

Abstract
Treatments with poxvirus vectors can have long-lasting immunological impact in the host, and thus they have been extensively studied to treat diseases and for vaccine development. More importantly, the oncolytic properties of poxviruses have led to their development as cancer therapeutics. Two poxviruses, vaccinia virus (VACV) and myxoma virus (MYXV), have been extensively studied as virotherapeutics with promising results. Vaccinia virus vectors have advanced to the clinic and have been tested as oncolytic therapeutics for several cancer types with successes in phase I/II clinical trials. In addition to oncolytic applications, MYXV has been explored for additional applications including immunotherapeutics, purging of cancer progenitor cells, and treatments for graft-versus-host diseases. These novel therapeutic applications have encouraged its advancement into clinical trials. To meet the demands of different treatment needs, VACVand MYXV can be genetically engineered to express therapeutic transgenes. The engineering process used in poxvirus vectors can be very different from that of other DNA virus vectors (e.g., the herpesviruses). This chapter is intended to serve as a guide to those wishing to engineer poxvirus vectors for therapeutic transgene expression and to produce viral preparations for preclinical studies.
Keywords: Poxvirus, Poxvirus genetic engineering, Vaccinia virus (VACV), Myxoma virus (MYXV), Infection/transfection, Transgene expression, Poxvirus transfer vector
1. Introduction
1.1. Overview of Recombinant Pox Viruses
This chapter provides general guidelines for researchers intending to engineer therapeutic recombinant poxviruses in a laboratory setting with the capacity to safely handle BSL-2 agents.
The United States Food and Drug Administration (FDA) considers any agent(s) which “...mediate their effects by transcription and/or translation of transferred genetic material and/or by integrating into the host genome and that are administered as nucleic acids, viruses, or genetically engineered microorganisms” [1] to be examples of gene therapeutic agents. By this definition, poxviruses have much to offer as delivery vectors for therapeutic genetic sequences. The poxviruses (family Poxviridae) are large, double-stranded DNA (dsDNA) viruses that have had an outsized impact upon human health and history, both positive and negative. Smallpox has been a scourge of humanity from prehistory until the close of the twentieth century, a highly contagious disease that killed between one-fourth and one-third of its victims and routinely left survivors with lifelong scarring [2, 3]. The causative agent of smallpox, the variola virus (VARV), was the impetus for Edward Jenner’s early vaccination experiments with the cowpox virus (CPXV). Jenner’s experiments and insight led to the practice of vaccination and, eventually, the global eradication of smallpox through a program of comprehensive human vaccination with the vaccinia virus (VACV) [2, 3]. Because of its great utility as a smallpox vaccine, the VACV is the prototype and the best characterized of all the poxviruses.
In addition to the VACV’s well-known role as a vaccine against smallpox, the ease with which VACV and other poxviruses can be engineered to express foreign proteins has made them valuable therapeutic vectors. Until the discovery of the mimivirus in 2003 [4], the poxviruses were thought to be the largest of the viruses and were the first viruses to be observed directly by light microscopy. Poxviruses possess large genomes and are capable of integrating at least 25 kilobases of foreign DNA [5]. Two of the most common poxvirus vectors used in therapy and gene delivery are VACV and myxoma virus (MYXV). Both can be easily manipulated to integrate therapeutic transgenes into the viral genome [6]. Because their replication cycle occurs entirely within the cytoplasm [7], there is no integration of viral DNA into the host genome and no splicing of viral transcripts [8]. This property minimizes the potential for genetic impact upon hosts that receive them as therapeutic agents. Finally, these poxviruses can also be prepared at high titers [9, 10] to meet the demand of escalated doses for treatment in preclinical models and in human patients.
Poxviruses, especially VACV and MYXV, along with other viruses are currently being intensively investigated as anticancer therapeutic agents [11]. Originally oncolytic viruses were envisioned as replication-competent viruses that would preferentially infect and destroy cancerous cells via cytolytic activity. The theoretical advantage of utilizing replication-competent viruses is that they can produce progeny viruses to amplify the therapeutic outcome, all the while leaving noncancerous cells unscathed. While the promise of a single efficacious viral agent to treat cancer has not yet been achieved, we now realize that successful oncolytic viruses are capable of shaping the host immune response to tumor cells [12] and even to alter the tumor microenvironment to facilitate the action of cytotoxic T cells [13]. Viruses can be further armed [14] with therapeutic genes (transgenes) including cytokines and chemokines [15, 16], prodrug activators [17], tumor-associated antigens [18], inducers of apoptosis [19], and ligands of the toll-like receptors (TLRs) [20, 21]. In addition, imaging enablers such as the sodium iodide symporter [22] and luciferase [23] can also be incorporated. More importantly, virotherapy can complement and even synergize with chemotherapy and immunotherapy [24–26]. Treatment benefits of some chemotherapeutic agents can be further improved by the prior or subsequent use of an oncolytic viral agent [27], while concurrent delivery of both virotherapy and chemotherapy [24, 27] or radiotherapy [28] can also be beneficial. Of the poxviruses, the VACV is by far the furthest advanced as an oncolytic virus, and several VACV-based oncolytic virus candidates have been tested in humans in Phase I/II clinical trials, including Pexa-Vec (JX-594, SillaJen, Inc.) [29, 30] and GLV-1h68 (GL-ONC1, Genelux Corporation) [17]. Pexa-Vec is a VACV modified by insertion of human granulocyte-monocyte colony stimulating factor (hGM-CSF) into the thymidine kinase (TK) gene [30, 31]. GLV-1h68 is a Lister strain of VACV modified via insertion of Renilla luciferase (RLuc-GFP) into the F14.5L ORF, the insertion of β-galactosidase into the viral TK gene, and the insertion of β-glucuronidase into the viral hemagglutinin ORF (A56R) [32].
