

Abstract
Cinnamycin is a lantibiotic peptide, which selectively binds to and permeabilizes membranes containing phosphatidylethanolamine (PE) lipids. As PE is a major component of many bacterial cell membranes, cinnamycin has considerable potential for destroying these. In this study, molecular dynamics simulations are used to elucidate the structure of a lipid–cinnamycin complex and the origin of selective lipid binding. The simulations reveal that cinnamycin selectively binds to PE by forming an extensive hydrogen-bonding network involving all three hydrogen atoms of the primary ammonium group of the PE head group. The substitution of a single hydrogen atom with a methyl group on the ammonium nitrogen destabilizes this hydrogen-bonding network. In addition to binding the primary ammonium group, cinnamycin also interacts with the phosphate group of the lipid through a previously uncharacterized phosphate-binding site formed by the backbone Phe10-Abu11-Phe12-Val13 moieties (Abu = 1-α-aminobutyric acid). In addition, hydroxylation of Asp15 at Cβ plays a role in selective binding of PE due to its tight interaction with the charged amine of the lipid head group. The simulations reveal that the position and orientation of the peptide in the membrane depend on the type of lipid to which it binds, suggesting a reason for why cinnamycin selectively permeabilizes PE-containing membranes.
1. Introduction
Antimicrobial peptides present a solution to the major problem that multiresistant bacteria pose to human health.1 Peptides such as nicin2,3 and ε-polylysine4 have long been used for food preservation due to their antimicrobial abilities, and related peptides are being explored for clinical applications.1,5 An exact relationship between the amino acid sequences of these peptides and their antimicrobial action(s) has yet to be established, hindering rational optimization of their properties for both clinical and preservative use. Therefore, it is of considerable importance to elucidate the relationship between peptide sequence/structure and their function and mechanism of action.
Cinnamycin is a 19-residue tetracyclic peptide (see Figure 1a), which selectively binds to lipid molecules with a phosphatidylethanolamine (PE) head group with a 1:1 stoichiometry,6 which, subsequently, results in permeation of PE-containing membranes.7 In contrast, cinnamycin shows a much lower binding affinity for, and limited permeabilization of, vesicles consisting of other lipids [e.g., phosphatidylcholine (PC), phosphatidylserine (PS), and N-methyl PE (mPE)].7,8 When cinnamycin binds to a membrane, it also induces nonselective lipid flip-flops, which may be related to its antimicrobial ability.8
Figure 1.

Primary structure of cinnamycin. (a) The sequence of cinnamycin showing the cross-links. (b) Chemical structure of the four uncommon amino acids present in cinnamycin: meso-lanthionine (Lan), (2S,3S,6R)-3-methyllanthionine (MeLan), (2S,8S)-lysinoalanine (LysAla), and erytho-3-hydroxy-l-aspartic acid (HyAsp). The two backbone moieties of Lan will be referred to as Ala(S)X and Ala(S)Y, while the corresponding moieties of MeLan will be referred to as Abu(S)X and Ala(S)Y, where X and Y specify the positions in the sequence. The two moieties of LysAla19/6 will be referred to as Lys19 and Ala(N)6. Abu = 1-α-aminobutyric acid.
Cinnamycin is a part of the lantibiotic family, which is defined by the presence of the thioether amino acids meso-lanthionine (Lan) and (2S,3S,6R)-3-methyllanthionine (MeLan) that are formed through side-chain cross-linking of a cysteine residue with either serine or threonine, respectively (see Figure 1b).9 Due to the presence of these amino acids, lantibiotics are highly cross-linked internally, and the presence of three such cross-links together with a (2S,8S)-lysinoalanine (LysAla) bridge makes cinnamycin one of the smallest peptides with a well-defined three dimensional structure.10 Based on NMR experiments, a pocket in the cinnamycin molecule formed by residues 7–14 has been suggested to be the binding site for PE molecules.6 Furthermore, cinnamycin has an erytho-3-hydroxy-l-aspartic acid (HyAsp) at residue 15, the function of which is unknown. Together with the highly homologous peptides, the duramycins and ancovenin, cinnamycin defines a family of cinnamycin-like peptides.11 In addition to their hemolytic12 and antimicrobial properties,13 cinnamycin-like peptides also inhibit angiotensin-I converting enzyme,14 the activity of phospholipase A2,15 and herpes simplex virus,16 and, thus, these peptides have potential applications in the treatment of blood pressure regulation, inflammation, and viral infection.17 Its ability to selectively bind to PE lipids makes cinnamycin a potential probe for determining the location of PE lipids in cell membranes and for disrupting PE-containing membranes such as those of cancer cells and of bacteria.10,18,19
Based on NMR studies, it has been suggested that the selectivity of cinnamycin for PE is due to the binding of the primary ammonium (i.e., charged amine) group of the PE head group into a small binding pocket on the peptide surface that cannot encompass larger head groups such as that of e.g., PC.20 However, some observations are difficult to accommodate within this model: e.g., preincubation of cinnamycin with alkyl-phosphopropanolamine cannot inhibit its hemolytic activity, indicating that molecules with a larger distance between the ammonium group and the phosphate group than that in the head group of PE fail to bind to cinnamycin.12 Cinnamycin and the related peptide duramycin, which only differ in a few residues, have been found to induce general lipid flip–flop in the presence of PE lipids8 and during vesicle tubulation,21 permeation,7 and fusion.8 The focus of the current study is, thus, to characterize the selectivity of cinnamycin for PE and to investigate how cinnamycin may perturb PE-containing membranes.
We use molecular dynamics (MD) simulations to optimize a previously published NMR-derived structural model of cinnamycin (PDB ID: 2DDE, model 1)20 and to characterize its interaction with different lipid species, thereby providing an understanding of its selectivity for PE. Simulations of cinnamycin with one molecule of 1,2-dilauroyl-sn-glycero-3-phosphoethanolamine (DLPE) bound in different solvents are analyzed, providing an optimized model of the PE–cinnamycin complex. Simulations of cinnamycin embedded in a bilayer of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and a range of different lipids reveal the effect of the lipid species on the stability of the cinnamycin–lipid complex. The function of the Cβ hydroxylation of Asp15 is investigated by simulating the HyAsp15Asp mutant. Finally, the accommodation of the cinnamycin–PE complex in both POPC and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE)/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) bilayers (the latter mimicking a bacterial membrane) is examined.
2. Results and Discussion
2.1. Cinnamycin–DLPE Complex Structure
An NMR structure of cinnamycin bound to a lysophosphatidylethanolamine (LPE)20 was used as a starting structure for our simulations. To evaluate the stability of this NMR structure, the fragment of LPE resolved in the PDB ID: 2DDE (model 1) coordinate set was converted into DLPE and the system was solvated in water, dimethyl sulfoxide (DMSO), or ethanol. The systems were then energy minimized, equilibrated, and simulated for at least 0.3 μs using atomistic resolution (Table 1). To ensure that we obtained a reasonable protein model, the ϕ and ψ angles of the amino acid residues of cinnamycin in DMSO were calculated (Figure S2) following the same protocol presented by Hosoda et al.,20 and a comparison of nuclear Overhauser effect (NOE) distances between NMR and the simulated structures was done (Figure S3) following the restraint distance presented by Hosoda et al.20
Table 1. Atomistic Simulations of DLPE Bound to Cinnamycin in Different solvents.
| simulation namea | simulation time (μs) | solvent | HyAsp15 rotatedb | NOE rest. eq.c |
|---|---|---|---|---|
| DNMR | 0.3 | DMSO | no | no |
| ENMR | 0.3 | ethanol | no | no |
| WNMR | 0.3 | water | no | no |
| Dunflip | 1.0 | DMSO | no | yes |
| Dflip | 1.0 | DMSO | yes | yes |
| Eunflip | 1.0 | ethanol | no | yes |
| Eflip | 1.0 | ethanol | yes | yes |
| Dflip_Wd | 0.65 | water | yes | yes |
The initial letter indicates the solvent used, while the subscript indicates the starting conformation and equilibration protocol used: NMR if the NMR model was used and no NOE restraints were applied; unflip if NOE restraints were used; and flip if restraints were applied and an initial rotation of the HyAsp15 side chain was performed.
Specifies whether or not the χ1 angle of HyAsp15 was rotated by 200° to position it in the gauche(+) orientation (specified as flip). In this orientation, the negatively charged HyAsp carboxylate points toward the LysAla19/6 secondary ammonium bridge, which allows for an attractive electrostatic interaction between them.
Specifies whether or not the NOE-derived restraints were applied during the equilibration period of the simulation.
The equilibrated complex from Dflip was resolvated in water and equilibrated with distance restraints.
