

Abstract
Mass spectrometry (MS) measurements are not inherently calibrated. Researchers use various calibration methods to assign meaning to arbitrary signal intensities and improve precision. Internal calibration (IC) methods use internal standards (IS) such as synthesized or recombinant proteins or peptides to calibrate MS measurements by comparing endogenous analyte signal to the signal from known IS concentrations spiked into the same sample. However, recent work suggests that using IS as IC introduces quantitative biases that affect comparison across studies because of the inability of IS to capture all sources of variation present throughout an MS workflow. Here, we describe a single-point external calibration strategy to calibrate signal intensity measurements to a common reference material, placing MS measurements on the same scale and harmonizing signal intensities between instruments, acquisition methods, and sites. We demonstrate data harmonization between laboratories and methodologies using this generalizable approach.
Graphical Abstract
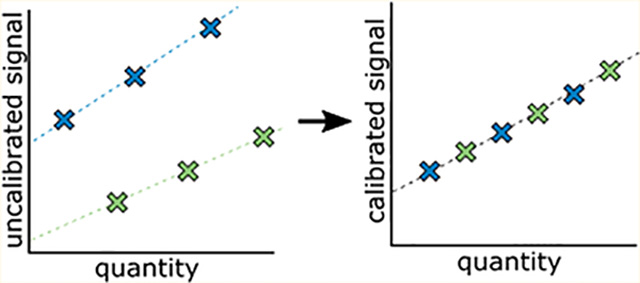
To convert signal from any analytical measurement into a more meaningful value, the signal is calibrated by scaling it relative to a reference standard. The goal of calibration is to put all measurements on the same scale, regardless of methodology, operator, instrumentation, or location. The bottom-up liquid chromatography-mass spectrometry (LCMS) field has approached the calibration of protein abundance in two ways: either through internal or external calibration.
Internal standards for MS can be unpaired or paired. Unpaired (also referred to as surrogate) standards typically consist of an exogenous protein or peptide spiked into the experimental sample itself, reviewed in greater detail elsewhere,1 while paired standards typically take the form of isotopically labeled peptides synthesized with heavy (15N, 13C, 18O) amino acids of the same sequence as the target analyte peptide. Although isotopically labeled synthetic peptides can serve as reasonable internal standards, this method suffers from several limitations. First, such peptides are not good calibrants because they do not necessarily reflect the level of the undigested protein2 and because methods for determining the amount of synthetic peptide in the standard often suffer from poor accuracy and precision.3 Second, this approach requires the enormous cost of synthesizing standards for every target in the experiment. Finally, a recent paper demonstrated lack of harmonized protein quantification when using stable isotope labeled peptides as internal calibrators.2 An alternative paired internal reference approach is “winged” peptides, where the measured peptide is flanked by some series of amino acids, such that the peptide standard is digested out of the wings. However, wings do not accurately capture the digestion conditions of the native protein sequence.2 Beyond winged peptides, researchers also attempt to use intact proteins as calibrants, but the inability to confirm that the standard protein has the same characteristics as the native protein (such as folding, PTMs, etc.) prevents this approach from being an ideal calibrant. In addition to protein-level internal standards, a final alternative approach, super-SILAC,4 pools experimental samples into a single master representative sample. The super-SILAC mixes can be used as internal standards, where the same master super-SILAC mix could be spiked into samples across experiments and laboratories as a calibrant. Because a super-SILAC mix includes all proteins in their endogenous states and respective matrixes, this approach to signal calibration would address many of the above-mentioned limitations. Although the super-SILAC approach is promising, it has not been demonstrated in the years following its proposal. Additionally, the SILAC method is applicable only to cell culturing experiments and is therefore limiting in scope. Because these internal standard approaches all suffer from known limitations, we propose to calibrate protein measurements relative to a common external reference material, which preserves all matrix and digestion properties of the protein measured.
In contrast to calibration by internal standard reference materials, external standard reference materials are separate samples whose acquisitions are interspersed among the experimental sample acquisitions. The external standard reference material is a representative matrix reflective of the experimental matrix; for example, an experiment measuring analytes in human cell lysates would use a pool of human cell culture, or in plasma would use a pool of plasma, or in yeast would use a bulk culture of yeast. The reference material is prepared alongside experimental samples in each sample processing batch, capturing all the conditions that the experimental samples experience from protein extraction to digestion kinetics and to instrument variation. Using this type of external calibration approach is common in clinical chemistry, where using a reference material such as normal human plasma for external calibration of patient samples improves precision and harmonization of measurements.5–8 Despite these successful implementations of calibration by external references in clinical MS experiments, the broader MS community, despite advances in label-free quantification,9 has not yet broadly adopted such an approach.
