

ABSTRACT
The clinical use of gentamicin over prolonged periods is limited because of dose and time-dependent nephrotoxicity, in which intracellular oxidative stress and heightened inflammation have been implicated. Macroautophagy/autophagy is an essential and highly conserved self-digestion pathway that plays important roles in the maintenance of cellular function and viability under stress. The aim of this study was to determine changes in autophagy in response to the antioxidant N-acetylcysteine (NAC), via its effects on oxidative stress, inflammation, apoptosis, and renal function, following treatment with gentamicin in mini pigs. Adult mini pigs were divided into isotonic saline solution, gentamicin, and gentamicin plus NAC combination treatment groups. Gentamicin-induced histopathological changes, including inflammatory cell infiltration and tubular necrosis, were attenuated by NAC. NAC ameliorated the gentamicin-induced decreases in the levels of autophagy-related proteins, such as LC3 (microtubule-associated protein 1 light chain 3), PINK1 (phosphatase and tensin homologue deleted on chromosome10-induced kinase 1), phospho-parkin, AMBRA1 (activatingmolecule in Beclin 1-regulated autophagy), p62/SQSTM1 (sequestosome protein 1), and polyubiquitinated protein aggregates. NAC also caused a significant reduction in oxidative damage markers, including 4-hydroxy-2-nonenal, protein carbonyls, γ-H2AX (gamma histone variant H2AX), and 8-hydroxy-2′-deoxyguanosine, in gentamicin-treated animals. These data show that the protective effects of NAC might be related, at least in part, to a reduced inflammatory response, as observed in animals treated with both gentamicin and NAC. These results suggest that autophagy could be a new therapeutic target for preventing gentamicin-induced kidney injury, and that NAC might ameliorate gentamicin-induced nephrotoxicity by autophagy.
Keywords: Autophagy, gentamicin, N-acetylcysteine, nephrotoxicity, oxidative damage
INTRODUCTION
Acute kidney injury (AKI) is encountered frequently in nephrology. Drug-induced AKI is a common complication of many diagnostic and therapeutic procedures, and can lead to renal impairment. Gentamicin is an aminoglycoside antibiotic that is used widely for the treatment of life-threatening gram-negative infections. However, its use is limited by the risk of serious side effects such as ototoxicity and nephrotoxicity; gentamicin induces nephrotoxicity in 10% to 20% of patients. The pathogenesis of gentamicin-induced nephrotoxicity is unclear. Previous studies revealed that antibiotics induce mitochondrial dysfunction and reactive oxygen species (ROS) overproduction in mammalian cells, which ultimately lead to oxidative damage in tissues (1).
N-acetylcysteine (NAC) is an antioxidant that scavenges ROS and alleviates oxidative injury (2, 3). NAC is a readily available and inexpensive amino acid derivative, the use of which that has been validated scientifically over 4 decades. Its potential role as a frontline defense against some of today's most deadly public health threats, including acetaminophen toxicity, influenza, chronic obstructive pulmonary disease (COPD), and Helicobacter pylori, has well been documented (4–7).
Autophagy is a lysosomal degradation pathway that allows cells to self-digest their own components, and thus remove damaged organelles and misfolded proteins, in well-defined structures called autophagosomes. This degradation process provides nutrients to maintain cellular functions under environmental stress, thus allowing survival of cancer cells under stressful conditions (8). Autophagy plays an essential role in stress adaption during renal injury by removing protein aggregates and damaged organelles such as mitochondria (9). Although autophagy can regulate many critical aspects of normal and diseased conditions in the kidneys (10), the role of autophagy in gentamicin-induced AKI and the effects of NAC on renal injury are unknown.
Autophagic flux refers to all autophagic processes, including the formation of autophagosomes, the fusion of autophagosomes with lysosomes, and their subsequent breakdown. LC3 is the mammalian homolog of yeast Atg (Autophagy-related gene)8, and is essential for autophagosome formation. Precursor LC3 can be digested by Atg4 to form LC3-I, which is then activated by Atg7 to form membrane-bound LC3-II. LC3-II is a membrane marker of autophagosomes, and the LC3-II/I ratio is associated with autophagic flow. LC3 levels depend on the balance between autophagosome formation and degradation; i.e., autophagic flux. P62/SQSTM1 is an ubiquitin-binding protein that interacts with LC3 to mediate degradation of polyubiquitinated protein aggregates (poly UB) and mitochondria in mammalian cells, via the autophagy–lysosome pathway (11). This enhances cell survival because p62/SQSTM1 is sequestered in autophagosomes after interacting directly with LC3. Therefore, p62/SQSTM1 and poly UB might reflect autophagic degradation activity. Increased levels of p62/SQSTM1 and poly UB in the kidneys reflects decreased autophagic degradation activity.
PINK1 spans the outer mitochondrial membrane with its kinase domain facing the cytosol, and is degraded rapidly in healthy mitochondria; therefore, it is a sensor of damaged mitochondria. When mitochondria are damaged PINK1 can phosphorylate parkin, causing the latter to be translocated from the cytoplasm to the mitochondrial membrane. Phosphorylated (activated) parkin (p-parkin) then ubiquitinates proteins in the outer mitochondrial membrane, leading to the isolation and degradation of ubiquitinated damaged mitochondria via the autophagy–lysosome pathway, in which P62/SQSTM1 serves as an adaptor protein between the ubiquitinated mitochondria and LC3 (12). Therefore, PINK1 and parkin are involved in removing damaged mitochondria via mitophagy (13).
The autophagy-promoting protein AMBRA1 (activating molecule in Beclin1-regulated autophagy) binds to depolarized mitochondria in a parkin-dependent manner. The overexpression of AMBRA1 enhances the removal of depolarized mitochondria, but only in the presence of parkin (14).
8-hydroxy-2′-deoxyguanosine (8-OHdG) is a sensitive biomarker of mitochondrial DNA oxidative damage (15). γ-H2AX is a core histone protein that becomes phosphorylated when DNA is damaged (16). In addition, carbonylated proteins and 4-hydroxy-2-nonenal (4-HNE) are products of oxidative damage-induced protein and lipid peroxidation (17).