The distance between the ammonium group of the lipid and the back of the binding pocket, defined by the sulfur atom of MeLan5/11, was used to monitor the interactions of DLPE with cinnamycin (Figure 2). It was seen that in the simulations started from the NMR model equilibrated without NOE restraints (DNMR, ENMR, and WNMR), the ammonium group of the lipid always moved out of the binding pocket during the initial equilibration of the simulations, as indicated by the distance, which is much greater than the initial value of 3.4 Å. The initial complex was, thus, not stable, regardless of the solvent environment (the NMR structure was determined in DMSO) as the lipid always moved out of the binding pocket during the energy minimization or within a few picoseconds of MD simulation [also see Figure S4a,b in the Supporting Information (SI)].
Figure 2.
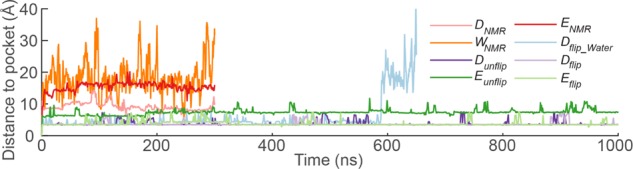
Distance between the binding pocket and the ammonium group of the lipid as a function of time for simulations of DLPE bound to cinnamycin in different solvents. See Table 1 in Section 4 for details of the simulations.
We attempted to optimize the model to form a stable lipid–peptide complex. A series of equilibration simulations of the DLPE–cinnamycin complex were conducted. Distance restraints between atoms of the lipid and the peptide molecules were derived from the published NMR data6 (see methods, Figure 11), and simulations with decreasing force constants were conducted to obtain a well-relaxed structure obeying the NOE measurements. Two starting conformations varying in the orientation of the HyAsp15 rotamer were employed to ensure that both rotamers were sampled adequately. These two rotamers were: (1) the NMR model (simulations marked unflip) and (2) a model in which the HyAsp15 side chain was oriented into the binding pocket (simulations marked flip). Simulations of both models were undertaken in three different solvents (DMSO, water, and ethanol). These simulations were intended to elucidate which orientation of the HyAsp side chain leads to the most stable complex.
Figure 11.
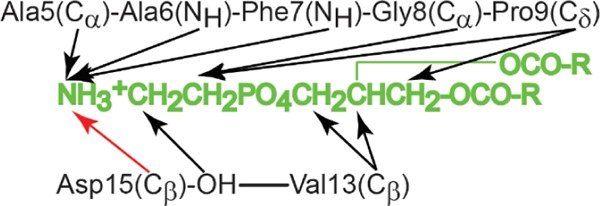
Distance restraints between cinnamycin and the lipid corresponding to the reported NOEs6 were applied to obtain a stable complex. The black arrows are restraints applied to keep the distance below 5 Å, while the red arrow specifies a restraint applied to keep that distance below 4 Å. These distances were chosen on the basis of the distances observed in the 10 NMR models (PDB code 2DDE) and their relative intensity described in the manuscript.
In simulations of Dunflip, the HyAsp15 side chain rotated into the binding pocket. As a result, both of the simulated structures in DMSO (Dunflip and Dflip) yielded similar conformations at the end of the restrained simulations with a backbone root-mean-square deviation (RMSD) of 0.7 Å (see Figure 3). When changing the solvent to ethanol, however, this flip was not observed, resulting in the structures yielded by Eunflip and Eflip being quite different at the end of the simulations, with a backbone RMSD of 2.2 Å. In Eunflip, cinnamycin became distorted after a few hundred nanoseconds of unrestrained simulation indicating that this complex was not stable. Cinnamycin from Eflip had a low backbone RMSD with respect to both Dunflip and Dflip (below 0.8 Å) indicating that the differences between the two solvents did not affect the structure of the complex much. In water, the restrained simulations did not result in complex formation, as the lipid did not remain bound during the equilibration (Figure S4c in the SI). To assess the stability of a cinnamycin–DLPE complex in water, the complex formed in DMSO (from the Dflip-simulation) was resolvated in water (named Dflip_W). The lipid, however, diffused away from the binding site after ∼590 ns (as shown in Figure 2) suggesting that the hydrophobic environment of the membrane is needed for a stable interaction between cinnamycin and the lipid. This may explain why long-tailed lipids bind more strongly to cinnamycin than short-tailed lipids in a membrane environment,22 as lipids with shorter tails are more likely to be outside a membrane bilayer environment because they are more soluble in water, as previously stated by Gao et al.23 We conclude that the complexes formed in Dunflip, Dflip, and Eflip represent the same conformation, as also observed from the overlay in Figure 3a, and we will from now on refer to this as the MD model. However, the model does not match the NMR model, as we will discuss next.
Figure 3.
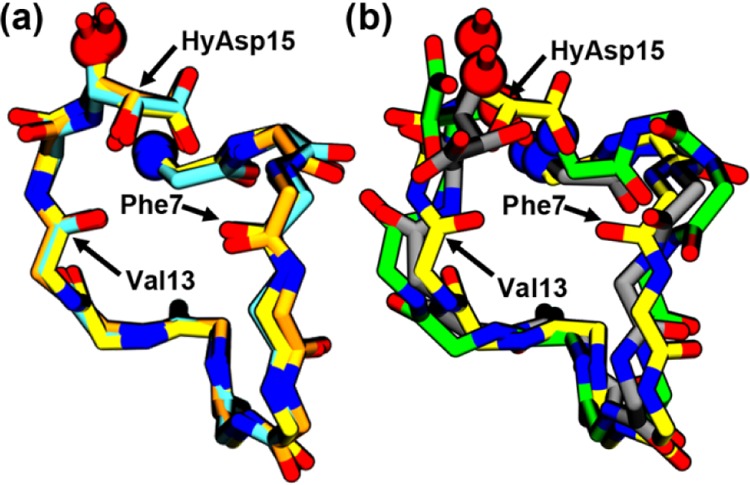
Backbone alignment (aligned on Cα, N, C, and O atoms) of (a) the structures obtained after the equilibration of simulations Dflip (yellow), Dunflip (orange), and Eflip (cyan) is compared with (b) an alignment of the Dflip (yellow), Eunflip (green), and NMR (gray) structures. Only the nonhydrogen atoms of the backbone of residues 5–15 and the side chain of HyAsp15 are shown. The locations where the N- and the C-termini have been truncated from the images for clarity are shown as blue and red spheres, respectively.
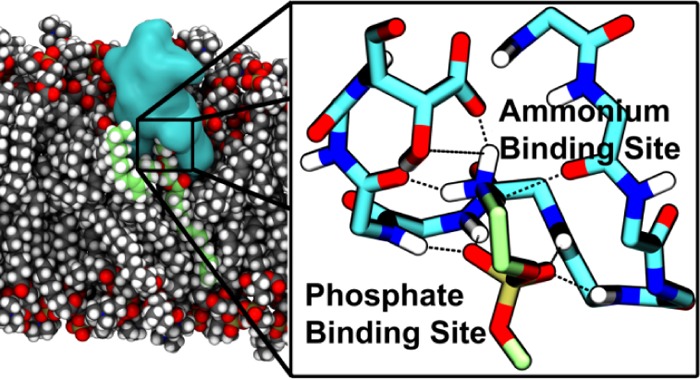
Although the lowest-energy NMR model (2DDE, model 1) fulfilled most of the NMR restraints applied in this study, careful comparison revealed that fewer close interactions were observed between the peptide and the lipid in the NMR model (Figure 4a) than in the MD model (Figure 4b). In particular, in the NMR model, the phosphate group of the lipid was not in contact with the peptide and the hydroxyl group was the only part of HyAsp15 interacting with the lipid ammonium group, while the carboxylate of HyAsp15 pointed away from the binding pocket. Furthermore, no hydrogen bonds were observed between the peptide and the lipid. A slightly different binding pattern was found in the MD models, as the lipid phosphate group remained bound in a pocket in the MD model (Figure 4b,c), making it possible for the amide hydrogens of residues 10–13 to form hydrogen bonds with oxygen atoms of the lipid phosphate. Furthermore, the ammonium group of the lipid formed stable hydrogen bonds to the backbone carbonyl groups of Phe7 and Val13 as well as a bifurcated hydrogen bond to the carboxylate and hydroxyl groups of HyAsp15. This led to a much tighter binding than in the NMR model and, thus, a more stable complex, as indicated from the much shorter and less variable distance between the lipid ammonium group and the binding pocket observed in Figure 3 for Dunflip (3.7 ± 0.8 Å), Dflip (3.6 ± 0.7 Å), and Eflip (3.8 ± 0.9 Å) compared to DNMR (9.1 ± 1.4 Å), ENMR (15.4 ± 1.7 Å), and WNMR (18.1 ± 6.4 Å). Another indication of the validity of the rotated HyAsp15 form is the location of the carboxylate group and hydroxyl group in this conformation. The negatively charged carboxylate of HyAsp15 was positioned much closer to the positively charged ammonium group of the lipid, which again is a clear indication of a stronger peptide–lipid interaction. The hydroxyl group was at the same time positioned close to the ethyl part of the ethanolamine group (e.g., 3.5 ± 0.7 Å for Dflip), thus still fitting well with the observed NMR cross-peaks between these two groups.6 Interestingly, one of the 10 NMR models (model 10) showed a similar orientation of the HyAsp15 side chain, thus confirming that the orientation also matches the NMR restraints used by Hosoda et al. to develop the NMR model.20 To conclude, this optimized conformation in which the HyAsp15 side chain as well as Phe7(O) and Val13(O) are pointing into the binding pocket seems to represent a more likely model of the PE–cinnamycin complex and was, thus, used for the subsequent simulations in lipid bilayers.