Here, we describe a generalized approach for calibration by external reference to correct for sample preparation batch variance and instrument-to-instrument variance in not only selected reaction monitoring/multiple reaction monitoring (SRM/MRM) experiments but also any LC-MS experiment. With this external reference approach, the most robust calibrators and reference materials will be stable over time—just as with all other reference materials. We demonstrate this approach in yeast, employing the BY4741 strain as the external reference. The BY4741 strain is particularly useful as a reference material because the copies per cell for many proteins have been estimated,10 enabling not only harmonization of the MS signal but also conversion of the signal into a biologically meaningful quantity.
EXPERIMENTAL SECTION
Sample Preparation.
The data regenerated in this work used yeast strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (Dharmacon) cultured in yeast extract peptone dextrose (YEPD) to mid log phase and then treated with NaCl to a final concentration of 0.4 M NaCl. Cell pellets were harvested and lysed individually with 8 M urea buffer solution and bead beating (seven cycles of 4 min beating with 1 min rest on ice). Cell lysates were reduced, alkylated, digested for 16 h, and desalted with a mixed-mode method.
Selected Reaction Monitoring Mass Spectrometry (SRM-MS).
Data were acquired using selected reaction monitoring (SRM) on a Proxeon EasyLC coupled to a Thermo Altis triple quadrupole mass spectrometer. Peptides were separated by reverse phase liquid chromatography using pulled tip columns created from 75 μm inner diameter fused silica capillary (New Objectives, Woburn, MA) in-house using a laser pulling device and packed with 3 μm ReproSil-Pur C18 beads (Dr. Maisch GmbH, Ammerbuch, Germany) to 30 cm. Trap columns were created from 150 μm inner diameter fused silica capillary fritted with Kasil on one end and packed with the same C18 beads to 3 cm. Solvent A was 0.1% formic acid in water (v/v), and solvent B was 0.1% formic acid in 80% acetonitrile (v/v). For each injection, approximately 1 μg total protein was loaded and eluted using a 90 min gradient from 5 to 40% B in 25 min, 40 to 60% B in 5 min, followed by a 15 min wash and then 15 min equilibration back to initial conditions. Total analytical run time was 45 min. Thermo RAW files were imported into Skyline11 (Skyline-daily version 4.1.1.18151) for processing and Total Area Fragment results were exported using a Custom Report.
Data-Independent Acquisition Mass Spectrometry (DIA-MS).
Data were acquired using data-independent acquisition (DIA) on a Waters NanoAcquity UPLC coupled to a Thermo Q-Exactive HF orbitrap mass spectrometer. Peptides were separated by reverse phase liquid chromatography using pulled tip columns created from 75 μm inner diameter fused silica capillary (New Objectives, Woburn, MA) in-house using a laser pulling device and packed with 3 μm ReproSil-Pur C18 beads (Dr. Maisch GmbH, Ammerbuch, Germany) to 30 cm. Trap columns were created from 150 μm inner diameter fused silica capillary fritted with Kasil on one end and packed with the same C18 beads to 3 cm. Solvent A was 0.1% formic acid in water (v/v), and solvent B was 0.1% formic acid in 98% acetonitrile (v/v). For each injection, approximately 1 μg of total protein was loaded and eluted using a 90 min separating gradient starting at 5 and increasing to 35% B, followed by a 40 min wash and equilibration (total 130 min method). DIA methods followed the chromatogram library workflow, described in greater detail elsewhere.12 Briefly, the untreated (reference) samples and osmotic shocked peptide samples were pooled 1:0.33:0.33:0.33 to create a library sample, and a Thermo Q-Exactive HF was configured to acquire six gas-phase fractions, each with 4 m/z DIA spectra using an overlapping window pattern from narrow mass ranges. For quantitative samples, the Thermo Q-Exactive HF was configured to acquire 25 × 24 m/z DIA spectra using an overlapping window pattern from 388.43 to 1012.70 m/z. The specific windowing schemes for both the chromatogram library construction and quantitative experiments are described in Table 1 in the Supporting Information. All DIA spectra were programmed with a normalized collision energy of 27 and an assumed charge state of +2.