Myxoma virus (MYXV) has recently been recognized for its oncolytic potential in treating cancerous conditions in preclinical models [33] and has proven to be a valuable candidate for virotherapy [11]. The discovery of MYXVoncolytic potential was serendipitous [34], as MYXV has a strict tropism for European rabbits and is not known to cause disease in any other organism [35]. Myxoma virus (genus Leporipoxvirus) is antigenically distinct from VACV (genus Orthopoxvirus), making it a viable alternative to VACV virotherapy; it is often an efficacy concern when patients have been previously vaccinated with VACV against smallpox, as the aging population is most likely to experience cancer. Despite its narrow host tropism for pathogenesis, MYXV productively infects neoplastic cells derived from humans [36, 37], mice [24, 27, 38, 39], canines [40], and felines [41]. Although not yet tested in the clinic, MYXV has shown promise in preclinical animal models of neoplasms that are particularly difficult to treat and cure, including pancreatic cancer [24], gliomas [36], medulloblastoma [42], and ovarian cancer [27]. In addition to conventional oncolytic applications, MYXV has been examined for its immunotherapeutic potential and effectiveness in combinatorial treatments with chemotherapy [24, 27]. It has also been shown that MYXV efficiently binds to and subsequently destroys CD138+ cells derived from human multiple myeloma patients. The binding of MYXV to these multiple myeloma cells induced apoptosis efficiently via the extrinsic pathway [43]. This allows for a potential ex vivo therapy by efficiently purging neoplastic cells from autologous stem cell grafts before re-implantation into the host [44, 45].
Based on our understanding of poxvirus virology, we provide a general guideline for the engineering, isolation, and purification of recombinant poxviruses suitable for in vitro and preclinical testing.
Each protocol (in Subheading 3) refers to reagents, consumables (such as tissue culture vessels), and equipment necessary to fully execute the described procedure. These referenced materials are detailed in Subheading 2 entitled “Materials.”
1.2. Genetic Engineering of Recombinant Poxviruses
During poxvirus infection, recombination occurs in the virus genome [6]. This is a universal characteristic of poxviruses that has shaped the development of the methods used to produce recombinant poxviruses. The activities of viral enzymes, including the 3′-to-5′ exonuclease activity of the virally encoded DNA polymerase [46], topoisomerase I [47–49] and Holliday junction resolvase [50] play roles in recombination during infection. The poxvirus infection that is needed to generate the recombinant virus can be from the destination parental poxvirus or a helper poxvirus [51]. Nevertheless, to engineer a recombinant poxvirus, the first step is to produce the transfer plasmids that contain the gene of interest with its expression driven by a poxvirus-specific promoter. In a transfer plasmid the expression cassette for the gene of interest is flanked by approximately 500–1000 base pairs (bp) of dsDNA homologous to the sequence in the parental virus genome. This design will lead to integration of the transgene into the poxvirus genome in a site-specific manner. Initial attempts to insert and subsequently express foreign genes in VACV made use of the VACV thymidine kinase gene as an insertion site [52] and inserted the herpesvirus thymidine kinase, providing its own selection mechanism [53]. In the VACV, the viral thymidine kinase gene [53] and the VACV growth factor gene [54] loci have both been successfully used as insertion sites for foreign genetic material without reducing infectivity. If a wild-type (WT) MYXV vector is desirable for the study, insertion at the locus between M135R and M136R gene can be considered, as it will not affect either viral infectivity in vitro or pathogenesis in rabbits [55]. The MYXV vector with a single gene knockout of either M063R [56, 57] or M135R [45] is also commonly used as the backbone vector for therapeutic testing in preclinical models. Because M063R-null [56] and M135R-null [58] MYXV do not cause pathogenic diseases in European rabbits, this accommodates the need for a replicating MYXV vector without the possibility of endangering other animals in the process. Other genes in MYXV, including M153R [59], M062R [27], and Serp2 [40], can also be individually deleted to enhance its immunotherapeutic effect. In MYXV deletion of M062R alone results in a replication-defective viral vector in cells from all species tested [60]. However, treatments using M062R-null MYXV can provide immunological benefits in preclinical models and in some cases it has surpassed the therapeutic effect of WT MYXV (unpublished data J Liu).
