

Genetic reassortment of influenza virus plays a key role in virus evolution and the emergence of pandemic strains. The reassortment occurs extensively within either FluA or FluB viruses but never between them. Here, we bioinformatically compared available promoter sequences of FluA and FluB viruses and confirmed the presence of the type-specific promoter elements. Our in vivo and in vitro mutagenesis studies showed that a type-specific promoter element—the nucleotide at position 5 in the 3′ end of vRNA promoters—plays key roles in modulating polymerase activity. Interestingly, FluA and FluB viruses showed different tolerances upon key promoter element swapping in the context of virus infections. We concluded that the nucleotide at position 5 in the 3′ end of the vRNA promoters of FluA and FluB viruses is a critical type-specific determinant. This work has implications for further elucidating the mechanisms of the intertypic exclusion of reassortment between FluA and FluB viruses.
KEYWORDS: FluA and FluB viruses, type-specific, promoter element, polymerase
ABSTRACT
Type A and type B influenza viruses (FluA and FluB viruses) are two major human pathogens that share common structural and functional features. FluA and FluB viruses can reassort within each type but never between the types. Here, we bioinformatically analyzed all promoter sequences of FluA and FluB viruses and confirmed the presence of the type-specific promoter elements. We then studied the promoter elements with cell-based in vivo assays and an in vitro replication initiation assay. Our results identified, for the first time, a type-specific promoter element—the nucleotide at position 5 in the 3′ end of the viral RNA (vRNA)—that plays a key role(s) in modulating polymerase activity in a type-specific manner. Interestingly, swapping the promoter element between FluA and FluB recombinant viruses showed different tolerances: the replacement of FluA virus-specific U5 with FluB virus-specific C5 in influenza virus A/WSN/33 (H1N1) could be reverted to U5 after 2 to 3 passages, while the replacement of FluB virus-specific C5 with FluA virus-specific U5 in influenza virus B/Yamagata/88 could be maintained, but with significantly reduced replication efficiency. Therefore, our findings indicate that the nucleotide variation at position 5 in the 3′ end of the vRNA promoter between FluA and FluB viruses contributes to their RNP incompatibility, which may shed new light on the mechanisms of intertypic exclusion of reassortment between FluA and FluB viruses.
IMPORTANCE Genetic reassortment of influenza virus plays a key role in virus evolution and the emergence of pandemic strains. The reassortment occurs extensively within either FluA or FluB viruses but never between them. Here, we bioinformatically compared available promoter sequences of FluA and FluB viruses and confirmed the presence of the type-specific promoter elements. Our in vivo and in vitro mutagenesis studies showed that a type-specific promoter element—the nucleotide at position 5 in the 3′ end of vRNA promoters—plays key roles in modulating polymerase activity. Interestingly, FluA and FluB viruses showed different tolerances upon key promoter element swapping in the context of virus infections. We concluded that the nucleotide at position 5 in the 3′ end of the vRNA promoters of FluA and FluB viruses is a critical type-specific determinant. This work has implications for further elucidating the mechanisms of the intertypic exclusion of reassortment between FluA and FluB viruses.
INTRODUCTION
Influenza virus belongs to the family Orthomyxoviridae, which is characterized by a segmented, negative-sense, single-stranded RNA genome. Influenza viruses are classified into types A, B, C, and D by antigenic differences in the nucleoprotein (NP) and matrix protein 1 (M1) (1). Among these types, only the influenza A (FluA) and influenza B (FluB) viruses can cause human seasonal epidemics (2, 3). Both FluA and FluB viruses can lead to severe infections in the human upper respiratory tract and even trigger significant morbidity and mortality; thus, they are considered a major public health concern (1). The FluA viruses are further classified, based on their viral surface proteins (hemagglutinin [HA] and neuraminidase [NA]), into at least 16 HA subtypes and 9 NA subtypes, while the FluB viruses are less genetically diverse and have only 1 type. The FluB viruses mutate at a rate 2 to 3 times lower than that of the FluA viruses and are predominantly restricted to humans, whereas the FluA viruses infect a huge variety of animals as well as humans and are also responsible for human pandemics (3, 4).
FluA and FluB viruses share many common features, especially their similar genome compositions and the homologous proteins encoded (1). The genomes of both FluA and FluB viruses contain eight RNA segments, designated PB2, PB1, PA, HA, NP, NA, M, and NS. Each RNA segment of FluA or FluB viruses is encapsidated into a rod-shaped ribonucleoprotein (RNP) complex. The eight RNPs are selectively packaged into progeny virions (5, 6). The packaging signals of each RNA segment have been identified for the FluA H1N1 virus (WSN or PR8); they are composed of the noncoding regions (NCRs) and the terminal coding regions (6, 7). FluA and FluB viruses have the ability to reassort intratypically but not intertypically (1). The molecular mechanisms underlying this phenomenon have not been clearly characterized. However, mounting evidence suggests that the inability to reassort between the two types is due mainly to inefficient formation of intertypic polymerase complex and incompatible packaging signals (8, 9).
Each RNP of FluA and FluB viruses comprises a heterotrimeric RNA-dependent RNA polymerase (RdRp) (containing PA, PB1, and PB2) and a viral RNA segment that intertwines onto multiple copies of the viral NP (1). The RNP complexes are the minimal components required for viral RNA (vRNA) transcription (vRNA → mRNA) and replication (vRNA ↔ cRNA) to occur in the nuclei of infected cells (1). The initiation of vRNA → mRNA synthesis is primer dependent, involving a cap-snatching process, whereas the initiation of vRNA ↔ cRNA syntheses is primer independent, and the mechanisms for the initiation of vRNA synthesis and cRNA synthesis differ (1, 10). The NCRs of each RNA segment of FluA and FluB viruses are composed of the highly conserved promoter regions and the segment-specific noncoding regions (ssNCRs) at both the 3′ and 5′ ends (1). The nucleotides in the promoter regions play essential roles in initiating viral RNA transcription and replication (1). The vRNPs use terminal initiation on their vRNA promoters, while the cRNPs use internal dinucleotide initiation and realignment on their cRNA promoters (10–12).
Although the lengths and sequences of the ssNCRs differ significantly among FluA and FluB virus RNAs, which can be attributed to type specificity, the sequences of the promoter regions are very similar (13, 14). The nucleotides in the promoter region are highly conserved across all viral strains and genomic segments among FluA and FluB viruses (1, 15, 16). In FluA viruses, there are 12 and 13 highly conserved nucleotides at the 3′ and 5′ termini, respectively, with the exception of a natural variation at position 4 (C4 or U4 promoter) in the 3′ end (1, 17, 18). In FluB viruses, there are only 9 and 10 highly conserved nucleotides at the 3′ and 5′ termini, respectively, except for the nucleotide at position 6 (A/U) in the 5′ end, which differs in a segment-specific manner (1, 16). Various secondary-structure models (e.g., panhandle, RNA fork, corkscrew, and hook structures) have been proposed for the influenza viral RNA promoters over time (19–23). However, more-recent evidence favors the corkscrew model for both the vRNA and cRNA promoters (24). The main features of the corkscrew are the two terminal short hairpin loop structures, each with a stem of 2 bp and a tetraloop at both the 3′ and 5′ ends; the subsequent nucleotides bring the two ends together by forming a partial duplex region with Watson-Crick base pairings. The first 3 bp of the duplex region are highly conserved among all segments in FluA viruses, while they are segment specific in FluB viruses (25). Recently, the high-resolution crystal structures of the vRNA promoter-bound bat FluA and human FluB virus polymerases showed the presence of the 5′ hairpin loop and the duplex region (23, 26, 27). The vRNA promoter-bound RdRp complexes are found in “open” and “transcription-ready” conformations that are compatible with cap snatching and transcription initiation, while a cRNA promoter-bound “resting” RdRp adopts a “closed” conformation that is not compatible with cap snatching (23, 26–28). Although the 3-dimensional (3D) structures of the promoter-bound FluA and FluB virus polymerases showed no obvious structural differences, there are nucleotide differences between the FluA and FluB vRNA promoters (25). However, the biological significance of the differences between the FluA and FluB viral RNA promoters remains unclear.
