the absence of reovirus, IFN-λ had no effect on MDA-MB-231 cell viability, while IFN-β decreased cell viability in a concentration-dependent manner. However, MDA-MB-231 cells responded to type I and type III IFN treatment, indicating that these cells have functional receptors to detect and activate signaling pathways downstream of ligand engagement. MDA-MB-231 cells can express low basal levels and are responsive to type I IFNs (81–83). The high levels of type III IFN detected in MDA-MB-231 cells, and the lack of type I IFN, indicate that the STAT activation observed in these cells is likely in response to the interaction of IFN-λ with its receptors.
Despite the robust induction of type III IFN in response to infection, high levels of activated STAT1 and STAT2 were detected only in the presence of topotecan. Low levels of activated STAT1 were observed in infected cells in the absence of topoisomerase inhibitors, but no STAT activation was observed in the presence of doxorubicin or epirubicin. It is possible that the low levels of activated STAT1 and STAT2 in infected MDA-MB-231 cells are a result of impaired sensing of IFN-λ due to low-level expression of the IFN-λ receptor. It is also possible that treatment of cells with topotecan may sensitize cells to IFN-λ through the upregulation of the IFN-λ receptor. Surprisingly, despite high levels of activated STAT1 and STAT2 following reovirus infection of topotecan-treated cells, reovirus infectivity and replication remained unimpaired.
In this study, we generated reoviruses with unique infective and cytotoxic properties by forward genetics following coinfection with reoviruses of three different serotypes. The novel genetic composition of the reassortant viruses could inform future studies on viral factors that promote infection and killing of cells by reovirus. Through high-throughput screening, we identified topoisomerase inhibitors as a class of drugs that enhances infection and the cytotoxic properties of reovirus in the context of TNBC. We also show that infection of a breast cancer cell line leads to the robust production of type III, but not type I, IFN. This study presents evidence for the pairing of reassortant reoviruses generated by forward genetics with topoisomerase inhibitors identified by high-throughput screening as a promising therapeutic against TNBC.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
MDA-MB-231 cells (gift from Jennifer Pietenpol, Vanderbilt University) and MDA-MB-436 cells (ATCC HTB-130) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Life Technologies) and 100 U per ml penicillin and streptomycin (Life Technologies). Spinner-adapted L929 cells (Terry Dermody, University of Pittsburgh) were grown in Joklik’s modified minimal essential medium (MEM) with 5% FBS, 2 mM l-glutamine (Life Technologies), penicillin and streptomycin, and 0.25 mg per ml amphotericin B (Life Technologies).
Working stocks of reovirus strains type 1 Lang (T1L) and type 3 Dearing (T3D) were prepared following rescue with reovirus cDNAs in BHK-T7 cells (gift from Terry Dermody, University of Pittsburgh), followed by plaque purification and passage in L929 cells (84). Reovirus type 2 Jones (T2J) is a laboratory strain, and type 3 Cashdollar (T3C$) is a distinct type 3 reovirus (85). Purified virions were prepared using second-passage L929 cell lysate stocks. Virus was purified from infected cell lysates by Vertrel XF (TMC Industries Inc.) extraction and CsCl gradient centrifugation as described previously (86). The band corresponding to the density of reovirus particles (1.36 g/cm3) was collected and dialyzed exhaustively against virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]). The reovirus particle concentration was determined from the equivalence of 1 unit of optical density at 260 nm to 2.1 × 1012 particles (87). Viral titers were determined by a plaque assay using L929 cells (88). Reovirus virions were labeled with succinimidyl ester Alexa Fluor 488 (A488) (Life Technologies) as described previously (43, 89).
Reovirus polyclonal rabbit antiserum raised against reovirus strains T1L and T3D was purified as described previously (90) and cross-adsorbed for MDA-MB-231 cells. Secondary IRDye 680 and 800 antibodies (Li-Cor Biosciences) and goat anti-rabbit A488 (Life Technologies) were used.
Serial passage of T1L, T2J, and T3D in MDA-MB-231 cells.
MDA-MB-231 cells were adsorbed with T1L, T2J, and T3D at a multiplicity of infection (MOI) of 1 PFU/cell for 1 h at room temperature and incubated for 48 h at 37°C in MDA-MB-231 cell medium. Cells were freeze-thawed three times, and fresh MDA-MB-231 cells were infected with 500 μl of the freeze-thawed cell supernatant and incubated for 48 h at 37°C. Serial passage was repeated 20 times, and individual viral titers were obtained by plaque isolation following a plaque assay in L929 cells.
Electrophoretic mobility of reovirus.