1.2.1. Construction of the Transfer Plasmid Vector
Once a poxvirus viral vector has been chosen for the study and the insertion site in the viral genomic backbone has been determined, the promoters to drive gene expression also need to be identified. The actual methods used to construct the transfer vector are not in any way specific to poxvirus biology, and researchers wishing to make transfer vectors have a number of options.
New DNA manipulation and cloning technologies, as well as inexpensive whole-gene synthesis [61] mean that any researcher wishing to insert novel sequences into poxvirus transfer vectors has a wealth of choices. A systematic survey of the history and current state of DNA manipulation and molecular cloning methodologies is beyond the scope of this publication, and many reviews exist to guide the research in their choice of cloning procedures. One of such guides and current, detailed advice can be found published by the iGEN/TU Eindhoven Community [62].
Gateway cloning [63, 64] has been extensively utilized to engineer poxvirus transfer plasmids [55, 65, 66] and is the methodology of choice in our laboratory. The Gateway cloning system allows for the simple, rapid, and highly efficient insertion of “att” bounded sequences into a destination vector using a recombinase derived from lambda phage. Smallwood et al. has provided an excellent and detailed protocol [67].
1.2.2. Promoters for Transgene Expression in the Recombinant Poxvirus
Poxvirus genes can be categorized into three temporal classes corresponding to the kinetics of their transcription: early, intermediate, or late [68, 69]. Viral gene transcription occurs in a cascade fashion. During the infection, each kinetic class of genes encodes factors necessary for the transcription of the subsequent temporal class [69]. Poxvirus-specific promoters are required to drive transgene expression in the context of a poxvirus infection. Early promoters operate before viral DNA replication, while intermediate and late promoters operate only after DNA replication [70]. Across the Poxviridae, promoters of a particular temporal class differ slightly but have highly conserved core regions [68]. The promoter structures of each temporal class have been well characterized in the VACV [71, 72]. Because transcription factors are highly conserved across the Chordopoxvirinae, promoters from one poxvirus can often be used to drive transcription during infections caused by poxviruses of different genera [68]. For example, the VACV p7.5 early/late promoter drives transcription in the fowlpox virus (FWPV, genus Avipoxvirus) [73], the VACV p11 late promoter drives transcription in MYXV (genus Leporipoxvirus) [55], and the synthetic early/late promoter (vvSyn E/L) drives transcription in the tanapox virus (TANV, genus Yatapoxvirus) [21]. Poxvirus promoters combining the temporal classes have been made by ligating regions from temporally distinct promoters and have been extensively utilized to drive robust transgene expression for both virology studies and therapeutic applications [55, 74].
The researcher wishing to express exogenous genes in the context of a poxvirus infection should be aware of the constraints imposed by each type of poxvirus promoter used. For example, to engineer poxviruses for high levels of transgene production (e.g., vaccine antigens [75]) it is appropriate to choose a promoter such as the early/late VACV p7.5 [76] or the vvSyn E/L [39, 74, 77] promoter to drive continuous expression throughout the course of an infection. In cases where the gene product must be expressed late in the replicative process due to toxicity or as a selection control for transgene expression only during a productive infection, a late promoter such as the poxvirus p11 late promoter [55] may be the appropriate choice. Table 1 shows several poxvirus promoters including both native and synthetic promoters from different temporal classes.
Table 1.
Promoter sequences can be used to drive transgene expression
| Name | Kinetic class | Sequence | References |
|---|---|---|---|
| VVACV TK | Early | GAATAAAGTGAACAATAATTAATTCTTTATT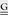 TCATCATG TCATCATG |
[78] |
| 7.5 kd | Early | CGTAAAAGTAGAAAATATATTCTAATTTATTCA |
[71] |
| GG8R | Intermediate | CATTTAACTTTAAATAATTTACAAAAATTTAAAATA | [79] |
| vvD6R | Late | ATATATGCTCATATATTTATAGAAGATATCACATATC G G |
[80] |
| vvP11 | Late | GAATTTCATTTTGTTTTTTTCTATGCT G G |
[81] |
| vvSynE/L | Early/late | AAAAATTGAAATTTTATTTTTTTTTTTTGGAATATAAATA | [74] |
| pSynE/L | Early/late | AATTGGATCAGCTTTTTTTTTTTTTTTTTTGGCATATAAATAAGGTC-GAAGCTTGGTACCAAAAATTGAAAAACTATTCTAATTTATTGCACG | [82] |
The transcription start sites are indicated by double underline where known. Start codons are shown highlighted in gray
1.3. Engineering Recombinant Poxviruses
After construction of the transfer plasmid (or shuttle vector), recombinant poxvirus is generated via homologous recombination in poxvirus infected cells. Generally a length of 500–1000 bp on each flank of the insertion DNA fragment is sufficient to guide homologous recombination in a site-specific manner into the viral genome [6].