Thermo RAW files were converted to .mzML format using the ProteoWizard package (version 3.0.10106), where they were centroided using vendor provided file reading libraries. Converted acquisition files were processed using Encyclope-DIA (version 0.7.0) configured with default settings (10 ppm precursor and fragment tolerances, considering only Y ions, and trypsin digestion was assumed). EncyclopeDIA features were submitted to Percolator (version 3.1) for validation at 1% FDR.
Calibration to an External Reference Sample.
The process of calibrating to an external reference material is straightforward (Figure 1). The percent change of an experimental sample (E) relative to the reference material (C) is calculated from the peak area (A) of a given peptide as
where the relative change is analogous to the delta notation used in isotopic composition chemistry13,14 but expressed as a percentage (%) instead of per mille (‰). To illustrate, consider a peptide with the same abundance (peak area) in the experimental and reference material. In this case, the R value is 0%:
Figure 1.

Scheme for calibration by global reference and by working reference. (a) Signal from samples collected at one site (experimental, EA; local reference, C′A; global reference, CA) are on a different scale compared to those collected at another site (experimental, EB; local reference, C′B; global reference, CB). (b) To harmonize the signals, we set a common scale relative to the global reference material (CA and CB). While the signal may not be measured on the same absolute scale (1a), as long as these reference materials are the same sample they should represent the same quantity. (c) Signals measured for batch A and batch B are calibrated by reporting their signal relative to the reference material signal. In cases necessitating a local working reference material (C′), experimental samples can be calibrated to their respective local working reference and then secondarily calibrated to the global reference.
In an alternate example, where the peptide is 2 times more abundant in the experimental sample than in the reference sample, the R value reflects that the abundance of the peptide in the experimental sample is 2 times (100% change) the abundance of the peptide in the reference material:
We note that R values are in the form of a percent relative to the reference but can also be converted to more meaningful units when those values are known in the reference material. Assuming that mass spectrometry response and analyte abundance are linear, if we quantify the analyte through any other method besides mass spectrometry, we can equate the unitage of the new method to the mass spectrometry signal. Converting the relative mass spectrometry signal from a percentage of the reference material to a relevant unitage such as concentrations (e.g., fmol/μL, μg/mL) makes interpretation of the measured values easier across different scientific fields and also enables transfer of the measurements between different lots or batches of reference material. To illustrate this point using our example in yeast, Ghememaghami et al. quantified the molecules-per-cell abundance of nearly all proteins expressed in the yeast strain BY4741 under laboratory standard conditions (YEPD media, 37C incubation, mid log growth phase) using a TAP-tag and quantitative Western blot approach. In expressing our measured mass spectrometry signals relative to the same reference material (BY4741 grown in laboratory standard conditions), we can associate the R of a reference material signal from a given protein to itself with a multiplier M from Ghememaghami et al.
To demonstrate, consider the example above where the abundance of a peptide in the experimental sample is twice (100% change) the abundance of a peptide in the reference material, and assume the peptide is unique to its protein of origin. With use of the Ghememaghami et al. molecules-per-cell multiplier for that protein in the BY4741 reference material, the equation becomes
where the 100% increase is now converted to the units held by M. We imagine a reference material may have a multiplier based on any quantitative assay, including enzyme-linked immunosorbent assay (ELISA), GFP-tagged fluorescence, or protein-specific colorimetric assays, if that assay and the MS assay is performed on the same reference material. However, we note that using a multiplier is not required for single-point calibration by an external reference material as we describe here because the purpose of the calibration is to place experimental measures relative to a reference material which is reported in the R value.
In the above scenarios, the reference material is the same for all experimental samples. However, we can imagine situations where this is not practical, for example, when experimental samples must be batched or where experimental samples are acquired longitudinally. In these situations, we introduce a local working reference material (C′). Here, three steps are required: (1) the peak area of a given peptide measured in an experimental sample in one batch is calibrated to its respective working reference material; (2) the experimental samples in another batch are calibrated to their respective working reference material; and (3) the working reference materials in turn are calibrated to each other through the global master reference (C).15 In this scenario, the peak area of a given peptide measured in one experimental sample can be expressed as
in which the peak area for a given peptide in the experimental sample relative to the master sample is a value equivalent to the multiplication of the experimental standard relative to the working standard and the working standard relative to the master standard. To demonstrate, assume an experiment in which the abundance of analyte in the local working reference (AC′) is threefold greater than that in the global master reference (AC), for example, , and assume that the abundance of analyte in the experimental sample (AE) is threefold greater than that in the local working reference, for example, . With use of these values and the equation above,
we find that the experimental analyte, relative to the global master reference, is 900% more abundant.
