

Glycoprotein K (gK) is an essential and highly conserved HSV-1 protein. Previously, we reported that gK binds to SPP, an endoplasmic reticulum (ER) protein, and blocking this binding reduces virus infectivity in vitro and also affects gK and UL20 subcellular localization. To evaluate the function of gK binding to SPP in vivo, we generated SPP-inducible knockout mice and observed the following in the absence of SPP: (i) that significantly less HSV-1 replication was seen in ocularly infected mice than in control mice; (ii) that expression of various HSV-1 genes and cellular infiltrates in the eye and trigeminal ganglia of infected mice was less than that in control mice; and (iii) that latency was significantly reduced in infected mice. Thus, blocking of gK binding to SPP may be a useful tool to control HSV-1-induced eye disease in patients with herpes stromal keratitis (HSK).
KEYWORDS: inducible knockout, tamoxifen, ocular infection, virus replication, viral transcripts, infiltrates, HSV-1, SPP
ABSTRACT
We previously reported that herpes simplex virus (HSV) glycoprotein K (gK) binds to signal peptide peptidase (SPP), also known as minor histocompatibility antigen H13. Binding of gK to SPP is required for HSV-1 infectivity in vitro. SPP is a member of the γ-secretase family, and mice lacking SPP are embryonic lethal. To determine how SPP affects HSV-1 infectivity in vivo, the SPP gene was deleted using a tamoxifen-inducible Cre recombinase driven by the ubiquitously expressed ROSA26 promoter. SPP mRNA was reduced by more than 93% in the cornea and trigeminal ganglia (TG) and by 99% in the liver of tamoxifen-injected mice, while SPP protein expression was reduced by 90% compared to the level in control mice. Mice lacking SPP had significantly less HSV-1 replication in the eye as well as reduced gK, UL20, ICP0, and gB transcripts in the cornea and TG compared to levels in control mice. In addition, reduced infiltration of CD45+, CD4+, CD8+, F4/80+, CD11c+, and NK1.1+ T cells was observed in the cornea and TG of SPP-inducible knockout mice compared to that in control mice. Finally, in the absence of SPP, latency was significantly reduced in SPP-inducible knockout mice compared to that in control mice. Thus, in this study we have generated SPP-inducible knockout mice and shown that the absence of SPP affects virus replication in the eye of ocularly infected mice and that this reduction is correlated with the interaction of gK and SPP. These results suggest that blocking this interaction may have therapeutic potential in treating HSV-1-associated eye disease.
IMPORTANCE Glycoprotein K (gK) is an essential and highly conserved HSV-1 protein. Previously, we reported that gK binds to SPP, an endoplasmic reticulum (ER) protein, and blocking this binding reduces virus infectivity in vitro and also affects gK and UL20 subcellular localization. To evaluate the function of gK binding to SPP in vivo, we generated SPP-inducible knockout mice and observed the following in the absence of SPP: (i) that significantly less HSV-1 replication was seen in ocularly infected mice than in control mice; (ii) that expression of various HSV-1 genes and cellular infiltrates in the eye and trigeminal ganglia of infected mice was less than that in control mice; and (iii) that latency was significantly reduced in infected mice. Thus, blocking of gK binding to SPP may be a useful tool to control HSV-1-induced eye disease in patients with herpes stromal keratitis (HSK).
INTRODUCTION
Herpes simplex virus 1 (HSV-1) encodes at least 80 genes, 12 of which encode glycoproteins (1–6), and one of these 12 glycoproteins is glycoprotein K (gK) (1, 3, 7). gK is a highly hydrophobic protein, with approximately 84% amino acid homology between HSV-1 and HSV-2 (1, 8, 9). HSV-1 UL20 binds to gK, and this binding is required for cell surface expression of gK (10). We have previously demonstrated that immunization of mice with gK, but not with any other HSV-1 glycoprotein, exacerbated corneal scarring (CS) and herpetic dermatitis following ocular HSV-1 infection of immunized mice (4, 6, 11). This exacerbation of eye disease did not depend on the strain of mice or strain of virus. We also reported that individuals with herpes stromal keratitis (HSK) had higher anti-gK antibody titers than seropositive individuals with no history of HSK despite having similar neutralizing antibody and anti-gD antibody titers (12).
We have also reported that a recombinant HSV-1 expressing two additional copies of the gK gene exacerbated CS in two different mouse strains (13). An 8-amino-acid peptide (ITAYGLVL) within the gK signal sequence functions as an immunodominant T-cell stimulatory sequence in vitro and in vivo (14). The eyes of ocularly infected BALB/c mice, C57BL/6 mice, and NZW rabbits displayed significantly more virus replication and greater virus-induced CS following peptide administration in an eye drop (15). Moreover, in HSV-infected humanized HLA-A*0201 transgenic mice, this gK 8-mer epitope induced strong gamma interferon (IFN-γ)-producing cytotoxic CD8+ T-cell responses (15). This 8-mer upregulates CD8+ CD25+ T-cell responses in the cornea of infected mice, leading to exacerbation of CS (14–16).
In genome-wide screening of both HSV-1 (17) and HSV-2 (18), gK elicited CD8+ IFN-γ responses in mice and humans, respectively, and depletion of CD8+ T cells in gK-immunized mice reduced the exacerbation of gK-induced CS in ocularly infected mice (19). Thus, in addition to being an essential HSV-1 gene, gK also plays a pathogenic role in primary and latent HSV infection. gK binding to HSV-1 UL20 was previously shown using two-hybrid and pulldown assays, and we have now shown that gK, but no other HSV-1 genes, specifically binds to signal peptide peptidase (SPP) (20). SPP dominant negative mutants and a short hairpin RNA (shRNA) against SPP significantly reduced HSV-1 replication in vitro (20), while SPP inhibitors reduced virus infectivity in vivo (21).
SPP, also known as minor histocompatibility antigen H13, cleaves signal peptides after their release by signal peptidase (SP) (22, 23). SPP and SPP-like proteins are evolutionarily conserved among different species (24–27), and mouse and human SPPs have 96% amino acid homology (28). SPP is an endoplasmic reticulum (ER) protein (29) that can cleave signal peptides in the absence of protein cofactors (22, 30, 31) and also plays a major role in cellular signaling events such as in major histocompatibility complex class I (MHC-I) signal peptide processing (23). In addition to its role in intracellular and extracellular signaling, SPP is also implicated in pathogenic human conditions, including Alzheimer’s (32), cancers (33), and certain infectious diseases (34–38).
Based on our previously published study, SPP may also play a significant role in HSK (20, 21). However, SPP is an essential gene, and mice lacking SPP are embryonic lethal (39). Thus, to evaluate the role of SPP in HSV-1 infectivity, we generated an SPP-inducible knockout (KO) mouse strain using a tamoxifen-inducible Cre recombinase driven by the ubiquitously expressed Rosa26 promoter. Before and after tamoxifen treatments, these mice had no visible abnormalities. In the present study, we investigated the role of SPP in HSV-1 infectivity using SPP-inducible knockout mice. Here, we show that in the absence of SPP, the following were observed: (i) virus replication was significantly reduced in the eye of infected mice; (ii) viral transcripts in the cornea and trigeminal ganglia (TG) of infected mice were similar to those of control mice; (iii) the cornea and TG of SPP-inducible knockout mice had less cellular infiltrates than those of control mice; and (iv) latency in ocularly infected mice was significantly less than that in control mice. Thus, blocking SPP binding to gK may present a clinically effective and expedient approach to reducing viral replication as well as its resulting pathology.
