

Recent data indicate that intestinal bacteria promote intestinal infection of several enteric viruses. Here, we show that coxsackievirus, an enteric virus in the picornavirus family, also relies on microbiota for intestinal replication and pathogenesis. Relatively minor depletion of the microbiota was sufficient to decrease coxsackievirus infection, while poliovirus infection was unaffected. Surprisingly, a single dose of one antibiotic was sufficient to reduce coxsackievirus infection. Therefore, these data indicate that closely related viruses may differ with respect to their reliance on microbiota.
KEYWORDS: antibiotics, coxsackievirus, enteric viruses, microbiota, poliovirus, viral pathogenesis
ABSTRACT
Accumulating evidence suggests that intestinal bacteria promote enteric virus infection in mice. For example, previous work demonstrated that antibiotic treatment of mice prior to oral infection with poliovirus reduced viral replication and pathogenesis. Here, we examined the effect of antibiotic treatment on infection with coxsackievirus B3 (CVB3), a picornavirus closely related to poliovirus. We treated mice with a mixture of five antibiotics to deplete host microbiota and examined CVB3 replication and pathogenesis following oral inoculation. We found that, as seen with poliovirus, CVB3 shedding and pathogenesis were reduced in antibiotic-treated mice. While treatment with just two antibiotics, vancomycin and ampicillin, was sufficient to reduce CVB3 replication and pathogenesis, this treatment had no effect on poliovirus. The quantity and composition of bacterial communities were altered by treatment with the five-antibiotic cocktail and by treatment with vancomycin and ampicillin. To determine whether more-subtle changes in bacterial populations impact viral replication, we examined viral infection in mice treated with milder antibiotic regimens. Mice treated with one-tenth the standard concentration of the normal antibiotic cocktail supported replication of poliovirus but not CVB3. Importantly, a single dose of one antibiotic, streptomycin, was sufficient to reduce CVB3 shedding and pathogenesis while having no effect on poliovirus shedding and pathogenesis. Overall, replication and pathogenesis of CVB3 are more sensitive to antibiotic treatment than poliovirus, indicating that closely related viruses may differ with respect to their reliance on microbiota.
IMPORTANCE Recent data indicate that intestinal bacteria promote intestinal infection of several enteric viruses. Here, we show that coxsackievirus, an enteric virus in the picornavirus family, also relies on microbiota for intestinal replication and pathogenesis. Relatively minor depletion of the microbiota was sufficient to decrease coxsackievirus infection, while poliovirus infection was unaffected. Surprisingly, a single dose of one antibiotic was sufficient to reduce coxsackievirus infection. Therefore, these data indicate that closely related viruses may differ with respect to their reliance on microbiota.
INTRODUCTION
Enteric viruses are spread through the fecal-oral route and cause morbidity and mortality in humans worldwide (1–4). These viruses initiate infection in the gastrointestinal tract, which is home to a community of bacteria, fungi, and viruses termed the microbiota. The microbiota plays important roles in human development, metabolism, and immunity (5). We and others have shown that intestinal bacteria can enhance viral replication and pathogenesis of enteric viruses, including poliovirus, reovirus, rotavirus, mouse mammary tumor virus, and noroviruses (6–11). While the mechanisms underlying bacterial promotion of enteric virus replication are not entirely clear, several groups have shown that bacteria can enhance viral infection through effects on host immunity and/or effects on viral particles (12). For example, we have shown that bacterial surface polysaccharides bind to poliovirus, a member of the Picornaviridae, and enhance virion stability and cell attachment (13). However, it is not known whether bacteria influence the replication of other picornaviruses in vivo or whether different antibiotic regimens equally impact replication and pathogenesis of related viruses.
Coxsackievirus is an enteric virus in the Picornaviridae family that initiates infection in the gastrointestinal tract. Among enteric viruses, coxsackieviruses are very common and can cause hemorrhagic conjunctivitis; hand, foot, and mouth disease; and myocarditis (14–17). Coxsackievirus B3 (CVB3) shares 62.3% sequence identity with poliovirus, a closely related picornavirus. CVB3 is the most commonly isolated virus associated with viral myocarditis (18, 19), which can lead to heart disease and heart failure. Unfortunately, there are no vaccines or treatments for coxsackievirus infections.
Previously, we established an oral inoculation model for CVB3 to study viral replication within the gastrointestinal tract (20, 21), and here we examined whether bacteria influence coxsackievirus infection. Similarly to our observations described in a previously published study of poliovirus (9), we found that intestinal bacteria enhance CVB3 fecal shedding and pathogenesis. However, CVB3 and poliovirus differed with respect to their reliance on microbiota, since minimal depletion of the microbiota reduced replication and pathogenesis of CVB3 but not poliovirus. These data suggest that CVB3 is more sensitive than poliovirus to perturbations in the microbiota.
(This article was submitted to an online preprint archive [22].)
RESULTS
Intestinal bacteria enhance CVB3 replication and pathogenesis in vivo.