Utilizing a helper poxvirus such as fowlpox virus (FWPV) [83] or Shope fibroma virus (SFV) [84] to provide the necessary machinery for recombination has been shown to greatly improve the ease of downstream purification steps. In these modified protocols, instead of using live virus of the chosen poxvirus vector, purified poxvirus genomic DNA from the destination poxvirus vector is used along with the shuttle vector [83, 84]. In these systems significantly improved recombination efficiency between the transfer plasmid and the destination poxvirus vector has been reported. The marked improvement in abundance of recombinant virus is likely due to an effective inhibition of infection by the helper poxvirus in the subsequent purification process. By choosing a cell line for purification that is nonpermissive to the helper poxvirus but permissive to the chosen viral vector, one can significantly eliminate the presence of helper virus in subsequent steps. However, due to the need for additional reagents (live viruses such as FWPVor SFV) besides the chosen viral backbones (e.g., VACV or MYXV), in this chapter we will focus on the conventional methodology to generate recombinant poxviruses.
It is a common practice to transfect the vector into cells already hosting a poxvirus infection, typically referred to as an infection/transfection procedure [83]. This is usually accomplished by inoculating permissive cells with poxvirus (e.g., at a multiplicity of infection (MOI) of 1), followed by transfection of the transfer plasmid 1–2 h later. Because the majority of progeny virions will be the parental virus [85], it is necessary to apply a selection method to screen for recombinant viruses bearing the desired transgenes. Thus, a plaque or focus purification step is necessary to isolate the virus until a pure population of the recombinant virus is established.
There are many types of selection that allow the isolation of recombinant virus from the unaltered parental virus. Historically, selection was first conducted by probing of replica-plated plaques on nitrocellulose membranes followed by in situ hybridization to identify the successful recombinants [86]. Selection can be accomplished via biochemical means using selectable markers such as resistance gene to neomycin [86], hygromycin [87], or guanine phosphoriboxyltransferase (gpt) [88, 89]. However, the use of genotoxic selectable markers has been shown to cause secondary mutations elsewhere in the genome that are not immediately apparent without direct sequencing [83]. We thus advise caution if one of the above selection methods is used. Fluorescent reporters provide selection that can be used together with other selectable markers [90, 91] or used alone. Although we reference selection schemes for recombinant poxviruses based upon drugs or other biochemical properties, this protocol is written with fluorescence-based selection in mind.
1.4. Considerations for Clinical Application Using Poxvirus Vectors
When making recombinant viruses for purely research work it is usually acceptable to leave genes for selection proteins intact in the recombinant virus. If, however, a virus is intended for therapeutic use in a human clinical trial, a clinical-grade viral preparation must be made [92]. These recombinant viruses must also be free of expressible drug resistance proteins (such as the neomycin phosphotransferase used during neomycin selection), as there are potential safety concerns due to adventitious mutations in these viruses [93]. It is therefore necessary to excise genes encoding any drug resistance proteins used during the isolation of the virus. Such an excisable selection system was published by Rintoul et al. in 2011 [94]. The authors describe a selectable and excisable marker (SEM) system, which consists of a transfer plasmid, pSEM-1, which contains (a) a luciferase gene driven by a poxvirus early/late synthetic promoter and (b) a gene encoding yellow fluorescent protein (yfp) fused to the gene encoding the drug selection marker guanine phosphoribosyltransferase (gpt). The entire portion of pSEM-1 containing these two genes was flanked by sequence homologous to the viral thymidine kinase gene (dictating the site of insertion into the viral genome), while loxP sites flanked the portion containing the yfp-gpt gene. When the virus was inoculated onto U2OS cells, both YFP and luciferase activity were detected; when U2OS cells stably expressing the Cre recombinase were inoculated, only luciferase activity was detected. Thus, the yfp-gpt portion of the viral genome had been excised.
When live, replication-competent viruses (particularly VACV) are used as virotherapeutics, there exists a risk that productive viral infection within tumor cells in patients could spread to healthy tissue and cause adverse effects. To protect against this possibility, researchers should have access to antiviral drugs that can blunt any undesirable viral spread originating with the treated tumor tissue. VACV infection in research animals has been successfully treated with two investigational drugs, ST-246™ (SIGA Technologies, Inc) [95] and CMX-001™ (Chimerix, Inc) (Brincidofivir) [96, 97]. Although it has not been approved for use in poxvirus infections, Cidofovir (Vistide™ Gilead Sciences, Inc.) may also be used [98]. In one case, combinatorial treatment was provided to a pediatric patient with refractory atopic dermatitis who developed eczema vaccinatum from contact vaccinia infection and the patient recovered; the combinatorial treatment included intravenous vaccinia immune globulin, cidofovir, and ST-246 [99, 100]. ST-246 and CMX-001 are predicted to be able to control MYXV infection effectively. Although MYXV inoculation in a non-rabbit species is unlikely to cause infectious disease due to a strict species tropism, the availability of these drugs provides an extra measure of safety.