Data Analysis.
All raw data is publicly available on Panorama Public (https://panoramaweb.org/singlepointcal.url, individual file descriptions provided in Table 3 in the Supporting Information, ProteomeXchange ID PXD011297) along with Skyline documents for the SRM and DIA experiments performed in this work. Additionally, the processed quantitative data from this work is available in Table 2 in the Supporting Information. A Skyline-based tutorial for applying the method described in this work is provided along with open source code in the form of an annotated Python notebook at https://bitbucket.org/lkpino/single-point_calibration/wiki/Home.
RESULTS AND DISCUSSION
To demonstrate the proposed single-point calibration approach, we reproduced a portion of an osmotic shock experiment described by Selevsek et al.,16 in which cultures of S. cerevisiae strain BY4741 were grown unperturbed in YEPD media or shocked with 0.4 M NaCl. We evaluated the MS signal of proteins under osmotic shock with and without calibration to the reference material (unperturbed BY4741). First, we compared the effect of calibration on measurements made from identical biological samples prepared on different days by the same operator at the same site using the same instrument and acquisition method (Figure 2a). Because these two samples were highly comparable (same operator, same site, same instrument), we should not expect to see dramatically uncorrelated values, and indeed the raw signal shows improved agreement between days without calibration to the reference material. Applying calibration to the reference material in this case does not improve the agreement between the two samples but does assign biologically meaningful units (protein copies per cell) to the measurements (Figure 2b).
Figure 2.

External reference calibration harmonizes quantification across MS methods and laboratories. (a, c, e, g, i) Uncalibrated peak areas (log10) of shared precursors from between paired data sets are plotted across sites (site A, this work; site B, Selevsek et al.) and methodologies (DIA/SWATH and SRM). The bias of trends across MS methods reflects systematic differences in data acquisition and instrument platforms, as all data was bioinformatically processed using the same Skyline-based method. (b, d, f, h, j) Application of single-point external reference calibration and the biological unit multiplier10 harmonizes the majority of quantitative values and converts area ratios to meaningful units (protein molecules per cell).
On the basis of the precursors detected in these DIA data sets, we developed a targeted SRM method on a Thermo Altis triple quadrupole. We picked targets that spanned a range of signal response on the QEHF. We measured the two sample processing replicates using this scheduled SRM method and observe the same high agreement in both uncalibrated signals and calibrated signals that we see in measuring the two sample processing replicates by DIA (Figures 2a,b in the Supporting Information). Because these measurements were made in the same laboratory using the same method, the same chromatography column, the same instrument and the samples were acquired consecutively within 11 h of each other to minimize instrument performance variability, we might expect that the signals would be highly correlated even without calibration of each batch.
Next, we compared the signal from our DIA method on a Thermo Q Exactive-HF to the signal from the SRM method on a Thermo Altis triple quadrupole. The uncalibrated Orbitrap and triple quadrupole signals roughly follow the same trend, as might be expected from identical samples collected on two LCMS systems, but they also show a distinct bias away from the y = x line of equality because the raw signal from the Orbitrap is higher than that from the triple quadrupole (Figure 2c). After calibrating the osmotic shock signal to the reference material, we see the measurements falling tightly along the line of equality, indicating improved harmonization of the signals (Figure 2d). The amount of improvement is quantified by calculating the perpendicular offset, which is the distance of a point to the y = x line. The mean perpendicular offset drops from 0.5 to nearly zero by applying single-point calibration to the reference material (Figure 1 in the Supporting Information). There are three notable outliers falling to the right of the y = x line. Closer inspection reveals that these points are from low abundance peptides, where the signal is made irreproducible by interference (data not shown).