A total of 5 × 1010 particles of purified reovirus or freeze-thawed supernatants containing reovirus mixed with 2× SDS sample buffer (20% glycerol, 100 mM Tris-HCl [pH 6.8], 0.4% SDS, and 3 mg bromophenol blue) were separated by SDS-PAGE using 4-to-20% gradient polyacrylamide gels (Bio-Rad Laboratories) at 10 mA for 16 h. The gel was stained with 5 μg/ml ethidium bromide for 20 min and imaged using the ChemiDoc XRS+ system (Bio-Rad).
Next-generation sequencing of reovirus.
RNAs from viral preparations of T1L, T2J, T3D, r1Reovirus, and r2Reovirus were obtained using an RNeasy RNA purification kit (Qiagen). Ten nanograms of viral RNA was used as the input for cDNA synthesis using Clontech SMARTer stranded total RNA-Seq kit v2 (pico input, mammalian) according to the manufacturer’s instructions. Libraries were validated by capillary electrophoresis on an Agilent 4200 TapeStation, pooled, and sequenced on an Illumina HiSeq3000 instrument with 100-bp paired-end reads averaging 13 million reads/sample, yielding an average depth of coverage of >1,000 reads. Reads were trimmed of the adapter sequence using Trimmomatic (version 0.36) (http://www.usadellab.org/cms/?page=trimmomatic), using the TruSeq3-PE-2 paired-end adapter reference. Trimmed reads from each sample were aligned to all of the parental strain reference sequences using the Burrows-Wheeler Aligner (BWA) (version 0.7.10-r789) (http://bio-bwa.sourceforge.net/). Deduplication was performed with Picard tools (version 1.74[1243]) (https://broadinstitute.github.io/picard/), and variation was called, again for each sample against all the parental strain references, using the GATK pipeline’s (version 3.4) (https://software.broadinstitute.org/gatk/) HaplotypeCaller, with ploidy set to 1 and other default parameters. The resultant variant-call files (.vcf) were examined for sample similarity/variation from the parental reference strains.
Flow cytometric analysis of cell surface reovirus.
MDA-MB-231 cells were adsorbed with 5 × 103 to 5 × 104 particles per cell of A633-labeled virus for 1 h at room temperature. Cells were washed with phosphate-buffered saline (PBS), detached with Cellstripper (Cellgro) for 10 min at 37°C, quenched, and washed with PBS containing 2% FBS. Cells were fixed in 1% electron microscopy (EM)-grade paraformaldehyde (Electron Microscopy Sciences). The mean fluorescence intensity (MFI) was assessed using a CytoFLEX flow cytometer (Beckman Coulter) and quantified using FlowJo software.
Reovirus infectivity assay.
Reovirus infectivity was assessed by indirect immunofluorescence (91). MDA-MB-231 and L929 cells were adsorbed with reovirus at a range of MOIs for 1 h at room temperature, washed with PBS, and incubated in medium for 16 to 24 h at 37°C. To assess the effects of topoisomerase inhibitors on reovirus infectivity, cells were pretreated with topoisomerase inhibitors or E64-d for 1 h at 37°C, reovirus was added to cells, and the mixture was incubated for 18 to 24 h at 37°C. Cells were fixed with ice-cold methanol and stored at −20°C for at least 30 min. Methanol was removed, and cells were washed twice with PBS and blocked with PBS containing 1% bovine serum albumin (BSA) for 15 min at room temperature. Cells were stained with reovirus-specific polyclonal antiserum (1:2,000) for 1 h at room temperature, washed twice with PBS, stained with goat anti-rabbit Alexa Fluor 488 (1:1,000) for 1 h at room temperature, counterstained with 0.5 ng/ml 4′,6-diamidino-2-phenylindole (DAPI) for 5 min at room temperature, and washed twice with PBS. Immunofluorescence was detected using a Lionheart FX automated microscope (BioTek) with a 4× Plan Fluorite (PL FL) phase objective (numerical aperture [NA], 0.13), and percent infectivity was determined (reovirus-positive cells/DAPI-positive cells) using Gen5 software (BioTek).
Reovirus replication assay.
MDA-MB-231 cells were adsorbed with reovirus at an MOI of 10 PFU/cell for 1 h at room temperature, washed with PBS, and incubated for 0 to 3 days in MDA-MB-231 medium at 37°C. To determine the effects of topoisomerase inhibitors on reovirus replication, MDA-MB-231 cells were treated with the vehicle or topoisomerase inhibitors for 1 h at 37°C, medium was removed, and cells were adsorbed with reovirus at an MOI of 10 PFU/cell for 1 h at room temperature, washed with PBS, and incubated for 0 to 3 days with complete medium containing the vehicle or topoisomerase inhibitors at 37°C. Cells were freeze-thawed three times, and viral titers were determined by a plaque assay using L929 cells. Viral yields were calculated by dividing viral titers by the viral titer at day 0.