Figure 4.

NMR structure and the MD-optimized cinnamycin–PE complex. (a) The NMR and (b) the MD models of the PE–cinnamycin complex with the surface of the peptide shown in gray and the bound lipid colored green. For clarity, the peptide is visualized as its main chain and its modified side chains (excluding nonpolar hydrogens), while the lipid is shown without its hydrophobic tails (a figure with the tails shown is found in Figure S4d in the SI). (c) Schematic drawing of the optimized binding mode. The peptide is illustrated in black, the lipid head group in green, and the hydrogen bonds in red. Wavy lines indicate that backbone atoms have been omitted for clarity. All side chain atoms excluding HyAsp15 and LysAla19/6 have been omitted for clarity. Numbers on the Cα-carbons specify the residue numbers.
2.2. Binding of Different Lipids
The selectivity of cinnamycin for PE lipids has been suggested to be due to the exact fit between the size of the ammonium-binding pocket of cinnamycin and the substituents on the ammonium group of the lipid, making it impossible to fit larger groups into the binding pocket.20 However, the peptide is dynamic and it is possible that the binding pocket may adapt to the larger size of other lipid types. Furthermore, it is not known how the hydrogen-bonding network is affected by the presence of groups other than the primary ammonium group of PE lipids. We, therefore, conducted several simulations with POPE, N-methyl 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POmPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS), and POPC bound to a cinnamycin molecule embedded in a POPC bilayer to determine the source of the selective binding of PE lipids (Figure 5).
Figure 5.
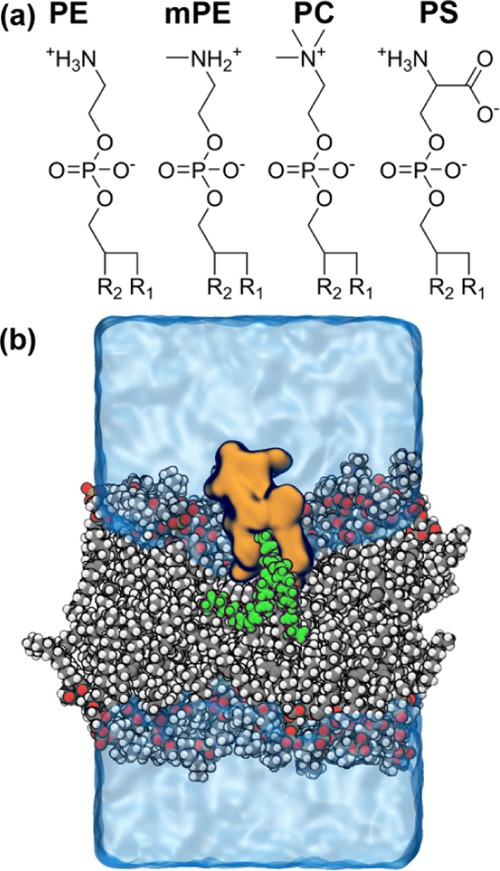
Simulations of cinnamycin–lipid complexes in a bilayer membrane. (a) The four lipids investigated in the binding study, R1 and R2, representing palmitoyl and oleoyl groups, respectively. (b) A snapshot from a simulation with cinnamycin (orange) bound to a PE lipid (green) embedded in a POPC bilayer (gray/white/red/bronze). Water and ions are shown as a transparent blue surface. The simulation box was approximately 6 × 6 × 10 nm3.
Initially, the dynamics of the PE–cinnamycin complex was investigated. It was clear from root mean square fluctuation of the backbone dihedral angles (Figure S5 in the SI) and from visual inspection of the simulation trajectories that the main flexibility in the binding pocket of cinnamycin was located around the side chain of HyAsp15 and the backbone near Phe7. Thus, the orientations of the HyAsp15 side chain, Phe7(O), and Phe7(NH) were plotted together with the distance between the lipid and the binding pocket and the number of hydrogen bonds between them (see the SI for details). Figure 6 shows the temporal evolution of these structural features during a simulation of the wild-type (wt) cinnamycin with the aspartate hydroxylated and POPE bound (PE_HyAsp). Plots for all of the simulations can be found in the Figure S6, Supporting Information. It is apparent from the figure that both the side chain of HyAsp15 (HyAsp in Figure 6) and the backbone of Phe7 (Phe(NH) and Phe(O)) were quite dynamic as they exchange between pointing into the pocket (gray) and out of the pocket (white) several times during the simulation. The figure shows a direct correlation between the orientation of Phe7(O) and the number of hydrogen bonds to the ammonium group of the lipid; when Phe7(O) rotates out of the pocket, the number of hydrogen bonds decreases (blue line) as one of the hydrogen bonds formed between cinnamycin and the ammonium is via this functional group. Although four hydrogen bonds were observed in the simulations, the maximum number of hydrogen bonds on the plot never reached this level due to the time averaging. Most often, the rotation of the Phe7(O) and the loss of the related hydrogen bond were also related to the lipid ammonium moving out of the binding pocket as exemplified by the increased distance to the binding pocket (red curve) e.g., observed at 50 ns in Figure 6. Histograms showing the distance to the binding pocket observed over the last 400 ns of all simulations are plotted in Figure 7. By fitting Gaussian functions to these data, two populations of distances can be characterized: a tightly bound mode located at a distance of 3.5 ± 0.2 Å from the back of the binding pocket and a more loosely bound conformation at 4.3 ± 0.4 Å, as indicated by the larger standard deviation (see the dark purple line in Figure 7a). These two populations probably represent two submodes of binding, as also observed from the red line in Figure 6.
Figure 6.

Temporal evolution of a simulation of cinnamycin bound to one POPE molecule embedded in a POPC bilayer (simulations named PE_HyAsp). The plot shows the change in the orientation of the HyAsp15 side chain, Phe7(O), and Phe7(NH) over time (gray when the given group points into the binding pocket and otherwise white), the distance between the ammonium group of the lipid and the binding pocket (red line), and the number of hydrogen bonds between the ammonium group of the lipid and cinnamycin (blue line). The left y axis describes the gray and white coloring, while the right y axis describes the red and blue curves. Above the plot, snapshots of the complex are shown. It is interesting to notice how the HyAsp15 rotated in the 85 ns snapshot was followed by the Phe(NH) rotating out of the binding pocket, as shown in the snapshot at 120 ns. The lipid ammonium group has moved out of the ammonium-binding pocket at 60 ns and has rebound again in the snapshot at 160 ns.
Figure 7.
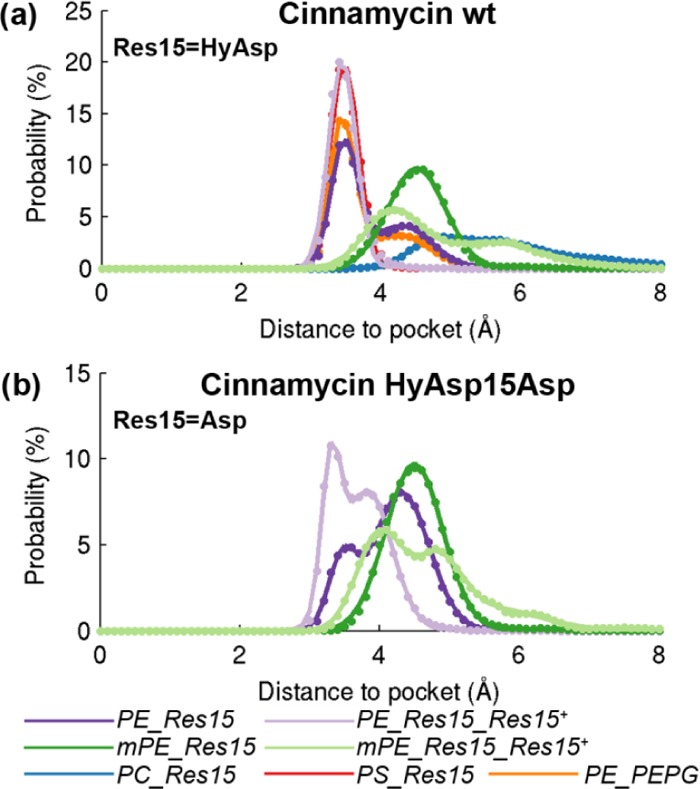
Distributions of the distance between the lipid head group and the binding pocket, measured from the nitrogen of the lipid ammonium to the sulfur of meLan5/11. The names refer to the simulations described in Table 2 in Section 4. Plotted for (a) the wild-type cinnamycin and (b) the hyAsp15Asp mutant.