We previously determined that intestinal bacteria enhance poliovirus replication and pathogenesis in orally inoculated mice (9). To determine if bacteria enhance infection of another closely related enteric virus, we examined CVB3 replication and pathogenesis in antibiotic-treated mice. To facilitate direct comparison of poliovirus and CVB3, we performed these experiments in mice that are orally susceptible to both viruses, due to expression of the human poliovirus receptor (PVR) and lack of the interferon alpha/beta receptor (IFNAR) (C57BL/6 PVR+/+ IFNAR−/− mice). Our previous work established that, similarly to the results seen with poliovirus, immunocompetent mice orally inoculated with CVB3 shed relatively small amounts of virus and do not develop disease. However, orally inoculated IFNAR−/− mice shed larger amounts of CVB3 and succumb to disease via liver failure, with increased serum levels of alanine aminotransferase and liver pathology (20, 21). Here, following weeklong administration of a combination of ampicillin, neomycin, metronidazole, vancomycin, and streptomycin, mice were perorally inoculated with 5 × 107 PFU of CVB3. Feces specimens were collected at 24, 48, and 72 h postinoculation, and CVB3 titers were determined by plaque assay. Similarly to our observations described in a previously published study of poliovirus (9), we found that mice treated with antibiotics showed reduced CVB3 shedding at 48 and 72 h postinoculation (Fig. 1A), reduced CVB3 titers in tissues at 72 h postinoculation (Fig. 1B), and reduced lethality (Fig. 1C) compared to untreated mice. These data suggest that intestinal bacteria enhance CVB3 replication and pathogenesis.
FIG 1.
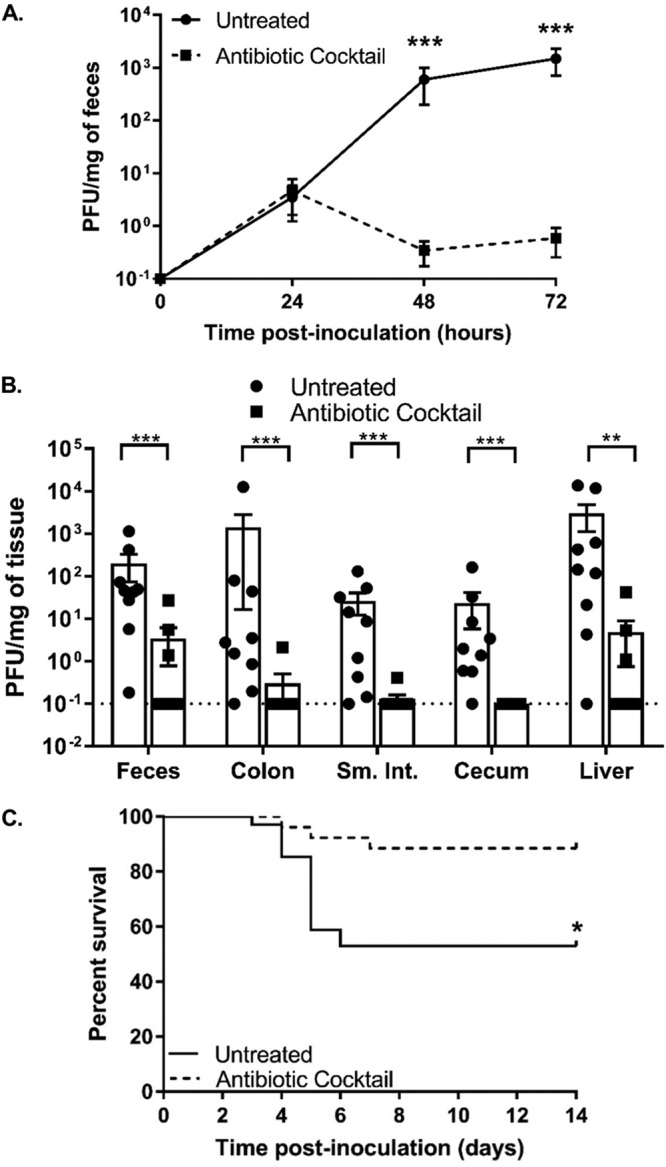
Treatment with a cocktail of five antibiotics reduced CVB3 shedding and lethality. Male C57BL/6 PVR+/+ IFNAR−/− mice were left treated or treated with a combination of 5 antibiotics (ampicillin, neomycin, streptomycin, metronidazole, and vancomycin) for 5 days by oral gavage followed by administration in drinking water available ad libitum for 9 days prior to oral inoculation with 5 × 107 PFU of CVB3. (A and B) Viral titers in feces specimens collected at 24, 48, and 72 h postinoculation (A) or tissues collected at 72 h postinoculation (B) were determined by plaque assay. Data represent means ± standard errors of the means (SEM). *, P < 0.05; **, P < 0.001; ***, P < 0.0001 (Mann-Whitney test). (C) Survival of untreated or antibiotic-treated mice orally inoculated with CVB3. *, P < 0.05 (log rank test). n = 9 to 25 mice per group from 3 to 7 independent experiments. Sm. Int, small intestine.