In this study, we first bioinformatically analyzed all available promoter sequences of the FluA and FluB vRNAs and compared them with the experimentally determined promoter sequences. We confirmed the presence of FluA and FluB virus-specific nucleotide variations in the 3′ and 5′ termini. The nucleotide at position 5 in the 3′ end of the promoter and the nucleotide at position 6′ (the prime is used to distinguish this nucleotide from the nucleotide in the 3′ end of the promoter) in the 5′ end of the promoter differ in a type-specific manner. We then performed mutagenesis analyses of the type-specific promoter elements with both in vivo and in vitro functional assays. We found, for the first time, that the nucleotide at position 5 in the 3′ end of the vRNA promoters of FluA and FluB viruses modulates polymerase activity in a type-specific manner. Swapping the nucleotide between a FluA and a FluB virus showed different tolerances during virus passages. This work may provide new insight into the mechanisms of the intertypic exclusion of reassortment between FluA and FluB viruses.
RESULTS
Bioinformatics confirmation of the presence of the FluA- and FluB-specific vRNA promoter elements.
It has been reported that there are differences within the highly conserved promoter regions of FluA and FluB viruses (25). In order to systematically compare the promoter sequences at the 3′ and 5′ ends of the viral RNAs for FluA and FluB viruses, we obtained all available promoter sequences of FluA and FluB viruses from the NCBI Influenza Virus Resource database on 22 September 2018. These promoter sequences of different segments were then analyzed bioinformatically (Fig. 1A). Representative promoter logos of the 3′ and 5′ ends of the eight segments of FluA and FluB viruses were then generated by a WebLogo application (http://weblogo.berkeley.edu/logo.cgi) (29). All the sequences are shown as the vRNA on the negative-sense strand (Fig. 1A). To validate these promoter logos, we also bioinformatically compared the logo sequences with the promoter sequences that were determined experimentally (30–37) (Fig. 1B). The comparison further confirmed that the promoter logos are representative.
FIG 1.

Bioinformatics analysis of the promoter sequences of FluA and FluB segments and confirmation of the presence of the type-specific promoter elements. (A) Promoter sequence logos of FluA and FluB viruses. All the sequences (on the negative-sense strand) of the eight RNA segments of FluA and FluB viruses in the NCBI Influenza Virus Resource database were aligned, and only the terminal 12 nucleotides in the 3′ end (left) and 13 nucleotides in the 5′ end (right) are shown based on relative frequencies. The number of sequences used for generating each promoter logo is given to the left or right of the logo. The height of each letter in the promoter logos is in proportion to the frequency of a given nucleotide at that position. Dashed lines represent gaps in the sequences. The numbers above the logos indicate the positions in the 3′ or 5′ end of the promoter. Nucleotides boxed in red represent type-specific promoter elements for FluA or FluB viruses. (B) Comparison between our bioinformatically analyzed FluA or FluB promoter logos (upper) and experimentally determined FluA or FluB promoter sequences (lower). The upper FluA or FluB promoter logos were derived from panel A. The lower FluA or FluB promoter logos were generated from the promoter sequences that were determined experimentally. (C) Model vRNA promoters of FluA (left) and FluB (right) viruses in the potential corkscrew configuration. The prime notation is used in the 5′ end to distinguish its nucleotides from those in the 3′ end of the promoter. Red nucleotides circled in blue indicate the type-specific promoter elements for FluA and FluB viruses.
According to the “corkscrew” secondary structure proposed for the vRNA promoter (Fig. 1C), aside from the natural variation at position 4 in the 3′ end of the FluA vRNA promoters, there are two type-specific elements within the 3′ and 5′ hairpin loop regions that differ between the FluA and FluB vRNA promoters. One element is the nucleotide at position 5 in the 3′ end. The FluA vRNA promoter contains 5U, while the FluB vRNA promoter contains 5C, without exception. The other element is the nucleotide at position 6′ in the 5′ end, which displays as 6′A in all eight segments of FluA vRNA promoters, while it displays as 6′U in segments HA, NA, and NS and as 6′A in segments PB1, PB2, PA, NP, and M in FluB vRNA promoters. In summary, the 5U and 6′A nucleotides in the FluA vRNA promoters and the 5C and 6′U nucleotides in the FluB vRNA promoters are representative type-specific promoter elements (Fig. 1C).
The FluA and FluB virus polymerases favor their homotypic NCRs in the RNP reconstitution systems.
It has been reported previously that the polymerases of FluA or FluB viruses can recognize heterotypic vRNAs (8, 38). Here, since the type specificities of heterotypic vRNAs are attributed mainly to their type-specific NCRs, we compared RNA synthesis capacities from templates containing either the FluA-specific NCRs of influenza virus A/WSN/33 (H1N1) (A/WSN) or the FluB-specific NCRs of influenza virus B/Yamagata/88 (B/Yam) in both the FluA and FluB RNP reconstitution systems (39). Model vRNA templates were constructed in POLI-driven RNA expression plasmids. They contained the same reporter gene encoding the A/WSN HA protein, flanked by the HA-NCRs of either A/WSN or B/Yam (Fig. 2A). The model vRNAs were then cotransfected with protein expression plasmids encoding the proteins (PA, PB1, PB2, NP) of either A/WSN or B/Yam into 293T cells. At 24 h posttransfection, the steady-state levels of mRNA, cRNA, and vRNA were examined by primer extension analysis. As can be seen in Fig. 2B and C, both A/WSN and B/Yam RNPs in combination with their own homotypic HA-NCR vRNAs showed steady-state levels of mRNA, cRNA, and vRNA significantly higher than those of the FluA and FluB virus RNPs in combination with the heterotypic HA-NCR vRNAs. Moreover, A/WSN and B/Yam with four protein expression plasmids (4P) showed significantly different ratios of mRNA to vRNA regardless of the homotypic or heterotypic templates used, which may reflect different intrinsic properties for FluA and FluB virus polymerases (Fig. 2B and C). In general, these results suggested that the FluA and FluB virus polymerases favor their homotypic NCRs in the RNP reconstitution systems.
FIG 2.
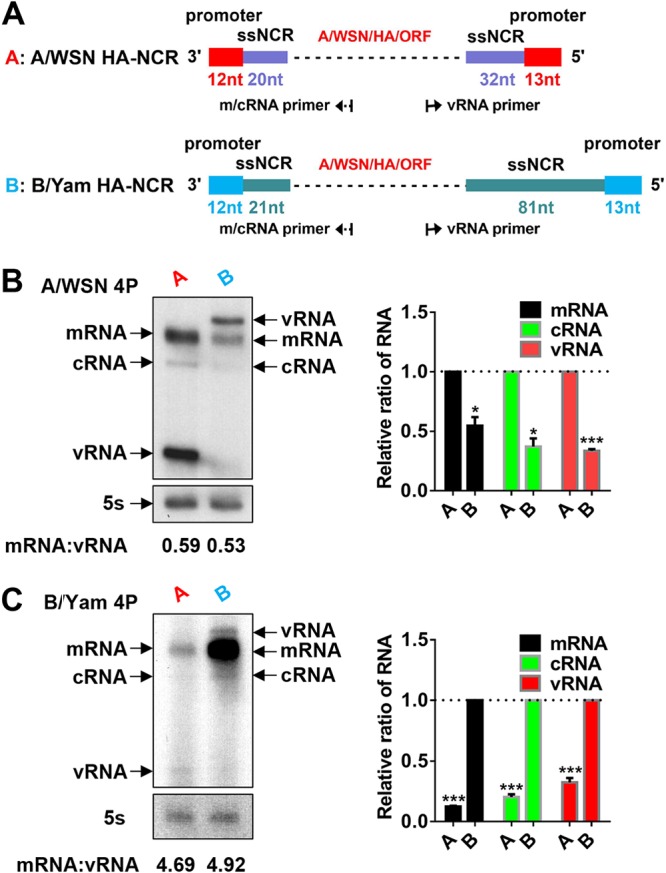
The FluA and FluB polymerases favor their homotypic NCRs in the RNP reconstitution systems. (A) Schematic representation of model FluA and FluB HA-NCR vRNA constructs. The model FluA and FluB HA-NCR vRNAs, which contain a reporter gene (A/WSN HA ORF) flanked by the HA-NCR of A/WSN (containing the promoter [red] and ssNCR [purple]) (abbreviated as A) or B/Yam (containing the promoter [blue] and ssNCR [green]) (abbreviated as B), respectively, were constructed in POLI-driven RNA expression plasmids. The lengths of the sequences of the promoters and ssNCRs are also shown in corresponding colors. Dashed lines represent the A/WSN HA ORF sequences. (B and C) Primer extension analysis to assess RNA levels derived from homo- or heterotypic model HA-NCR vRNA templates as shown in panel A in the A/WSN or B/Yam RNP reconstitution system, respectively. The homo- or heterotypic RNPs were reconstituted in 293T cells by cotransfecting four protein (4P) expression plasmids of A/WSN (pcDNA3.1-A/WSN-PB2, -PB1, -PA, and -NP) or B/Yam (pcDNA3.1-B/Yam-PB2, -PB1, -PA, and -NP) and corresponding POLI-derived model HA-NCR vRNA plasmids. The steady-state levels of mRNA, cRNA, and vRNA were detected by primer extension assays at 24 h posttransfection. The mRNA, cRNA, and vRNA levels shown in panels B and C were quantified by phosphorimager analysis. Values were normalized against those for cellular 5S rRNA. Normalized values obtained for wild-type A/WSN or B/Yam RNPs were set to 1. The average ratios of mRNA to vRNA are shown below the gels. Data are means ± standard errors of the means of results from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The type-specific promoter elements are involved in differentiating between FluA and FluB virus polymerases.