RESULTS
Generation of SPP knockout mice.
We previously reported that HSV-1 gK binds to SPP and that this binding is required for HSV-1 infectivity in vitro (20). In addition, we have shown that inhibitors of SPP reduced virus infectivity and eye disease in vivo (21). To determine the effect of SPP on HSV-1 infectivity in ocularly infected mice in vivo, we first obtained heterozygote SPP mice (known as H13) from the European mouse mutant cell repository and tried to generate SPP knockout mice by crossing them to generate homozygote SPP knockout mice. However, the breeding results showed that SPP−/− homozygous mice are embryonically lethal as reported previously (39). To address this issue, the SPP gene was knocked out in mice using a tamoxifen-inducible Cre recombinase driven by the ubiquitously expressed Rosa26 promoter, as described in Materials and Methods (Fig. 1A). Starting at 2 weeks after birth, pups were treated with tamoxifen as described in Materials and Methods, and SPP mRNA expression was measured by reverse transcription-PCR (RT-PCR) in the cornea, TG, and liver of treated mice at various times post-tamoxifen treatment (Fig. 1B). After three tamoxifen injections, SPP mRNA levels were 20% of that in control mice (data not shown). However, after seven tamoxifen injections, SPP mRNA levels were reduced to only 10% of control levels in TG, 15% of control levels in the cornea, and less than 1% of control levels in the liver of tamoxifen-treated mice (Fig. 1B). We also compared the effect of tamoxifen treatment on SPP protein expression by Western blotting in the TG and cornea of treated and control mice (Fig. 1C). We observed SPP expression in tamoxifen-treated mice that was less than 10% of that seen in untreated controls (Fig. 1C). At early times post-tamoxifen treatment, treated mice showed some stunted growth, but by 6 weeks of age SPP-inducible knockout and control untreated mice were similar in size (data not shown).
FIG 1.

Schematic diagram of development of SPP conditional mice using tamoxifen induction in vivo. (A) Schematic view of tamoxifen-inducible SPP knockout mice generation. SPPflox/flox mice were generated by crossing insertion deletion SPP KO (heterozygous SPP+/−) mice with FLPo mice to remove the FRT cassette. SPPflox/flox mice were then bred with CreERT2 mice to generate SPPflox/flox CreERT2+/+ mice, in which SPP can be knocked out by tamoxifen induction of Cre recombinase. (B) SPPflox/flox CreERT2+/+ mice were treated with tamoxifen or corn oil as a control. Cornea, TG, and liver were harvested after tamoxifen injection, and SPP mRNA levels were measured by qRT-PCR. (C) WT mice or SPPflox/flox CreERT2+/+ mice were treated as described above; TG and corneas were harvested, and SPP protein levels in cornea or TG were analyzed by Western blotting using anti-SPP antibody.
Reduced virus replication in the eyes of ocularly infected SPP-inducible knockout mice.
To determine if the absence of SPP affected the rate of infectious virus clearance from HSV-1-infected eye, SPP-inducible knockout mice (tamoxifen treated) and control (corn oil treated) mice were ocularly infected with HSV-1 strain McKrae. Tear films were collected from 56 eyes/group in two separate experiments on days 1, 2, 3, 4, and 5 postinfection (p.i.), and the presence of infectious virus was determined by plaque assays (Fig. 2). Virus titers in the eyes of the two groups of infected mice were similar on days 1 and 2 p.i.; however, from days 3 to 5 p.i., the amount of infectious virus in the eyes of control mice was significantly higher than that in the eyes of SPP-inducible knockout mice (Fig. 2) (P < 0.001). The similar levels of virus in wild-type (WT) and SPP KO mouse corneas on days 1 and 2 p.i. are likely a consequence of the input virus. The reduced level of virus in the eyes of SPP KO mice on days 3 and 4, on the other hand, is likely caused by the lack of SPP. Thus, the absence of SPP in SPP-inducible knockout mice was associated with reduced virus replication in the eyes of infected mice. This result is consistent with our previous work showing that when the binding of SPP to gK is blocked using SPP dominant negative mutants or shRNA against SPP, in vitro HSV-1 replication is significantly reduced (20).
FIG 2.
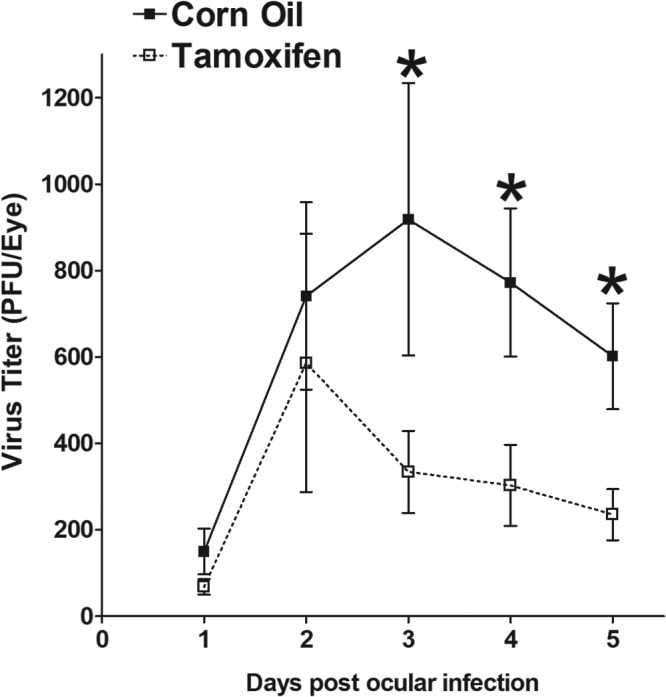
Virus titers in the eyes of SPP-inducible knockout mice. SPPflox/flox CreERT2+/+ mice were treated with tamoxifen or corn oil as described in Materials and Methods. Treated mice were ocularly infected with 2 × 105 PFU/eye of HSV-1 strain McKrae. Tear films were collected on days 1 to 5, and virus titers were determined by standard plaque assays. Each point represents the mean titer of 56 eyes from two separate experiments.
Expression of gK, UL20, ICP0, and gB transcripts is altered during primary ocular infection of SPP-inducible knockout mice.
Because gK binds to both SPP and UL20, we asked whether the absence of SPP affects the levels of gK and UL20 transcripts in vivo by infecting SPP-inducible knockout and control mice with 2 × 105 PFU/eye of HSV-1 strain McKrae. The corneas and TGs were collected on days 3 and 5 p.i., and total RNA was isolated and analyzed by TaqMan RT-PCR to determine the copy numbers for gK and UL20 mRNAs. We also determined the effect of inducible SPP knockout on ICP0 and gB viral transcripts as infection controls, using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA in each sample as an internal control. The results showed different levels of each transcript in corneas and TG of infected mice with and without SPP (Fig. 3). On day 3 p.i., levels of gK (Fig. 3A), UL20 (Fig. 3B), ICP0 (Fig. 3C), and gB (Fig. 3D) transcripts were significantly higher in corneas and TG of corn oil-treated control mice than in tamoxifen-treated SPP-inducible knockout mice (P < 0.01). However, by day 5 p.i., gK transcript levels in corneas and TG of mice with and without SPP were similar (Fig. 3A) (day 5, P > 0.05), while day 5 UL20 (Fig. 3B) and ICP0 (Fig. 3C) transcripts in the corneas and TG of mice with and without SPP had increased above the levels of day 3 p.i. (Fig. 3, compare day 3 versus day 5). However, levels of both transcripts in corneas but not TG of mice without SPP were significantly lower than those in control mice (Fig. 3) (P < 0.05). Finally, gB transcripts declined on day 5 p.i. in corneas and TG of both mouse groups, with no statistically significant difference between levels in mice with and without SPP (Fig. 3C) (P > 0.05).