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) is a method for detecting DNA fragmentation by labeling the terminal end of nucleic acids. Apoptotic signaling induces apoptosis via caspase-dependent pathways (18). In addition, caspase 3 cleavage is an indicator of caspase 3 activation.
Compared with rodents, miniature pigs are more similar to humans in terms of their anatomy, physiology, and genetics. The results obtained from pharmaceutical experiments in mice have led to various forms of damage to humans, whereas the results of experiments in miniature pigs resemble those in humans more closely. Therefore, in recent years, larger animals, such as miniature pigs, have been used to establish a variety of human disease models (19, 20). The use of miniature pigs also has ethical and financial advantages.
In the present study, gentamicin-induced AKI models were established using Chinese experimental miniature pigs, and the changes in oxidative damage and autophagy in the kidneys were assessed. In addition, the effects of NAC treatment on renal function, autophagy, apoptosis, and inflammation were investigated.
MATERIALS AND METHODS
Animals
Experiments were performed using 3-month-old male Chinese experimental mini pigs weighing ∼8 kg to 10 kg. The animals were bred by the China Agricultural University, and were derived from little swine from Guizhou Province, China, in 1985. Their characteristics include an inherently small size, early sexual maturity, rapid breeding, and ease of management. In addition, the genetic integrity and uniformity of the population is maintained. The animals were placed in individual metabolic cages and divided into 3 experimental groups, according to previous studies (21, 22). Pigs in the control group (CON; n = 6) received a daily intramuscular (i.m.) injection of 0.5 mL isotonic saline solution for 10 days. Animals in the gentamicin group (GEN; n = 6) received 80 mg/kg gentamicin sulfate i.m. daily for 10 days. Pigs in the gentamicin + NAC group (GEN + NAC; n = 6) were injected with 80 mg/kg gentamicin sulfate i.m. and treated with NAC (0.3 mg/kg) in drinking water for 10 days. The animals in the NAC group (NAC; n = 6) were treated with NAC (0.3 mg/kg) in drinking water for 10 days. In our preliminary study, the daily i.m. injection of 80 mg/kg gentamicin and NAC treatment (0.175 mg/kg) in drinking water for 10 days was insufficient to alleviate gentamicin-induced AKI, so those results are not shown here. The Institutional Animal Care and Use Committee of the Chinese PLA General Hospital approved all animal protocols.
Serum biochemical analysis
All mini pigs were euthanized on their scheduled termination day using pentobarbital. Blood samples were transferred to fresh 2 mL Eppendorf tubes without an anticoagulant and then kept at room temperature for about 20 min to allow the blood to clot. Clotted blood samples were centrifuged at 750×g force, for 10 min at room temperature. Serum was extracted for immediate use or storage at −20°C until use. Serum biochemical parameters were analyzed using an autoanalyzer (Cobas8000, Roche, Mannheim, Germany).
Light microscopy
Kidneys were removed after tying the renal pedicle, and then cut into halves via saggital sections. The tissues were then fixed by immersion in 10% formaldehyde for 1 day. After dehydration, the tissues were embedded in paraffin, cut into 4 μm, mounted on glass slides, counterstained with periodic acid–Schiff, and analyzed using light microscopy. A histopathological scoring system was designed by a veterinary pathologist and pathological changes were assessed in a single blinded manner using a light microscope at an original magnification of ×100 or ×400 (23). Assessed parameters included: glomerular necrosis, tubular degeneration and necrosis, tubular dilatation, epithelial sloughing and presence of protein casts, vascular congestion and extravasation, and infiltration of the interstitium by inflammatory polymorphonuclear cells. These parameters were assessed according to the degree of pathological changes based on a 0 to 5 grading system outlined below.
0—Normal with no noticeable histopathological change;
1—Minor: 0% to 9%;
2—Mild: 10% to 29%;
3—Moderate: 30% to 49%;
4—Marked: 50% to 69%; and
5—Severe: 70% to 100%.
For each section, at least 50 glomeruli and 100 proximal and distal convoluted tubules were examined. Histological scores were reported as mean ± standard error of the mean.
Immunohistochemical localization of 8-OHdG
8-OHdG, an index of DNA damage by oxygen-derived free radicals, was determined using immunohistochemistry. Ten days after gentamicin administration the kidneys were fixed in 10% buffered formaldehyde, and 4 μm sections were prepared from paraffin-embedded tissues. After deparaffinization, endogenous peroxidase was quenched using 0.3% H2O2 in 60% methanol for 30 min. After washing with PBS (Phosphate buffer saline), the sections were incubated with 1.5% normal goat serum for 20 min, followed by mouse monoclonal anti-8-OHdG antibodies (1:50; Santa Cruz Biotechnology Inc., sc-66036) overnight at 4°C. After 3 washes with PBS the samples were incubated with biotin-conjugated goat anti-mouse IgG (Immunoglobulin G) for 30 min at room temperature. The sections were washed with PBS, and incubated with streptavidin-conjugated peroxidase for 30 min at room temperature. Finally, the sections were washed with PBS, incubated with 1:100 DAB (Diaminobenzidine) solution for 5 min, and then examined under a microscope.
Terminal uridine nick-end-labeled staining
Paraffin sections (4 μm thick) were deparaffinized in toluene and dehydrated using a graded series of ethanol solutions. Renal tissue apoptosis was identified using TUNEL assays with an In Situ Cell Death Detection Kit (Roche, Mannheim, Germany, 11684817910) according to the manufacturer's instructions. For estimation of the level of apoptosis, the TUNEL positive renal tubular epithelial cells, as well as unstained cells, were scored in 10 different fields at ×400 magnification. The apoptotic index was expressed as the percentage of total cells scored. The results were expressed as the average number of TUNEL-positive cells for each group. All counting procedures were performed blindly.