Treatment with ampicillin and vancomycin reduces CVB3 infection but not poliovirus infection.
To determine the effect of specific antibiotics on CVB3 replication, we treated mice with antibiotics administered either individually or in different combinations prior to oral inoculation with 5 × 107 PFU of CVB3. Feces specimens were collected at 72 h postinoculation, and CVB3 titers were quantified by plaque assay. We found no difference between the CVB3 fecal titers from untreated mice and the titers from those treated with vancomycin (Fig. 2A). In contrast, CVB3 fecal titers were reduced in mice treated with ampicillin or ampicillin plus vancomycin compared to untreated mice (Fig. 2A). In fact, the fecal titers in mice treated with ampicillin and vancomycin were similar to the fecal titers in mice treated with the five-antibiotic cocktail. Additionally, we observed that mice treated with ampicillin and vancomycin prior to oral inoculation were protected from CVB3-induced lethality (Fig. 2B). Overall, these data suggest that treatment with ampicillin and vancomycin is sufficient to reduce both CVB3 shedding and pathogenesis.
FIG 2.

Treatment with two antibiotics, ampicillin and vancomycin, was sufficient to reduce CVB3 shedding and lethality. Male C57BL/6 PVR+/+ IFNAR−/− mice were left untreated or treated with various antibiotics for 5 days by oral gavage followed by administration in drinking water available ad libitum for 9 days prior to oral inoculation with 5 × 107 PFU of CVB3. (A) Viral titers in feces specimens collected at 72 h postinoculation were determined by plaque assay. Data represent means ± SEM. *, P < 0.05; **, P < 0.001; ***, P < 0.0001 (Mann-Whitney test). Abx cocktail, five-antibiotic cocktail. (B) Survival of untreated or ampicillin-and-vancomycin-treated mice. *, P < 0.05 (log rank test). n = 12 to 17 mice per group from 3 independent experiments.
Next, we examined whether treatment with ampicillin and vancomycin would be sufficient to reduce poliovirus replication and pathogenesis. Since our laboratory previously determined that depletion of intestinal bacteria with a four-antibiotic cocktail reduced poliovirus replication and pathogenesis (9) and since CVB3 and poliovirus are from the same viral family, we hypothesized that ampicillin and vancomycin treatment would be sufficient to reduce poliovirus infection. To address this hypothesis, we treated mice with ampicillin and vancomycin prior to oral inoculation with 2 × 107 PFU of poliovirus. Feces specimens were collected at 24, 48, and 72 h postinoculation, and poliovirus titers were quantified by plaque assay. We found no significant difference in poliovirus fecal titers of untreated mice compared to those treated with ampicillin and vancomycin (Fig. 3A). These data suggest that treatment with ampicillin and vancomycin does not alter poliovirus replication in the intestine. However, high fecal titers are not always indicative of poliovirus replication in the intestine (9, 20). Input inoculum virus can be shed for a few days following infection and can confound interpretation of viral titer data, particularly during antibiotic treatment, due to reduced peristalsis (9, 20). Therefore, to ensure that the fecal titers detected in poliovirus-infected mice would be due to replicated rather than input virus, we differentiated between inoculum and replicated viruses in mouse feces by the use of light-sensitive viruses. Poliovirus was propagated in the presence of neutral red dye to create a light-sensitive poliovirus stock. Neutral red dye within virions confers light sensitivity due to RNA cross-linking. Upon viral replication in the dark, such as in the gastrointestinal tract, progeny virions are light insensitive, which facilitates the assessment of replication by differentiation of inoculum virus from replicated virus (9, 13, 21). Following inoculation with light-sensitive, neutral red poliovirus, feces specimens were collected at 24, 48, and 72 h postinfection in the dark. Following processing, poliovirus replication was quantified by determining the ratio of light-exposed to dark-exposed fecal titers. Again, in contrast to CVB3 results, we found that poliovirus replication was not reduced in mice treated with ampicillin and vancomycin (Fig. 3B). Furthermore, treatment of mice with ampicillin and vancomycin did not alter the survival of mice inoculated with poliovirus (Fig. 3C). Overall, these data suggest that treatment with ampicillin and vancomycin is sufficient to reduce CVB3 shedding and pathogenesis while having no effect on poliovirus replication and pathogenesis.
FIG 3.