We assessed the agreement of quantitative measurements made on different instruments using different acquisition strategies (e.g., the Selevsek et al. SWATH-MS experiments versus Selevsek et al. SRM-MS experiments). Of note, although the 100 precursors targeted for SRM-MS were selected from detections and transitions derived from the SWATH-MS data, only 69 precursors had finite calibrated values to compare between the two. We found that although both platforms reported linear trends, the magnitude of the platforms’ signals correlated poorly (Figure 2e). Applying single-point calibration improved this agreement (Figure 2f), harmonizing the difference in signal intensities between the two platforms.
We then compared the agreement of quantitative measurements made from samples prepared by different operators on different instruments using the same acquisition style but different implementations (e.g., the Selevsek et al. SWATH-MS experiments performed on a Sciex 5600 tripleTOF versus our DIA-MS experiments performed on a Thermo Q Exactive HF). Although we refer to these two methods as SWATH and DIA, the methodological details of the two approaches are very similar (see Experimental Section and Table 1 in the Supporting Information). We found that 9932 shared precursors were measured with nonzero values between the two methods. The uncalibrated measurements of these 9932 precursors correlate poorly to each other and do not follow a y = x line of equality (Figure 2g). However, applying calibration using the reference materials improves agreement of the measurements from a mean perpendicular offset of 3.3 uncalibrated to an offset of 0.1 offset across sample preparations, operators, and instruments in these two studies (Figure 2h, Figure 1 in the Supporting Information). We calculated the Pearson product-moment correlation coefficient between the uncalibrated data and between the calibrated data. The uncalibrated DIA v SWATH correlation coefficient is 0.63, while the calibrated DIA v SWATH correlation coefficient is 0.92. For context, the correlation coefficient between the uncalibrated DIA-1 v DIA-2 data is 0.92, while the calibrated DIA-1 v DIA-2 correlation coefficient is 0.87. The improved correlation coefficient between DIA v SWATH data suggests that single-point calibration normalizes for experimental variations which may not affect all peptides in a systematic manner.
In addition to the global DIA and SWATH comparison, we compared targeted SRM methods between the two laboratory sites. Because we built our SRM method from our DIA detections, and site B built their SRM method from their SWATH detections, many of the precursors were not shared. Of the 11 precursors shared in the two SRM methods, we see a dramatic improvement in data harmonization by applying single-point calibration (Figure 2c,d in the Supporting Information).
Finally, we compared the quantitative measurements made at the different sites using different acquisition strategies on different instruments (e.g., the Selevesek et al. SRM-MS experiments versus our DIA-MS work experiments). Of the 100 Selevsek et al. SRM targeted peptides, 40 were also detected and measured by our DIA work. Similar to the poor agreement between different acquisition strategies on different instruments at the same site, we expected to see poor agreement when we looked between different sites (Figure 2i). We find that calibrating the measurements improved agreement slightly, and greatly improved the accuracy of the y = x model (Figure 2j). The complementary comparison between the Selevsek et al. targeted SRM experiment and our global DIA experiment was also performed (e.g., Selevsek et al. global SWATH experiment compared to our SRM experiment) with similar improvements to data harmonization (Figure 2e,f in the Supporting Information).
CONCLUSION
In summary, our analyses demonstrate that calibrating to an external reference material improves the harmonization of quantitative LC-MS proteomics data. The single-point calibration method, illustrated here in yeast, is generalizable to any proteomics experiment and is universally applicable across acquisition methods. To extrapolate from the various examples we show here, this approach is especially useful for longitudinal studies where samples are collected over extended time frames, consortium projects spanning multiple laboratories, and large-scale projects employing multiple instruments. We note that while removing internal standards from an experiment increases variance in instrument response, employing an external reference approach does not preclude the use of internal standards. Neither approach is perfect, and in the most ideal metrological scenario, the external reference approach illustrated here could be used together with internal standards. These approaches to ensure accurate and precise measurements come at the experimental cost of an additional sample acquisition (in the case of external reference calibration) or additional transitions monitored by the method (in the case of internal standards).
Following other analytical fields such as isotope ratio mass spectrometry,14 the proposed external reference material is a homogeneous pool of unprocessed material. We propose that one aliquot of this unprocessed material could be measured by another assay, and those measured values used as a multiplier commutable to all the other aliquots. We emphasize that the reference material is a predefined standard appropriate for the experimental system. Here, for yeast, we chose a reference material (pellets of BY4741 strain yeast grown under the same conditions as those in Ghememaghami et al.) with a useful unitage (protein copies-per-cell) established by TAP- and GFP-tagging methods described in the same work by Ghememaghami et al.