Western blot
Proteins were extracted from gentamicin-induced injured kidney tissues, and Bradford assays (Bio-Rad, Hercules, CA) were used to measure the protein concentration. Approximately 100 μg of each protein sample was resolved using SDS-PAGE and transferred to nitrocellulose membranes. The resulting membranes were blocked in skimmed milk powder (5% w/v) in phosphate buffer saline containing 0.1% Tween-20 (PBS-T) at 4°C overnight. They were then incubated with primary antibodies against LC3 (Sigma, St. Louis, MO, L7543; 1:1000), p62/SQSTM1 (Santa Cruz Biotechnology, Santa Cruz, Calif, sc-55603; 1:1000), parkin (Santa Cruz Biotechnology, Santa Cruz, Calif, sc-30130; 1:500), PINK1 (Santa Cruz Biotechnology, Santa Cruz, Calif, sc-33796; 1:500), p-parkin, AMBRA1 (Abcam, Cambridge, UK, ab72098; 1:500), γ-H2AX (Abcam, Cambridge, UK, ab11174; 1:500), 4-HNE (4-hydroxy-2-nonenal; Abcam, Cambridge, UK, ab46546; 1:500), carbonylated proteins (Cell Biolabs, San Diego, CA; STA-308; 1:500), cleaved caspase 3 (Cell Signaling Technology, Japan, K.K. #9661;1:500), and β-actin (Sigma, St. Louis, USA, A4700; 1:5000). Following 3 washes with tris buffered saline–Tween, the blots were probed with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, Calif, sc-2096 and sc-2963, respectively) at dilutions of 1:1000 to 5000. The final results were obtained by exposing the membranes to autoradiographic film (Kodak XAR film, Rochester, NY) after incubation with a chemiluminescent detection system (ECL Substrate Western Blot Detection System; Pierce, Rockford, Ill).
Transmission electron microscopy
Kidneys were cut into tissue blocks (1 mm3) and fixed in 2.5% glutaraldehyde in 0.01 mol/L phosphate buffer at 4°C, followed by 2% osmium tetroxide. They were then dehydrated in a series of graded ethanol solutions. Ethanol was then substituted with propylene oxide and the tissue was embedded in epoxy resin. Ultrathin sections were double stained with uranyl acetate and lead and examined under a JEM1200EX transmission electron microscope (JOEL, Tokyo, Japan) at 80 kV.
RNA isolation and real-time quantitative PCR
Total RNA was isolated from renal tissues using TRIzol (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Reverse transcription was performed using a TIANScript RT kit (Tiangen Biotech, Beijing, China, KR104). Amplification was performed in a 7500 real-time PCR System (Applied Biosystems, Carlsbad, Calif). Reaction contained 50 ng total cDNA, 0.2 μM primers, and 10 μL 2× SYBR green buffer (TaKaRa, Shiga, Japan, DRR820A) in a final volume of 20 μL. Primers were designed using the software package Primer Express 2.0 (Applied Biosystems, Carlsbad, Calif) based on GenBank nucleotide sequences as follows:
Chemokine (C-C motif) ligand (CCL)-5 (Accession: NM_001129946.1):
Forward 5’-GTGTGTGCCAACCCAGAGAA-3’,
Reverse 5’-GGACAAGAGCAAGAAGCAGTAGG-3’.
Intercellular adhesion molecule 1 (ICAM-1) (Accession: NM_213816.1):
Forward 5’-ACCCACCCACACCTTGCTAC-3’,
Reverse 5’-GCTGGGAACAGTCCATCCA-3’.
Interleukin (IL)-6 (Accession: JQ839263.1):
Forward 5’-GGGAAATGTCGAGGCTGTG-3’,
Reverse 5’-AGGGGTGGTGGCTTTGTCT-3’.
Monocyte chemoattractant protein-1 (MCP-1) (Accession: NM_213816.1):
Forward 5’-GGGTATTTAGGGCAAGTTAGAAGGA-3’,
Reverse 5’-CATAAGCCACCTGGACAAGAAAA-3’.
Chemokine (C-X-C motif) ligand 2 (CXCL2) (Accession: NM_001001861.1):
Forward 5’-CGGAAGTCATAGCCACTCTCAA-3’,
Reverse 5’-CAGTAGCCAGTAAGTTTCCTCCATC-3’.
Vascular cell adhesion molecule 1 (VCAM-1) (Accession: NM_213891.1):
Forward 5’-AGCACTTTCAGGGAGGACACA-3’,
Reverse 5’-AACGGCAAACACCATCCAA-3’.
GAPDH (Glyceraldehyde-3-phosphate dehydrogenase, Accession: NM_001206359.1):
Forward 5’-TCCCTGCTTCTACCGGCGCT-3’,
Reverse 5’-ACACGTTGGGGGTGGGGACA-3’.
PCR was performed using the following cycling conditions: 95°C for 30 s, 40 cycles of denaturation at 95°C for 15 s, and extension at 60°C for 30 s. All samples were run in triplicate. The relative abundance of target mRNA was determined with the comparative cycle threshold method.
Statistical analyses
All data were presented as mean ± SD for a minimum of 3 independent experiments in triplicate. All comparisons were made using either one-way ANOVA or a 2-tailed t test analysis depending on how many conditions were compared in each experiment. One-way ANOVA (Analysis of Variance) was followed by Tukey's post hoc test. A value of P < 0.05 was considered significant.
RESULTS
N-acetylcysteine relieves gentamicin-induced acute kidney injury in mini pigs
In a preliminary study, the daily i.m. injection of 80 mg/kg gentamicin for 10 days was sufficient to induce AKI. Blood urea nitrogen and serum creatinine levels were significantly higher in the GEN group than in the CON group, and significantly lower in the GEN + NAC group than the GEN group (Table 1). Pathological analysis revealed that gentamicin induced prominent renal lesions, including necrosis, tubular epithelial cell degeneration, protein casts in the tubular lumina, and interstitial lymphocytic infiltration. The NAC treatment inhibited gentamicin-induced acute tubular necrosis and cast formation (Fig. 1). The histological scores of the kidney sections demonstrated that the NAC intervention significantly decreased gentamicin-induced degeneration, necrosis, cell and protein casts, and inflammatory cell infiltration in the kidneys of mini pigs (Table 2).
Table 1.