Considering the key roles of the vRNA promoters of FluA and FluB viruses in initiating viral RNA synthesis and the presence of type-specific promoter elements (5U and 6′A in FluA viruses; 5C and 6′U in FluB viruses), we next studied whether the type-specific promoter element(s) of the HA-NCRs contributed to the differentiation between FluA and FluB virus polymerases. We first constructed a series of type-specific promoter element-swapping (either alone or in combination) mutants in the plasmids expressing either FluA or FluB HA-NCR vRNAs (Fig. 3A). The effects of the mutations on viral RNA synthesis and viral protein expression were then examined by primer extension and Western blot analyses in both the A/WSN and B/Yam RNP reconstitution systems. Within the A/WSN RNP reconstitution system (Fig. 3B), in comparison with wild-type FluA HA-NCR vRNA, the mutant FluA HA-NCR vRNAs implanted with FluB virus promoter elements (U5C/A6′U or U5C) showed significantly reduced levels of the three RNA species (mRNA, cRNA, and vRNA) and of protein expression, while the A6′U mutant showed RNA and protein levels similar to those with wild-type vRNA. Similar results were also obtained with the mutant FluB HA-NCR vRNAs implanted with FluA virus promoter elements (C5U/U6′A or C5U) in the B/Yam RNP reconstitution system (Fig. 3C). These results suggested that swapping the promoter elements between FluA and FluB HA-NCR vRNAs resulted in significantly reduced polymerase activity and that the nucleotide at position 5 in the 3′ end plays a key role in regulating type-specific polymerase activity.
FIG 3.

The type-specific promoter elements are involved in differentiating between FluA and FluB virus polymerases. (A) Schematic representation of FluA and FluB wild-type and mutant model HA-NCR vRNA constructs. The promoters specific to FluA (red) and FluB (blue) viruses are indicated. Model FluA HA-NCR vRNAs were implanted with FluB virus-specific promoter elements (A U5C/A6′U, A U5C, and A A6′U), while model FluB HA-NCR vRNAs were implanted with FluA virus-specific promoter elements (B C5U/U6′A, B C5U, and B U6′A). Asterisks denote the implanted type-specific promoter elements. Dashed lines represent A/WSN HA ORF sequences. The prime notation in the 5′ end is used to distinguish its nucleotides from those in the 3′ end of the promoter. (B and C) Effects of implanted type-specific promoter elements on viral RNA synthesis and protein expression in the FluA (B) and FluB (C) virus RNP reconstitution systems. (D and E) Effects of implanted type-specific promoter elements on viral RNA synthesis and protein expression in mismatched RNP reconstitution systems in which A/WSN 4P (D) or B/Yam 4P (E) was in combination with heterotypic model HA-NCR vRNAs. Steady-state levels of mRNA, cRNA, and vRNA were detected by a primer extension (PE) assay, and the levels of protein production were examined by Western blotting (WB) at 24 h posttransfection. The shorter-exposure bands from the PE assay are also shown. The HA proteins expressed by wild-type and mutant RNPs were examined by Western blotting with an anti-A/WSN HA antibody at 24 h posttransfection. β-Actin was detected as a loading control. The exposure time for panel E (including PE and WB assays) was longer than those for panels B through D. The effects shown in panels B, C, D, and E are representative results from three independent experiments.
To further confirm these results, we also examined the effects of the type-specific promoter elements with mismatched RNP reconstitution systems in which A/WSN 4P or B/Yam 4P was in combination with heterotypic HA-NCR vRNAs. When A/WSN 4P was combined with FluB HA-NCR vRNAs, the C5U U6′A double mutant and the C5U single mutant, which contain an implanted FluA virus promoter element(s), showed significantly increased levels of the three RNA species and protein expression, while the A6′U mutant showed RNA and protein levels comparable to those for wild-type vRNA (Fig. 3D). Similarly, when B/Yam 4P was combined with FluA HA-NCR vRNAs, the U5C A6′U double mutant and the U5C single mutant, which contain an implanted FluB virus promoter element(s), showed significantly increased levels of mRNA and cRNA (but not vRNA) and protein expression, while the A6′U mutant showed RNA and protein levels comparable to those of the wild type. The reason for the unchanged vRNA levels remains unknown (Fig. 3E). We also obtained similar results independently with homotypic or heterotypic HA-NCR or NA-NCR vRNA templates with RNP reconstitution systems derived from influenza virus A/Hong Kong/68 (H3N2) (A/HK68) and influenza virus A/Beijing/01/2009 (H1N1) (A/BJ09) (data not shown). Taking these results together, we concluded that the type-specific promoter elements are indeed involved in differentiating between FluA and FluB virus polymerases. Furthermore, we identified, for the first time, the nucleotide at position 5 in the 3′ end of FluA and FluB vRNA promoters as playing a critical role in regulating type-specific polymerase activity.
The identity of the nucleotide at position 5 in the 3′ end of the FluA or FluB vRNA promoter is critical in determining polymerase activity.
In order to further explore the identities of the nucleotides at position 5 in the 3′ ends of FluA and FluB vRNA promoters in the regulation of polymerase activity, we further mutated the nucleotides at position 5 in the 3′ ends of A/WSN and B/Yam HA-NCR vRNAs into other, alternative nucleotides and examined their effects on their respective RNP reconstitution systems. Figure 4A shows that within the FluA virus RNP reconstitution system, the U5C and U5A mutants exhibited RNA levels significantly lower than that of the wild-type template, whereas the U5G mutant showed only residual RNA and protein synthesis capacities. Similar results were obtained with similar mutations in the FluB virus RNP reconstitution system (Fig. 4B). We also examined the effects of the same identity mutations in the NA-NCR vRNAs in both the FluA and FluB virus RNP reconstitution systems by Western blot analysis. We obtained the same results as those shown with the identity mutants of HA-NCR vRNAs (Fig. 4C and D). Therefore, we concluded that the identities of the nucleotides at position 5 in the 3′ ends of both the FluA and FluB vRNA promoters are important in modulating polymerase activity. In particular, the alteration to G could diminish polymerase activity enormously. These results further emphasize the importance of the identity of residue 5 in the 3′ ends of the influenza vRNA promoters in regulating in vivo viral RNA polymerase activity.
FIG 4.

Effects of nucleotide identity mutants at position 5 in the 3′ ends of FluA or FluB model HA-NCR or NA-NCR vRNAs in the FluA and FluB virus RNP reconstitution systems. (A and B) Effects of nucleotide identity mutants at position 5 in the 3′ end of the A/WSN (A) or B/Yam (B) model HA-NCR vRNA promoter on viral RNA synthesis and protein expression in the A/WSN (A) or B/Yam (B) RNP reconstitution system. (C and D) Effects of nucleotide identity mutants at position 5 in the 3′ end of the A/WSN (C) or B/Yam (D) NA-NCR-containing vRNA promoter on reporter protein expression in the A/WSN (C) or B/Yam (D) RNP reconstitution system. The RNPs were reconstituted in 293T cells by cotransfecting 4P expression plasmids of A/WSN or B/Yam and wild-type or mutant POLI-derived model HA-NCR or NA-NCR vRNA plasmids. The steady-state levels of mRNA, cRNA, and vRNA were detected by primer extension (PE) analysis at 24 h posttransfection. The shorter-exposure bands from the PE assay are shown. The levels of mRNA, cRNA, and vRNA in panels A and B were quantified by phosphorimager analysis. The values were normalized against those for cellular 5S rRNA. HA protein production was examined by Western blotting (WB) with an anti-A/WSN HA antibody at 24 h posttransfection. The HA protein expression levels shown in panels A through D were quantified by densitometry analysis. β-Actin was detected as a loading control. Normalized values obtained in wild-type A/WSN or B/Yam RNPs (lanes 1) were set to 1. Data are means ± standard errors of the means of results from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Effects of identity substitutions at position 5 in the 3′ ends of Flu A and FluB vRNA promoters in an in vitro replication initiation assay.