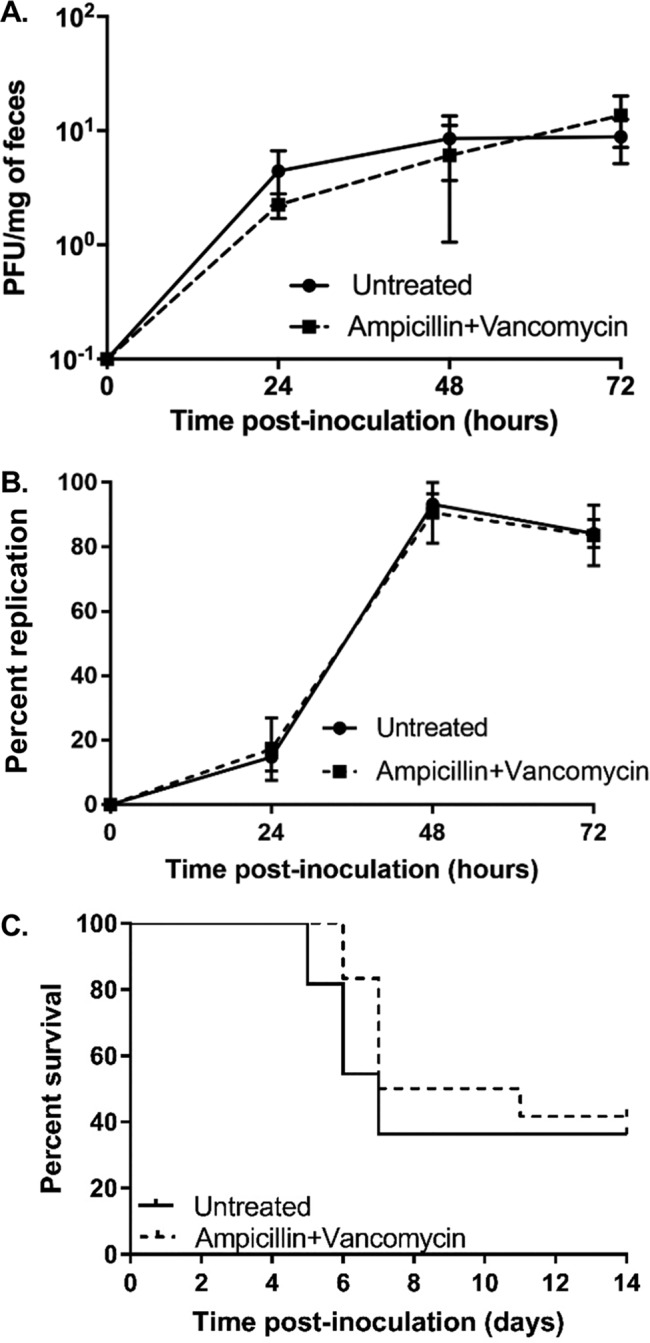
Treatment with ampicillin and vancomycin had no effect on poliovirus shedding and lethality. Male C57BL/6 PVR+/+ IFNAR−/− mice were left untreated or treated with various antibiotics for 5 days by oral gavage followed by administration in drinking water available ad libitum for 9 days prior to oral inoculation with 2 × 107 PFU of poliovirus. (A) Viral fecal titers at 24, 48, and 72 h postinoculation were determined by plaque assay. (B) Viral replication efficiency was examined using light-sensitive viruses. Mice were orally inoculated with neutral red-containing, light-sensitive poliovirus in the dark, and feces specimens were collected at 24, 48, and 72 h postinoculation in the dark. Replication status was determined by dividing the number of PFU per milliliter of light-exposed samples (representing replicated virus) by the number of PFU per milliliter of dark-exposed samples (representing input/nonreplicated virus plus replicated virus) and multiplying by 100%. Data represent means ± SEM. (C) Survival of untreated or ampicillin-and-vancomycin-treated mice following inoculation with 2 × 107 PFU of poliovirus. The log rank test was used to assess the results of comparisons of survival data. n = 11 to 12 mice per group from 4 independent experiments.
Microbiota composition of mice treated with different antibiotic regimens.
To examine the intestinal bacterial community of mice, we analyzed fecal samples using real-time quantitative PCR (qRT-PCR) and 16S rRNA sequencing. As expected, quantitative PCR (qPCR) experiments revealed that treatment with the five-antibiotic cocktail or vancomycin and ampicillin significantly reduced prevalences of Eubacteria (general bacterial population; Fig. 4A), Bacteroidetes (a predominant phylum of Gram-negative bacteria; Fig. 4B), and Firmicutes (a predominant phylum of Gram-positive bacteria; Fig. 4C). Treatment with the five-antibiotic cocktail reduced Eubacteria 16S rRNA gene copy numbers over 80,000-fold, and treatment with ampicillin and vancomycin reduced Eubacteria 16S rRNA gene copy numbers by over 1,200-fold, and trends were similar for Bacteroidetes and Firmicutes (Fig. 4A to C). As expected, the five-antibiotic cocktail depleted bacteria more extensively than the ampicillin and vancomycin combination. To identify the microbial communities in each treatment group, we performed 16S rRNA sequencing. We found that the overall diversity of bacteria decreased in both mice treated with the five-antibiotic cocktail and mice treated with ampicillin plus vancomycin (Fig. 4D). While the microbial communities in mice treated with the five-antibiotic cocktail differed from the microbial communities in those treated with ampicillin and vancomycin (Fig. 4E), the relative prevalences of distinct members of the microbiota were more similar in the two antibiotic treatment groups than they were in comparison to the untreated controls (Fig. 4F). For example, Bacteroidetes and Firmicutes were the most prevalent members in untreated mice, while Tenericutes predominated in the two different antibiotic treatment groups. Overall, these data suggest that both antibiotic treatment regimens alter host microbiota but that treatment with the cocktail of antibiotics depletes microbiota to a greater extent than treatment with ampicillin and vancomycin.