Cell viability assay.
Cell viability was assessed by measuring metabolic activity using Presto blue reagent (Invitrogen). L929, MDA-MB-231, and MDA-MB-436 cells were adsorbed with reovirus at a range of MOIs for 1 h at room temperature or treated with 1 μM staurosporine, washed with PBS, and incubated for 0 to 7 days at 37°C. To determine the effects of topoisomerase inhibitors on cell viability, cells were pretreated with increasing concentrations of topoisomerase inhibitors for 1 h at 37°C, reovirus was added to cells, and the cells were incubated in the presence of the inhibitors for 0 to 3 days. To determine the effect of recombinant IFNs on cell viability, MDA-MB-231 cells were treated with 10 to 5,000 IU/ml human IFN-β (Peprotech), 10 to 1,000 ng/ml IFN-λ (Peprotech), or 1 μM doxorubicin or infected with reovirus at an MOI of 100 PFU/cell and assessed for cell viability for 0 to 3 days. Presto blue was added at each time point for 30 min at 37°C, and fluorescence (540-nm excitation/590-nm emission) was measured with a Synergy HT plate reader (BioTek).
Screening of NIH Clinical Collection small-molecule inhibitors.
The NIH Clinical Collection was obtained from the NIH Roadmap Molecular Libraries Screening Centers Network. MDA-MB-231 cells were treated with DMSO, 4 μM E64-d, or 10 μM compounds from the NIH Clinical Collection for 1 h at 37°C. Medium (mock) or reovirus was added to cells at an MOI of 20 PFU/cell, and cells were incubated for 20 h at 37°C. Cells were fixed and scored for infectivity by indirect immunofluorescence as described above. Z-scores for each well were calculated using the formula Z-score = (a − b)/c, where a is the percent infectivity (infected cells/number of cells), b is the median percent infectivity for each ≤ and c is the standard deviation of the percent infectivity for each plate. Z-scores of ≤−2 or ≥2.0 were considered significant. Data for all compounds in the screen are provided in Table S2 in the supplemental material.
Immunoblotting for DNA damage response and innate immune molecules.
MDA-MB-231 cells were treated with DMSO or 2 μM topoisomerase inhibitors for 1 h at 37°C, infected with mock or reovirus at an MOI of 100 PFU/cell, and incubated for 0 to 2 days at 37°C. To assess the ability of IFNs to stimulate immune signaling, MDA-MB-231 cells were treated with 10 and 100 ng/ml of IFN-λ or 100 and 1,000 IU/ml IFN-β for 1 h at 37°C. Whole-cell lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% sodium dodecyl sulfate, 0.1% sodium deoxycholate) and a fresh protease inhibitor cocktail (catalog number P8340; Sigma-Aldrich), phosphatase inhibitor cocktail 2 (catalog number P5726; Sigma-Aldrich), 1 mM sodium vanadate, and 1 mM phenylmethylsulfonyl fluoride (PMSF), and the protein concentration was determined using the DC protein assay (Bio-Rad). Whole-cell lysates were resolved by SDS-PAGE in 4-to-20% gradient Mini-Protean TGX gels (Bio-Rad) and transferred to 0.2-μm-pore-size nitrocellulose membranes (Bio-Rad). Membranes were incubated for 1 h in blocking buffer (Tris-buffered saline [TBS] with 5% powdered milk) and then incubated with primary antibodies specific for phospho-STAT1 (Y701) (clone D4A7, catalog number 7649), phospho-STAT2 (Y690) (clone D3P2P, catalog number 88410), phospho-STAT3 (Y705) (clone D3A7, catalog number 9145), phospho-ATM (S1981) (clone 10H11.E12, catalog number 4526), phospho-p53 (clone S15, catalog number 9284), total STAT1 (clone D3A7, catalog number 9145), total STAT2 (clone D9J7L, catalog number 72604), total STAT3 (clone 124H6, catalog number 9139), total ATM (clone D2E2, catalog number 2873), total p53 (clone 1C12, catalog number 2524), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (clone GA1R, catalog number MA5-15738) and reovirus polyclonal antiserum overnight at 4°C. Antibodies were obtained from Cell Signaling Technology, except for GAPDH, which was obtained from Thermo Fisher. Membranes were washed with TBS-T (TBS with 0.1% Tween 20) and incubated with secondary antibodies conjugated to IRDye 680 or IRDye 800. Membranes were imaged using the LiCor Odyssey CLx system and processed in ImageStudio (Li-Cor Biosciences).
qPCR assessment of type I and III interferon transcript levels.