Since the vRNA promoter is critical in determining the efficiency of replication initiation, we then examined the effects of the identity mutants on de novo dinucleotide (pppApG) synthesis by a primer-independent in vitro replication initiation assay as described previously (11). We used partially TAP (tandem affinity purification)-purified recombinant polymerases of the FluA and FluB viruses in the in vitro assay (Fig. 5A and B). The 3′ and 5′ strands of the FluA and FluB model promoter vRNAs (14 or 15 nucleotides [nt] long, respectively) and the identity substitution mutants with mutations at position 5 in the 3′ strand were synthesized as described previously (39, 40). As shown in Fig. 5C and D, the G substitutions in both the vRNA promoters showed only residual levels of pppApG, and the A substitutions in both the vRNA promoters showed moderate reductions in de novo pppApG synthesis. These results are consistent with the in vivo results observed in the RNP reconstitution system (Fig. 4). However, differential effects were obtained with the swapping mutants. The U5C mutation in the FluA model vRNA showed a drastic reduction in pppApG synthesis with the FluA virus polymerase, while the C5U mutation in the FluB model vRNA with the FluB virus polymerase showed a pppApG synthesis capacity like that of the wild type or even higher, indicating that a divergence may exist between FluA and FluB virus polymerases upon de novo pppApG synthesis (compare lanes 2 in Fig. 5C and D).
FIG 5.

Effects of nucleotide identity substitutions at position 5 in the FluA and FluB virus promoters in an in vitro replication initiation assay. (A) Partially purified recombinant A/WSN polymerase proteins (3P) were prepared in 293T cells. 293T cells were cotransfected with plasmids expressing PB1, PA, and PB2-TAP of A/WSN and were purified by IgG Sepharose chromatography. The purified 3P complexes were analyzed by SDS-PAGE and silver staining. (B) Partially purified recombinant B/Yam polymerase proteins were prepared as described for panel A. (C) Autoradiograph showing pppApG synthesized de novo by partially purified A/WSN 3P complexes with the wild-type model 3′ vRNA (3′-UCGUUUUCGUCCGG-5′) (lane 1) or position 5 substitution mutants (lanes 2 to 4) in the presence of the wild-type model 5′ vRNA of FluA virus promoters (3′-CCGGAACAAAGAUGA-5′). (D) Autoradiograph showing pppApG synthesized de novo by partially purified B/Yam 3P complexes with the wild-type model 3′ vRNA (3′-UCGUCUUCGUCUGG-5′) (lane 1) or position 5 substitution mutants (lanes 2 to 4) in the presence of the wild-type model 5′ vRNA of FluB virus promoters (3′-CCAGAACAAUGAUGA-5′). (E) Autoradiograph showing pppApG synthesized de novo by partially purified A/WSN 3P complexes with the wild-type model 5′ cRNA (3′-CCGGACGAAAACGA-5′) (lane 1) or position 5 substitution mutants (lanes 2 to 4) in the presence of the wild-type model 3′ cRNA of FluA virus promoters (3′-UCAUCUUUGUUCCGG-5′). The pppA*pG products (where “*p” represents 32P) are indicated. The pound sign (#) after “A” or “B” is used to distinguish the model cRNA promoters from model vRNA promoters. (F) Autoradiograph showing pppApG synthesized de novo by partially purified B/Yam 3P complexes with the wild-type model 5′ cRNA (3′-CCAGACGAAGACGA-5′) (lane 1) or position 5 substitution mutants (lanes 2 to 4) in the presence of the wild-type model 3′ cRNA of FluB promoters (3′-UCAUCAUUGUUCUGG-5′). Statistical analyses of pppA*pG levels in the autoradiographs in panels C, D, E, and F were performed with a phosphorimager. Data are means ± standard errors of the means of results from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Since the cRNA and vRNA promoters are complementary to each other, a mutation in the 3′ end of the vRNA promoter would be copied into the 5′ end of the cRNA promoter to generate more vRNAs in vivo. Therefore, we also examined the effects of the identity substitution mutations at position 5 in the 5′ end of the cRNA promoter by the in vitro replication initiation assay. As shown in Fig. 5E and F, the nucleotide substitutions at position 5 in the 5′ strand of the FluA and FluB virus promoters, combined with their respective wild-type 3′ strand cRNA promoters, showed differential effects with their homotypic polymerases on de novo pppApG synthesis. For the FluA virus polymerase, the FluA A5′G swapping mutant model cRNA promoter showed significantly reduced pppApG synthesis, while the other two identity mutants showed pppApG synthesis capacities similar to that of the wild-type 5′ cRNA promoter (Fig. 5E). In contrast, for the FluB virus polymerase, the pppApG synthesis capacity of the FluA G5′A swapping mutant was similar to that of the wild type, while those of the other two identity mutants were significantly reduced (Fig. 5F). These results indicate that the identity of the nucleotide at position 5 in the 5′ cRNA promoter could also regulate replication initiation and that the swapping mutant caused a more significant effect on FluA virus polymerase than on FluB virus polymerase. Taking the in vitro results together, we concluded that the identity of the nucleotide at position 5 in the 3′ ends of the vRNA promoters could interfere with the normal functions of both the 3′ vRNA and the 5′ cRNA in a type-specific manner. Our results also implied that the FluA and FluB virus polymerases might have different tolerances for swapping of the type-specific position 5 nucleotide between the FluA and FluB viral promoters with regard to replication initiation.
Effects of the type-specific promoter element mutations in the context of virus infection systems.
Considering the significant effects of the type-specific promoter element mutations in modulating the polymerase activity observed in the RNP reconstitution assay, we were interested in studying the biological significance of these mutations in virus infection systems. With the A/WSN and B/Yam (a gift from Adolfo García-Sastre) reverse genetic systems, we attempted to rescue recombinant viruses with the promoter element mutations in the HA segments of both viruses.
In the case of A/WSN virus rescue, we transfected human embryonic kidney 293T and Madin-Darby canine kidney (MDCK) cell mixtures with either wild-type or mutated pHW2000-A/WSN-HA plasmids containing the U5C A6′U, U5C, A6′U, U5A, or U5G mutation, together with seven other rescue plasmids, and collected the supernatants at 3 days posttransfection. We found that plaques could be formed with the supernatants (P0) for the A6′U and U5A mutations only. However, when we passaged the P0 supernatants of the U5C, U5C A6′U, and U5G mutants for 2 to 3 passages, we obtained plaques for the U5C and U5C A6′U mutants, but not for the U5G mutant. After sequencing the whole genomes of the U5C A6′U and U5C mutant viruses, we found that in both mutant viruses, the U5C point mutation had reverted to the wild-type nucleotide U, while the A6′U mutation was maintained (Fig. 6A and B). Subsequently, we measured the replication efficiencies for the rescued mutant (A6′U and U5A) viruses by traditional plaque assays with plaque-purified P2 viruses at a multiplicity of infection (MOI) of 0.001. As seen in Fig. 6C and D, the A6′U mutant virus replicated at levels similar to those of the wild-type virus, whereas the U5A mutant virus showed a ∼1.5-log reduction. In order to examine the effects of these mutations on virus RNA synthesis, we also measured the viral RNA levels (mRNA, cRNA, and vRNA) of the HA segment by primer extension analysis with an MOI of 5 at 6 and 9 h postinfection. As expected, Fig. 6E shows that the effects of the mutations on the three viral RNA species were similar to the effects shown in the RNP reconstitution assay. These results confirmed that the mutations introduced into the promoter of the HA segment indeed affected virus replication efficiency by affecting HA viral RNA synthesis.
FIG 6.