Effects of N-acetylcysteine on serum creatinine (Scr) and blood urea nitrogen (BUN) levels in gentamicin-induced AKI model mini pigs
| CON | GEN | GEN + NAC | NAC | |
| BUN (mmol/L) | 3.55 ± 0.61 | 28.90 ± 9.90* | 3.95 ± 0.72† | 3.98 ± 0.61 |
| Scr (μmol/L) | 78.03 ± 10.39 | 365.50 ± 117.59* | 83.55 ± 28.81† | 77.80 ± 12.21 |
Values are mean ± SD (n = 6).
*P < 0.05, vs. CON.
†P < 0.05, vs. GEN.
BUN, blood urea nitrogen; CON, control group; GEN, gentamicin group; GEN + NAC, gentamicin + N-acetylcysteine group; NAC, N-acetylcysteine group; Scr, serum creatinine.
Fig. 1.
Effects of N-acetylcysteine on renal histology in gentamicin-induced AKI model mini pigs.
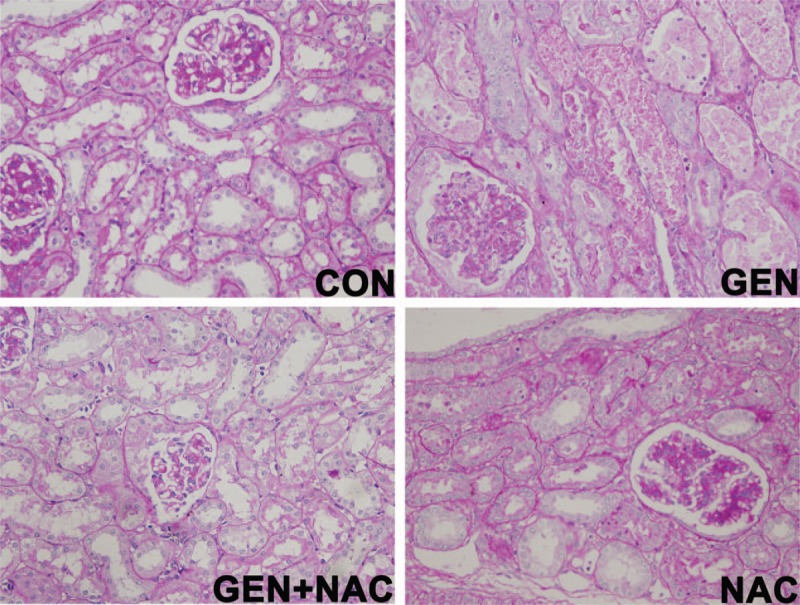
Renal damage was assessed semiquantitatively. Images of periodic acid–Schiff-stained kidney sections from mini pigs treated with gentamicin or/and N-acetylcysteine (400× magnification). AKI, Acute kidney injury; CON, control group; GEN, gentamicin group; GEN + NAC, gentamicin + N-acetylcysteine group; NAC, N-acetylcysteine group (n = 6 per group).
Table 2.
Effects of N-acetylcysteine on histological and morphometric scores in mini pigs with gentamicin-induced nephrotoxicity
| GEN | GEN + NAC | |
| Tubular necrosis | 3.67 ± 0.65 | 1.92 ± 0.90* |
| Interstitial ICI | 3.17 ± 0.58 | 1.58 ± 0.67* |
| Tubular degeneration | 2.17 ± 0.72 | 1.25 ± 0.45* |
| Tubular dilation | 2.50 ± 0.52 | 1.58 ± 0.51* |
| Protein cast | 1.50 ± 0.52 | 0.67 ± 0.49* |
Data are mean ± standard error of the microscopic score (n = 6). The scoring scale ranged from 0 to 5 according to the proportion of renal tubules and glomeruli that presented with degeneration, necrosis, cell and protein casts, and inflammatory cell infiltration (ICI).
*Significant reduction in renal damage score as a result of coadministering gentamicin with N-acetylcysteine compared with administering gentamicin alone, P < 0.05.
GEN, gentamicin group; GEN + NAC, gentamicin + N-acetylcysteine group; ICI, inflammatory cell infiltration.
Effects of N-acetylcysteine on autophagy in the kidneys of gentamicin-treated mini pigs
The expression of p62/SQSTM1 and poly UB increased significantly in the kidneys of pigs in the GEN group compared with those in the CON group, but decreased significantly in the GEN + NAC group compared with the GEN group (Fig. 2, A and B).
Fig. 2.
Effects of N-acetylcysteine on autophagy in the kidneys of gentamicin-induced AKI mini pigs.
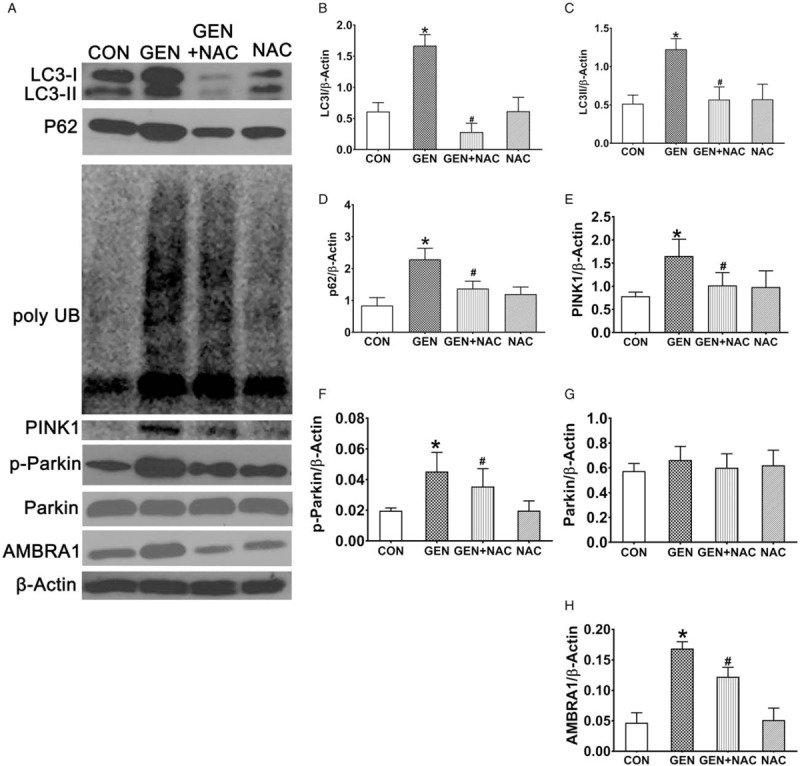
(A) The expression of p62/SQSTM1, poly UB, LC3, PINK1, parkin, p-parkin, and AMBRA1 in the kidneys of mini pigs treated with gentamicin and gentamicin + N-acetylcysteine was analyzed by western blotting. (B–J) Quantification of LC3, p62/SQSTM1, PINK1, p-parkin, parkin, and AMBRA1 levels. All data are presented as mean ± SD (n = 6). ∗P < 0.05 vs. CON, #P < 0.05 vs. GEN. AKI, Acute kidney injury; AMBRA1, activating molecule in Beclin1-regulated autophagy; CON, control group; GEN, gentamicin group; GEN + NAC, gentamicin + N-acetylcysteine group; NAC, N-acetylcysteine group; poly UB, polyubiquitinated protein aggregates; p-parkin, phosphorylated parkin.