After the ammonium moved out of the pocket, the HyAsp15 rotated at ∼70 ns, such that the carboxylate pointed toward the lipid ammonium, followed by the Phe(NH) orienting out of the binding pocket at ∼90 ns in the simulation shown in Figure 6. The flip of the HyAsp15 was likely induced by the attraction between the negatively charged carboxylate of HyAsp15 and the positively charged ammonium group of the lipid. In the MD model, the carboxylate of HyAsp15 makes a hydrogen bond with Phe7(NH) (see Figure 4). This hydrogen bond stabilizes the orientation of both HyAsp15 and Phe7 backbone, and the movement of one or the other may, therefore, make the orientation observed in the MD model for these residues less stable. This is likely why Phe7(NH) rotates after the HyAsp15 has rotated. It should be noted that the order of the movements of the residues varies in individual simulations (see Figure S6 in the SI), so only the correlation between these movements can be deducted but not the cause–effect relation. Furthermore, rotating the HyAsp15, such that the carboxylate points toward the lipid ammonium, does not preclude the lipid binding as observed at ∼150 ns where the lipid rebinds to the ammonium-binding pocket.
In the published NOE measurements, cross-peaks were observed between the hydroxyl group of HyAsp15 and the ethanolamine group of the lipid (see Figure 11).6 Upon rotation of the HyAsp15 side chain in the MD model, the carboxyl group points toward the lipid (e.g., at 85 ns in Figure 6) leading to the hydroxyl group pointing away from the ethanolamine. We, therefore, restrained the χ1 angle of HyAsp15 such that its carboxylate pointed toward the LysAla19/6 ammonium bridge (as observed in the MD model) to get a better sampling of the state fitting the NMR data the best (simulations named PE_HyAsp_HyAsp+). Interestingly, this resulted in a much more stable interaction between cinnamycin and the lipid, since the lipid stayed tightly bound at a distance of 3.4 ± 0.2 Å in these simulations (the light purple line in Figure 7a). A figure equivalent to Figure 6 is shown in Figure 8a for one of these simulations where hyAsp15 is restrained. It is apparent from the figure that both Phe7(NH) and Phe7(O) remained in their initial orientations, and the number of hydrogen bonds was high throughout the simulation. This indicates that fixing the orientation of the HyAsp15 side chain stabilizes both the orientation of Phe7 and lipid binding.
Figure 8.

Relation between the dynamic behavior of the binding pocket and the stability of the lipid binding over time shown for a representative simulation of cinnamycin with (a) POPE bound with the HyAsp15 side chain restrained (PE_HyAsp_HyAsp+), (b) POmPE bound (mPE_HyAsp), (c) POmPE bound with HyAsp15 side chain restrained (mPE_HyAsp_HyAsp+), (d) POPS bound (PS_HyAsp), and (e) POPC bound (PC_HyAsp). Color coding and axes are the same as in Figure 6.
To investigate whether the stabilization was due to some very specific interactions related to the HyAsp15 or if it was mainly due to an overall stabilization of the binding pocket, simulations were conducted restraining the rotation of either Phe7(NH) (simulation named PE_HyAsp_Ala(N)6) or Phe7(O) (simulation named PE_HyAsp_Gly8). Both these restraints lead to a stabilization of the binding although to a slightly lower extent than when restraining HyAsp15 (see Figure S7a,e and Figure S7b,f in the SI, respectively). When the bound lipid did move out of the binding pocket with either Phe7(O) or Phe7(NH) restrained, it was always correlated to the rotation of both the HyAsp15 side chain and the Phe7 backbone angle not retrained. These findings indicate that the orientation of the Phe7 backbone is important and that HyAsp15 stabilizes the optimal binding conformation when its carboxylate group points toward the lysinoalanine. To test this hypothesis, simulations were conducted where the HyAsp15 side chain was restrained away from this conformation (by inverting the potential used to restrain the HyAsp15 into the position) to investigate if the stabilization was related to this specific conformation (simulation PE_HyAsp_HyAsp–). These simulations did not lead to a stable binding mode, as indicated by the fact that the lipid moved in and out of the binding pocket (see Figure S7c,g in the SI). However, simulations where the orientation of both Phe7(NH) and Phe7(O) were restrained to point into the pocket while the HyAsp15 side chain was kept away from its MD model orientation lead to a quite stable binding of the lipid (simulation PE_HyAsp_Ala(N)6Gly8HyAsp– shown in Figure S7d,h in the SI). Thus, orienting the HyAsp15 carboxylate toward the LysAla19/6 bridge is likely to be important for stable binding of PE lipids. Furthermore, it may be possible to improve the binding stability by decreasing the flexibility near Phe7. This may be done experimentally by directly reducing the flexibility of the backbone, e.g., by mutating Gly8 to a d-amino acid, or by fixating the HyAsp15 orientation, which also reduces the flexibility of the Phe7 backbone.
Having characterized the dynamic nature of the interactions of cinnamycin with its optimal binding partner, PE, binding of other lipid species may also be investigated and compared (Table 2). As cinnamycin-producing organisms modify their PE lipids to mPE for immunity, the interaction with mPE is of particular interest to study.24 Simulations of cinnamycin with POmPE in the binding pocket (mPE_HyAsp) revealed only the less-stable binding mode characterized by a rather long binding distance of 4.5 ± 0.4 Å (Figure 7a, dark green curve). Another difference, apparent from Figure 8b, is that the correlation observed for POPE, between the number of hydrogen bonds to the lipid ammonium and the orientation of Phe7(O), was not found for POmPE (see Figure 8b, blue curve). This suggests that Phe7 probably forms the weakest hydrogen bond with the lipid since it is broken when changing the type of the lipid bound from POPE to POmPE, while the hydrogen bonds with Val13(NH) and the HyAsp15 side chain were all conserved throughout the simulations (see Figure S8 in the SI). Restraining HyAsp15 (simulations named mPE_HyAsp_HyAsp+) did not lead to a stable binding of POmPE (Figure 8c).
Table 2. Atomistic Simulations of Cinnamycin–Lipid Complexes in a Bilayer.
| simulation namea | simulation time (μs) | lipid bound | bilayer type | mutation | restrained residuesb |
|---|---|---|---|---|---|
| PE_HyASP | 4 × 0.5 | POPE | POPC | ||
| PE_HyASP_HyASP | 2 × 0.5 | POPE | POPC | HyAsp15 | |
| PE_HyASP_Ala(N)6Gly8HyAsp | 2 × 0.5 | POPE | POPC | Ala(N)6, Gly8, HyAsp15 | |
| PE_HyAsp_HyAsp | 2 × 0.5 | POPE | POPC | HyAsp15 | |
| PE_Asp | 2 × 0.5 | POPE | POPC | Asp15 | |
| PE_Asp_Asp | 2 × 0.5 | POPE | POPC | Asp15 | Asp15+ |
| PE_HyAsp_Ala(N)6 | 2 × 0.5 | POPE | POPC | Ala(N)6 | |
| PE_HyAsp_Gly8 | 2 × 0.5 | POPE | POPC | Gly8 | |
| mPE_HyAsp | 6 × 0.5 | POmPE | POPC |
Simulations are named lipid_residue_setup, in which the lipid refers to the lipid in complex with cinnamycin, the residue denotes the amino acid at position 15 of cinnamycin, and the setup reflects either the restraints applied or changes in the bilayer.
Restraints applied in the simulations. Restraints specified with a + or – superscript denoting whether the side chain χ1 angle is restrained in or away from the gauche(+) conformation, respectively, while the Ala(N)6 and Gly8 labels specify restraints put on the backbone to fix the backbone orientation of Phe7(HN) and Phe7(O) to fit the optimized structure (see details in the SI).
Cinnamycin with POPS bound (PS_HyAsp) (Figure 9d) was remarkably stable at a distance of 3.5 ± 0.2 Å (red in Figure 7a), similar to the distance observed for the tightly bound mode of POPE. This suggests that the observed selectivity for PE over PS is not due to steric clash between the peptide and the carboxylate group of the bound PS lipid head group. Instead the selectivity may be due to differences in the orientation and interaction with the membrane (see Section 2.4). It should be noted that bacterial membranes typically contain insignificant amounts of PS,25,26 whereas mammalian plasma membranes contain PS primarily in the inner leaflet but is present at the surface of the cell during apoptosis.27
Figure 9.