FIG 4.

Profiling the bacterial populations present in mice treated with different antibiotic regimens. Male C57BL/6 PVR+/+ IFNAR−/− mice were left untreated or treated with various antibiotics for 5 days by oral gavage followed by administration in drinking water available ad libitum for 9 days. DNA was extracted from fecal pellets, bacteria were quantified by 16S rRNA qRT-PCR (A to C), and population diversity was determined by 16S rRNA gene sequencing (D to F). Group qRT-PCR (copies per milligram of feces) was performed on fecal genomic DNA collected from mice that were left untreated, treated with a cocktail of five antibiotics (Abx), or treated with ampicillin and vancomycin (Amp+Vanc). (A) Eubacteria (general bacterial population). (B) Bacteroidetes (a predominant phylum of Gram-negative bacteria). (C) Firmicutes (a predominant phylum of Gram-positive bacteria). Panel A data are derived from mice that were subsequently infected with poliovirus (Fig. 1) and CVB3 (Fig. 3); the bacterial profiles for the two viral infection groups were similar. Data represent means ± SEM. *, P < 0.05; **, P < 0.001; ***, P < 0.0001; ****, P < 0.00001 (Mann-Whitney test). The V3-V4 region was amplified and sequenced using Illumina sequencing, and analysis was performed on forward reads using Qiime. (D) Faith’s phylogenetic diversity (intrasample) data determined using a rarefaction cutoff of 10,000 reads/sample. (E) A principal-component-analysis plot generated using Emporer (33). (F) Bacterial taxonomy was assigned and visualized at the level of class.
CVB3, but not poliovirus, is sensitive to relatively minor perturbations of the microbiota.
Given that treatment with just ampicillin and vancomycin was sufficient to inhibit CVB3 infection, we hypothesized that microbiota enhancement of CVB3 infection may require relatively high numbers of bacteria. To test this, we treated mice with milder antibiotic regimens. First, we treated immunocompetent C57BL/6 mice with 1/10 the normal dose (0.1×) of the five-antibiotic cocktail and then orally infected them with either 5 × 107 PFU of CVB3 or 2 × 107 PFU of poliovirus. Feces specimens were harvested at 72 h postinoculation and quantified via plaque assay. We found that treatment with the 0.1× five-antibiotic cocktail was sufficient to significantly reduce CVB3 shedding while having no effect on poliovirus shedding (Fig. 5A). Importantly, this experiment was performed with immunocompetent mice, indicating that the enhanced reliance of CVB3 on microbiota occurs in both immunocompetent and immune-deficient (IFNAR knockout) animals. To further examine the relative levels of dependence of CVB3 and poliovirus on microbiota, we treated C57BL/6 PVR+/+ IFNAR−/− mice with a single dose of one antibiotic, streptomycin, 1 day prior to oral inoculation with 5 × 107 PFU of CVB3 or 2 × 107 PFU of poliovirus. Feces specimens were harvested at 72 h postinoculation, and viral titers were quantified via plaque assay. We found that mice treated with one dose of streptomycin had significantly reduced levels of CVB3 shedding, while poliovirus shedding was unaffected (Fig. 5B). Furthermore, a single dose of streptomycin was sufficient to completely rescue mice from CVB3-associated lethality (Fig. 5C), while there was no effect on poliovirus-associated lethality (Fig. 5D). These data suggest that CVB3 is more sensitive than poliovirus to perturbations in the microbiota and that a single dose of one antibiotic can dramatically alter the outcome of CVB3 infection. Overall, our data suggest that these two closely related enteric viruses have different microbiota requirements.
FIG 5.

CVB3 replication is more sensitive to antibiotic treatment than poliovirus replication. (A) Male C57BL/6 PVR+/+ mice were left untreated or treated with a combination of 5 antibiotics (ampicillin, neomycin, streptomycin, metronidazole, and vancomycin) at a 0.1× concentration (one-tenth the standard concentration) for 5 days by oral gavage followed by a 0.1× concentration administered in drinking water available ad libitum for 9 days prior to oral inoculation with either 5 × 107 PFU of CVB3 or 2 × 107 of poliovirus. (A) Fecal titers at 72 h postinoculation were determined by plaque assay. (B to D) Male C57BL/6 PVR+/+ IFNAR−/− mice were left untreated or treated with a single gavage of streptomycin the day prior to oral inoculation with either 5 × 107 PFU CVB3 or 2 × 107 PFU poliovirus. (B) CVB3 and poliovirus fecal titers at 72 h postinoculation determined by plaque assay. (C and D) Survival of untreated or streptomycin-treated mice orally inoculated with CVB3 (C) or poliovirus (D). Data represent means ± SEM. *, P < 0.05 (Mann-Whitney test or log rank test). n = 6 to 9 mice per group from 2 independent experiments.