In the current study, LC3 expression was measured using western blotting. The expression of LC3-I and LC3-II increased significantly in the kidneys of pigs in the GEN group compared with the CON group, but decreased significantly in the GEN + NAC group compared with the GEN group (Fig. 2, A–C).
PINK1 expression increased significantly in kidneys from the GEN group compared with the CON group, but decreased significantly in the GEN + NAC group compared with the GEN group (Fig. 2E). Although parkin levels were similar in the kidneys of pigs in the CON, GEN, and GEN + NAC groups, p-parkin levels increased significantly in the GEN group compared with the CON group, but decreased significantly in the GEN + NAC group compared with the GEN group (Fig. 2, F and G).
AMBRA1 expression increased significantly in the kidneys of mini pigs in the GEN group compared with the CON group, but decreased significantly in the GEN + NAC group compared with the GEN group (Fig. 2H).
N-acetylcysteine ameliorates gentamicin-induced oxidative damage in mini pig kidneys
To assess the intracellular accumulation of oxidative damage, the levels of γ-H2AX, protein carbonyls, and 4-HNE were measured in the kidneys of pigs in each group. The levels of these markers increased significantly after 10 days of gentamicin treatment. However, the levels were clearly lower in the kidneys of pigs in the GEN + NAC than the GEN group (Fig. 3, A–C). Immunohistochemical staining for 8-OHdG demonstrated that levels were decreased in the kidneys of pigs in the GEN + NAC group compared with the GEN group (Fig. 3D).
Fig. 3.
Effects of N-acetylcysteine on oxidative damage in the kidneys of gentamicin-induced AKI mini pigs.
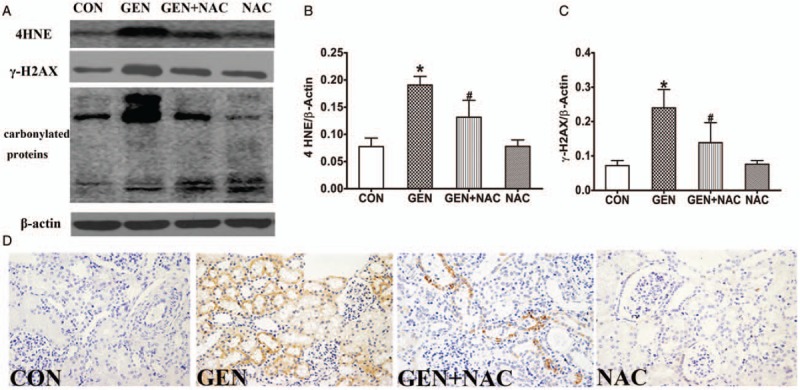
(A) The levels of γ-H2AX, 4-HNE, and protein carbonyls in the kidney tissues of gentamicin-treated pigs were measured using western blotting. (B and C) Quantitative analysis of γ-H2AX and 4-HNE expression presented as mean ± SD (n = 6). ∗P < 0.05 vs. CON, #P < 0.05 vs. GEN. (D) Immunohistochemical staining for 8-OHdG in the kidneys of gentamicin-treated pigs (magnification, 400×). 4-HNE, 4-hydroxy-2-nonenal; 8-OHdG, 8-hydroxy-2′-deoxyguanosine; AKI, Acute kidney injury; CON, control group; GEN, gentamicin group; GEN + NAC, gentamicin + N-acetylcysteine group; NAC, N-acetylcysteine group; poly UB, polyubiquitinated protein aggregates.
NAC inhibited mitochondrial structural damage, including swelling and disintegration of cristae caused by gentamicin (Fig. 4).
Fig. 4.
Transmission electron microscopy (TEM) images of mitochondrial structures in renal tissues of gentamicin-treated pigs.
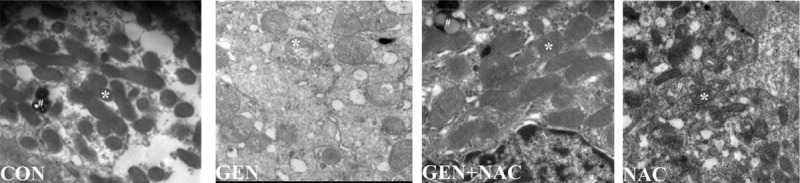
White asterisk indicates a mitochondrion. (20,500× magnification), CON, control group; GEN, gentamicin group; GEN + NAC, gentamicin + N-acetylcysteine group; NAC, N-acetylcysteine group.
N-acetylcysteine decreases gentamicin-induced apoptosis and the inflammatory response in the kidneys of mini pigs
The number of TUNEL-positive cells and cleaved caspase 3 levels were increased significantly in the kidneys of pigs in the GEN group compared with the CON group, but were decreased significantly in the kidneys of the GEN + NAC group compared with the GEN group (Fig. 5). Compared with the CON group, the levels of inflammatory cytokines, including MCP-1/CCL2, macrophage inflammatory protein-2/CXCL2, CCL-5, VCAM-1, IL-6, and ICAM-1/cluster of differentiation-54, were increased significantly in the kidneys of the GEN group. However, NAC intervention decreased the levels of these inflammatory cytokines significantly compared with the GEN group (Fig. 5).