For all experiments, single-point calibration by external reference improves data harmonization the most when exact physical samples serve as global reference materials, suggesting that laboratories should preserve aliquots of their local working reference materials for future calibration to global reference materials such as the NIST yeast standard or commercially available pooled biofluid products like plasma or CSF.7 Even in the absence of an exact global reference, we harmonized LCMS data by using a thoroughly described reference material and following well-described procedures to approximate the previous local reference. Going forward, we propose that LCMS experimental design should include the selection of an appropriate reference material to support data harmonization.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported in part by National Institutes of Health Grants F31 AG055257 (to L.K.P.), F31 GM119273 (to B.C.S.), P41 GM103533 (to M.J.M.), R01 GM121696 (to M.J.M.), U54 HG008097 (to M.J.M.) and RF1 AG053959 (to M.J.M.).
ABBREVIATIONS
- IC
internal calibration
- IS
internal standards
- EC
external calibration
- DIA
data-independent acquisition
- SRM
selected reaction monitoring
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.8b04581.
Additional figures describing effect of calibration on signal responses and demonstrations of external calibration for data harmonization (PDF)
Tables describing isolation window schemes using DIA and SWATH methods (XLSX)
Tables for processed quantitative data from DIA and SRM experiments (XLSX)
Descriptions of raw files and data repository locations (XLSX)
The authors declare the following competing financial interest(s): The MacCoss Lab at the University of Washington has a sponsored research agreement with Thermo Fisher Scientific, the manufacturer of the instrumentation used in this research. Additionally, M.J.M. is a paid consultant for Thermo Fisher Scientific. The Hoofnagle laboratory receives instrument and grant support from Waters.
All raw data is publicly available on Panorama Public (https://panoramaweb.org/singlepointcal.url) along with Skyline documents for the SRM and DIA experiments performed in this work.
REFERENCES
- 1.Domon B; Aebersold R Nat. Biotechnol 2010, 28 (7), 710–721. [DOI] [PubMed] [Google Scholar]
- 2.Shuford CM; Walters JJ; Holland PM; Sreenivasan U; Askari N; Ray K; Grant RP Anal. Chem 2017, 89 (14), 7406–7415. [DOI] [PubMed] [Google Scholar]
- 3.Hoofnagle AN; Whiteaker JR; Carr SA; Kuhn E; Liu T; Massoni SA; Thomas SN; Townsend RR; Zimmerman LJ; Boja E; et al. Clin. Chem 2016, 62 (1), 48–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geiger T; Cox J; Ostasiewicz P; Wisniewski JR; Mann M Nat. Methods 2010, 7 (5), 383–385. [DOI] [PubMed] [Google Scholar]
- 5.Hoofnagle AN; Becker JO; Wener MH; Heinecke JW Clin. Chem 2008, 54 (11), 1796–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agger SA; Marney LC; Hoofnagle AN Clin. Chem 2010, 56 (12), 1804–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grant RP; Hoofnagle AN Clin. Chem 2014, 60 (7), 941–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Netzel BC; Grant RP; Hoofnagle AN; Rockwood AL; Shuford CM; Grebe SK G. Clin. Chem 2016, 62 (1), 297–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Mol. Cell. Proteomics 2014, 13 (9), 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghaemmaghami S; Huh W-K; Bower K; Howson RW; Belle A; Dephoure N; O’Shea EK; Weissman JS Nature 2003, 425 (6959), 737–741. [DOI] [PubMed] [Google Scholar]
- 11.MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26 (7), 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Searle BC; Pino LK; Egertson JD; Ting YS; Lawrence RT; Villen J; MacCoss MJ Nat. Commun 2018, submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Craig H Science 1961, 133 (3465), 1702–1703. [DOI] [PubMed] [Google Scholar]
- 14.Craig H Science 1961, 133 (3467), 1833–1834. [DOI] [PubMed] [Google Scholar]
- 15.Thienpont LM; Van Uytfanghe K; De Leenheer AP Clin. Chim. Acta 2002, 323 (1–2), 73–87. [DOI] [PubMed] [Google Scholar]
- 16.Selevsek N; Chang C-Y; Gillet LC; Navarro P; Bernhardt OM; Reiter L; Cheng L-Y; Vitek O; Aebersold R Mol. Cell. Proteomics 2015, 14 (3), 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.