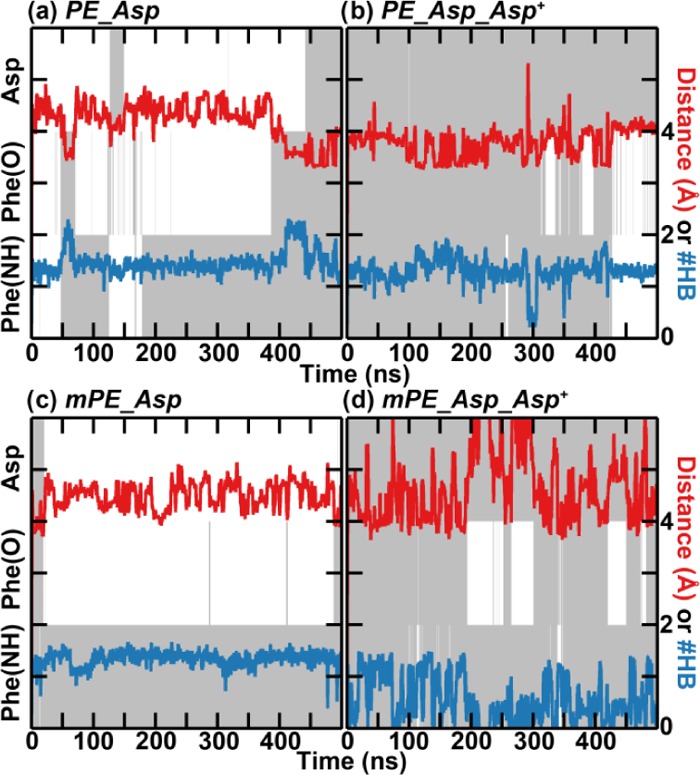
Relation between the orientation of specific functional groups and the stability of the binding over time for cinnamycin with the HyAsp15Asp mutation. The simulations were conducted with (a) POPE bound to cinnamycin HyAsp15Asp (PE_Asp), (b) as (a) but with the orientation of the side chain of Asp restrained (PE_Asp_Asp+), (c) POmPE bound to cinnamycin HyAsp15Asp (mPE_Asp), and (d) as (c) but with the orientation of Asp restrained (mPE_Asp_Asp+). Color coding and axes are the same as in Figure 6.
Starting the simulation with POPC in the binding pocket of cinnamycin (PC_HyAsp) led to an even less-stable binding than observed with POmPE bound, and the lipid was released after ∼300 ns in the simulation shown in Figure 8e. The release of the lipid indicates that the lipids need to interact with the ammonium-binding site to exhibit a high affinity for cinnamycin.
2.3. Function of HyAsp15
Cinnamycin has five post-translational modifications: one Lan, two MeLan, one LysAla, and one HyAsp. The Lan, MeLan, and LysAla residues crosslink cinnamycin, which results in a relatively rigid peptide with a well-defined structure, while the function of HyAsp15 is not similarly clear. HyAsp15 was observed in the simulations to make a hydrogen bond with the ammonium of the PE lipid. However, it is uncertain if this is needed for a stable binding of the lipids. To investigate this further, HyAsp15 was mutated in silico to an Asp, and simulations were conducted with POPE bound (PE_Asp), with POPE bound and the orientation of Asp15 restrained (PE_Asp_Asp+), with POmPE bound (mPE_Asp), and with POmPE bound and the orientation of Asp15 restrained (mPE_Asp_Asp+). A plot equivalent to Figure 6 of the correlation between the orientation of Phe7(O), Phe7(NH), and the side chain of Asp15, the number of hydrogen bounds to the ammonium group of the lipid, and the distance between the ammonium and the binding pocket are given in Figure 9 for representative simulations.
With POPE located in the binding site of the mutated cinnamycin (PE_Asp), the distance between the lipid ammonium and the binding pocket was found to change substantially indicating that the interaction was unstable (Figure 9a). Rather the lipid moved between the bound state located at a distance of 3.5 ± 0.2 Å and a weakly bound state at 4.3 ± 0.4 Å similar to what was observed for the wild-type (wt) cinnamycin (dark purple lines in Figure 7b). Unlike the wt cinnamycin, two peaks, located at 3.3 ± 0.1 and 3.8 ± 0.3 Å, were still observed in the distance histogram after restraining Asp15 (PE_Asp_Asp+, the light purple line in Figure 7b). The conformation located at 3.8 Å was not observed for the wt cinnamycin (Figure 7a) indicating that the hydroxyl group hinders the accessibility of this state hereby stabilizing the binding of PE lipids when compared to the cinnamycin HyAsp15Asp mutant. Furthermore, while restraining the HyAsp15 in the wt cinnamycin stabilized the orientation of Phe7 backbone (PE_HyAsp_HyAsp+), it was still flexible in the mutant even with Asp15 restrained (PE_Asp_Asp+, Figure 9b). The increased flexibility of the Phe7 in the mutant indicates that the tight binding of PE in the wt cinnamycin decreases the flexibility of the backbone near Phe7. Likewise, the number of hydrogen bonds is decreased compared to wt cinnamycin (compare the blue line in Figures 9b and 7a), which destabilized the binding. With POmPE bound (mPE_Asp), a single peak was observed at 4.5 ± 0.4 Å (the dark green line in Figure 7b) and the Phe7 was quite flexible (Figure 9c) equivalent to what was observed for the wt peptide (mPE_HyAsp). Restraining the Asp15 side chain also failed to generate a stable bound conformation (mPE_Asp_Asp+, Figure 9d), and the number of hydrogen bonds decreased when compared to both the wt cinnamycin and the unrestrained HyAsp15Asp mutant. This is because mPE can only bind in the less-stable binding mode exemplified for PE at the 60 and 85 ns snapshots in Figure 6. Thus, when the Asp15 side chain orientation is restrained, no hydrogen bonds can be made with Asp15 and the lipid ammonium only forms a hydrogen bond to Val13.
Overall, it may be concluded that the function of the hydroxyl group mainly is to fix the PE lipid in the binding pocket, which stabilizes the binding site, and to act as a hydrogen bond acceptor to form a hydrogen-bonding network fitting the tetrahedral shape of the ammonium group, as illustrated in Figure 4c. Furthermore, the small ammonium-binding pocket only allows the PE head group to bind tightly while disallowing the binding of larger head groups such as mPE, independent of whether the Asp15 is hydroxylated or not. Finally, the binding mode of POPS is very similar to that of POPE indicating that the selectivity for POPE over POPS cannot be explained by the interactions between cinnamycin and the lipid head group. Instead, the selectivity could be related to the orientation of the peptide in the membrane, which we will be explaining next.
2.4. Orientation of Cinnamycin Relative to the Membrane
Interactions of cinnamycin with different lipid species also affected the orientation and the insertion depth of the peptide in the membrane, as apparent from Figure 10 (the roll angle and the distance to the membrane center are defined in Figure S9 in the SI).
Figure 10.
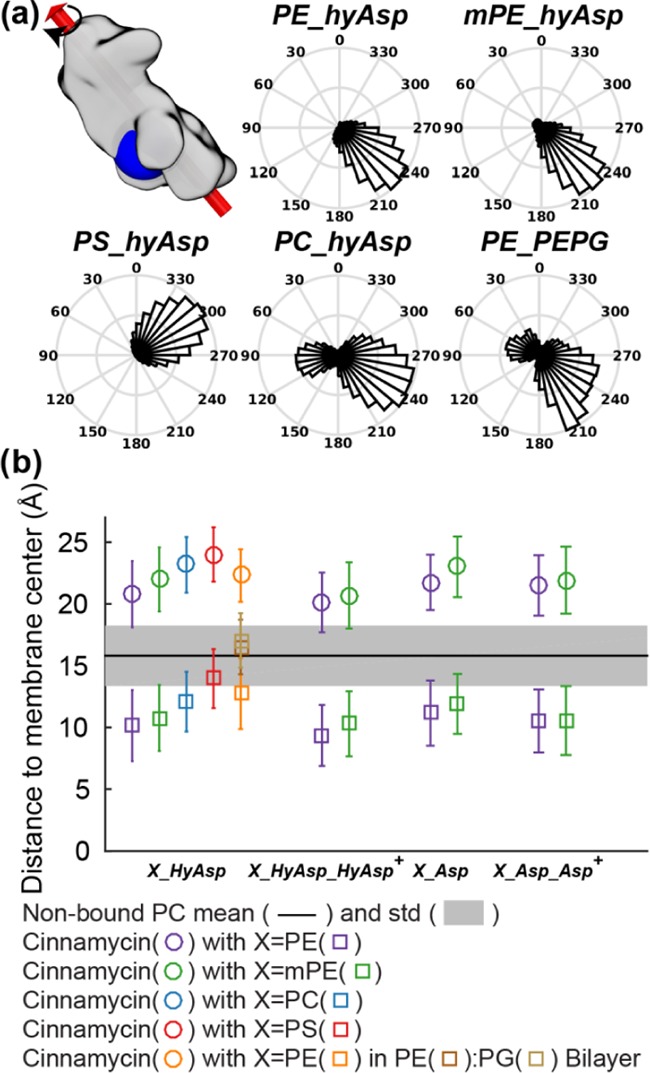
Position and orientation of cinnamycin relative to the membrane are dependent on the lipid bound and the composition of the membrane. (a) The roll angle (see the text for definition) with respect to the bilayer of the peptide when POPE (PE_HyAsp), POmPE (mPE_HyAsp), POPS (PS_HyAsp), or POPC (PC_HyAsp) is bound to cinnamycin in a POPC bilayer and with POPE bound to cinnamycin in a 4:1 POPE/POPG bilayer (PE_PEPG). An angle of ∼0° indicates that the binding pocket is oriented into the solvent, while an angle between 90 and 270° indicates that the binding pocket is oriented toward the membrane. (b) The distance above the bilayer center of: the center of mass (COM) of cinnamycin (circles), ester oxygens of the bound lipid and nonbound PE and PG lipids (squares), and ester oxygens of the nonbound PC lipids (the black line is mean and the shaded area is standard deviation). Error bars specify standard deviation of the measured distances.