Fig. 5.
Effects of N-acetylcysteine on apoptosis and the inflammatory response in gentamicin-injected AKI mini pigs.
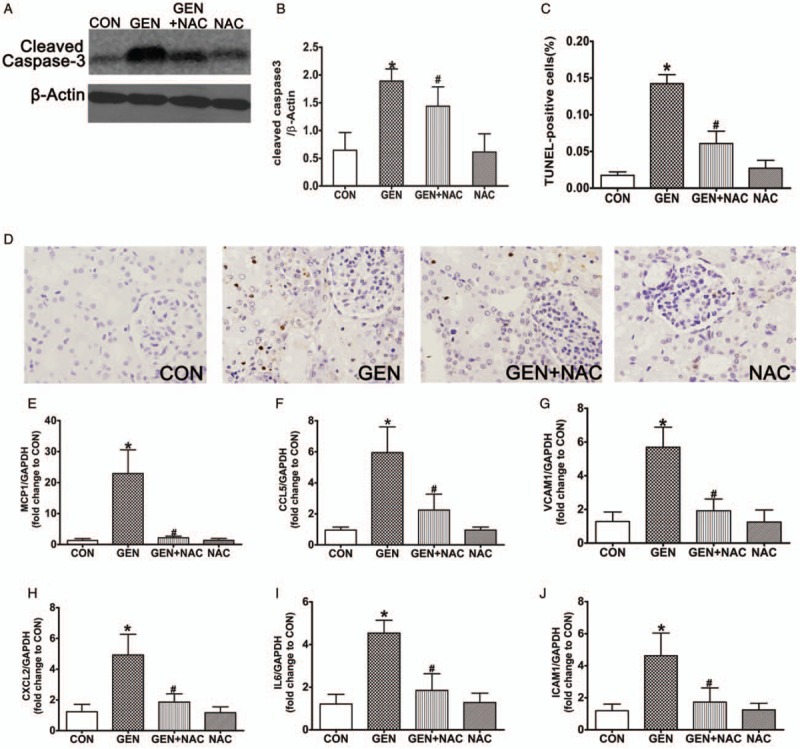
(A) The levels of cleaved caspase 3 were analyzed in kidney extracts from gentamicin-treated pigs using western blotting. (B) Quantitation of cleaved caspase-3 expression presented as mean ± SDs (n = 6). ∗P < 0.05 vs. CON, #P < 0.05 vs. GEN. (C) The number of terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) (+) cells was scored as described in the Methods section. ∗P < 0.05 vs. CON, #P < 0.05 vs. GEN. (D) TUNEL staining in kidney cortical sections (400×). (E–J) The mRNA expression levels of MCP-1, CCL-5, VCAM-1, CXCL2, IL-6, and ICAM-1 in mini pig kidneys were measured by quantitative PCR. Data are presented as mean ± SD (n = 6). ∗P < 0.05 vs. CON, #P < 0.05 vs. GEN. AKI, Acute kidney injury; CON, control group; GEN, gentamicin group; GEN + NAC, gentamicin + N-acetylcysteine group; NAC, N-acetylcysteine group; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling.
DISCUSSION
The exact mechanism of aminoglycoside antibiotic-induced nephrotoxicity, such as that caused by gentamicin, is unclear. Oxidative stress plays a critical role in the pathophysiology of gentamicin-induced AKI (24). Autophagy, a lysosomal degradation pathway, plays a crucial role in removing protein aggregates and damaged organelles to maintain renal pathophysiology and intracellular homeostasis. The role of autophagy in gentamicin-induced AKI and the effects of the antioxidant NAC on renal injury are unclear. In this study, we first assessed the changes in renal function, autophagy (particularly mitophagy), oxidative damage, apoptosis, and the inflammatory response in the kidneys of the gentamicin-injected mini pigs, and then investigated the effects of NAC treatment on gentamicin-induced nephrotoxicity.
Gentamicin induced AKI in the mini pigs successfully. Specifically, the expression of p62/SQSTM1 and polyubiquitin aggregates was increased significantly in the kidneys of gentamicin-treated mini pigs. This suggests that the autophagic degradation capacity was decreased markedly by gentamicin, although there was a compensatory increase in the levels of the autophagosome formation-related proteins LC3-I and LC3-II. In the current study, NAC relieved gentamicin-induced AKI. In the NAC-treated pigs, the expressions of p62/SQSTM1, polyubiquitin aggregates, LC3-I, LC3-II, and the LC3-II/I ratio were decreased significantly compared with the GEN group. These results suggest that the kidney autophagic degradation capacity was increased by NAC intervention.
Mitophagy is the only pathway that removes damaged mitochondria from cells. Therefore, inhibiting mitophagy leads to the accumulation of the damaged mitochondria. Because mitochondria are the main intracellular sites of ROS production, the accumulation of damaged mitochondria increases ROS production and further aggravates cellular oxidative injury (25). After mitochondrial damage, mitochondria-localized PINK1 recruits parkin (an E3 ligase) from the cytoplasm to mitochondria, where it becomes phosphorylated. Activated p-parkin can then ubiquitinate outer mitochondrial membrane proteins such as voltage-dependent anion channel 1. Subsequently, p62 binds to ubiquitinated voltage-dependent anion channel 1 on the mitochondria and LC3 on the developing autophagosome, which stimulates mitochondrial sequestration, fusion with lysosomes, and removal by the autophagic machinery (26). Thus, parkin regulates mitochondrial quality by translocating to depolarized (injured) mitochondria and inducing their selective autophagic removal via mitophagy. In addition, p-parkin interacts with AMBRA1, a protein that promotes mitophagy in the vertebrate central nervous system (14). Prolonged mitochondrial depolarization increases the interaction between parkin with AMBRA1. In the current study, the levels of the mitophagy formation markers PINK1, phospho-parkin, and AMBRA1 were increased markedly in gentamicin-treated mini pig kidneys, suggesting that mitochondrial damage induced a significant mitophagy response. Conversely, NAC administration decreased the levels of the above mitophagy markers.