MDA-MB-231 cells were treated with DMSO or 2 μM topoisomerase inhibitors for 1 h at 37°C, infected with mock or r2Reovirus at an MOI of 100 PFU/cell, and incubated for 0, 8, 12, 24, and 48 h. RNA was isolated using a Qiagen RNeasy kit with on-column DNase digestion. cDNAs were generated using 500 ng of RNA and random primers with a high-capacity cDNA reverse transcription kit (Thermo Fisher) in a SimpliAmp thermal cycler (Thermo Fisher). cDNA was diluted 1:5 in nuclease-free water, and quantitative PCRs (qPCRs) were performed in MicroAmp fast optical 96-well reaction plates (Applied Biosystems) using PrimeTime qPCR assays (IDT) for IFNB1, IFNL1, and HPRT1 and a custom assay for the reovirus S1 gene segment (probe, 5′-56-FAM-TCAATGCTG-ZEN-TCGAACCACGAGTTGA-3IABkFQ-3′; primer 1, 5′-CGAGTCAGGTCACGCAATTA-3′; primer 2, 5′-GGATGTTCGTCCAGTGAGATTAG-3′), using a 7500 fast real-time PCR system (Applied Biosystems) and accompanying software to analyze qPCR data.
IFN-λ ELISA.
MDA-MB-231 cells were treated with DMSO or 2 μM topoisomerase inhibitors for 1 h at 37°C, infected with mock or r2Reovirus at an MOI of 100 PFU/cell, and incubated for 0, 8, 12, 24, and 48 h. Cell supernatants were collected, and levels of IFN-λ were determined with the IFN-lambda 1/3 DuoSet ELISA kit (R&D Systems). Plates were read on a Synergy HT plate reader (BioTek) using 450 nm for sample detection and 540 nm for wavelength correction.
Statistical analysis.
Mean values for quadruplicate experiments were compared using one- or two-way analysis of variance (ANOVA) with Dunnett’s or Tukey’s multiple-comparison test (GraphPad Prism). P values of <0.05 were considered statistically significant.
Data availability.
Individual mutations identified in reassortant viruses are listed in Table S1. The read files for this study have been deposited with the NCBI Sequence Read Archive (SRA) and are available under accession number PRJNA561538.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from Children’s Healthcare of Atlanta and the Pediatric Research Institute and Winship Comprehensive Cancer Institute (IRG-14-188-01) from the American Cancer Society (B.A.M.). Flow cytometry experiments were performed in the Emory Pediatrics Flow Cytometry Core (UL1TR002378). Imaging was performed at the Emory Integrated Cellular Imaging Core (2P30CA138292-04 and the Emory Pediatrics Institute). The Yerkes NHP Genomics Core is supported in part by NIH grant P51 OD011132. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01411-19.
REFERENCES
- 1.Abramson VG, Lehmann BD, Ballinger TJ, Pietenpol JA. 2015. Subtyping of triple-negative breast cancer: implications for therapy. Cancer 121:8–16. doi: 10.1002/cncr.28914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. 2011. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wahba HA, El-Hadaad HA. 2015. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med 12:106–116. doi: 10.7497/j.issn.2095-3941.2015.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeichner SB, Terawaki H, Gogineni K. 2016. A review of systemic treatment in metastatic triple-negative breast cancer. Breast Cancer (Auckl) 10:25–36. doi: 10.4137/BCBCR.S32783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dock G. 1904. The influence of complicating diseases upon leukemia. Am J Med Sci 127:563–592. doi: 10.1097/00000441-190404000-00001. [DOI] [Google Scholar]
- 6.Cattaneo R, Miest T, Shashkova EV, Barry MA. 2008. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol 6:529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell J, McFadden G. 2014. Viruses for tumor therapy. Cell Host Microbe 15:260–265. doi: 10.1016/j.chom.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips MB, Stuart JD, Rodriguez Stewart RM, Berry JT, Mainou BA, Boehme KW. 2018. Current understanding of reovirus oncolysis mechanisms. Oncolytic Virother 7:53–63. doi: 10.2147/OV.S143808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, Bigner DD, Gromeier M, Nair SK. 2017. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci Transl Med 9:eaan4220. doi: 10.1126/scitranslmed.aan4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Sherbiny YM, Holmes TD, Wetherill LF, Black EV, Wilson EB, Phillips SL, Scott GB, Adair RA, Dave R, Scott KJ, Morgan RS, Coffey M, Toogood GJ, Melcher AA, Cook GP. 2015. Controlled infection with a therapeutic virus defines the activation kinetics of human natural killer cells in vivo. Clin Exp Immunol 180:98–107. doi: 10.1111/cei.12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelly KR, Espitia CM, Zhao W, Wu K, Visconte V, Anwer F, Calton CM, Carew JS, Nawrocki ST. 2018. Oncolytic reovirus sensitizes multiple myeloma cells to anti-PD-L1 therapy. Leukemia 32:230–233. doi: 10.1038/leu.2017.