Effects of the type-specific promoter element mutants in the context of the A/WSN virus infection system. (A and B) Sequence traces (positive sense) of cDNA amplified by a vRNA-specific reverse transcription-PCR from A/WSN HA U5C and U5C A6′U mutants in MDCK cells. Extracted RNAs were treated with RppH and were circularized with T4 RNA ligase prior to reverse transcription-PCR amplification across the junction and amplicon cloning. Vertical lines represent the junction sites. Nucleotides boxed in red indicate mutations at the promoter region of vRNA. Red arrows indicate the position of the corrected 3′-terminal residue (C5 → U5) of the vRNA. The corrected U5 residues in the negative sense would be displayed as A5 in the sequence traces (positive sense). (C and D) Growth curves of wild-type (WT) and mutant A/WSN viruses in MDCK cells. MDCK cells were infected with viruses at an MOI of 0.001. At 0, 12, 24, 36, 48, 60, and 72 h postinfection, the virus titers in the supernatants were determined by plaque assays in MDCK cells. Data are means ± standard errors of the means of results from three independent experiments. (E) Primer extension analysis examining the levels of viral RNAs in 293T cells after infection at an MOI of 5. Total RNAs were extracted from 293T cells infected at an MOI of 5 with wild-type or mutant A/WSN viruses for 6 or 9 h. The shorter-exposure bands in primer extension analysis are shown. The levels of mRNA, cRNA, and vRNA in wild-type or mutant virus-infected cells were quantified by phosphorimager analysis. The values were normalized against those for cellular 5S rRNA. Normalized values obtained for wild-type A/WSN-infected cells were set to 1. Data are means ± standard errors of the means of results from two to three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In the case of B/Yam virus rescue, the same experiments as those described above were performed, and we were able to rescue all mutant (C5U U6′A, C5U, U6′A, and C5A) viruses except for the C5G mutant at P0. However, we found that the C5U swapping mutations in the mutant viruses were maintained during serial passages (Fig. 7A and B). Measurement of virus growth curves showed that the U6′A mutation did not affect virus replication efficiency, while the other three mutant (C5U U6′A, C5U, and C5A) viruses replicated at significantly lower efficiencies (reduced about 1 to 1.5 log units) than the wild-type virus (Fig. 7C and D). Primer extension analysis also confirmed the effects of the mutations on HA viral RNA synthesis (Fig. 7E). Therefore, these results further confirmed the role of the type-specific promoter elements at position 5 in the 3′ ends of the FluA and FluB virus promoters in differentiating the FluA and FluB virus polymerases in the context of the virus infection systems. Moreover, we also observed that the FluA and FluB virus polymerases behave differently upon promoter element swapping. The U5C mutation in the A/WSN virus could be reverted to the wild-type nucleotide efficiently during virus passages, while the C5U mutation in the B/Yam virus could be maintained through a series of passages, but it reduced virus replication efficiency significantly. This result was also supported, to a certain extent, by the results for the swapping mutants in in vitro replication initiation assays. This finding highlights the different tolerances of the FluA and FluB virus polymerases upon nucleotide substitution in the viral RNA promoters, suggesting that the selection pressure and/or error-correcting abilities of the FluA and FluB virus polymerases differ significantly.
FIG 7.

Effects of the type-specific promoter element mutants in the context of the B/Yam virus infection system. (A and B) Sequence traces (positive sense) of cDNA amplified from circularized RNA templates by the vRNA-specific reverse transcription-PCR from B/Yam HA C5U and C5U U6′A mutants in MDCK cells. Vertical lines represent the junction sites. Nucleotides boxed in red indicate the maintained mutations at the promoter region of vRNA. (C and D) Growth curves of wild-type and mutant B/Yam viruses in MDCK cells. MDCK cells were infected with viruses at an MOI of 0.001. At 0, 12, 24, 36, 48, 60, and 72 h postinfection, the virus titers in the supernatants were determined by plaque assays in MDCK cells. Data are means ± standard errors of the means of results from three independent experiments. (E) Primer extension analysis examining the levels of viral RNAs in 293T cells after infection at an MOI of 5. Total RNAs were extracted from 293T cells infected at an MOI of 5 with wild-type or mutant B/Yam viruses for 6 or 9 h. The levels of mRNA, cRNA, and vRNA in wild-type or mutant virus-infected cells were quantified by phosphorimager analysis. The values were normalized against those for cellular 5S rRNA. Normalized values obtained for wild-type B/Yam-infected cells were set to 1. Data are means ± standard errors of the means of results from two to three independent experiments.
DISCUSSION
The ability to undergo reassortment is one of the hallmarks of all segmented RNA viruses. Thus, the segmented negative-strand RNA viruses readily reshuffle RNA segments in progeny viruses during coinfection in the same cells (1). Although FluA and FluB viruses are known to cocirculate in human populations, heterotypic reassortants between the two types have never been observed in nature or in vitro. Mounting evidence suggests that the inability to reassort between the two types is due mainly to inefficient formation of intertypic polymerase complexes and incompatible packaging signals (8, 9). In this investigation, we studied the type-specific promoter elements of FluA and FluB viruses, and we report here, for the first time, that the identities of the nucleotides at position 5 in the 3′ ends of the FluA and FluB virus promoters are key type-determined features that play a critical role in modulating type-specific polymerase activity. This finding indicates that the promoter divergence between FluA and FluB viruses contributes to their RNP incompatibilities, which may provide new insight into the mechanisms of heterotypic exclusion of reassortments between the FluA and FluB viruses. Since the promoter plays a key role in the initiation of viral RNA synthesis, which is the first rate-limiting step during influenza virus replication, we speculate that the homotypic preference between influenza viral RNA promoters and polymerases might be an evolutionary mechanism not only to save cell resources for efficient virus production but also to reduce heterotypic interference upon coinfection of the same cell with different virus types.
Structural studies of the vRNA promoter bound to the FluA or FluB virus polymerase complex showed that the promoter adopts a structure similar to the “corkscrew” model, in which the 5′ end forms a stem-loop, while the 3′ vRNA extremity (nucleotides 1 to 9) is single stranded and highly dynamic, switching between preinitiation and initiation states. The two different 3′ extremity vRNA binding sites have been reported previously (23, 26, 27, 41). Interestingly, a very recent costructure of FluD virus polymerase and the vRNA promoter obtained by cryo-electron microscopy (cryo-EM) revealed another 3′ vRNA extremity binding site on the polymerase (42). Therefore, the type-specific residue at position 5 in the 3′ end of the vRNA promoter may affect various 3′ vRNA extremity binding statuses on the polymerases. In addition, our in vitro replication initiation assay results indicated that this residue, when copied into cRNA, could also affect the function of the 5′ cRNA promoter (Fig. 5E and F). Regarding the homotypic preference between the promoters and polymerases reported here, we have made great efforts to identify type-specific amino acids in FluA and FluB virus polymerases that mediate the homotypic preference. Unfortunately, we failed to identify such a type-specific amino acid(s). We speculate that the homotypic preference may be mediated by the subtle global structure differences between the FluA and the FluB virus polymerases. Alternatively, the different nucleotide identity at position 5 in either 3′ vRNA or 5′ cRNA may cause differing steric hindrances upon promoter binding and thus may change the initiation site on the template. Furthermore, we do not exclude the possibility that a host factor(s) might be involved in mediating the homotypic preference. Anyway, the key evidence that a type-specific promoter element at position 5 in the 3′ end of vRNA plays a role in differentiating between FluA and FluB virus polymerases, together with the observation that the G substitution at position 5 dramatically destroyed polymerase activity, requires more-elaborate structure studies to reveal the underlying mechanisms.
Within the FluA viruses, the biological significance of the natural variations at position 4 (the C4 or U4 promoter) in the 3′ end of the vRNA promoter has been studied extensively (17, 18). It has been shown that the C4 promoter can downregulate viral RNA transcription (vRNA → mRNA) and activate viral RNA replication (vRNA ↔ cRNA) (17). The structures of the C4 and U4 promoters obtained by nuclear magnetic resonance (NMR) spectroscopy exhibited a large RNA structural change and implicated differential viral RNA synthesis by RdRp (18). It has also been shown that the 3′ U1 and U4 of the FluA vRNA template are important in regulating realignment during viral RNA transcription in order to generate sufficient viral mRNAs (43). Neumann and Hobom showed previously that the triple 3-5-8 mutations in the 3′ arm of the vRNA promoter could considerably enhance polymerase activity in the chloramphenicol acetyltransferase (CAT) reporter assay (44). Such enhancements have also been linked to host restriction effects conferred by PB2 residue 627 (45). In addition, Leahy et al. observed that the identity of residue 5 in the 3′ end of the vRNA promoter could also affect endonuclease activity (46). Interestingly, we observed here that the A-type U5 and B-type C5 promoters confer homotypic preference between the promoter and the polymerases. We also showed, by an in vitro assay, that identity substitutions of residue 5 in 3′ vRNA or in 5′ cRNA could differentially affect viral RNA replication initiation. Our data emphasize, for the first time, that the residue at position 5 in the 3′ end of the vRNA promoter might play multiple roles during viral RNA synthesis, leading to differentiation between FluA and FluB viruses, and thus has great biological significance during influenza virus replication.