As the principal sites of ROS production, mitochondria can activate and regulate autophagy (27). However, the chronic impairment of mitochondrial function can lead to high levels of ROS production, thereby shifting the role of mitochondria from bulk autophagy inducers to self-removal signals for mitochondria via mitophagy. This represents a negative feedback mechanism by which autophagy eliminates the source of oxidative stress and protects cells from oxidative damage (28).
In humans, it is likely that bactericidal antibiotic-induced oxidative stress and related oxidative cellular damage underlie many of the adverse side effects associated with these drugs (3, 29). A previous study that characterized the effects of bactericidal antibiotics on mammalian cells demonstrated that they cause widespread cellular oxidative damage (3). In the current study, the levels of oxidative damage markers including 4-HNE, protein carbonyls, γ-H2AX and 8-OHdG, inflammatory cytokines, and apoptosis were elevated significantly in the kidneys of gentamicin-induced AKI in mini pigs. However, NAC decreased the levels of gentamicin-induced oxidative damage products, inflammation, and apoptosis significantly. Therefore, the present study found that NAC reduced gentamicin-induced oxidative damage in the kidney and increased the autophagy degradation activity. Thus, we speculate that NAC treatment might mitigate the renal injury by the increased autophagic degradation of damaged mitochondria and biological macromolecules and reduce ROS, in addition to its direct antioxidative effects.
NAC, which is the acetylated variant of the amino acid L-cysteine, is both a thiol-containing antioxidant and a mucolytic agent. NAC breaks the disulfide bridges of high-molecular weight mucoglycoproteins, resulting in decreased viscosity. In addition, NAC scavenges ROS and SH-groups. NAC not only has an antioxidant function, but also inhibits neutrophil activation, vasodilation, and microbial attachment. NAC may induce changes that aid the action of antibiotics in chronically inflamed stomach tissue, but is unable to reduce H. pylori burden or severe inflammation (30).
A meta-analysis assessed the evidence for NAC efficacy in preventing exacerbations of chronic bronchitis or COPD. NAC may reduce exacerbation frequency by acting at a number of target sites: it inhibited attachment of bacteria to the epithelium by disrupting bacterial receptor sites on the epithelial surface and mucus, as well as transmigration of neutrophils via suppression of IL-8 and ICAM-1. Furthermore, NAC improved small airway function by decreasing epithelial thickening and reducing secretory cell hyperplasia. This is why NAC influences lung hyperinflation and reduces emphysema. NAC reduced the concentration of lysozyme and lactoferrin, activated neutrophils and macrophages, and restored the host innate antiviral response by preventing suppression of oxidant-sensitive retinoic acid-inducible gene-I (31).
NAC neutralized influenza A virus-mediated oxidative stress, and NAC treatment and sod1 (Superoxide dismutase 1) transfection considerably diminished viral polymerase activity (32). In addition, NAC treatment reduced the severity of H9N2 swine influenza virus-induced acute lung injury by inhibiting TLR4 (Toll-like receptor) (33).
In conclusion, NAC acts as a precursor for the substrate cysteine during synthesis of GSH (Glutathione). Its role is to deliver sulfhydryl moieties for utilization in biological processes. NAC acts as a mucolytic, antioxidant, and anti-inflammatory agent to prevent COPD, H. pylori, and influenza.
However, our study also has some limitations. First, the dosages of NAC and gentamicin may only apply to mini pigs, so these results need to be verified in humans. Second, we did not focus whether cotreatment of gentamicin with NAC effect on the MIC and killing of bacteria.
Nevertheless, it is important to extend the current investigation to human subjects to confirm these findings, demonstrate their relevance to humans, and maximize the translational value of this work. In humans, it is likely that oxidative stress and the related oxidative cellular damage induced by bactericidal antibiotics, such as gentamicin, underlies many of the adverse side effects associated with bactericidal antibiotics (34, 35). It will be intriguing to explore these possibilities using appropriate clinical trials with the goal of developing effective antibacterial therapies with minimal adverse side effects.
AUTHOR CONTRIBUTIONS
J.C., L.T., X.Y.-B., G.Y.-C., and X.M.-C. conceived and designed the experiments. J.C. and H.Q. performed the experiments. Q.H. and J.C. analyzed the data. Q.H., S.P.-L., and X.Y.-B. contributed reagents/materials/analysis tools. J.C. and X.M.-C. wrote the paper.
Footnotes
Authors J.C. and L.T. contributed equally to this work.
This work was supported by grants (No.81830060,81570659, and 81770663) from the National Natural Science Foundation of China, a grant from the National Key R&D Program of China (2018YFA0108803), a grant (No. 2016YFA0101002) from the National Key Research and Development Program of China, and a grant (No. 20158332) from the Natural Science Foundation of Hainan Province.
The authors report no conflicts of interest.