DISCUSSION
Work performed in several laboratories has shown that enteric viruses from four families benefit from intestinal bacteria, but the precise microbiota requirements for different viruses are unclear (6–9). Here, we examined the role of bacteria on intestinal replication and pathogenesis of coxsackievirus, an enteric virus from the Picornaviridae family, using an oral inoculation mouse model. Not surprisingly, we found that CVB3 infection is sensitive to antibiotic-mediated depletion of microbiota (Fig. 1). This suggests that, similarly to poliovirus, bacteria promote CVB3 replication in the intestine.
Interestingly, we found that CVB3 was sensitive to relatively minor perturbations of the microbiota whereas poliovirus was unaffected by these conditions and was able to replicate and cause disease. For example, levels of CVB3 replication and pathogenesis were dramatically diminished by treatment with ampicillin and vancomycin (Fig. 2), by treatment with a lower concentration of antibiotics (Fig. 5A), or by treatment with even a single dose of a single antibiotic (Fig. 5B to D). Poliovirus replication and pathogenesis were not affected by any of these treatment regimens. This suggests that even closely related enteric viruses may utilize different bacteria and/or mechanisms to enhance intestinal replication.
It is possible that CVB3 replication is dependent on the presence of a certain minimum number of bacteria or on the presence of a particular bacterial strain or constellation of bacterial strains or on a combination of these factors. We examined bacterial prevalence by 16S rRNA qPCR analysis, and we examined bacterial diversity by 16S rRNA sequence analysis. We found that the overall abundance and diversity of the microbiota were altered during antibiotic treatment (Fig. 4). The levels of diversity of bacterial phyla in mice administered the whole cocktail of antibiotics and in those administered just ampicillin and vancomycin were similar (Fig. 4D to F), whereas the relative bacterial abundances were more dramatically altered between those treatment groups (Fig. 4A to C). Thus, it is possible that bacterial prevalence matters more than bacterial community composition for CVB3 replication, although future experiments will be needed to test this hypothesis.
While the precise mechanisms underlying the different microbiota requirements of CVB3 and poliovirus are unclear, it is possible that CVB3 is more reliant on microbiota for dampening host immune responses or enhancing the infectivity of viral particles. Treatment with ampicillin and vancomycin was sufficient to reduce CVB3 infection, and Baldridge et al. have shown that ampicillin and vancomycin are sufficient to regulate the lambda interferon (IFN-λ) pathway and to reduce the persistence of another enteric virus, murine norovirus (7). It remains unclear if the IFN-λ pathway plays a role in bacterial enhancement of CVB3 intestinal replication. Additionally, antibiotics can regulate the host interferon response (23) and can have noncanonical effects on enteric viruses (24).
In conclusion, we found that, similarly to poliovirus and other enteric viruses, bacteria enhance CVB3 replication and virus-associated lethality. Interestingly, a single dose of a clinically relevant broad-spectrum antibiotic, streptomycin, was sufficient to reduce CVB3 replication and completely abolish CVB3-associated lethality while having no effect on poliovirus. This result could have implications for how standard antibiotic treatment regimens for bacterial infections could also impact the course of enteric virus infections.
MATERIALS AND METHODS
Cells and viruses.
HeLa cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% calf serum. Poliovirus work was performed in areas meeting or exceeding biosafety level 2 standards (BSL2+) in accordance with practices recommended by the World Health Organization. Poliovirus (serotype 1 virulent Mahoney) infections and plaque assays were performed using HeLa cells according to previously published methods (25). The CVB3-Nancy infectious clone was obtained from Marco Vignuzzi (Pasteur Institute, Paris, France). Stocks of CVB3 were prepared as previously described (20). CVB3 titers were determined by plaque assay with HeLa cells (20, 21).
To determine whether fecal viruses were input/inoculum virus or virus that had undergone replication, we used neutral-red-labeled, light-sensitive poliovirus as described previously (25). Light inactivation was performed by exposing neutral red-poliovirus stocks to a fluorescent light bulb for 10 min. The ratio of light-insensitive PFU to light-sensitive PFU in the virus stock was 1 × 105:1.5 × 105. Following oral inoculation, to determine the percentage of replicated virus in the intestine, samples were processed in the dark and a portion was light exposed. The percentage of replicated virus was calculated by dividing the light-exposed number of PFU per milliliter by the non-light-exposed number of PFU per milliliter and multiplying by 100.
Mouse experiments.