FIG 3.

Expression of gK, UL20, ICP0, and gB transcripts in cornea and TG of SPP-inducible knockout and ocularly infected mice. Tamoxifen- or corn oil-treated SPPflox/flox CreERT2+/+ mice were ocularly infected with 2 × 105 PFU/eye of HSV-1 strain McKrae. The levels of gK, UL20, ICP0, and gB transcripts in the cornea and TG were determined on days 3 and 5 p.i. by quantitative RT-PCR. In each experiment, the estimated relative copy numbers of HSV-1 UL20, gK, and gB were calculated using standard curves generated from pUL20 (54), pAC-gB1 (75), pAc-gK1 (3), and pcDNA3.1-ICP0 (76). Briefly, a DNA template, serially diluted 10-fold such that 5 μl contained from 103 to 1011 copies of each plasmid, was subjected to TaqMan PCR with the same primer set. The copy number for each reaction product was determined by comparing the CT of each sample to the threshold cycle of the standard. GAPDH expression was used to normalize the relative expression of each transcript in the cornea and TG of infected mice. Each bar represents the mean ± standard errors of the mean from five mouse corneas or TG, as follows: gK (A), UL20 (B), ICP0 (C), and gB (D).
Collectively, these results indicated that in the absence of SPP, gK and UL20 transcripts were time and tissue independent, whereas ICP0 and gB transcripts were time and tissue dependent.
Absence of SPP in SPP-inducible knockout mice alters cellular transcript expression in cornea and TG of infected mice.
We have previously shown that virus replication directly correlates with the presence of immune infiltrates in corneas of infected mice (40–43). This result suggests that mice lacking SPP have less virus replication and fewer viral transcripts in their corneas and TG (Fig. 2 and 3). To determine whether lower virus replication in the absence of SPP affects cellular transcript levels in corneas and TG of infected mice, mice with and without SPP were infected ocularly as described above. Corneas and TG were collected on days 3 and 5 p.i., total RNA extract was isolated, and relative levels of CD45, F4/80, CD11c, CD4, CD8, and NK1.1 were determined using TaqMan RT-PCR. The results are presented as fold increase (or decrease) compared to baseline mRNA levels in corneas and TG of uninfected naive mice for each group. GAPDH mRNA in each sample was used as an internal control. On day 3 p.i., levels of CD45, CD11c, CD8, and NK1.1 were similar in mice with and without SPP (Fig. 4A) (P > 0.05), while mice without SPP (tamoxifen induced) had lower levels of F4/80 and CD4 expression than corn oil-treated control mice (Fig. 4A) (P < 0.01). In TG of infected mice on day 3 p.i., no differences were detected in CD45, F4/80, or CD4 expression levels (Fig. 4B) (P > 0.05), while mice without SPP (tamoxifen induced) had significantly lower levels of CD11c, CD8, and NK1.1 transcripts (Fig. 4B) (P < 0.01).
FIG 4.

Effect of SPP depletion on various transcripts in corneas and TG of infected mice. Tamoxifen- or corn oil-treated SPPflox/flox CreERT2+/+ mice were ocularly infected with 2 × 105 PFU/eye of HSV-1 strains McKrae. Corneas and TG from infected mice on days 3 and 5 p.i. were isolated, and quantitative RT-PCR was performed using total RNA, as described in Materials and Methods. CD45, CD4, CD8, CD11c, F4/80, and NK1.1 expression in corneas and TG of naive mice was used as a baseline to estimate the relative expression of each transcript in corneas and TG of infected mice. GAPDH expression was used to normalize the relative expression of each transcript. Each point represents the mean ± standard error of the mean from five mouse corneas or TG. Data represent fold change in CD45, CD4, CD8, CD11c, F4/80, and NK1.1 expression levels in corneas and TG of infected mice on day 3 p.i. (panels A and B, respectively) and on day 5 p.i. (panels C and D, respectively).
In corneas of infected mice by day 5 p.i., CD45 and F4/80 transcript levels were similar in mice with and without (tamoxifen induced) SPP (Fig. 4C) (P > 0.05), while mice without SPP (tamoxifen induced) had significantly lower levels of CD11c, CD4, CD8, and NK1.1 transcripts than corn oil-treated control mice (Fig. 4C) (P < 0.01). On day 5 p.i., levels of CD45, F4/80, CD11c, CD4, CD8, and NK1.1 transcripts in TG of SPP-inducible knockout mice were significantly lower than those in corn oil-treated control mice (Fig. 4D) (P < 0.01). These results suggest that the level of all cellular transcripts increases from day 3 p.i. to day 5 p.i. and that this increase is more pronounced in control mice than in mice lacking SPP (tamoxifen induced).
To confirm our RT-PCR results with regard to the presence of CD45, F4/80, CD11c, CD4, and CD8 transcripts in corneas and TG of infected mice, mice lacking SPP (tamoxifen induced) and corn oil-treated control mice were infected as described above. Corneas and TG from four mice per group were isolated on day 5 p.i., combined, and analyzed by fluorescence-activated cell sorting (FACS) for the presence of CD45, F4/80, CD11c, CD4, and CD8 infiltrates (Fig. 5). Except for the level of CD8 infiltrates in corneas of infected mice that was not similar to the level of CD8 mRNA, the levels of CD45, F4/80, CD11c, and CD4 infiltrates were lower in TG (Fig. 5) and corneas (Fig. 5) of mice lacking SPP (tamoxifen) than in corn oil-treated control mice. These results suggest that the absence of SPP significantly reduced the recruitment of cellular infiltrates into corneas and TG of infected mice in a time-dependent manner and that this reduction directly correlated with lower virus replication in mice lacking SPP (tamoxifen induced).
FIG 5.

Effect of SPP depletion on various infiltrates in corneas and TG of infected mice on day 5 p.i. Tamoxifen- or corn oil-treated SPPflox/flox CreERT2+/+ mice were ocularly infected with HSV-1 McKrae. Corneas and TG from four mice were harvested on day 5 p.i., combined, dissociated to single cells, and then stained with anti-CD4, anti-CD8, anti-CD11c, anti-CD45, or anti-F4/80 antibodies. Positive cell populations were analyzed by flow cytometry, as indicated.
Latency is reduced in the TG of latently infected mice lacking SPP.
In these experiments, mice lacking SPP (tamoxifen induced) and corn oil-treated control mice were infected ocularly with 2 × 105 PFU/eye of HSV-1 strain McKrae as described above, and individual TG from the surviving mice were isolated on day 28 p.i. Total DNA was isolated, and TaqMan qPCR was used to quantify gB DNA copy number. Cellular GAPDH DNA was used as an internal control. The amount of gB DNA during latency in mice lacking SPP (tamoxifen induced) was significantly lower than that in corn oil-treated control mice (Fig. 6) (P = 0.04, Student's t test). These results suggest that in the absence of SPP, latency in the TG of HSV-1-infected mice is reduced, which correlates with lower virus replication in infected mice.