REFERENCES
- 1.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int 82:516–524, 2012. [DOI] [PubMed] [Google Scholar]
- 2.Johnson ST, Bigam DL, Emara M, Obaid L, Slack G, Korbutt G, Jewell LD, Van Aerde J, Cheung PY. N-acetylcysteine improves the hemodynamics and oxidative stress in hypoxic newborn pigs reoxygenated with 100% oxygen. Shock 28:484–490, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Kalghatgi S, Spina CS, Costello JC, Liesa M, Morones-Ramirez JR, Slomovic S, Molina A, Shirihai OS, Collins JJ. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci Transl Med 5:192ra85, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pyo CW, Shin N, Jung KI, Choi JH, Choi SY. Alteration of copper-zinc superoxide dismutase 1 expression by influenza A virus is correlated with virus replication. Biochem Biophys Res Commun 450:711–716, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Fowdar K, Chen H, He Z, Zhang J, Zhong X, Zhang J, Li M, Bai J. The effect of N-acetyl cysteine on exacerbations of chronic obstructive pulmonary disease: a meta-analysis and systematic review. Heart Lung 46:120–128, 2017. [DOI] [PubMed] [Google Scholar]
- 6.Jang S, Bak EJ, Cha JH. N-acetylcysteine prevents the development of gastritis induced by Helicobacter pylori infection. J Microbiol 55:396–402, 2017. [DOI] [PubMed] [Google Scholar]
- 7.Teriaky A. The role of N-acetylcysteine in the treatment of non-acetaminophen acute liver failure. Saudi J Gastroenterol 23:131–132, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, Van Moer K, Kreis S, Guerin C, Buart S, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A 114:E9271–E9279, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S, Sugimoto T, Haneda M, Kashiwagi A, Koya D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 120:1043–1055, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He L, Livingston MJ, Dong Z. Autophagy in acute kidney injury and repair. Nephron Clin Pract 127:56–60, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linares JF, Cordes T, Duran A, Reina-Campos M, Valencia T, Ahn CS, Castilla EA, Moscat J, Metallo CM, Diaz-Meco MT. ATF4-induced metabolic reprograming is a synthetic vulnerability of the p62-deficient tumor stroma. Cell Metab 26:817–829, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hattori N, Saiki S, Imai Y. Regulation by mitophagy. Int J Biochem Cell Biol 53:147–150, 2014. [DOI] [PubMed] [Google Scholar]
- 13.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107:378–383, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Humbeeck C, Cornelissen T, Vandenberghe W. Ambra1: a Parkin-binding protein involved in mitophagy. Autophagy 7:1555–1556, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakovcevic D, Dedic-Plavetic N, Vrbanec D, Jakovcevic A, Jakic-Razumovic J. Breast cancer molecular subtypes and oxidative DNA damage. Appl Immunohistochem Mol Morphol 23:696–703, 2015. [DOI] [PubMed] [Google Scholar]
- 16.Sánchez-Flores M, Pásaro E, Bonassi S, Laffon B, Valdiglesias V. γH2AX assay as DNA damage biomarker for human population studies: defining experimental conditions. Toxicol Sci 144:406–413, 2015. [DOI] [PubMed] [Google Scholar]
- 17.Calamaras TD, Lee C, Lan F, Ido Y, Siwik DA, Colucci WS. Lipid peroxidation product 4-hydroxy-trans-2-nonenal (HNE) causes protein synthesis in cardiac myocytes via activated mTORC1-P70S6K-RPS6 signaling. Free Radic Biol Med 82:137–146, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ 22:526–539, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee TF, Liu JQ, Li YQ, Nasim K, Chaba T, Bigam DL, Cheung PY. Improved renal recovery with postresuscitation N-acetylcysteine treatment in asphyxiated newborn pigs. Shock 35:428–433, 2011. [DOI] [PubMed] [Google Scholar]
- 20.Aigner B, Renner S, Kessler B, Klymiuk N, Kurome M, Wünsch A, Wolf E. Transgenic pigs as models for translational biomedical research. J Mol Med (Berl) 88:653–664, 2010. [DOI] [PubMed] [Google Scholar]
- 21.Plotnikov EY, Pavlenko TA, Pevzner IB, Zorova LD, Manskikh VN, Silachev DN, Sukhikh GT, Zorov DB. The role of oxidative stress in acute renal injury of newborn rats exposed to hypoxia and endotoxin. FEBS J 284:3069–3078, 2017. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu MH, Gois PH, Volpini RA, Canale D, Luchi WM, Froeder L, Heilberg IP, Seguro AC. N-acetylcysteine protects against star fruit-induced acute kidney injury. Ren Fail 39:193–202, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dam VP, Scott JL, Ross A, Kinobe RT. Inhibition of cystathionine gamma-lyase and the biosynthesis of endogenous hydrogen sulphide ameliorates gentamicin-induced nephrotoxicity. Eur J Pharmacol 685:165–173, 2012. [DOI] [PubMed] [Google Scholar]
- 24.Morales AI, Detaille D, Prieto M, Puente A, Briones E, Arévalo M, Leverve X, López-Novoa JM, El-Mir MY. Metformin prevents experimental gentamicin-induced nephropathy by a mitochondria-dependent pathway. Kidney Int 77:861–869, 2010. [DOI] [PubMed] [Google Scholar]
- 25.Xiao B, Deng X, Lim GG, Xie S, Zhou ZD, Lim KL, Tan EK. Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis 8:e3097, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Apostolova N, Blas-Garcia A, Esplugues JV. Mitochondria sentencing about cellular life and death: a matter of oxidative stress. Curr Pharm Des 17:4047–4060, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Xiao B, Goh JY, Xiao L, Xian H, Lim KL, Liou YC. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J Biol Chem 292:16697–16708, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22:377–388, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kent A, Turner MA, Sharland M, Heath PT. Aminoglycoside toxicity in neonates: something to worry about? Expert Rev Anti Infect Ther 12:319–331, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Jang S, Bak EJ, Cha JH. N-acetyl-cysteine prevents the development of gastritis induced by Helicobacter pylori infection. J Microbiol 55:396–402, 2017. [DOI] [PubMed] [Google Scholar]
- 31.Matera MG, Calzetta L, Cazzola M. Oxidation pathway and exacerbations in COPD: the role of NAC. Expert Rev Respir Med 10:89–97, 2016. [DOI] [PubMed] [Google Scholar]
- 32.Pyo CW, Shin N, Jung KI, Choi JH, Choi SY. Alteration of copper–zinc superoxide dismutase 1 expression by influenza A virus is correlated with virus replication. Biochem Biophys Res Commun 450:711–716, 2014. [DOI] [PubMed] [Google Scholar]
- 33.Zhang RH, Li CH, Wang CL, Xu MJ, Xu T, Wei D, Liu BJ, Wang GH, Tian SF. N-acetyl-L-cystine (NAC) protects against H9N2 swine influenza virus-induced acute lung injury. Int Immunopharmacol 22:1–8, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ural MŞ. Chlorpyrifos-induced changes in oxidant/antioxidant status and haematological parameters of Cyprinus carpio carpio: ameliorative effect of lycopene. Chemosphere 90:2059–2064, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Trujillo J, Chirino YI, Molina-Jijón E, Andérica-Romero AC, Tapia E, Pedraza-Chaverrí J. Renoprotective effect of the antioxidant curcumin: recent findings. Redox Biol 1:448–456, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]