All animals were handled according to the Guide for the Care of Laboratory Animals of the National Institutes of Health. All mouse studies were performed at the University of Texas Southwestern Medical Center (UT Southwestern) (Animal Welfare Assurance no. A3472-01) using protocols approved by the local Institutional Animal Care and Use Committee in a manner designed to minimize pain, and any animals that exhibited severe disease were euthanized immediately with isoflurane. C57BL/6 PVR+/+ IFNAR−/− mice were obtained from S. Koike (Tokyo, Japan) (26). We used IFNAR−/− mice for these experiments since orally inoculated immunocompetent animals shed small amounts of virus following oral inoculation and do not develop disease (9, 13, 21). Wild-type, immunocompetent C57BL/6 PVR+/+ mice were used in Fig. 5A experiments. Throughout, littermate controls and/or cage randomization of different litters were used to limit variability from microbiota differences. For example, the mice from each cage were split into two groups—one treated with antibiotics and one not treated with antibiotics. For the experiments whose results are presented in Fig. 5, where subtle changes in microbiota could impact results, we split mice from each cage and infected with CVB3 or poliovirus to allow a direct comparison of the two viruses. Mice were administered either a combination of 5 antibiotics (ampicillin, neomycin, streptomycin, metronidazole, and vancomycin; 10 mg of each antibiotic per day) or individual antibiotics (10 mg per antibiotic per day) for 5 days by oral gavage followed by administration in drinking water available ad libitum (for ampicillin, neomycin, streptomycin, and metronidazole, 1 g/liter; for vancomycin: 500 mg/liter) for the duration of the experiment. Mice were treated with antibiotics for 9 days prior to peroral inoculation with poliovirus or CVB3. For oral inoculations, 8-to-12-week-old male mice were perorally inoculated with either 5 × 107 PFU of CVB3 or 2 × 107 PFU of poliovirus. Male mice were used since CVB3 was shown to have a sex-dependent enhancement of replication and lethality (20). Disease was monitored until day 14 postinoculation for survival experiments. Mice were euthanized upon severe disease onset. For viral shedding and replication experiments, feces specimens were collected at indicated time points postinoculation and processed as previously described (9). Tissues were aseptically removed from mice and homogenized in phosphate-buffered saline (PBS) using 0.9-to-2.0-mm-diameter stainless steel beads in a Bullet blender (Next Advance) and freeze-thawed 3 times in liquid nitrogen to release intracellular virus. Cellular debris was removed by centrifugation at 13,000 rpm for 3 min. The amount of virus in supernatant was determined by plaque assay in HeLa cells, and titers were normalized to the weight of each tissue.
qRT-PCR and sequencing.
DNA was extracted from fecal pellets using PowerFecal DNA (Qiagen). The V3-V4 region of the 16S rRNA gene was amplified using primers 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐[CCTACGGGNGGCWGCAG] (forward primer) and 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG‐[GACTACHVGGGTATCTAATCC] (reverse primer) and sequenced on an Illumina MiSeq v3 sequencer using paired-end (PE) sequencing and 300 cycles. Analysis was performed on forward reads using Qiime 1.9.1 (27). Primer sequences were trimmed from each read, and all reads were quality filtered (Phred score > 25) using the multiple_split_libraries_fastq.py command. Operational taxonomic units (OTUs) were assigned using open-reference picking (SILVA 128 release) (28, 29) with default parameters, except for 0.1% subsampling, as follows: for filter_alignment.py, gap filter threshold, 0.8; lane mask filtering, true; entropy threshold, 0.10. This yielded a minimum of 134,043 and median of 238,345 sequence counts per sample. Bacterial taxonomy was assigned (30, 31) and visualized at the level of class. Intrasample and between-sample diversity levels were calculated using core_diversity.py workflow (32). Faith’s phylogenetic diversity (intrasample) used a rarefaction cutoff of 10,000 reads/sample. A principal-coordinate-analysis (PCoA) plot was visualized using Emporer (33).
Statistical analysis.
The differences between groups were examined by the use of the unpaired two-tailed Student's t test. Error bars in figures represent standard errors of the means. A P value of <0.05 was considered significant. All analyses of data were performed using GraphPad Prism version 7.00 for Windows (GraphPad Software, La Jolla, CA).
ACKNOWLEDGMENTS
This work was funded by a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases award (J.K.P.), R01 AI74668 (J.K.P.), and K01 DK110216 (C.M.R.). J.K.P. is a Howard Hughes Medical Institute Faculty Scholar. M.A.W.A. was supported in part by NIH, NIAID, grant T32 AI007520. B.T.M. was supported by NIH, NIAID, grants T32 AI007520 and F32 AI138392.