Previously, Lee and Seong attempted to rescue a FluA virus containing an NA U5C, U5A, or U5G mutation with transfected RNP in the presence of helper viruses, but they failed to rescue any of these mutant viruses (17). However, Bergmann and Muster successfully rescued the NA U5C mutant and found that it could revert to the wild type after passaging of the virus in embryonated chicken eggs (47), a finding in agreement with our observations of revertants during the HA U5C and NA U5C virus rescues in this study. In contrast, we found that FluB viruses could maintain the HA C5U or NA C5U mutation after serial passages. A previous study of promoter mutagenesis by an in vitro transcription assay showed that FluB virus polymerase appears to be more tolerant of mismatched (according to the panhandle structure) mutations (25). These results are actually in agreement with the following facts: (i) a greater degree of natural heterogeneity is observed in the 3′-terminal nucleotides of FluB virion RNA, and (ii) FluB viruses mutate at a rate 2 to 3 times lower than that of FluA viruses and consequently are less genetically diverse, as demonstrated by the fact that FluB viruses have only one serotype. All this evidence demonstrates that the tolerances of nucleotide variations and the error-correcting abilities of FluA and FluB virus polymerases differ significantly. However, the underlying mechanisms remain unclear, and follow-up studies would be of great interest.
In summary, we report here that the identities of the nucleotides at position 5 in the 3′ ends of the promoters of FluA and FluB viruses are key type-determined features that play critical roles in determining the type-specific polymerase activities both in vivo and in vitro. Our results demonstrated that incompatibility between the promoter and the polymerase exists among different influenza virus types and thus may contribute to heterotypic exclusion of reassortments. Our discoveries here not only broaden our basic knowledge of influenza virus type specificities that may preclude heterotypic reassortments but also provide new insight into the biological significance of the influenza virus promoters.
MATERIALS AND METHODS
Bioinformatics analysis of the promoter sequences of all segments of FluA and FluB viruses.
All DNA sequences of each segment of the FluA and FluB viruses were obtained from the NCBI Influenza Virus Resource database on 22 September 2018 as described previously (48). For the HA and NA segments of FluA viruses, only H3 and N2 were used. We used highly conserved promoter motifs as signatures for selecting NCR-containing sequences. The NCR-containing sequences were aligned using MUSCLE software (49) with an additional manual check. After the alignment, positions with an 80% or higher ratio of gaps were removed. Promoter sequence logos were generated based on nucleotide frequencies by WebLogo (http://weblogo.berkeley.edu/logo.cgi) (29). To validate these promoter logos, we also bioinformatically compared the logo sequences with promoter sequences that had been determined experimentally (30–37). The accession numbers for the sequences used in this paper are available upon request; no new sequences were determined in this study.
Cells and antibodies.
Madin-Darby canine kidney (MDCK) and human embryonic kidney 293T cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C. The rabbit anti-β-actin monoclonal antibody (catalog no. 4970) was purchased from Cell Signaling Technology. The rabbit anti-influenza virus HA monoclonal antibody (catalog no. 86001-RM01) was purchased from Sino Biological Inc. (China).
Plasmids.
The eight plasmids of the influenza virus A/WSN/33 (H1N1) (A/WSN) reverse genetic system (pHW2000-A/WSN-PB2, -PB1, -PA, -HA, -NP, -NA, -M, and -NS) have been described previously (48). The eight plasmids of the influenza virus B/Yamagata/88 (B/Yam) reverse genetic system (pDZ-B/Yam-PB2, -PB1, -PA, -HA, -NP, -NA, -M, and -NS) were kindly provided by Adolfo García-Sastre (Icahn School of Medicine at Mount Sinai, New York, NY). The series of pcDNA3.1 protein expression plasmids expressing individual A/WSN 4P and B/Yam 4P were constructed by PCR with specific primers, followed by ligation into a pcDNA3.1-myc/his empty plasmid. The plasmids expressing the wild-type FluA HA-NCR and FluB HA-NCR model vRNAs (pHW2000-muta-A/HA-NCR and pHW2000-muta-B/HA-NCR) used for the RNP reconstitution assay were constructed by PCR, followed by ligation into a modified pHW2000 plasmid with a deleted cytomegalovirus (CMV) promoter. All the plasmids expressing mutant HA-NCR model vRNA were generated by a site-directed mutagenesis kit (New England BioLabs [NEB]). The pHW2000-A/WSN-HA and pDZ-B/Yam-HA plasmids used for virus rescue were also generated by site-directed mutagenesis. The tandem affinity purification (TAP)-tagged pcDNA plasmids encoding PB2 derived from A/WSN/33 (pcDNA3.0-A/WSN-PB2-TAP) have been described previously (50). The TAP-tagged or nontagged pCAGGS plasmids expressing PB2 or PB1 and PA derived from B/Yam were constructed by PCR with appropriate primers, followed by ligation of the PCR products into an empty pCAGGS plasmid.
Virus rescue and viral growth curves.
Recombinant viruses (wild-type A/WSN, wild-type B/Yam, and their corresponding mutant viruses) were rescued as described previously (35, 51). The rescued viruses were plaque purified. The purified viruses were then passaged for 2 to 3 generations on MDCK cells, and their genomes were sequenced. The growth kinetics of the wild-type and mutant viruses were performed in MDCK cells at a multiplicity of infection (MOI) of 0.001, and PFU titers were determined by a standard plaque assay in MDCK cells.
Viral RNA sequencing.
The 3′ and 5′ termini of viral RNA transcripts were sequenced as described previously (52). Briefly, RNAs extracted from transfected 293T cells or from supernatants of each virus stock were treated with RppH (NEB) according to the manufacturer′s instructions. The RNAs were then circularized with T4 RNA ligase (NEB). cDNA copies of the ligated 3′ and 5′ termini were synthesized by reverse transcription with specific primers. The cDNAs were amplified by PCR using Q5 High-Fidelity polymerase (NEB) and specific primers and were cloned as described in the pClone007 Blunt Simple Vector kit (TsingKe Biotech, China). Then the plasmids were sent for sequencing (Ruibiotech, China). The viral RNA open reading frame (ORF) was sequenced by traditional methods (48).
RNP reconstitution, primer extension, and Western blotting.
To reconstitute the virus RNPs, approximately 1 × 106 293T cells in 35-mm dishes were transfected with 0.5 μg of protein expression plasmids encoding PB2, PB1, PA, and NP of A/WSN (pcDNA3.1-A/WSN-PB2, -PB1, -PA, and -NP) or B/Yam (pcDNA3.1-B/Yam-PB2, -PB1, -PA, and -NP) together with 0.5 μg of a plasmid expressing wild-type or mutant model vRNA in the presence of 5 μl Lipofectamine 2000 (Invitrogen). Cells were harvested at 24 h posttransfection. The total RNAs and proteins were then analyzed by primer extension and Western blotting. For the single-cycle infection assays, 293T cells were infected with a wild-type or mutant A/WSN or B/Yam virus at an MOI of 5. Total RNA was then extracted at 6 and 9 h postinfection and was subjected to primer extension analysis (see below).
Primer extension analysis was performed as described previously (39). Primer 5′-CACTGCCACATTCTTCTCGAG-3′ was used to detect A/WSN HA-specific mRNA and cRNA. Primer 5′-AGTTCACTGGTGCTTTTGGT-3′ was used to detect A/WSN HA-specific vRNA. Primer 5′-AGTGGTATCACACCAGTCAC-3′ was used to detect B/Yam HA-specific mRNA and cRNA. Primer 5′-GGTCTCCAGAGACAATGTTTC-3′ was used to detect B/Yam HA-specific vRNA. Primer 5′ -TCCCAGGCGGTCTCCCATCC-3′ was used to detect 5S rRNA as an internal control. The RNA signals were quantified by phosphorimager analysis using ImageQuant TL software (GE Healthcare).