FIG 6.

Effect of SPP deficiency on HSV-1 latency in TG of latently infected mice. Tamoxifen- or corn oil-treated SPPflox/flox CreERT2+/+ mice were ocularly infected with HSV-1 strain McKrae as described in the legend of Fig. 3. TG were harvested during latency on day 28 p.i., and qPCR was performed on individual TG for gB DNA. The estimated relative copy number of gB was calculated using standard curves generated from pAC-gB1, as described above. GAPDH expression was used to normalize relative expression in the TG. Each point represents the mean ± standard error of the mean from 28 TG.
DISCUSSION
HSV-1 causes a serious eye infection that can lead to blindness (44–47). Ocular HSV-1-infection can cause HSK, also referred to as CS (6, 47, 48). HSV-1-induced HSK is the result of immune responses triggered by the virus (48–51). The increase in cellular infiltrates induced by viral gene expression in the eye contributes to the pathology associated with ocular HSV-1 infection. Our published studies have shown that the HSV-1 gK gene, one of more than HSV-1 80 genes, contributes to this pathology, independent of virus or mouse strain (4, 6, 12–15). Thus, as we previously reported, gK might exert its pathogenic functions by interacting with SPP (20, 21). We previously demonstrated that gK binds to SPP and that SPP is required for HSV-1 infectivity in vitro (20). In addition to SPP, gK also binds to HSV-1 gB and UL20 (52, 53). However, our previous work ruled out the possibility of gK binding to any cellular protein other than SPP and of SPP binding to any other HSV-1 gene (21). We have also shown that, similar to the interaction of gK and SPP, UL20, another gK binding partner, binds to a host protein encoded by the zinc finger DHHC-type containing 3 (ZDHHC3) gene (also known as Golgi-specific DHHC zinc finger protein [GODZ]) (54). The binding of gK and GODZ is required for cell surface expression of gK, and blocking the interaction of UL20 to GODZ affected virus replication in vitro. Thus, binding of gK to UL20 and SPP is required for proper localization and virus infectivity (21, 54).
Considering the interaction between gK and SPP, we have shown that gK overexpression in mice infected with recombinant HSV-1 expressing two additional copies of gK exhibit more severe disease than mice infected with parental virus carrying one copy of gK (13). Our current study focused on SPP in order to understand the importance of SPP interaction with gK and the pathology associated with gK. Our previous work using SPP dominant negative constructs and SPP shRNA demonstrated that SPP is essential for viral replication in vitro (21). However, in the absence of SPP-deficient mice, we could not extend our in vitro results to an in vivo study. To address the effect of SPP on virus replication in vivo, here we generated SPP-inducible knockout mice. Direct deletion of SPP was not possible because the SPP gene is essential, and mice lacking it are embryonically lethal (39). Thus, we used a tamoxifen-inducible Cre recombinase driven by the ubiquitously expressed Rosa26 promoter to delete the SPP gene in mice. Tamoxifen treatment had no obvious side effects on the SPP-inducible knockout mice at the dosage used. Mice lacking SPP showed early stunted growth, but by 6 weeks of age, no significant differences were detected between mice lacking SPP (tamoxifen induced), control mice, and wild-type C57BL/6 mice.
In this study, we investigated the in vivo effects of SPP using ocularly infected mice lacking SPP (tamoxifen induced). The absence of SPP significantly reduced virus replication in the eye. We also found significantly lower levels of gK, UL20, ICP0, and gB transcripts in the corneas and TG of infected mice lacking SPP (tamoxifen induced) than in control mice on days 3 and 5 p.i. Because SPPflox/flox CreERT2+/+ mice are in a C57BL/6 background, we included WT C57BL/6 mice as controls in some of our studies and found that WT mice behaved identically to untreated or corn oil-treated SPPflox/flox CreERT2+/+ mice (data not shown). Thus, without tamoxifen treatment, SPPflox/flox CreERT2+/+ mice behaved like WT C57BL/6 mice. Similarly, we recently reported that GODZ−/− infected mice, lacking GODZ expression, had less virus replication in the eye and less gK, UL20, and gB transcripts in the cornea than controls (42). Consistent with lower levels of viral transcripts in corneas and TG of mice lacking SPP (tamoxifen induced) than those of control mice, we also detected lower levels of immune infiltrates in corneas of mice lacking SPP (tamoxifen induced). These differences in lower levels of immune infiltrates in corneas of SPP-deficient mice are a result of a lack of SPP rather than a lower degree of infection in the eyes of infected mice. Similar to results in SPP-inducible knockout mice, following infection of GODZ−/− mice, low numbers of infiltrates were detected in corneas of infected mice compared with levels in control mice. Also similar to previous studies (11, 55), lower virus replication in the eye and TG of mice lacking SPP (tamoxifen induced) correlated with lower levels of immune infiltrates. Following ocular HSV-1 infection, the level of immune infiltration in the cornea is associated with the severity of eye disease; however, the exact identity of the immune responses that contribute to eye disease is an area of intense controversy (56–58). Thus, CS is most likely associated with the influx of infiltrates into the eye following ocular infection. Depending on the literature referenced, one can conclude that CS is caused by T cells (CD4+ and CD8+) (59–63) or macrophages, NK cells, or dendritic cells (64–69). Here, we found a direct correlation between reduced virus replication in the eye of mice lacking SPP (tamoxifen induced) and reduced cellular infiltration into cornea of infected mice. Less infiltration into the TG was also associated with lower latency.
The absence of SPP in infected mice lacking SPP (tamoxifen induced) did not affect their susceptibility to ocular infection with the virulent HSV-1 strain McKrae. We have previously shown a direct relationship between virus load and the duration of primary virus replication in the eye with the severity of CS in ocularly infected BALB/c mice (3, 4, 6). In contrast to BALB/c mice, which are highly susceptible to infection with the virulent HSV-1 strain McKrae, mice in the C57BL/6 background used in this study are refractory to CS. However, the level of eye disease following infection of these mice lacking SPP (tamoxifen induced) was similar to that in control mice. Overall, despite the importance of SPP in embryogenesis, the absence of SPP did not have any side effects in infected mice. However, the levels of latency differed significantly between mice lacking SPP (tamoxifen induced) and control mice. Lower levels of latency in the absence of SPP correlated with lower virus replication and lower infiltration into the eye of mice lacking SPP (tamoxifen induced) than in control mice. This study confirms our previous results showing a direct correlation between reduced virus replication in the eye and reduced latency in infected mice (70). Although by using seven injections of tamoxifen in this study we have reduced SPP mRNA and SPP protein expression in the corneas and TG of treated mice by more than 90%, we were not able to completely block SPP expression. Residual SPP expression may have contributed to mouse survival. In addition, we were not able to obtain sterile immunity in infected mice. To overcome the presence of residual SPP in SPP-inducible knockout mice, we are currently generating conditional SPP knockout mice in peripheral nerves and eyes rather than using inducible knockout mice.
In summary, we have provided the first evidence that the absence of SPP reduces virus replication and minimizes the influx of cell infiltrates into the corneas and TG of infected mice, thus reducing the pathology associated with gK and SPP interaction. We conclude that pathology associated with gK is due to its interaction with SPP. Consequently, blocking this gK-SPP interaction may have therapeutic potential in patients suffering from the devastating effects of HSK.