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajani K, Parrish C, Kottke T, Thompson J, Zaidi S, Ilett L, Shim KG, Diaz RM, Pandha H, Harrington K, Coffey M, Melcher A, Vile R. 2016. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol Ther 24:166–174. doi: 10.1038/mt.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duncan MR, Stanish SM, Cox DC. 1978. Differential sensitivity of normal and transformed human cells to reovirus infection. J Virol 28:444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kemp V, Hoeben RC, van den Wollenberg DJ. 2015. Exploring reovirus plasticity for improving its use as oncolytic virus. Viruses 8:E4. doi: 10.3390/v8010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shatkin AJ, Sipe JD, Loh P. 1968. Separation of ten reovirus genome segments by polyacrylamide gel electrophoresis. J Virol 2:986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dermody TS, Parker J, Sherry B. 2013. Orthoreoviruses, p 1304–1346. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 17.Sabin AB. 1959. Reoviruses. A new group of respiratory and enteric viruses formerly classified as ECHO type 10 is described. Science 130:1387–1389. doi: 10.1126/science.130.3386.1387. [DOI] [PubMed] [Google Scholar]
- 18.Ouattara LA, Barin F, Barthez MA, Bonnaud B, Roingeard P, Goudeau A, Castelnau P, Vernet G, Paranhos-Baccalà G, Komurian-Pradel F. 2011. Novel human reovirus isolated from children with acute necrotizing encephalopathy. Emerg Infect Dis 17:1436–1444. doi: 10.3201/eid1708.101528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tai JH, Williams JV, Edwards KM, Wright PF, Crowe JE Jr, Dermody TS. 2005. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J Infect Dis 191:1221–1224. doi: 10.1086/428911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouziat R, Hinterleitner R, Brown JJ, Stencel-Baerenwald JE, Ikizler M, Mayassi T, Meisel M, Kim SM, Discepolo V, Pruijssers AJ, Ernest JD, Iskarpatyoti JA, Costes LM, Lawrence I, Palanski BA, Varma M, Zurenski MA, Khomandiak S, McAllister N, Aravamudhan P, Boehme KW, Hu F, Samsom JN, Reinecker HC, Kupfer SS, Guandalini S, Semrad CE, Abadie V, Khosla C, Barreiro LB, Xavier RJ, Ng A, Dermody TS, Jabri B. 2017. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 356:44–50. doi: 10.1126/science.aah5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berger AK, Danthi P. 2013. Reovirus activates a caspase-independent cell death pathway. mBio 4:e00178-13. doi: 10.1128/mBio.00178-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berger AK, Hiller BE, Thete D, Snyder AJ, Perez E Jr, Upton JW, Danthi P. 2017. Viral RNA at two stages of reovirus infection is required for the induction of necroptosis. J Virol 91:e02404-16. doi: 10.1128/JVI.02404-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown JJ, Short SP, Stencel-Baerenwald J, Urbanek K, Pruijssers AJ, McAllister N, Ikizler M, Taylor G, Aravamudhan P, Khomandiak S, Jabri B, Williams CS, Dermody TS. 2018. Reovirus-induced apoptosis in the intestine limits establishment of enteric infection. J Virol 92:e02062-17. doi: 10.1128/JVI.02062-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarke P, Beckham JD, Leser JS, Hoyt CC, Tyler KL. 2009. Fas-mediated apoptotic signaling in the mouse brain following reovirus infection. J Virol 83:6161–6170. doi: 10.1128/JVI.02488-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clarke P, Meintzer SM, Spalding AC, Johnson GL, Tyler KL. 2001. Caspase 8-dependent sensitization of cancer cells to TRAIL-induced apoptosis following reovirus-infection. Oncogene 20:6910–6919. doi: 10.1038/sj.onc.1204842. [DOI] [PubMed] [Google Scholar]