With POPE or POmPE lipids bound (simulations PE_HyAsp and mPE_HyAsp, respectively), the roll angle of the peptide shown in Figure 10a was ∼230° indicating that the binding pocket was pointing into the membrane. However, with POPS bound (PS_HyAsp), the binding pocket was rotated to an angle of 300° such that it was oriented toward the solvent. The difference in the orientation is a consequence of the charged carboxylate group of the POPS head group; the carboxylate projects out of the binding pocket and its highly hydrophilic character makes it favorable to position itself in the solvent instead of being embedded in the membrane, which is fulfilled by rotating the peptide. As previously stated, the POPC–cinnamycin complex was not stable, and in one of the simulations, the lipid fully dissociated (Figure 8e). The release also affected the orientation of the peptide in the membrane since two populations are observed: one at 90° and the other at about 240°. The one at 240° is approximately the same orientation as observed for POPE and POmPE, while the other at 90° occurred due to the release of the lipid from the binding pocket. The release resulted in the negatively charged HyAsp15 side chain flipping out of the binding pocket because the charged carboxylate group was no longer interacting with the positively charged ammonium group of the lipid. The flip hereby made it necessary for the peptide to rotate in the membrane to position the carboxylate group of HyAsp15 toward the solvent and the positively charged head groups of the membrane.
Besides the roll of the peptide, its insertion depth was also found to depend on the lipid-type bound (see circles in Figure 10b). The peptide was situated deepest in the membrane with POPE bound (PE_HyAsp). Increasing the size of the head group from PE (PE_HyAsp) over mPE (mPE_HyAsp) to PC (PC_HyAsp) led to a gradual increase in the distance between the center of mass (COM) of the peptide and the hydrophobic interface of the bilayer. Restraining the HyAsp15 side chain to point into the pocket as in the MD model led to a deeper insertion of the peptide (comparing X_HyAsp_HyAsp+ with X_HyAsp in Figure 10b), while mutating HyAsp15 to Asp (comparing X_HyAsp with X_Asp in Figure 9b) resulted in the peptide being located higher in the membrane. These data correlate with the stability of peptide–lipid interactions: when the peptide was in complex with a lipid that showed less-stable binding, the peptides seemed to be located higher in the membrane. An explanation for this correlation could be that if the lipids do not fulfill the hydrogen bond donors and acceptors of cinnamycin, the peptide is located higher in the membrane to satisfy these missing hydrogen bonds by interaction with the polar part of the lipids or the solvent. Cinnamycin with POPS bound (PS_HyAsp) was located highest in the membrane possibly due to the hydrophilic character of the carboxyl group of the POPS head group, which was, consequently, observed to be solvated in the simulations.
Investigating the position of the bound lipid along the bilayer normal (squares in Figure 9b) also shows that, when POPE was bound (PE_HyAsp), the ester oxygens of the bound lipid were pushed further into the membrane compared to the nonbound lipids (the shaded area in Figure 9b). This shift of the bound lipid compared to the other lipids demonstrates that deep insertion of the peptide also led the hydrophilic part of the bound lipid being placed far into the hydrophobic part of the membrane. This effect was much smaller with POPS bound (the red square in Figure 9b) in accordance with the need for placing the carboxylate group of the PS lipid head group in the solvent. Similar to the peptide position, the distance between the bound lipid and the bilayer center increased when going from PE over mPE to PC signifying that the higher positioning of the peptide in the bilayer results in less insertion of the polar head group of the bound lipid in the hydrophobic part of the bilayer.
A bilayer with a lipid composition of 20:80 POPG/POPE was also modeled, to mimic a bacterial membrane.28 In these simulations, two different populations of orientations were observed (Figure 10a), one at about 60° (i.e., the pocket pointing toward the solvent) and the other at approximately 230° (i.e., the pocket oriented into the membrane). The conformations giving rise to the peak at 60° all originate from the same simulation in which interactions between POPG lipids in the bilayer and the two positively charged residues, Arg2 and LysAla19/6, are observed. These two residues are placed at opposite sides of the peptide with respect to the binding pocket. The electrostatic interactions, therefore, orient the peptide such that these residues point toward the bilayer, thus exposing the binding pocket and the bound head group of the bound lipid to the solvent. The simulation with the pocket oriented toward the solvent furthermore resulted in two peaks in the pocket distance histogram located at 3.5 ± 0.4 and 4.3 ± 0.8 Å (the black curve in Figure S10d in the SI) indicating that orienting the binding pocket into the solvent leads to a much less-stable binding of the bound lipid compared to when the pocket is oriented toward the bilayer center. For comparison, only one was observed at 3.5 Å ± 0.4 Å in the two other repeats wherein the binding pocket is oriented into the bilayer (orange and light blue lines in Figure S10d in the SI). The electrostatic interaction observed between Arg2 and LysAla19/6 is likely reduced in vivo by the higher ion concentration present, and we, thus, regard the single simulation showing these interactions to be less abundant in vivo and, therefore, do not expect the orientation of the peptide to be significantly affected by the PG lipids. Furthermore, the ester oxygens specifying the hydrophobic interface are located further from the bilayer center for the POPE/POPG bilayer as compared to the POPC bilayers (brown squares compared to the black line in Figure 10b), indicating a higher hydrophobic thickness. This is consistent with the higher lipid tail order and lower head group area previously found for POPE bilayers compared to POPC bilayers.29−32 This could be one of the reasons for cinnamycin being positioned higher in the PE/PG bilayer compared to that in the POPC bilayer in the simulations.
2.5. Membrane Effects
Within the timescale simulated, with only one cinnamycin molecule present, we do not observe any changes in the biophysical properties of the membrane that is indicative of early stages of membrane disruption, even after we extended three simulations to 1 μs (one PE_HyAsp, one PE_HyAsp_HyAsp+, and one PS_HyAsp. Changes in water penetration and membrane thickness are shown in Figure S11). It is likely that both a higher concentration of cinnamycin is required as well as an extended simulation time. This is, however, beyond the scope of this paper.
2.6. Discussion
We have shown that cinnamycin binds to PE lipids via a tight hydrogen-bonding network formed between the lipid ammonium and the backbone carbonyls of Val13 and Phe7 alongside the side chain of HyAsp15. Further stabilization of the lipid binding is obtained through binding of the lipid phosphate to a binding site formed by the backbone amides of residues 10–13. Besides showing more extensive favorable interactions between the lipid and the peptide than observed in the DMSO-solution NMR structure,20 this new model can explain previously puzzling experimental findings. First of all, two peaks were observed in the NMR spectrum for the LysAla19/6 side chain ammonium when the peptide was lipid-bound, while no peaks were present when the peptide was studied in the solution.6 Such a difference indicates that the binding of a lipid results in changes in the protein structure or dynamics that causes the two hydrogen atoms to experience different environments and which, furthermore, slows the hydrogen exchange with water. These findings were initially thought to be due to interactions with the phosphate group of the lipid,6 however, with the publication of the NMR model, it became apparent that the phosphate group is too far away to affect the environment of the LysAla19/6 side chain and no obvious explanation was evident from the NMR model.20 Our new model shows that the HyAsp15 side chain needs to be oriented into the pocket for optimal lipid binding. Such orientation of the carboxylate group of HyAsp15 results in hydrogen bond formation between the HyAsp15 and the ammonium of the LysAla19/6, the lipid ammonium, as well as the backbone amide of Ser6 and Phe7 (see Figure 4). This interaction between the HyAsp15 and the LysAla19/6 fixes the LysAla19/6 bridge, hereby slowing the hydrogen exchange and making the environment of the two ammonium hydrogen atoms different, thus explaining how two individual peaks can appear in the NMR measurements upon lipid binding.