REFERENCES
- 1.Parashar UD, Hummelman EG, Bresee JS, Miller MA, Glass RI. 2003. Global illness and deaths caused by rotavirus disease in children. Emerg Infect Dis 9:565–572. doi: 10.3201/eid0905.020562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrmann JE, Taylor DN, Echeverria P, Blacklow NR. 1991. Astroviruses as a cause of gastroenteritis in children. N Engl J Med 324:1757–1760. doi: 10.1056/NEJM199106203242501. [DOI] [PubMed] [Google Scholar]
- 3.Division of Viral Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention. 2011. Updated norovirus outbreak management and disease prevention guidelines. MMWR Recomm Rep 60:1–18. [PubMed] [Google Scholar]
- 4.Montes M, Artieda J, Pineiro LD, Gastesi M, Diez-Nieves I, Cilla G. 2013. Hand, foot, and mouth disease outbreak and coxsackievirus A6, northern Spain, 2011. Emerg Infect Dis 19:676–678. doi: 10.3201/eid1904.121589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clemente JC, Ursell LK, Parfrey LW, Knight R. 2012. The impact of the gut microbiota on human health: an integrative view. Cell 148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinje J, Tibbetts SA, Wallet SM, Karst SM. 2014. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, Diamond MS, Ivanova Y, Artyomov M, Virgin HW. 2015. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 347:266–269. doi: 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kane M, Case LK, Kopaskie K, Kozlova A, MacDearmid C, Chervonsky AV, Golovkina TV. 2011. Successful transmission of a retrovirus depends on the commensal microbiota. Science 334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, Dermody TS, Pfeiffer JK. 2011. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson CM, Pfeiffer JK. 2014. Viruses and the microbiota. Annu Rev Virol 1:55–69. doi: 10.1146/annurev-virology-031413-085550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchiyama R, Chassaing B, Zhang B, Gewirtz AT. 2014. Antibiotic treatment suppresses rotavirus infection and enhances specific humoral immunity. J Infect Dis 210:171–182. doi: 10.1093/infdis/jiu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfeiffer JK, Virgin HW. 2016. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 351:aad5872. doi: 10.1126/science.aad5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson CM, Jesudhasan PR, Pfeiffer JK. 2014. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 15:36–46. doi: 10.1016/j.chom.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kosrirukvongs P, Kanyok R, Sitritantikorn S, Wasi C. 1996. Acute hemorrhagic conjunctivitis outbreak in Thailand, 1992. Southeast Asian J Trop Med Public Health 27:244–249. [PubMed] [Google Scholar]
- 15.He SJ, Han JF, Ding XX, Wang YD, Qin CF. 2013. Characterization of enterovirus 71 and coxsackievirus A16 isolated in hand, foot, and mouth disease patients in Guangdong, 2010. Int J Infect Dis 17:e1025–e1030. doi: 10.1016/j.ijid.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Andreoletti L, Leveque N, Boulagnon C, Brasselet C, Fornes P. 2009. Viral causes of human myocarditis. Arch Cardiovasc Dis 102:559–568. doi: 10.1016/j.acvd.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Strikas RA, Anderson LJ, Parker RA. 1986. Temporal and geographic patterns of isolates of nonpolio enterovirus in the United States, 1970–1983. J Infect Dis 153:346–351. doi: 10.1093/infdis/153.2.346. [DOI] [PubMed] [Google Scholar]
- 18.Kearney MT, Cotton JM, Richardson PJ, Shah AM. 2001. Viral myocarditis and dilated cardiomyopathy: mechanisms, manifestations, and management. Postgrad Med J 77:4–10. doi: 10.1136/pmj.77.903.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gebhard JR, Perry CM, Harkins S, Lane T, Mena I, Asensio VC, Campbell IL, Whitton JL. 1998. Coxsackievirus B3-induced myocarditis: perforin exacerbates disease, but plays no detectable role in virus clearance. Am J Pathol 153:417–428. doi: 10.1016/S0002-9440(10)65585-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson CM, Wang Y, Pfeiffer JK. 13 March 2017, posting date Sex-dependent intestinal replication of an enteric virus. J Virol doi: 10.1128/JVI.02101-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Pfeiffer JK. 2016. Emergence of a large-plaque variant in mice infected with coxsackievirus B3. mBio 7:e00119. doi: 10.1128/mBio.00119-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson CM, Woods Acevedo MA, McCune BT, Pfeiffer JK. 13 August 2019, posting date Related enteric viruses have different requirements for host microbiota in mice. BioRxiv 10.1101/733766. [DOI] [PMC free article] [PubMed]
- 23.Gopinath S, Kim MV, Rakib T, Wong PW, van Zandt M, Barry NA, Kaisho T, Goodman AL, Iwasaki A. 2018. Topical application of aminoglycoside antibiotics enhances host resistance to viral infections in a microbiota-independent manner. Nat Microbiol 3:611–621. doi: 10.1038/s41564-018-0138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woods Acevedo MA, Erickson AK, Pfeiffer JK. 2019. The antibiotic neomycin enhances coxsackievirus plaque formation. mSphere 4:e00632-18. doi: 10.1128/mSphere.00632-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuss SK, Etheredge CA, Pfeiffer JK. 2008. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog 4:e1000082. doi: 10.1371/journal.ppat.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, Yoneyama M, Fujita T, Taya C, Yonekawa H, Koike S. 2005. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol 79:4460–4469. doi: 10.1128/JVI.79.7.4460-4469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 29.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. 2013. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2:16. doi: 10.1186/2047-217X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]