- 26.Danthi P, Coffey CM, Parker JS, Abel TW, Dermody TS. 2008. Independent regulation of reovirus membrane penetration and apoptosis by the mu1 phi domain. PLoS Pathog 4:e1000248. doi: 10.1371/journal.ppat.1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pruijssers AJ, Hengel H, Abel TW, Dermody TS. 2013. Apoptosis induction influences reovirus replication and virulence in newborn mice. J Virol 87:12980–12989. doi: 10.1128/JVI.01931-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon EJ, Howells MA, Stuart JD, Boehme KW. 2017. Serotype-specific killing of large cell carcinoma cells by reovirus. Viruses 9:E140. doi: 10.3390/v9060140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connolly JL, Barton ES, Dermody TS. 2001. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J Virol 75:4029–4039. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danthi P, Hansberger MW, Campbell JA, Forrest JC, Dermody TS. 2006. JAM-A-independent, antibody-mediated uptake of reovirus into cells leads to apoptosis. J Virol 80:1261–1270. doi: 10.1128/JVI.80.3.1261-1270.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tyler KL, Squier MK, Rodgers SE, Schneider BE, Oberhaus SM, Grdina TA, Cohen JJ, Dermody TS. 1995. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J Virol 69:6972–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodríguez Stewart RM, Berry JTL, Berger AK, Yoon SB, Hirsch AL, Guberman JA, Patel NB, Tharp GK, Bosinger SE, Mainou BA. 2019. Enhanced killing of triple-negative breast cancer cells by reassortant reovirus and topoisomerase inhibitors. bioRxiv doi: 10.1101/644815. [DOI] [PMC free article] [PubMed]
- 33.Nibert ML, Margraf RL, Coombs KM. 1996. Nonrandom segregation of parental alleles in reovirus reassortants. J Virol 70:7295–7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wenske EA, Chanock SJ, Krata L, Fields BN. 1985. Genetic reassortment of mammalian reoviruses in mice. J Virol 56:613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 36.Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. 2008. Structure of reovirus sigma1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog 4:e1000235. doi: 10.1371/journal.ppat.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konopka-Anstadt JL, Mainou BA, Sutherland DM, Sekine Y, Strittmatter SM, Dermody TS. 2014. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell Host Microbe 15:681–691. doi: 10.1016/j.chom.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reiss K, Stencel JE, Liu Y, Blaum BS, Reiter DM, Feizi T, Dermody TS, Stehle T. 2012. The GM2 glycan serves as a functional coreceptor for serotype 1 reovirus. PLoS Pathog 8:e1003078. doi: 10.1371/journal.ppat.1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. 2011. Crystal structure of reovirus attachment protein sigma1 in complex with sialylated oligosaccharides. PLoS Pathog 7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baer GS, Dermody TS. 1997. Mutations in reovirus outer-capsid protein sigma3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J Virol 71:4921–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oberhaus SM, Smith RL, Clayton GH, Dermody TS, Tyler KL. 1997. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J Virol 71:2100–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. 2012. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 149:780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mainou BA, Zamora PF, Ashbrook AW, Dorset DC, Kim KS, Dermody TS. 2013. Reovirus cell entry requires functional microtubules. mBio 4:e00405-13. doi: 10.1128/mBio.00405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashbrook AW, Lentscher AJ, Zamora PF, Silva LA, May NA, Bauer JA, Morrison TE, Dermody TS. 2016. Antagonism of the sodium-potassium ATPase impairs chikungunya virus infection. mBio 7:e00693-16. doi: 10.1128/mBio.00693-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mainou BA, Ashbrook AW, Smith EC, Dorset DC, Denison MR, Dermody TS. 2015. Serotonin receptor agonist 5-nonyloxytryptamine alters the kinetics of reovirus cell entry. J Virol 89:8701–8712. doi: 10.1128/JVI.00739-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das S, Tripathi N, Siddharth S, Nayak A, Nayak D, Sethy C, Bharatam PV, Kundu CN. 2017. Etoposide and doxorubicin enhance the sensitivity of triple negative breast cancers through modulation of TRAIL-DR5 axis. Apoptosis 22:1205–1224. doi: 10.1007/s10495-017-1400-4. [DOI] [PubMed] [Google Scholar]
- 47.Sherry B, Torres J, Blum MA. 1998. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol 72:1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stuart JD, Holm GH, Boehme KW. 2018. Differential delivery of genomic double-stranded RNA causes reovirus strain-specific differences in interferon regulatory factor 3 activation. J Virol 92:e01947-17. doi: 10.1128/JVI.01947-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, Jakobsen MR, Nevels MM, Bowie AG, Unterholzner L. 2018. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-kappaB signaling after nuclear DNA damage. Mol Cell 71:745–760.e1–e5. doi: 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. 2005. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res 65:5054–5062. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- 51.Banerjee K, Resat H. 2016. Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer 138:2570–2578. doi: 10.1002/ijc.29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peterson ST, Kennedy EA, Brigleb PH, Taylor GM, Urbanek K, Bricker TL, Lee S, Shin H, Dermody TS, Boon ACM, Baldridge MT. 28 August 2019. Disruption of type III interferon genes Ifnl2 and Ifnl3 recapitulates loss of the type III IFN receptor in the mucosal antiviral response. J Virol doi: 10.1128/JVI.01073-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, Nice TJ, Virgin HW. 2017. Expression of Ifnlr1 on intestinal epithelial cells is critical to the antiviral effects of interferon lambda against norovirus and reovirus. J Virol 91:e02079-16. doi: 10.1128/JVI.02079-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 55.Cui H, Lin Y, Yue L, Zhao X, Liu J. 2011. Differential expression of the alpha2,3-sialic acid residues in breast cancer is associated with metastatic potential. Oncol Rep 25:1365–1371. doi: 10.3892/or.2011.1192. [DOI] [PubMed] [Google Scholar]
- 56.McSherry EA, McGee SF, Jirstrom K, Doyle EM, Brennan DJ, Landberg G, Dervan PA, Hopkins AM, Gallagher WM. 2009. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int J Cancer 125:1343–1351. doi: 10.1002/ijc.24498. [DOI] [PubMed] [Google Scholar]
- 57.Naik MU, Naik TU, Suckow AT, Duncan MK, Naik UP. 2008. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res 68:2194–2203. doi: 10.1158/0008-5472.CAN-07-3057. [DOI] [PubMed] [Google Scholar]
- 58.Ebert DH, Deussing J, Peters C, Dermody TS. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem 277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- 59.Noble S, Nibert ML. 1997. Characterization of an ATPase activity in reovirus cores and its genetic association with core-shell protein lambda1. J Virol 71:2182–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Drayna D, Fields BN. 1982. Activation and characterization of the reovirus transcriptase: genetic analysis. J Virol 41:110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coombs KM, Mak SC, Petrycky-Cox LD. 1994. Studies of the major reovirus core protein sigma 2: reversion of the assembly-defective mutant tsC447 is an intragenic process and involves back mutation of Asp-383 to Asn. J Virol 68:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Becker MM, Goral MI, Hazelton PR, Baer GS, Rodgers SE, Brown EG, Coombs KM, Dermody TS. 2001. Reovirus sigmaNS protein is required for nucleation of viral assembly complexes and formation of viral inclusions. J Virol 75:1459–1475. doi: 10.1128/JVI.75.3.1459-1475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tyler KL, Squier MK, Brown AL, Pike B, Willis D, Oberhaus SM, Dermody TS, Cohen JJ. 1996. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J Virol 70:7984–7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodgers SE, Barton ES, Oberhaus SM, Pike B, Gibson CA, Tyler KL, Dermody TS. 1997. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J Virol 71:2540–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thete D, Snyder AJ, Mainou BA, Danthi P. 2016. Reovirus mu1 protein affects infectivity by altering virus-receptor interactions. J Virol 90:10951–10962. doi: 10.1128/JVI.01843-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boehme KW, Hammer K, Tollefson WC, Konopka-Anstadt JL, Kobayashi T, Dermody TS. 2013. Nonstructural protein sigma1s mediates reovirus-induced cell cycle arrest and apoptosis. J Virol 87:12967–12979. doi: 10.1128/JVI.02080-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Phillips MB, Stuart JD, Simon EJ, Boehme KW. 2018. Nonstructural protein sigma1s is required for optimal reovirus protein expression. J Virol 92:e02259-17. doi: 10.1128/JVI.02259-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arcamone F, Penco S, Vigevani A, Redaelli S, Franchi G, DiMarco A, Casazza AM, Dasdia T, Formelli F, Necco A, Soranzo C. 1975. Synthesis and antitumor properties of new glycosides of daunomycinone and adriamycinone. J Med Chem 18:703–707. doi: 10.1021/jm00241a013. [DOI] [PubMed] [Google Scholar]
- 69.Burris HA III, Hanauske AR, Johnson RK, Marshall MH, Kuhn JG, Hilsenbeck SG, Von Hoff DD. 1992. Activity of topotecan, a new topoisomerase I inhibitor, against human tumor colony-forming units in vitro. J Natl Cancer Inst 84:1816–1820. doi: 10.1093/jnci/84.23.1816. [DOI] [PubMed] [Google Scholar]