1.5. Summary
It is an exciting time to be in the laboratory working with poxviruses as they continue to be developed as virotherapeutic agents for the treatment of diseases, both in humans and in companion animals [101, 102]. Although VACV has held center stage in the ongoing oncolytic poxviruses story, MYXV is steadily establishing itself as a valuable addition to the virotherapeutic armamentarium. VACVand MYXVare, however, only the beginning of the poxvirus story. Within the Chordopoxvirinae, the International Committee on Taxonomy of Viruses (ICTV) currently lists 12 genera containing 40 recognized poxviruses. The therapeutic potential of most of these poxviruses remains to be characterized and developed. In addition to vector administration, we continue to discover novel gene functions encoded by poxviruses that modulate the cellular innate immune state, any of which could prove important for the development of new therapeutics. Genetic manipulation of poxvirus genome provides a powerful tool for future investigations.
2. Materials
This section contains a detailed list of cells, media, materials, consumables, apparatus, and equipment used in the subsequent protocols.
2.1. Cell Lines
BSC-40, ATCC (catalog CRL-2761). Derived from Cercopithecus aethiops, epithelial, adherent, from kidney. BSL-1.
CV1, ATCC (catalog CCL-70). Derived from Cercopithecus aethiops, fibroblast, adherent, from kidney. BSL-1.
Vero, ATCC (catalog CRL-812). Derived from Cercopithecus aethiops, epithelial, adherent, from kidney. BSL-1.
2.2. Tissue Culture Medium, Reagents, and Solutions
DMEM, with 4.5 g/L glucose and L-glutamine.
Penicillin/streptomycin (Pen Strep).
Fetal bovine serum (FBS).
Tris–HCl 10 mM, pH 8.0, (sterile).
Trypsin 10×, without phenol red.
Sucrose (D-Sucrose, saccharose), molecular biology grade.Individual solutions are made to 24, 28, 32, 36 and 40% W/V and filtered through a 0.22 μm membrane.
Low-melting-temperature agarose.
PBS, with calcium and magnesium, sterile.
ViaFect Transfection Reagent, Promega (catalog E4982).
2.3. Consumables
150 mm cell culture dish, treated, DNase/RNase free, sterile.
24 well tissue culture-treated plate, sterile.
35 ×12 mm style tissue culture dish, tissue culture-treated, sterile.
25 cm cell scraper, sterile, individually wrapped.
Centrifuge tube, 50 mL, conical, sterile.
Ultra-Clear Centrifuge Tubes 1 × 3½ in (25 × 89 mm).
2.4. Apparatus and Equipment
Dounce-style homogenizer, 40 mL, with “loose” pestle, Wheaton.
Dismembrator (sonicator), with inverted “cup” and sound-deadening enclosure.
Ultracentrifuge, from ThermoScientific/Sorvall, model WX-Ultra 80, used with swinging bucket rotor, model AH-629.
3. Methods
3.1. Infection/Transfection of Permissive Cells
Seed permissive cells (e.g., BSC-40) into a 35 mm tissue culture dish at a density that will produce 80–90% confluency on the next day.
- Inoculation of poxviruses onto the monolayer:
-
(a)Use a poxvirus viral stock with sufficient plaque-forming units (pfu) to achieve an MOI of 1 in a minimal volume of growth medium (just sufficient to cover the bottom of the tissue culture dish). For a 35 mm culture dish 0.5 mL is sufficient. Allow viral attachment to the cells for 1 h, which can be performed at 4 °C. Every 10 min gently rock the plate to ensure complete exposure of the monolayer to the inoculum.
-
(b)After 1 h of incubation, remove the inoculum from the dish and wash three times with 1 mL of PBS each. Immediately add 2 mL of fresh growth medium to the cells.
-
(a)
Incubate the infected cell monolayer at 37 °C with 5% CO2 for 1 h before transfection with the transfer vector.
Transfect the infected cells with complexes formed from the transfection reagent of choice (e.g., ViaFect, see above) and the transfer vector, according to the manufacturer’s protocol. Remove the transfection complexes if so directed by the manufacturer’s protocol, as prolonged exposure may be toxic and cause cell death. Allow the infection to proceed following transfection until a complete cytopathic effect (CPE) in all cells is observed (48–72 h post infection).
When the infection is suitably advanced, collect the cell lysate together with the culture medium by scraping the monolayer with a sterile cell scraper. At this point the crude viral preparation may be stored at 80 °C until further processing.
3.2. Selection and Purification of Recombinant Viruses
Before purification, subject the cell lysate to 3 cycles of freeze (−80 °C)-thaw (37 °C water bath) followed by sonication in an ice bath for 1 min using a cup sonicator (e.g., Fisher Scientific sonicator with the amplitude set to 50, see above, Subheading 2.4).
Purification of the recombinant virus is conducted on a monolayer of BSC-40 cells seeded in 35 mm dishes or in 6-well plates. Serially dilute the original cell lysate and transfer an appropriate amount of inoculum into each well for an incubation period of 1 h. During the 1-hour incubation gently rock the dish every 10 min to ensure that sufficient culture medium is covering the monolayer cells. After 1 h, aspirate the inoculum and overlay an appropriate volume of medium containing low-melting agarose (0.67% in normal growth medium). Allow 20 min for the agarose overlay to solidify at room temperature before transferring the plates into the incubator. An alternative is to use methylcellulose to replace low-melting agarose. You may plate one dish of cells at each dilution for this step. Once a suitable dilution is identified, more plates can be seeded until a successful isolation is achieved.