MATERIALS AND METHODS
Cells, viruses, and mice.
A rabbit skin (RS) cell line was cultured in minimal essential medium plus 5% fetal bovine serum and maintained as described previously (4). Triple-plaque-purified HSV-1 strain McKrae was grown in RS cell monolayers as described previously (71). HSV-1 strain McKrae is a highly virulent strain of virus that infects mice efficiently without corneal scarification. The SPP+/− mouse strain was generated at Cedars-Sinai using embryonic stem (ES) cells (ES cell clone identifier EPD0867_5_D04) from the Wellcome Trust Sanger Institute (Hinxton, Cambridge, U.K.). The SPP+/− parental strain was C57BL/6 mice. SPP−/− mice were embryonic lethal. SPPflox/flox mice were generated by crossing SPP+/− mice with Rosa26 FLPo mice (catalog no. 009086; Jackson Laboratory). SPPflox/flox mice were then crossed with Rosa26 CreERT2 mice (no. 008463; Jackson Laboratory) to generate SPPflox/flox CreERT2+/+ mice. WT C57BL/6 mice were used as controls and were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in-house at Cedars-Sinai Medical Center. All animal procedures were performed in strict accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research (https://www.arvo.org/About/policies/statement-for-the-use-of-animals-in-ophthalmic-and-vision-research/) and the NIH Guide for the Care and Use of Laboratory Animals (72). The animal research protocol was approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center (protocol no. 5374).
Primers for screening SPP knockout mice.
The following primers were used to screen SPP knockout mice: SPP forward primer, TGCCTCCCGTTTAAGAGACC; SPP reverse primer, GACTCATTCTCCCCGCTCTG. The wild-type allele product length is 605 bp, and the floxed allele product length is 688 bp. A CAS reverse primer, TCGTGGTATCGTTATGCGCC, was paired with the SPP forward primer to detect the SPP mutant allele. Primers used for CreERT2 were CTGGCTTCTGAGGACCG, CCGAAAATCTGTGGGAAGTC, CGTGATCTGCAACTCCAGTC, and AGGCAAATTTTGGTGTACGG, with product sizes of 198 bp (WT) and 150 bp (mutant), according to The Jackson Laboratory.
SPP depletion using tamoxifen.
To deplete SPP, SPPflox/flox CreERT2+/+ mice were injected with tamoxifen (Sigma-Aldrich, Saint Louis, MO) three times before weaning (days 14, 16, and 18 after birth) and four times after weaning (days 35, 36, 37, and 38 after birth). Tamoxifen was prepared at a concentration of 20 mg/ml in corn oil (Spectrum Chemical MFG Group, Gardena, CA) and used within 1 week of preparation. Tamoxifen treatment permanently cleaves flox-SPP, thus knocking out the SPP gene.
Ocular infection.
SPPflox/flox CreERT2+/+ mice (7 to 8 weeks old) treated with tamoxifen or corn oil were infected ocularly in both eyes with 2 × 105 PFU of HSV-1 strain McKrae per eye in 2 μl of tissue culture medium as an eye drop without corneal scarification, as we described previously (41, 42).
RNA extraction from corneas and TG.
SPPflox/flox CreERT2+/+ mice treated with tamoxifen or corn oil were ocularly infected with 2 × 105 PFU/eye of HSV-1 strain McKrae. Corneas and TG from infected mice were collected on days 3 and 5 postinfection. Isolated tissues were immersed in TRIzol reagent and stored at −80°C until they were processed. The corneas or TG from each animal were processed for RNA extraction as we described previously (41, 42). Isolated total RNA was reverse transcribed with random hexamer primers, and murine leukemia virus reverse transcriptase was provided in a High Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s recommendations.
TaqMan qRT-PCR.
The sequences of the gB, gK, UL20, and ICP0 custom-made TaqMan primer sets used in this study were as follows: for gB, 5′-AACGCGACGCACATCAAG-3′ (forward), 5′-CTGGTACGCGATCAGAAAGC-3′ (reverse), and the probe FAM-CAGCCGCAGTACTACC-3′ (where FAM is 6-carboxyfluorescein); for gK, 5′--3′ (forward), 5′-CAGGCGGGTAATTTTCGTGTAG-3′ (reverse), and the probe 5′-FAM-CAGGCCGCATCGTATC-3′; for UL20, 5′-CCATCGTCGGCTACTACGTTAC-3′ (forward), 5′-CGATCCCTCTTGATGTTAACGTACA-3′ (reverse), and the probe 5′-FAM-CCCGCACCGCCCAC-3′; and for ICP0, 5′CGGACACGGAACTGTTCGA-3′ (forward), 5′-CGCCCCCGCAACTG-3′ (reverse), and the probe 5′-FAM-CCCCATCCACGCCCTG-3′. GAPDH primers from Applied Biosystems (assay identifier [ID], m999999.15_G1) were used as an internal control. Other TaqMan primers from Applied Biosystems used in this study were CD4 (Mm00442754_m1), CD8 (Mm01182107_g1), CD45 (MMCD45-34EX), CD11c (Mm00498698_m1), F4/80 (Mm00802529_m1), NK1.1 (Mm00493202_m1), and SPP (Mm00468786_m1).
Quantitative PCR (qPCR) was performed using a TaqMan gene expression assay kit in 384-well plates on an ABI QuantStudio 5 system (Applied Biosystems, Foster City, CA). Copy numbers for gB, gK, UL20, and ICP0 were calculated using standard curves generated from pAc-gB1 (for gB), pGem-gK1040 (for gK), pcDNA-UL20 (for UL20), and pcDNA-ICP0 (for latency-associated transcript [LAT]). Briefly, each plasmid DNA template was serially diluted 10-fold so that 1 μl contained from 103 to 108 copies of the desired gene and was then subjected to TaqMan PCR with the same set of primers as the test samples. By comparing the normalized threshold cycle (CT) of each sample to the threshold cycle of the standard curve, the copy number for each reaction product was determined. For fold change of expression analysis, the 2−ΔΔCT method was used to calculate gene expression fold change compared to expression in uninfected controls.
Titration of viruses in tears.
Tear films were collected from both eyes of a mouse from days 1 to 5 postinfection, using a cotton applicator. Each swab was placed in 1 ml of tissue culture medium and stored at –80°C until processing. The amount of virus in the medium was determined by a standard plaque assay using RS cells as we described previously (41, 73).
DNA extraction and PCR analysis for HSV-1 latency.
SPPflox/flox CreERT2+/+ mice were treated and infected as described above. DNA was isolated from homogenized individual TG on day 28 p.i. using a commercially available DNeasy Blood and Tissue kit (catalog no. 69506; Qiagen, Stanford, CA), according to the manufacturer’s instructions. PCR analyses were done using gB-specific primers (forward, 5′-AACGCGACGCACATCAAG-3′; reverse, 5′-CTGGTACGCGATCAGAAAGC-3′; and probe, 5′-FAM-CAGCCGCAGTACTACC-3′). The amplicon length for this primer set is 72 bp. Relative gB DNA copy numbers were calculated using standard curves generated from the plasmid pAc-gB1. In all experiments GAPDH was used to normalize transcripts.
Flow cytometric analysis of infected cornea and TG.