Another result from the current study is that, in the MD model, the lipid phosphate makes hydrogen bonds with the amide hydrogens of residues 10–13. These interactions are in accordance with several studies: Wakamatsu et al. used NMR to show that Val13 loses its contacts with Gly8 and Pro9 when a lipid binds.6 This is consistent with phosphate binding at this site. Furthermore, studies have shown that phosphatidylethanolamine binds to cinnamycin, while phosphatidylpropanolamine does not.12 The fact that the major difference between these two lipids is the distance between the ammonium and the phosphate group indicates that both functional groups bind to the peptide and that the distance between them is important. Both of these experimental findings indicate the existence of a phosphate-binding site in cinnamycin.
How cinnamycin kills bacterial cells remains poorly understood. The simulations presented here indicate that cinnamycin inserts itself deep in the membrane when PE is bound thereby drawing the bound PE head group far into the hydrophobic part of the membrane. This is consistent with the high electron density inside the hydrophobic region of the bilayer observed by small-angle X-ray scattering measurements.21 While the phosphoethanolamine moiety of the lipid is screened by interactions with cinnamycin, the glycerol group is not. The embedding of this hydrophilic group may, therefore, introduce a local polar environment in the hydrophobic part of the lipid bilayer, which can be speculated to be necessary for permeabilization, since the glycerol linker group has been found essential for permeabilization of vesicles but not for lipid binding.7 Since cinnamycin has been observed to transport not only PE lipids across the membrane but also other lipids present in the membrane, such as PC lipids,8 it is unlikely that it is only the bound lipid that moves over the membrane during these lipid flip-flops.
The binding of POPS to cinnamycin was remarkably stable in the simulations; however, the peptide was at the same time located much higher in the membrane compared to having PE bound. Experimentally, the binding affinity of cinnamycin-like peptides for PS lipids has been found to be much lower than for PE but higher than for other lipids such as PC.8,19 An explanation for this difference in binding affinity may be that by forcing cinnamycin to be positioned higher in the membrane, PS hinders the peptide from forming interactions with the hydrophobic core of the membrane equally favorable as when PE is bound.8,19 Furthermore, isothermal titration calorimetry data show that hydrophobic interactions contribute significantly in the binding of cinnamycin to PE-containing membranes indicating a deep insertion in the membrane.22,33
PC lipids were found to interact only with the phosphate-binding site, because the choline group was too large to fit in the amine binding site in addition to being unable to form hydrogen bonds at this site. The binding affinity of cinnamycin for POPC, which is rather low,33 may, therefore, be attributed to hydrophobic interactions and the interaction with the phosphate-binding site.
3. Conclusions
In summary, the binding conformation of the PE–cinnamycin complex was optimized, the binding of different lipids was characterized, the function of the hydroxyl group of HyAsp15 was investigated, and the location of the peptide/lipid complex in a membrane was examined. The selectivity of cinnamycin for PE lipids is suggested to be caused by a hydrogen-bonding network formed between the lipid ammonium and the backbone carbonyl of Phe7 and Val13 in addition to the hydroxyl and the carboxylate groups of HyAsp15 (Figure 4c). Beside this ammonium-binding site, the backbone amide hydrogens of residues 10–13 were found to interact with the lipid phosphate. Mutating HyAsp15 to Asp in silico resulted in an unstable binding conformation of PE indicating that the hydroxylation of Asp15 is also important for lipid binding.
Cinnamycin was found to bind deeply in the bilayer when PE was present, while binding of PS leads the peptide being positioned higher in the membrane and with the binding pocket oriented into the solvent instead of into the membrane (see Figure 10a). The selectivity for PE over PS may, therefore, be attributed to the stronger hydrophobic interaction obtained between cinnamycin and the bilayer when PE is bound since no significant difference in interactions was observed between cinnamycin and the bound PS or PE lipid head group.
In accordance with experiments, cinnamycin and mPE were found to form a less stable complex compared to PE interacting with cinnamycin. This selectivity for PE over mPE was mainly due to the secondary ammonium group of mPE not being able to fit in the binding pocket and the hydrogen bond with the Phe7 amide being lost. The loss of this specific hydrogen bond is in accordance with the observation that it is the most dynamic area of the binding pocket due to the flexibility of the Gly8 backbone placed next to it (see Figure 1). Restricting the motion around Phe7, e.g., by mutating Gly8 to a d-amino acid, could, therefore, potentially stabilize PE lipid binding further, increasing the efficiency of the peptide.
In summary, our results shed further light on the lipid–cinnamycin complexes, and the insights obtained from this study may be used to improve the lipid binding affinity of the peptide or even in the design of novel peptides with increased lipid binding affinity that could aid in the targeting of mPE-rich membranes. Furthermore, this study represents an initial step toward elucidating the membrane perturbation mechanism, since the structure obtained can be used to simulate how multiple peptides will interact with and perturb the membrane.
4. Methods
Simulations of the cinnamycin–DLPE complex in different solvents (see Table 1) were used to elucidate the optimal structure of the complex. DLPE was chosen to allow for efficient sampling of the bound conformation by keeping the size of the simulations to a minimum, as it has short acyl tails and a PE head group (and is represented in the CHARMM36 force field.34) Because none of the simulations initiated directly from the NMR model resulted in stable complex formation (see Section 2.1), simulations were equilibrated with restraints derived from the NMR data (see Figure 11) as specified in Table 1, and the production runs were conducted without restraints.
Besides the simulations of a cinnamycin–lipid complex in different solvents, we also simulated cinnamycin bound to lipids and embedded in a lipid bilayer membrane environment to investigate the origin of the selectivity of cinnamycin for PE lipids. The lipids bound to cinnamycin, thus, explored were 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), N-methyl 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POmPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS), and POPC. Coarse-grained (CG) simulations with the MARTINI CG force field22,23 were used to assemble lipid bilayers around cinnamycin, thus generating configurations from which the atomistic simulations were initiated (see Table 2). The production runs were conducted without the use of the NMR restraints. All time-evolution plots derived from these simulations are running averages with a window size of 100 frames (1 ns). A fuller description of the methods used is provided in the SI.
Acknowledgments
This research was supported by the Danish Council for Independent Research | Technology and Production Sciences (FTP-0602-01778B), the Danish National Research Foundation (DNRF59) and the Danish Centre for Scientific Computing (CSCAA). H.K. was supported by an Alfred Benzon Fellowship. C.S. was supported by a Marie Curie Intra European Fellowship within the 7th European Community Framework Program. Research in MS’s group is supported by grants from the Wellcome Trust, BBSRC, EPSRC, and the Leverhulme Trust.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b02949.
Methods section including the used simulation parameters; bond parameters, angle parameters, dihedral parameters; atom types and charges of the residues; CG parameters; distribution of ϕ and ψ; NOE analysis; DLPE; root-mean-square fluctuation of cinnamycin; relation between different interactions and the stability of the binding over time; PE–cinnamycin complex with different residues restrained; hydrogen bonds between the lipid amine and the binding pocket; definition of the orientation of the peptide with respect to the bilayer; distance histogram; plot comparing early and final mass densities (Tables S1–S4 and Figures S1–S11) (PDF)
Author Present Address
⊥ Schrödinger, 120 West 45th Street, 17th Floor, New York, New York 10036, United States (H.K.).
Author Present Address
∥ Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, People’s Republic of China (C.S.).
Author Present Address
§ Center for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, People’s Republic of China (C.S.).
The authors declare no competing financial interest.