Once well-separated viral foci or plaques are detected, proceed to isolate the virus from them for further purification. For each round of purification, first identify a discrete, well-separated plaque of viral infection and isolate it for amplification and analysis (see Note 1 for further explanation.)
Repeat the process of plaque (focus) purification until the resulting stock contains a uniform population of recombinant virus (see Note 2). We typically do a minimum of four rounds of plaque purification.
At each round of purification a sample of recombinant virus is isolated for viral DNA extraction and PCR verification for purity. For PCR verification, one can use a primer set that recognizes sequences surrounding the insertion site in the parental virus. It is also helpful to design a different set of primers to recognize sequences only present in the recombinant virus. Use viral DNA from your parental virus as a control. Once you confirm the purity of the recombinant virus, proceed with further confirmation by sequencing. We typically sequence the insert and several hundred bases surrounding the insertion site to ensure that the gene(s) of interest have integrated correctly and that no mutations have been introduced.
3.3. Poxvirus Amplification and Purification
Before testing in cells or animal models, it is necessary to produce a large quantity of poxvirus with high titer. Viral titer is usually expressed as plaque-forming units (pfu) per milliliter of virus preparation. Cell lines for the replication of VACV and other poxviruses include BSC-40 [103], CV-1 [104], and Vero [105]. These cells are obvious first choices for high-efficiency replication of poxviruses, and BSC-40 cells are most frequently used. A protocol for small-to medium-scale VACV and MYXV preparation is detailed below, which is modified from methods reported previously[106, 107].
Use cells that are fully permissive to the virus for amplification, such as BSC-40. Start with 20–40 tissue culture-treated dishes of size of 150 mm (each dish has 176.7 cm2 growth area for adherent cells) and allow them to reach full confluence (approximately 2.0 ×107 cells/dish). Cells should not be over-confluent before infection.
- Inoculate each dish with a low MOI (e.g., 0.001 for VACV).
-
(a)For infection, prepare an inoculum in a low volume of growth medium per dish (e.g., no more than 10 mL) with an appropriate amount of virus. Replace the medium from each plate with the inoculum.
-
(b)Incubate the inoculum at 37 °C for 1 h with occasional gentle rocking of the dish. This is to ensure sufficient exposure of all cells to the inoculum and to avoid drying of the monolayer.
-
(c)Add an additional volume of complete growth medium to a total of 30 mL to each dish after 1 h of incubation.
-
(a)
Incubate and allow the infection to develop until cells reach full production capacity (e.g., 72 h for MYXV, and up to 5 days for VACV). Monitor the progress of the infection daily. For VACV stock preparation, monitor the cells until a CPE is apparent but cells have not started to lyse.
Harvest the infected cells by scraping the monolayer and transferring the scraped cells along with the medium into a 50 mL centrifuge tube. Recover as much of the cells as possible.
Centrifuge cells at 750 × g for 10 min at 4 °C. Carefully discard the supernatant and continue to collect scraped cells using the same 50 mL conical tubes. In each tube, a pellet typically contains 4–6 dishes of infected cells. For temporary storage, discard as much medium after centrifugation as possible and store the tubes at −80° C until purification.
Resuspend briefly thawed cell pellets in ice-cold sterile 10 Mm Tris–HCl, pH 8.0. Use a volume approximately 6 times the volume of the cell pellet. For example, cells from 5 of the 150 mm dishes make up approximately 1 mL of volume and thus 6 mL of 10 mM Tris–HCl pH 8.0 is needed.
Freeze (−80 °C) and thaw (37 °C water bath) the resuspended cell pellet two more times. Sonicate the suspension for one min in an ice bath-equipped cup sonicator (see Subheading 3.2, step 1 and Note 3).
In a biosafety cabinet, transfer the sonicated cell pellet to a sterile 40 mL prechilled glass Dounce homogenizer (Wheaton) on ice and use the loose pestle for 20 strokes slowly. Collect the homogenized cell suspension into a 50 mL conical tube and centrifuge at 1500 × g for 15 min at 4 °C.
Save the supernatant in a fresh 50 mL conical tube on ice and resuspend the remaining cell pellet in 4 mL of ice-cold 10 mM Tris–HCl, pH 8.0. Repeat the homogenization step (step 8, above).
Centrifuge the homogenized pellet suspension at 1500 ×g for 15 min at 4 °C. Combine the supernatant with that from step 9 and discard the pellet.
Carefully layer the homogenate (no more than 20 mL of crude cell extract per centrifugation tube) over a 36% sucrose cushion in 10 mM Tris–HCl pH 8.0 (No less than 17 mL of volume) in an Ultra-Clear centrifuge tube.
Centrifuge at 44,500 × g for 80 min at 4 C (see Note 4).