SPPflox/flox CreERT2+/+ mice were treated and infected as described above. On day 5 p.i., mice were euthanized, and corneas and TG were harvested, cut into pieces, and digested with 2 mg/ml of Liberase (catalog no. 5401119001; Sigma-Aldrich) in Hanks’ balanced salt solution at 37°C for 1 h, as we described previously (74). The digested tissues were filtered through a 70-μm-pore-size filter to obtain a single-cell suspension. Cells were washed with 1× phosphate-buffered saline (PBS) and stained with Live/Dead Fixable Red Dye (catalog no. L34971; Thermo Fisher Scientific) on ice for 15 min in dark. After cells were washed with cell staining buffer (catalog no. 420201; BioLegend), they were blocked with Fc blocking antibody (catalog no. 553141; BD) on ice for 15 min, followed by incubation with antibody cocktails on ice, in the dark, for 30 min with Brilliant stain buffer (catalog no. 563794; BD Biosciences). The antibody cocktail used in this study includes following antibodies: (i) BV570 anti-mouse CD4 (clone RM4-5, catalog no. 100541; BioLegend); (ii) BV785 anti-mouse CD8 (clone 53-6.7, no. 100749); (iii) BV421 anti-mouse CD11c (clone N418, no. 117329; BioLegend); (iv) Alexa Fluor 532 anti-mouse CD45 (clone 30-F11, no. 58-0451-80; Thermo Fisher Scientific); and (v) BV711 anti-mouse F4/80 (clone BM8, no. 123147; BioLegend). After incubation, cells were washed twice with cold 1× PBS and subjected to flow cytometry analysis using an SA3800 Sony Spectral Analyzer (Sony Biotechnology, San Jose, CA). The flow analysis gate setting was as follows: the first gate of forward scatter (FSC) and side scatter (SSC) for size of lymphocyte population (spleen cells were used to setup gate of lymphocyte population) and the second gate for live cells.
Statistical analysis.
A Student's t test and Fisher exact test were performed using the computer program Instat (GraphPad, San Diego, CA). Results were considered statistically significant at a P value of <0.05.
ACKNOWLEDGMENT
This work was supported by NIH grant EY13615.
REFERENCES
- 1.McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol 69:1531–1574. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- 2.Spear PG, Eisenberg RJ, Cohen GH. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1–8. doi: 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- 3.Ghiasi H, Slanina S, Nesburn AB, Wechsler SL. 1994. Characterization of baculovirus-expressed herpes simplex virus type 1 glycoprotein K. J Virol 68:2347–2354. doi: 10.1007/BF01379126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghiasi H, Kaiwar R, Nesburn AB, Slanina S, Wechsler SL. 1994. Expression of seven herpes simplex virus type 1 glycoproteins (gB, gC, gD, gE, gG, gH, and gI): comparative protection against lethal challenge in mice. J Virol 68:2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghiasi H, Kaiwar R, Slanina S, Nesburn AB, Wechsler SL. 1994. Expression and characterization of baculovirus expressed herpes simplex virus type 1 glycoprotein L. Arch Virol 138:199–212. doi: 10.1007/bf01379126. [DOI] [PubMed] [Google Scholar]
- 6.Ghiasi H, Bahri S, Nesburn AB, Wechsler SL. 1995. Protection against herpes simplex virus-induced eye disease after vaccination with seven individually expressed herpes simplex virus 1 glycoproteins. Invest Ophthalmol Vis Sci 36:1352–1360. [PubMed] [Google Scholar]
- 7.Hutchinson L, Goldsmith K, Snoddy D, Ghosh H, Graham FL, Johnson DC. 1992. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J Virol 66:5603–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGeoch DJ, Cunningham C, McIntyre G, Dolan A. 1991. Comparative sequence analysis of the long repeat regions and adjoining parts of the long unique regions in the genomes of herpes simplex viruses types 1 and 2. J Gen Virol 72:3057–3075. doi: 10.1099/0022-1317-72-12-3057. [DOI] [PubMed] [Google Scholar]
- 9.Dolan A, Jamieson FE, Cunningham C, Barnett BC, McGeoch DJ. 1998. The genome sequence of herpes simplex virus type 2. J Virol 72:2010–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster TP, Melancon JM, Olivier TL, Kousoulas KG. 2004. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J Virol 78:13262–13277. doi: 10.1128/JVI.78.23.13262-13277.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koujah L, Suryawanshi RK, Shukla D. 2019. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell Mol Life Sci 76:405–419. doi: 10.1007/s00018-018-2938-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mott KR, Osorio Y, Maguen E, Nesburn AB, Wittek AE, Cai S, Chattopadhyay S, Ghiasi H. 2007. Role of anti-glycoproteins D (anti-gD) and K (anti-gK) IgGs in pathology of herpes stromal keratitis in humans. Invest Ophthalmol Vis Sci 48:2185–2193. doi: 10.1167/iovs.06-1276. [DOI] [PubMed] [Google Scholar]
- 13.Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J Virol 81:12962–12972. doi: 10.1128/JVI.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osorio Y, Mott KR, Jabbar AM, Moreno A, Foster TP, Kousoulas KG, Ghiasi H. 2007. Epitope mapping of HSV-1 glycoprotein K (gK) reveals a T cell epitope located within the signal domain of gK. Virus Res 128:71–80. doi: 10.1016/j.virusres.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mott KR, Chentoufi AA, Carpenter D, Benmohamed L, Wechsler SL, Ghiasi H. 2009. The role of a glycoprotein K (gK) CD8+ T-cell epitope of herpes simplex virus on virus replication and pathogenicity. Invest Ophthalmol Vis Sci 50:2903–2912. doi: 10.1167/iovs.08-2957. [DOI] [PubMed] [Google Scholar]
- 16.Allen SJ, Mott KR, Ljubimov AV, Ghiasi H. 2010. Exacerbation of corneal scarring in HSV-1 gK-immunized mice correlates with elevation of CD8+ CD25+ T cells in corneas of ocularly infected mice. Virology 399:11–22. doi: 10.1016/j.virol.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.St Leger AJ, Peters B, Sidney J, Sette A, Hendricks RL. 2011. Defining the herpes simplex virus-specific CD8+ T cell repertoire in C57BL/6 mice. J Immunol 186:3927–3933. doi: 10.4049/jimmunol.1003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jing L, Haas J, Chong TM, Bruckner JJ, Dann GC, Dong L, Marshak JO, McClurkan CL, Yamamoto TN, Bailer SM, Laing KJ, Wald A, Verjans GM, Koelle DM. 2012. Cross-presentation and genome-wide screening reveal candidate T cells antigens for a herpes simplex virus type 1 vaccine. J Clin Invest 122:654–673. doi: 10.1172/JCI60556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osorio Y, Cai S, Hofman FM, Brown DJ, Ghiasi H. 2004. Involvement of CD8+ T cells in exacerbation of corneal scarring in mice. Curr Eye Res 29:145–151. doi: 10.1080/02713680490504632. [DOI] [PubMed] [Google Scholar]