For Western blotting, the cells were lysed with CytoBuster lysis buffer (Novagen), and the samples were separated by SDS–PAGE and immunoblotted with the primary HA antibody and an IRDye-conjugated secondary antibody (Li-Cor Biosciences, Lincoln, NE). Protein expression levels were visualized by the Odyssey Infrared Imaging System (Li-Cor Biosciences, USA). The quantification of the signals was analyzed with the integrated software of the Odyssey system.
Preparation of the recombinant RNA polymerases of FluA and FluB viruses.
293T cells were transfected with pcDNA3.1-A/WSN-PB1, pcDNA3.1-A/WSN-PA, and pcDNA3.0-A/WSN-PB2-TAP to generate recombinant RdRp of A/WSN and with pCAGGS-B/Yam-PB1, pCAGGS-B/Yam-PA, and pCAGGS-B/Yam-PB2-TAP to generate recombinant RdRp of B/Yam using Lipofectamine 2000 (50, 53). At 48 h posttransfection, the cells were lysed, and the TAP-tagged RdRp’s were first purified by affinity purification with immunoglobulin G-Sepharose and then released by tobacco etch virus protease as described previously (50). The partially purified RdRp’s in tobacco etch virus cleavage buffer (0.15 M NaCl, 0.01 M HEPES [pH 8.0], 0.1% NP-40, 1 mM dithiothreitol [DTT], 10% glycerol) were stored at –20°C in 40% glycerol.
In vitro dinucleotide replication initiation assay.
We performed an in vitro dinucleotide (pppApG) replication initiation assay as described previously (11). Briefly, we used a 3-μl reaction mixture containing 1.5 μl of partially purified recombinant polymerases, 5 mM MgCl2, 1 mM DTT, 0.83 mM ATP, 0.05 mM [α-32P]GTP (3,000 Ci/mmol; Amersham), 2 U/μl RNasin, and 0.5 μM each 3′ end (FluA virus, 3′-UCGUUUUCGUCCGG-5′; FluB virus, 3′-UCGUCUUCGUCUGG-5′) and 5′ end (FluA virus, 3′-CCGGAACAAAGAUGA-5′; FluB virus, 3′-CCAGAACAAUGAUGA-5′) of wild-type or mutant vRNA promoters (Ruibiotech, China). The wild-type 3′ ends (FluA virus, 3′-UCAUCUUUGUUCCGG-5′; FluB virus, 3′-UCAUCAUUGUUCUGG-5′) and 5′ ends (FluA virus, 3′-CCGGACGAAAACGA-5′; FluB virus, 3′-CCAGACGAAGACGA-5′) or mutant 3′ ends and 5′ ends of the model cRNA promoters used in the assay are complementary to their corresponding model vRNA promoters. The nucleotides in the duplex region (positions 10 to 12 in the 3′ end and positions 11′ to 13′ in the 5′ end) of the FluB model vRNA and cRNA promoters represent the sequences of the HA, NA, and NS segments of FluB viruses (25). Both FluA and FluB model vRNA and cRNA promoters contained the conserved sequences plus 2 additional residues to extend the duplex region. The samples were incubated at 30°C for 1 h. Formamide-bromophenol blue-EDTA was added to the RNA transcription products, and the mixture was heated at 95°C for 2 min. The products were then analyzed on 25% polyacrylamide gels containing 6 M urea in Tris-borate-EDTA (TBE) buffer, visualized by autoradiography, and quantified by a Typhoon Trio Plus system (GE Healthcare).
Statistics.
GraphPad Prism software, version 6, was used for statistical analysis. A two-tailed Student t test was used for two-group comparisons. P values of <0.05 and <0.01 were considered to be significant.
ACKNOWLEDGMENTS
We thank Adolfo García-Sastre (Icahn School of Medicine at Mount Sinai, New York, NY) for providing us with the influenza virus B/Yamagata/88 reverse genetic system. We thank George Brownlee and Ervin Fodor (Sir William Dunn School of Pathology, Oxford University), as well as A. J. te Velthuis (University of Cambridge, Department of Pathology, Division of Virology) for very helpful discussions. We thank Hongjie Zhang (Core Facility for Protein Research, Institute of Crystal Structure of Biophysics, Chinese Academy of Sciences) for technical support with autoradiography.
This work was supported by grants from the CAMS Innovation Fund for Medical Sciences (award 2016-12M-1-014), the National Science and Technology Major Project (award 2018ZX10101001-004), the National Natural Science Foundation of China (award 31070152), and the Hunan Provincial Natural Science Foundation of China (award 2018JJ3039).
REFERENCES
- 1.Palese P, Shaw M. 2013. Orthomyxoviridae, p 1151–1185. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, Palese P, Shaw ML, Treanor J, Webster RG, García-Sastre A. 2018. Influenza. Nat Rev Dis Primers 4:3. doi: 10.1038/s41572-018-0002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hay AJ, Gregory V, Douglas AR, Lin YP. 2001. The evolution of human influenza viruses. Philos Trans R Soc Lond B Biol Sci 356:1861–1870. doi: 10.1098/rstb.2001.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoon SW, Webby RJ, Webster RG. 1992. Evolution and ecology of influenza A viruses. Curr Top Microbiol Immunol 56:152–179. [DOI] [PubMed] [Google Scholar]
- 5.Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. 2010. Genome packaging in influenza A virus. J Gen Virol 91:313–328. doi: 10.1099/vir.0.017608-0. [DOI] [PubMed] [Google Scholar]
- 6.Fujii Y, Goto H, Watanabe T, Yoshida T, Kawaoka Y. 2003. Selective incorporation of influenza virus RNA segments into virions. Proc Natl Acad Sci U S A 100:2002–2007. doi: 10.1073/pnas.0437772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisfeld AJ, Neumann G, Kawaoka Y. 2015. At the centre: influenza A virus ribonucleoproteins. Nat Rev Microbiol 13:28–41. doi: 10.1038/nrmicro3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwatsuki-Horimoto K, Hatta Y, Hatta M, Muramoto Y, Chen H, Kawaoka Y, Horimoto T. 2008. Limited compatibility between the RNA polymerase components of influenza virus type A and B. Virus Res 135:161–165. doi: 10.1016/j.virusres.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker SF, Nogales A, Finch C, Tuffy KM, Domm W, Perez DR, Topham DJ, Martínez-Sobrido L. 2014. Influenza A and B virus intertypic reassortment through compatible viral packaging signals. J Virol 88:10778–10791. doi: 10.1128/JVI.01440-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Te Velthuis AJ, Fodor E. 2016. Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis. Nat Rev Microbiol 14:479–493. doi: 10.1038/nrmicro.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng T, Vreede FT, Brownlee GG. 2006. Different de novo initiation strategies are used by influenza virus RNA polymerase on its cRNA and viral RNA promoters during viral RNA replication. J Virol 80:2337–2348. doi: 10.1128/JVI.80.5.2337-2348.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jorba N, Coloma R, Ortín J. 2009. Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog 5:e1000462. doi: 10.1371/journal.ppat.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desselberger U, Racaniello VR, Zazra JJ, Palese P. 1980. The 3′ and 5′-terminal sequences of influenza A, B and C virus RNA segments are highly conserved and show partial inverted complementarity. Gene 8:315–328. doi: 10.1016/0378-1119(80)90007-4. [DOI] [PubMed] [Google Scholar]
- 14.Stoeckle MY, Shaw MW, Choppin PW. 1987. Segment-specific and common nucleotide sequences in the noncoding regions of influenza B virus genome RNAs. Proc Natl Acad Sci U S A 84:2703–2707. doi: 10.1073/pnas.84.9.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson JS. 1979. 5′ and 3′ terminal nucleotide sequences of the RNA genome segments of influenza virus. Nucleic Acids Res 6:3745–3757. doi: 10.1093/nar/6.12.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou B, Lin X, Wang W, Halpin RA, Bera J, Stockwell TB, Barr IG, Wentworth DE. 2014. Universal influenza B virus genomic amplification facilitates sequencing, diagnostics, and reverse genetics. J Clin Microbiol 52:1330–1337. doi: 10.1128/JCM.03265-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee KH, Seong BL. 1998. The position 4 nucleotide at the 3′ end of the influenza virus neuraminidase vRNA is involved in temporal regulation of transcription and replication of neuraminidase RNAs and affects the repertoire of influenza virus surface antigens. J Gen Virol 79:1923–1934. doi: 10.1099/0022-1317-79-8-1923. [DOI] [PubMed] [Google Scholar]