After centrifugation, the virus will form a tight pellet on the bottom of the centrifuge tube. Carefully discard the supernatant and resuspend the pellet in no more than 7 mL of ice-cold 10 mM Tris–HCl pH 8.0.
Sonicate the viral preparation 2 times for 60 s each in the ice bath using the settings in Subheading 3.2, step 1.
Load the sonicated viral preparation on top of the sucrose step gradient, made with 6 mL each of 24, 28, 32, 36, and 40% sucrose that have been dissolved in 10 mM Tris–-HCl, pH 8.0 (see Note 5). Continue with purification by centrifugation of the resuspension on the gradient for 40 min at 24,000 × g at 4 °C.
Centrifugation should result in 1 distinct band near the middle of the gradient. Carefully remove the top layers of gradient material. Collect the band with a pipette tip and discard the remaining liquid (see Note 6).
Resuspend the collected band in 10 mM Tris–HCl, pH 8.0 and further dilute the band to fill up the centrifugation tube.
Centrifuge the diluted band for 60 min at 31,500 × g, 4 °C.
Carefully discard the supernatant and resuspend the pellet in1 mL of 10 mM Tris–HCl, pH 8.0.
Aliquot and store the purified virus preparation at −80 °C. Viral titer is evaluated by titration on BSC-40 cells. Proper execution of this protocol results in viral yields of 1–5 × 109 pfu/mL for MYXV, and approximately 1 × 1010 pfu/mL for replicating VACV.
For testing the recombinant virus in vitro and in animal models, stop at the step 13; virus preparation is of sufficient purity for this purpose. When generating recombinant virus to express cytokine or other therapeutic molecules, further purification through the remaining of the steps is necessary to further eliminate trace amount of transgene product which may remain with the virus pellet at step 13.
3.4. Determination of Viral Titer
If titration is conducted on a crude cell lysate which has not previously been processed, then freeze (at − 80 °C) and thaw (37 °C water bath) the sample 3 times followed by sonication before proceeding to step 2 for serial dilution of the virus stock. If a viral preparation is purified from a sucrose cushion (see Subheading 3.3, step 13) or sucrose gradient purification (see Subheading 3.3, step 15), sonicate the virus aliquot in an ice bath for 30 s before proceeding to step 2.
Make serial dilutions of your sample in PBS with Mg+2 and Ca+2 and 2% heat inactivated FBS (dilution medium). Use 1.5 mL Eppendorf tubes and make the initial 10−2 dilution by adding 10 μL of sample to 990 μL dilution medium, and all subsequent serial 10−1 dilutions by adding 100 μL of diluted virus to 900 μL of dilution medium. For samples of unknown titer, we suggest dilutions up to 10−8 (see Note 7).
Use 24-well tissue culture-treated plates with a monolayer of permissive cells (e.g., BSC-40 cells) seeded the day before for titration. Remove the growth medium from each well, vortex the dilution tube before adding 200 μL of the dilution tube into three wells. Start with the 10−8 wells and proceed from the highest dilution to the lowest dilution (10−2).
Place the dish(es) in a 37 °C with 5% CO2 incubator for 60 min. Return every 10 min to gently rock the plate(s).
Prepare the overlay medium (as instructed in Subheading 3.2, step 2 using low-melting agarose or methylcellulose) and warm it to 37 °C in a water bath.
After the 1-hour inoculation period, aspirate the inoculum off each well before adding 0.5 mL overlay medium.
Incubate the plate(s) for 2–3 days, or until plaques are clearly visible. For viruses expressing a fluorescent reporter gene, you will have to count the number of foci under a fluorescence microscope. Calculate the virus titer and express in units of pfu/mL.
4. Notes
The goal of this step is to find a dilution that produces well-separated regions of viral CPE so that you may isolate a pure clone of the virus. A suitable plaque can be marked using a sharpie under the microscope on the bottom of the dish. To identify a focus or plaque that expresses a fluorescent protein, simply search for and mark discrete foci of fluorescence on the bottom of the dish under a fluorescent microscope prior to isolating virus from plaques.
We recommend collecting viral DNA during each round of purification to check the purity. It is useful to pick many candidate plaques in the first round, then focus on your most promising samples in subsequent rounds. Each round is a potentially unique mixture or single clone of virus and should be stored at −80 °C until further purification or characterization.
We use a Fisher Scientific cup sonicator set to an amplitude of 50.
An ultracentrifuge and a rotor equipped with swinging buckets are needed.
After the sucrose solutions are made, they are sterilized by filtration through a 0.2 μm membrane using negative pressure. The sucrose gradient is set up the day before and left at 4 °C undisturbed overnight for the gradient formation.
Ten percent bleach or disinfectant is needed to disinfect the liquid before it is taken outside of the biosafety cabinet. Further disinfection (such as autoclaving) may be required before disposal.
Be sure to vortex the sample thoroughly before taking out a portion for the next dilution. You must use a new pipette tip each time to prevent carryover.