- 20.Allen SJ, Mott KR, Matsuura Y, Moriishi K, Kousoulas KG, Ghiasi H. 2014. Binding of HSV-1 glycoprotein K (gK) to signal peptide peptidase (SPP) is required for virus infectivity. PLoS One 9:e85360. doi: 10.1371/journal.pone.0085360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen SJ, Mott KR, Ghiasi H. 2014. Inhibitors of signal peptide peptidase (SPP) affect HSV-1 infectivity in vitro and in vivo. Exp Eye Res 123:8–15. doi: 10.1016/j.exer.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B. 2002. Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296:2215–2218. doi: 10.1126/science.1070925. [DOI] [PubMed] [Google Scholar]
- 23.Lemberg MK, Martoglio B. 2002. Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol Cell 10:735–744. doi: 10.1016/S1097-2765(02)00655-X. [DOI] [PubMed] [Google Scholar]
- 24.Sun H, Liu J, Ding F, Wang X, Liu M, Gu X. 2006. Investigation of differentially expressed proteins in rat gastrocnemius muscle during denervation-reinnervation. J Muscle Res Cell Motil 27:241–250. doi: 10.1007/s10974-006-9067-4. [DOI] [PubMed] [Google Scholar]
- 25.Bolhuis A, Matzen A, Hyyrylainen HL, Kontinen VP, Meima R, Chapuis J, Venema G, Bron S, Freudl R, van Dijl JM. 1999. Signal peptide peptidase- and ClpP-like proteins of Bacillus subtilis required for efficient translocation and processing of secretory proteins. J Biol Chem 274:24585–24592. doi: 10.1074/jbc.274.35.24585. [DOI] [PubMed] [Google Scholar]
- 26.Yan G, Zhang G, Fang X, Zhang Y, Li C, Ling F, Cooper DN, Li Q, Li Y, van Gool AJ, Du H, Chen J, Chen R, Zhang P, Huang Z, Thompson JR, Meng Y, Bai Y, Wang J, Zhuo M, Wang T, Huang Y, Wei L, Li J, Wang Z, Hu H, Yang P, Le L, Stenson PD, Li B, Liu X, Ball EV, An N, Huang Q, Zhang Y, Fan W, Zhang X, Li Y, Wang W, Katze MG, Su B, Nielsen R, Yang H, Wang J, Wang X, Wang J. 2011. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol 29:1019–1023. doi: 10.1038/nbt.1992. [DOI] [PubMed] [Google Scholar]
- 27.Golde TE, Wolfe MS, Greenbaum DC. 2009. Signal peptide peptidases: a family of intramembrane-cleaving proteases that cleave type 2 transmembrane proteins. Semin Cell Dev Biol 20:225–230. doi: 10.1016/j.semcdb.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Urny J, Hermans-Borgmeyer I, Gercken G, Schaller HC. 2003. Expression of the presenilin-like signal peptide peptidase (SPP) in mouse adult brain and during development. Gene Expr Patterns 3:685–691. doi: 10.1016/S1567-133X(03)00094-2. [DOI] [PubMed] [Google Scholar]
- 29.Grigorenko AP, Moliaka YK, Korovaitseva GI, Rogaev EI. 2002. Novel class of polytopic proteins with domains associated with putative protease activity. Biochemistry (Mosc) 67:826–835. doi: 10.1023/A:1016365227942. [DOI] [PubMed] [Google Scholar]
- 30.Sato T, Nyborg AC, Iwata N, Diehl TS, Saido TC, Golde TE, Wolfe MS. 2006. Signal peptide peptidase: biochemical properties and modulation by nonsteroidal antiinflammatory drugs. Biochemistry 45:8649–8656. doi: 10.1021/bi060597g. [DOI] [PubMed] [Google Scholar]
- 31.Narayanan S, Sato T, Wolfe MS. 2007. A C-terminal region of signal peptide peptidase defines a functional domain for intramembrane aspartic protease catalysis. J Biol Chem 282:20172–20179. doi: 10.1074/jbc.M701536200. [DOI] [PubMed] [Google Scholar]
- 32.Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, Wolfe MS. 2002. Activity-dependent isolation of the presenilin-gamma-secretase complex reveals nicastrin and a gamma substrate. Proc Natl Acad Sci U S A 99:2720–2725. doi: 10.1073/pnas.052436599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taniguchi Y, Kim SH, Sisodia SS. 2003. Presenilin-dependent “gamma-secretase” processing of deleted in colorectal cancer (DCC). J Biol Chem 278:30425–30428. doi: 10.1074/jbc.C300239200. [DOI] [PubMed] [Google Scholar]
- 34.Heimann M, Roman-Sosa G, Martoglio B, Thiel H-J, Rümenapf T. 2006. Core protein of pestiviruses is processed at the C terminus by signal peptide peptidase. J Virol 80:1915–1921. doi: 10.1128/JVI.80.4.1915-1921.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Chen H, Bahamontes-Rosa N, Kun JF, Traore B, Crompton PD, Chishti AH. 2009. Plasmodium falciparum signal peptide peptidase is a promising drug target against blood stage malaria. Biochem Biophys Res Commun 380:454–459. doi: 10.1016/j.bbrc.2009.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLauchlan J, Lemberg MK, Hope G, Martoglio B. 2002. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J 21:3980–3988. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okamoto K, Moriishi K, Miyamura T, Matsuura Y. 2004. Intramembrane proteolysis and endoplasmic reticulum retention of hepatitis C virus core protein. J Virol 78:6370–6380. doi: 10.1128/JVI.78.12.6370-6380.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loureiro J, Lilley BN, Spooner E, Noriega V, Tortorella D, Ploegh HL. 2006. Signal peptide peptidase is required for dislocation from the endoplasmic reticulum. Nature 441:894–897. doi: 10.1038/nature04830. [DOI] [PubMed] [Google Scholar]
- 39.Aizawa S, Okamoto T, Sugiyama Y, Kouwaki T, Ito A, Suzuki T, Ono C, Fukuhara T, Yamamoto M, Okochi M, Hiraga N, Imamura M, Chayama K, Suzuki R, Shoji I, Moriishi K, Moriya K, Koike K, Matsuura Y. 2016. TRC8-dependent degradation of hepatitis C virus immature core protein regulates viral propagation and pathogenesis. Nat Commun 7:11379. doi: 10.1038/ncomms11379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee DH, Ghiasi H. 2018. An M2 rather than a TH2 response contributes to better protection against latency reactivation following ocular infection of naive mice with a recombinant herpes simplex virus 1 expressing murine interleukin-4. J Virol 92:e00051-18. doi: 10.1128/JVI.00051-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang S, Ljubimov AV, Jin L, Pfeffer K, Kronenberg M, Ghiasi H. 2018. Herpes simplex virus 1 latency and the kinetics of reactivation are regulated by a complex network of interactions between the herpesvirus entry mediator, its ligands (gD, BTLA, LIGHT, and CD160), and the latency-associated transcript. J Virol 92:e01451-18. doi: 10.1128/JVI.01451-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S, Mott KR, Cilluffo M, Kilpatrick CL, Murakami S, Ljubimov AV, Kousoulas KG, Awasthi S, Luscher B, Ghiasi H. 2018. The absence of DHHC3 affects primary and latent herpes simplex virus 1 infection. J Virol 92:e01599-17. doi: 10.1128/JVI.01599-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee DH, Jaggi U, Ghiasi H. 2019. CCR2+ migratory macrophages with M1 status are the early-responders in the cornea of HSV-1 infected mice. PLoS One 14:e0215727. doi: 10.1371/journal.pone.0215727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barron BA, Gee L, Hauck WW, Kurinij N, Dawson CR, Jones DB, Wilhelmus KR, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA, for the Hepatic Eye Disease Study Group. 1994. Herpetic eye disease study. A controlled trial of oral acyclovir for herpes simplex stromal keratitis. Ophthalmology 101:1871–1882. doi: 10.1016/s0161-6420(13)31155-5. [DOI] [PubMed] [Google Scholar]