- 70.Hsiang YH, Hertzberg R, Hecht S, Liu LF. 1985. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260:14873–14878. [PubMed] [Google Scholar]
- 71.Ross WE, Bradley MO. 1981. DNA double-stranded breaks in mammalian cells after exposure to intercalating agents. Biochim Biophys Acta 654:129–134. doi: 10.1016/0005-2787(81)90145-3. [DOI] [PubMed] [Google Scholar]
- 72.Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. 2008. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol 82:161–172. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Danthi P, Pruijssers AJ, Berger AK, Holm GH, Zinkel SS, Dermody TS. 2010. Bid regulates the pathogenesis of neurotropic reovirus. PLoS Pathog 6:e1000980. doi: 10.1371/journal.ppat.1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang X, Wu H, Liu C, Tian J, Qu L. 2015. PI3K/Akt/p53 pathway inhibits reovirus infection. Infect Genet Evol 34:415–422. doi: 10.1016/j.meegid.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang L, Wei J, He S. 2010. Integrative genomic analyses on interferon-lambdas and their roles in cancer prediction. Int J Mol Med 25:299–304. [PubMed] [Google Scholar]
- 76.Onoguchi K, Yoneyama M, Takemura A, Akira S, Taniguchi T, Namiki H, Fujita T. 2007. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem 282:7576–7581. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- 77.Schafer SL, Lin R, Moore PA, Hiscott J, Pitha PM. 1998. Regulation of type I interferon gene expression by interferon regulatory factor-3. J Biol Chem 273:2714–2720. doi: 10.1074/jbc.273.5.2714. [DOI] [PubMed] [Google Scholar]
- 78.Stanifer ML, Kischnick C, Rippert A, Albrecht D, Boulant S. 2017. Reovirus inhibits interferon production by sequestering IRF3 into viral factories. Sci Rep 7:10873. doi: 10.1038/s41598-017-11469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pervolaraki K, Rastgou Talemi S, Albrecht D, Bormann F, Bamford C, Mendoza JL, Garcia KC, McLauchlan J, Hofer T, Stanifer ML, Boulant S. 2018. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog 14:e1007420. doi: 10.1371/journal.ppat.1007420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bowen JR, Quicke KM, Maddur MS, O’Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, Suthar MS. 2017. Zika virus antagonizes type I interferon responses during infection of human dendritic cells. PLoS Pathog 13:e1006164. doi: 10.1371/journal.ppat.1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang JX, Liu J, Wang J, He C, Li FP. 2005. The extract of huanglian, a medicinal herb, induces cell growth arrest and apoptosis by upregulation of interferon-beta and TNF-alpha in human breast cancer cells. Carcinogenesis 26:1934–1939. doi: 10.1093/carcin/bgi154. [DOI] [PubMed] [Google Scholar]
- 82.Lindner DJ, Kolla V, Kalvakolanu DV, Borden EC. 1997. Tamoxifen enhances interferon-regulated gene expression in breast cancer cells. Mol Cell Biochem 167:169–177. doi: 10.1023/a:1006854110122. [DOI] [PubMed] [Google Scholar]
- 83.Yoon N, Park MS, Shigemoto T, Peltier G, Lee RH. 2016. Activated human mesenchymal stem/stromal cells suppress metastatic features of MDA-MB-231 cells by secreting IFN-beta. Cell Death Dis 7:e2191. doi: 10.1038/cddis.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kobayashi T, Antar AA, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cashdollar LW, Chmelo R, Esparza J, Hudson GR, Joklik WK. 1984. Molecular cloning of the complete genome of reovirus serotype 3. Virology 133:191–196. doi: 10.1016/0042-6822(84)90438-0. [DOI] [PubMed] [Google Scholar]
- 86.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 88.Virgin HW, Dermody TS, Tyler KL. 1998. Cellular and humoral immunity to reovirus infection. Curr Top Microbiol Immunol 233:147–161. doi: 10.1007/978-3-642-72095-6_8. [DOI] [PubMed] [Google Scholar]
- 89.Mainou BA, Dermody TS. 2012. Transport to late endosomes is required for efficient reovirus infection. J Virol 86:8346–8358. doi: 10.1128/JVI.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Virgin HW IV, Bassel-Duby R, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol 62:4594–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Berger AK, Yi H, Kearns DB, Mainou BA. 2017. Bacteria and bacterial envelope components enhance mammalian reovirus thermostability. PLoS Pathog 13:e1006768. doi: 10.1371/journal.ppat.1006768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual mutations identified in reassortant viruses are listed in Table S1. The read files for this study have been deposited with the NCBI Sequence Read Archive (SRA) and are available under accession number PRJNA561538.