Supplementary Material
References
- Dischinger J.; Chipalu S. B.; Bierbaum G. Lantibiotics: Promising Candidates for Future Applications in Health Care. Int. J. Med. Microbiol. 2014, 304, 51–62. 10.1016/j.ijmm.2013.09.003. [DOI] [PubMed] [Google Scholar]
- European Economic Community Commission Directive. Official J. European Union 1983, 255, 1–−6. 83/463/EEC. [Google Scholar]
- Cotter P. D.; Hill C.; Ross R. P. Bacteriocins: Developing Innate Immunity for Food. Nat. Rev. Microbiol. 2005, 3, 777–788. 10.1038/nrmicro1273. [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration GRAS Notice No. GRN 000135 Agency Response Letter, 2004. [Google Scholar]
- Cotter P. D.; Ross R. P.; Hill C. Bacteriocins - a Viable Alternative to Antibiotics?. Nat. Rev. Microbiol. 2013, 11, 95–105. 10.1038/nrmicro2937. [DOI] [PubMed] [Google Scholar]
- Wakamatsu K.; Choung S. Y.; Kobayashi T.; Inoue K.; Higashijima T.; Miyazawa T. Complex Formation of Peptide Antibiotic Ro09-0198 with Lysophosphatidylethanolamine:1H NMR Analyses in Dimethyl Sulfoxide Solution. Biochemistry 1990, 29, 113–118. 10.1021/bi00453a013. [DOI] [PubMed] [Google Scholar]
- Choung S.-Y.; Kobayashi T.; Takemoto K.; Ishitsuka H.; Inoue K. Interaction of a Cyclic Peptide, Ro09-0198, with Phosphatidylethanolamine in Liposomal Membranes. Biochim. Biophys. Acta, Biomembr. 1988, 940, 180–187. 10.1016/0005-2736(88)90193-9. [DOI] [PubMed] [Google Scholar]
- Makino A.; Baba T.; Fujimoto K.; Iwamoto K.; Yano Y.; Terada N.; Ohno S.; Sato S. B.; Ohta A.; Umeda M.; et al. Cinnamycin (Ro 09-0198) Promotes Cell Binding and Toxicity by Inducing Transbilayer Lipid Movement. J. Biol. Chem. 2003, 278, 3204–3209. 10.1074/jbc.M210347200. [DOI] [PubMed] [Google Scholar]
- Ökesli A.; Cooper L. E.; Fogle E. J.; van der Donk W. A. Nine Post-Translational Modifications during the Biosynthesis of Cinnamycin. J. Am. Chem. Soc. 2011, 133, 13753–13760. 10.1021/ja205783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M. Lantibiotics as Probes for Phosphatidylethanolamine. Amino Acids 2011, 41, 1071–1079. 10.1007/s00726-009-0386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierbaum G.; Sahl H.-G. Lantibiotics: Mode of Action, Biosynthesis and Bioengineering. Curr. Pharm. Biotechnol. 2009, 10, 2–18. 10.2174/138920109787048616. [DOI] [PubMed] [Google Scholar]
- Choung S.-Y.; Kobayashi T.; Inoue J.; Takemoto K.; Ishitsuka H.; Inoue K. Hemolytic Activity of a Cyclic Peptide Ro09-0198 Isolated from Streptoverticillium. Biochim. Biophys. Acta, Biomembr. 1988, 940, 171–179. 10.1016/0005-2736(88)90192-7. [DOI] [PubMed] [Google Scholar]
- Lindenfelser L. A.; Pridham T. G.; Kemp C. E. Antibiotics against Plant Disease. V. Activity of Cinnamycin against Selected Microorganisms. Antibiot. Chemother. 1959, 9, 690–695. [PubMed] [Google Scholar]
- Kido Y.; Hamakado T.; Yoshida T.; Anno M.; Motoki Y. Isolation and Characterization of Ancovenin, a New Inhibitor of Angiotensin-i Converting Enzyme, Produced by Actinomycetes. J. Antibiot. 1983, 36, 1295–1299. 10.7164/antibiotics.36.1295. [DOI] [PubMed] [Google Scholar]
- Märki F.; Hänni E.; Fredenhagen A.; van Oostrum J. Mode of Action of the Lanthionine-Containing Peptide Antibiotics Duramycin, Duramycin B and C, and Cinnamycin as Indirect Inhibitors of Phospholipase A2. Biochem. Pharmacol. 1991, 42, 2027–2035. 10.1016/0006-2952(91)90604-4. [DOI] [PubMed] [Google Scholar]
- Naruse N.; Tenmyo O.; Tomita K.; Konishi M.; Miyaki T.; Kawaguchi H.; Fukase K.; Wakamiya T.; Shiba T. Lanthiopeptin, a New Peptide Antibiotic - Production, Isolation and Properties of Lanthiopeptin. J. Antibiot. 1989, 42, 837–845. 10.7164/antibiotics.42.837. [DOI] [PubMed] [Google Scholar]
- Cotter P. D.; Hill C.; Ross R. P. Bacterial Lantibiotics: Strategies to Improve Therapeutic Potential. Curr. Protein Pept. Sci. 2005, 6, 61–75. 10.2174/1389203053027584. [DOI] [PubMed] [Google Scholar]
- Yao S.; Hu K.; Tang G.; Liang X.; Du K.; Nie D.; Jiang S.; Zang L. Positron Emission Tomography Imaging of Cell Death with [18F]FPDuramycin. Apoptosis 2014, 19, 841–850. 10.1007/s10495-013-0964-x. [DOI] [PubMed] [Google Scholar]
- Hou S.; Johnson S. E.; Zhao M. A One-Step Staining Probe for Phosphatidylethanolamine. ChemBioChem 2015, 16, 1955–1960. 10.1002/cbic.201500127. [DOI] [PubMed] [Google Scholar]
- Hosoda K.; Ohya M.; Kohno T.; Maeda T.; Endo S.; Wakamatsu K. Structure Determination of an Immunopotentiator Peptide, Cinnamycin, Complexed with Lysophosphatidylethanolamine by 1H-NMR1. J. Biochem. 1996, 119, 226–230. 10.1093/oxfordjournals.jbchem.a021226. [DOI] [PubMed] [Google Scholar]
- Iwamoto K.; Hayakawa T.; Murate M.; Makino A.; Ito K.; Fujisawa T.; Kobayashi T. Curvature-Dependent Recognition of Ethanolamine Phospholipids by Duramycin and Cinnamycin. Biophys. J. 2007, 93, 1608–1619. 10.1529/biophysj.106.101584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaidze G.; Seelig J. Specific Binding of Cinnamycin (Ro 09-0198) to Phosphatidylethanolamine. Comparison between Micellar and Membrane Environments. Biochemistry 2003, 42, 12570–12576. 10.1021/bi035225b. [DOI] [PubMed] [Google Scholar]
- Gao J.; Zheng H. Illuminating the Lipidome to Advance Biomedical Research: Peptide-Based Probes of Membrane Lipids. Future Med. Chem. 2013, 5, 947–959. 10.4155/fmc.13.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke S.; Widdick D.; Bibb M. A Novel Mechanism of Immunity Controls the Onset of Cinnamycin Biosynthesis in Streptomyces cinnamoneus DSM 40646. J. Ind. Microbiol. Biotechnol. 2017, 44, 563–572. 10.1007/s10295-016-1869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-M.; Rock C. O. Membrane Lipid Homeostasis in Bacteria. Nat. Rev. Microbiol. 2008, 6, 222–233. 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- Epand R. M.; Epand R. F. Bacterial Membrane Lipids in the Action of Antimicrobial Agents. J. Pept. Sci. 2011, 17, 298–305. 10.1002/psc.1319. [DOI] [PubMed] [Google Scholar]
- Frey B.; Gaipl U. The Immune Functions of Phosphatidylserine in Membranes of Dying Cells and Microvesicles. Semin. Immunopathol. 2011, 33, 497–516. 10.1007/s00281-010-0228-6. [DOI] [PubMed] [Google Scholar]
- Epand R. F. R. M.; Epand R. F. R. M. Domains in Bacterial Membranes and the Action of Antimicrobial Agents. Mol. Biosyst. 2009, 5, 580–587. 10.1039/b900278m. [DOI] [PubMed] [Google Scholar]
- Lafleur M.; Cullis P. R.; Bloom M. Modulation of the Orientational Order Profile of the Lipid Acyl Chain in the Lα Phase. Eur. Biophys. J. 1990, 19, 55–62. 10.1007/BF00185086. [DOI] [PubMed] [Google Scholar]
- Fenske D. B.; Jarrell H. C.; Guo Y.; Hui S. W. Effect of Unsaturated Phosphatidylethanolamine on the Chain Order Profile of Bilayers at the Onset of the Hexagonal Phase Transition. A Deuterium NMR Study. Biochemistry 1990, 29, 11222–11229. 10.1021/bi00503a010. [DOI] [PubMed] [Google Scholar]
- Kučerka N.; van Oosten B.; Pan J.; Heberle F. A.; Harroun T. A.; Katsaras J. Molecular Structures of Fluid Phosphatidylethanolamine Bilayers Obtained from Simulation-to-Experiment Comparisons and Experimental Scattering Density Profiles. J. Phys. Chem. B 2015, 119, 1947–1956. 10.1021/jp511159q. [DOI] [PubMed] [Google Scholar]
- Kučerka N.; Nieh M.-P.; Katsaras J. Fluid Phase Lipid Areas and Bilayer Thicknesses of Commonly Used Phosphatidylcholines as a Function of Temperature. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 2761–2771. 10.1016/j.bbamem.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Machaidze G.; Ziegler A.; Seelig J. Specific Binding of Ro 09-0198 (Cinnamycin) to Phosphatidylethanolamine: A Thermodynamic Analysis. Biochemistry 2002, 41, 1965–1971. 10.1021/bi015841c. [DOI] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.