- 45.Wilhelmus KR, Dawson CR, Barron BA, Bacchetti P, Gee L, Jones DB, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA. 1996. Risk factors for herpes simplex virus epithelial keratitis recurring during treatment of stromal keratitis or iridocyclitis. Herpetic Eye Disease Study Group. Br J Ophthalmol 80:969–972. doi: 10.1136/bjo.80.11.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liesegang TJ. 1999. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 18:127–143. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 47.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 48.Dix RD. 2013. Pathogenesis of herpes simplex ocular disease, chapter 89, p 1–20. In Tasman W, Jaeger EA (ed), Duane’s foundations of clinical ophthalmology, 2nd ed Lippincott, Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 49.Metcalf JF, Kaufman HE. 1976. Herpetic stromal keratitis-evidence for cell-mediated immunopathogenesis. Am J Ophthalmol 82:827–834. doi: 10.1016/0002-9394(76)90057-x. [DOI] [PubMed] [Google Scholar]
- 50.Hendricks RL, Tumpey TM. 1990. Contribution of virus and immune factors to herpes simplex virus type I- induced corneal pathology. Invest Ophthalmol Vis Sci 31:1929–1939. [PubMed] [Google Scholar]
- 51.Brandt CR. 2005. The role of viral and host genes in corneal infection with herpes simplex virus type 1. Exp Eye Res 80:607–621. doi: 10.1016/j.exer.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 52.Chouljenko VN, Iyer AV, Chowdhury S, Kim J, Kousoulas KG. 2010. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J Virol 84:8596–8606. doi: 10.1128/JVI.00298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foster TP, Chouljenko VN, Kousoulas KG. 2008. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J Virol 82:6310–6323. doi: 10.1128/JVI.00147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang S, Mott KR, Wawrowsky K, Kousoulas KG, Luscher B, Ghiasi H. 2017. Binding of HSV-1 UL20 to GODZ affects its palmitoylation and is essential for infectivity and proper targeting and localization of UL20 and gK. J Virol 91:e00945-17. doi: 10.1128/JVI.00945-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaggi U, Wang S, Tormanen K, Matundan H, Ljubimov AV, Ghiasi H. 2018. Role of herpes simplex virus type 1 (HSV-1) glycoprotein K (gK) pathogenic CD8+ T cells in exacerbation of eye disease. Front Immunol 9:2895. doi: 10.3389/fimmu.2018.02895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Banerjee K, Deshpande S, Zheng M, Kumaraguru U, Schoenberger SP, Rouse BT. 2002. Herpetic stromal keratitis in the absence of viral antigen recognition. Cell Immunol 219:108–118. doi: 10.1016/s0008-8749(02)00601-9. [DOI] [PubMed] [Google Scholar]
- 57.Huster KM, Panoutsakopoulou V, Prince K, Sanchirico ME, Cantor H. 2002. T cell-dependent and -independent pathways to tissue destruction following herpes simplex virus-1 infection. Eur J Immunol 32:1414–1419. doi:. [DOI] [PubMed] [Google Scholar]
- 58.Zhao ZS, Granucci F, Yeh L, Schaffer PA, Cantor H. 1998. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science 279:1344–1347. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- 59.Hendricks RL, Janowicz M, Tumpey TM. 1992. Critical role of corneal Langerhans cells in the CD4- but not CD8- mediated immunopathology in herpes simplex virus-1-infected mouse corneas. J Immunol 148:2522–2529. [PubMed] [Google Scholar]
- 60.Ghiasi H, Roopenian DC, Slanina S, Cai S, Nesburn AB, Wechsler SL. 1997. The importance of MHC-I and MHC-II responses in vaccine efficacy against lethal herpes simplex virus type 1 challenge. Immunology 91:430–435. doi: 10.1046/j.1365-2567.1997.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mercadal CM, Bouley DM, DeStephano D, Rouse BT. 1993. Herpetic stromal keratitis in the reconstituted scid mouse model. J Virol 67:3404–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Doymaz MZ, Rouse BT. 1992. Herpetic stromal keratitis: an immunopathologic disease mediated by CD4+ T lymphocytes. Invest Ophthalmol Vis Sci 33:2165–2173. [PubMed] [Google Scholar]
- 63.Hendricks RL, Epstein RJ, Tumpey T. 1989. The effect of cellular immune tolerance to HSV-1 antigens on the immunopathology of HSV-1 keratitis. Invest Ophthalmol Vis Sci 30:105–115. [PubMed] [Google Scholar]
- 64.Tumpey TM, Cheng H, Cook DN, Smithies O, Oakes JE, Lausch RN. 1998. Absence of macrophage inflammatory protein-1α prevents the development of blinding herpes stromal keratitis. J Virol 72:3705–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tumpey TM, Cheng H, Yan XT, Oakes JE, Lausch RN. 1998. Chemokine synthesis in the HSV-1-infected cornea and its suppression by interleukin-10. J Leukoc Biol 63:486–492. doi: 10.1002/jlb.63.4.486. [DOI] [PubMed] [Google Scholar]
- 66.Oakes JE, Monteiro CA, Cubitt CL, Lausch RN. 1993. Induction of interleukin-8 gene expression is associated with herpes simplex virus infection of human corneal keratocytes but not human corneal epithelial cells. J Virol 67:4777–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brandt CR, Salkowski CA. 1992. Activation of NK cells in mice following corneal infection with herpes simplex virus type-1. Invest Ophthalmol Vis Sci 33:113–120. [PubMed] [Google Scholar]
- 68.Ghiasi H, Cai S, Perng GC, Nesburn AB, Wechsler SL. 2000. The role of natural killer cells in protection of mice against death and corneal scarring following ocular HSV-1 infection. Antiviral Res 45:33–45. doi: 10.1016/S0166-3542(99)00075-3. [DOI] [PubMed] [Google Scholar]
- 69.Allen SJ, Mott KR, Chentoufi AA, BenMohamed L, Wechsler SL, Ballantyne CM, Ghiasi H. 2011. CD11c controls herpes simplex virus 1 responses to limit virus replication during primary infection. J Virol 85:9945–9955. doi: 10.1128/JVI.05208-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matundan HH, Mott KR, Allen SJ, Wang S, Bresee CJ, Ghiasi YN, Town T, Wechsler SL, Ghiasi H. 2016. Interrelationship of Primary virus replication, level of latency, and time to reactivation in the trigeminal ganglia of latently infected mice. J Virol 90:9533–9542. doi: 10.1128/JVI.01373-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 73.Wang S, Hirose S, Ghiasi H. 2019. The absence of lymphotoxin-alpha, an HVEM ligand, affects HSV-1 infection in vivo differently than the absence of other HVEM cellular ligands. J Virol 93:e00707-19. doi: 10.1128/JVI.00707-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1992. Expression of herpes simplex virus type 1 glycoprotein B in insect cells. Initial analysis of its biochemical and immunological properties. Virus Res 22:25–39. doi: 10.1016/0168-1702(92)90087-P. [DOI] [PubMed] [Google Scholar]
- 76.Matundan H, Ghiasi H. 2018. Herpes simplex virus 1 ICP22 suppresses CD80 expression by murine dendritic cells. J Virol 93:e01803-18. doi: 10.1128/JVI.01803-18. [DOI] [PMC free article] [PubMed] [Google Scholar]