- 18.Lee MK, Bae SH, Park CJ, Cheong HK, Cheong C, Choi BS. 2003. A single-nucleotide natural variation (U4 to C4) in an influenza A virus promoter exhibits a large structural change: implications for differential viral RNA synthesis by RNA-dependent RNA polymerase. Nucleic Acids Res 31:1216–1223. doi: 10.1093/nar/gkg214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu MT, Parvin JD, Gupta S, Krystal M, Palese P. 1987. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc Natl Acad Sci U S A 84:8140–8144. doi: 10.1073/pnas.84.22.8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fodor E, Pritlove DC, Brownlee GG. 1994. The influenza virus panhandle is involved in the initiation of transcription. J Virol 68:4092–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flick R, Neumann G, Hoffmann E, Neumeier E, Hobom G. 1996. Promoter elements in the influenza vRNA terminal structure. RNA 2:1046–1057. [PMC free article] [PubMed] [Google Scholar]
- 22.Flick R, Hobom G. 1999. Interaction of influenza virus polymerase with viral RNA in the ‘corkscrew’ conformation. J Gen Virol 80:2565–2572. doi: 10.1099/0022-1317-80-10-2565. [DOI] [PubMed] [Google Scholar]
- 23.Pflug A, Guilligay D, Reich S, Cusack S. 2014. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 516:355–360. doi: 10.1038/nature14008. [DOI] [PubMed] [Google Scholar]
- 24.Tomescu AI, Robb NC, Hengrung N, Fodor E, Kapanidis AN. 2014. Single-molecule FRET reveals a corkscrew RNA structure for the polymerase-bound influenza virus promoter. Proc Natl Acad Sci U S A 111:3335–3342. doi: 10.1073/pnas.1406056111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee YS, Seong BL. 1996. Mutational analysis of influenza B virus RNA transcription in vitro. J Virol 70:1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reich S, Guilligay D, Pflug A, Malet H, Berger I, Crépin T, Hart D, Lunardi T, Nanao M, Ruigrok RWH, Cusack S. 2014. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 516:361–366. doi: 10.1038/nature14009. [DOI] [PubMed] [Google Scholar]
- 27.Reich S, Guilligay D, Cusack S. 2017. An in vitro fluorescence based study of initiation of RNA synthesis by influenza B polymerase. Nucleic Acids Res 45:3353–3368. doi: 10.1093/nar/gkx043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thierry E, Guilligay D, Kosinski J, Bock T, Gaudon S, Round A, Pflug A, Hengrung N, El Omari K, Baudin F, Hart DJ, Beck M, Cusack S. 2016. Influenza polymerase can adopt an alternative configuration involving a radical repacking of PB2 domains. Mol Cell 61:125–137. doi: 10.1016/j.molcel.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Wit E, Bestebroer TM, Spronken MI, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2007. Rapid sequencing of the non-coding regions of influenza A virus. J Virol Methods 139:85–89. doi: 10.1016/j.jviromet.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Lee CW. 2009. Sequencing and mutational analysis of the non-coding regions of influenza A virus. Vet Microbiol 135:239–247. doi: 10.1016/j.vetmic.2008.09.067. [DOI] [PubMed] [Google Scholar]
- 32.Wang R, Taubenberger JK. 2014. Characterization of the noncoding regions of the 1918 influenza A H1N1 virus. J Virol 88:1815–1818. doi: 10.1128/JVI.03098-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaw MW, Lamb RA, Erickson BW, Briedis DJ, Choppin PW. 1982. Complete nucleotide sequence of the neuraminidase gene of influenza B virus. Proc Natl Acad Sci U S A 79:6817–6821. doi: 10.1073/pnas.79.22.6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Londo DR, Davis AR, Nayak DP. 1983. Complete nucleotide sequence of the nucleoprotein gene of influenza B virus. J Virol 47:642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffmann E, Mahmood K, Yang CF, Webster RG, Greenberg HB, Kemble G. 2002. Rescue of influenza B virus from eight plasmids. Proc Natl Acad Sci U S A 99:11411–11416. doi: 10.1073/pnas.172393399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Briedis DJ, Lamb RA. 1982. Influenza B virus genome: sequences and structural organization of RNA segment 8 and the mRNAs coding for the NS1 and NS2 proteins. J Virol 42:186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deborde DC, Donabedian AM, Herlocher ML, Naeve CW, Maassab HF. 1988. Sequence comparison of wild-type and cold-adapted B/Ann Arbor/1/66 influenza virus genes. Virology 163:429–443. doi: 10.1016/0042-6822(88)90284-x. [DOI] [PubMed] [Google Scholar]
- 38.Crescenzo-Chaigne B, Naffakh N, van der Werf S. 1999. Comparative analysis of the ability of the polymerase complexes of influenza viruses type A, B and C to assemble into functional RNPs that allow expression and replication of heterotypic model RNA templates in vivo. Virology 265:342–353. doi: 10.1006/viro.1999.0059. [DOI] [PubMed] [Google Scholar]
- 39.Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, Brownlee GG. 2002. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol 76:8989–9001. doi: 10.1128/jvi.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brownlee GG, Sharps JL. 2002. The RNA polymerase of influenza A virus is stabilized by interaction with its viral RNA promoter. J Virol 76:7103–7113. doi: 10.1128/jvi.76.14.7103-7113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robb NC, Te Velthuis AJ, Wieneke R, Tampe R, Cordes T, Fodor E, Kapanidis AN. 2016. Single-molecule FRET reveals the pre-initiation and initiation conformations of influenza virus promoter RNA. Nucleic Acids Res 44:10304–10315. doi: 10.1093/nar/gkw884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng Q, Liu Y, Peng R, Wang M, Yang W, Song H, Chen Y, Liu S, Han M, Zhang X, Wang P, Yan J, Zhang B, Qi J, Deng T, Gao GF, Shi Y. 2019. Structural insight into RNA synthesis by influenza D polymerase. Nat Microbiol doi: 10.1038/s41564-019-0487-5. [DOI] [PubMed] [Google Scholar]
- 43.Te Velthuis AJW, Oymans J. 2018. Initiation, elongation and realignment during influenza virus mRNA synthesis. J Virol 92:e01775-17. doi: 10.1128/JVI.01775-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neumann G, Hobom G. 1995. Mutational analysis of influenza virus promoter elements in vivo. J Gen Virol 76:1709–1717. doi: 10.1099/0022-1317-76-7-1709. [DOI] [PubMed] [Google Scholar]
- 45.Crescenzo-Chaigne B, van der Werf S, Naffakh N. 2002. Differential effect of nucleotide substitutions in the 3′ arm of the influenza A virus vRNA promoter on transcription/replication by avian and human polymerase complexes is related to the nature of PB2 amino acid 627. Virology 303:240–252. doi: 10.1006/viro.2002.1637. [DOI] [PubMed] [Google Scholar]
- 46.Leahy MB, Dobbyn HC, Brownlee GG. 2001. Hairpin loop structure in the 3′ arm of the influenza A virus virion RNA promoter is required for endonuclease activity. J Virol 75:7042–7049. doi: 10.1128/JVI.75.15.7042-7049.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergmann M, Muster T. 1995. The relative amount of an influenza A virus segment present in the viral particle is not affected by a reduction in replication of that segment. J Gen Virol 76:3211–3215. doi: 10.1099/0022-1317-76-12-3211. [DOI] [PubMed] [Google Scholar]
- 48.Zhao L, Peng Y, Zhou K, Cao M, Wang J, Wang X, Jiang T, Deng T. 2014. New insights into the nonconserved noncoding region of the subtype-determinant hemagglutinin and neuraminidase segments of influenza A viruses. J Virol 88:11493–11503. doi: 10.1128/JVI.01337-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng T, Sharps J, Fodor E, Brownlee GG. 2005. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J Virol 79:8669–8674. doi: 10.1128/JVI.79.13.8669-8674.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A 96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szymkowiak C, Kwan WS, Qin S, Toner TJ, Shaw AR, Youil R. 2003. Rapid method for the characterization of 3′ and 5′ UTRs of influenza viruses. J Virol Methods 107:15–20. doi: 10.1016/S0166-0934(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 53.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]