References
- 1.FDA guidance for industry: gene therapy clinical trials-observing subjects for delayed adverse events (2006) [Google Scholar]
- 2.Fenner F, Henderson D, Arita I et al. (1988) Smallpox and its eradication In: Smallpox and its eradication, vol 6 World Health Organization, Geneva [Google Scholar]
- 3.Stewart AJ, Devlin PM (2006) The history of the smallpox vaccine. J Infect 52(5):329–334. 10.1016/j.jinf.2005.07.021 [DOI] [PubMed] [Google Scholar]
- 4.La Scola B, Audic S, Robert C et al. (2003) A giant virus in amoebae. Science 299 (5615):2033 10.1126/science.1081867 [DOI] [PubMed] [Google Scholar]
- 5.Smith GL, Moss B (1983) Infectious poxvirus vectors have capacity for at least 25,000 base pairs of foreign DNA. Gene 25(1):21–28. 10.1016/0378-1119(83)90163-4 [DOI] [PubMed] [Google Scholar]
- 6.Evans DH, Stuart D, McFadden G (1988) High levels of genetic recombination among cotransfected plasmid DNAs in poxvirus infected mammalian cells. J Virol 62 (2):367–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schramm B, Locker JK (2005) Cytoplasmic organization of POXvirus DNA replication. Traffic 6(10):839–846. 10.1111/j.1600-0854.2005.00324.x [DOI] [PubMed] [Google Scholar]
- 8.Yang Z, Martens CA, Bruno DP et al. (2012) Pervasive initiation and 3’-end formation of poxvirus postreplicative RNAs. J Biol Chem 287(37):31050–31060. 10.1074/jbc.M112.390054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kotwal GJ, Abrahams M-R (2004) Growing poxviruses and determining virus titer In: Vaccinia virus and poxvirology. Springer, Berlin, pp 101–112 [DOI] [PubMed] [Google Scholar]
- 10.Smallwood SE, Rahman MM, Smith DW et al. (2010) Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol Chapter 14:Unit 14A 11 10.1002/9780471729259.mc14a01s17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan WM, Rahman MM, McFadden G (2013) Oncolytic myxoma virus: the path to clinic. Vaccine 31(39):4252–4258. 10.1016/j.vaccine.2013.05.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bell J (2014) Oncolytic viruses: immune or cytolytic therapy? Mol Ther 22 (7):1231–1232. 10.1038/mt.2014.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devaud C, John LB, Westwood JA et al(2013) Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology 2(8): e25961. 10.4161/onci.25961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirn DH, Thorne SH (2009) Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer 9(1):64–71. 10.1038/nrc2545 [DOI] [PubMed] [Google Scholar]
- 15.Kirn DH, Wang Y, Le Boeuf F et al. (2007) Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med 4(12):e353. 10.1371/journal.pmed.0040353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, O’Malley M, Urban J et al. (2011) Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther 19(4):650–657. 10.1038/mt.2010.312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seubert CM, Stritzker J, Hess M et al. (2011) Enhanced tumor therapy using vaccinia virus strain GLV-1h68 in combination with a beta-galactosidase-activatable prodrug seco-analog of duocarmycin SA. Cancer Gene Ther 18 (1):42–52. 10.1038/cgt.2010.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zajac P, Oertli D, Marti W et al. (2003) PhaseI/II clinical trial of a nonreplicative vaccinia virus expressing multiple HLA-A0201-restricted tumor-associated epitopes and cost-imulatory molecules in metastatic melanoma patients. Hum Gene Ther 14 (16):1497–1510. 10.1089/104303403322495016 [DOI] [PubMed] [Google Scholar]
- 19.Loya SM, Zhang X (2015) Enhancing the bystander killing effect of an oncolytic HSV by arming it with a secretable apoptosis activator. Gene Ther 22(3):237–246. 10.1038/gt.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rojas JJ, Sampath P, Bonilla B et al. (2016) Manipulating TLR signaling increases the anti-tumor T cell response induced by viral cancer therapies. Cell Rep 15(2):264–273. 10.1016/j.celrep.2016.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conrad SJ, El-Aswad M, Kurban E et al. (2015) Oncolytic tanapoxvirus expressing FliC causes regression of human colorectal cancer xenografts in nude mice. J Exp Clin Cancer Res 34(1):19 10.1186/s13046-015-0131-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mansfield DC, Kyula JN, Rosenfelder N et al. (2016) Oncolytic vaccinia virus as a vector for therapeutic sodium iodide symporter gene therapy in prostate cancer. Gene Ther 23 (4):357–368. 10.1038/gt.2016.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Q, Yu YA, Wang E et al. (2007) Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res 67(20):10038–10046. 10.1158/0008-5472.can-07-0146 [DOI] [PubMed] [Google Scholar]
- 24.Wennier ST, Liu J, Li S et al. (2012) Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther 20(4):759–768. 10.1038/mt.2011.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wennier ST, Liu J, McFadden G (2012) Bugs and drugs: oncolytic virotherapy in combination with chemotherapy. Curr Pharm Bio-technol 13(9):1817–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu F, Wang X, Guo ZS et al. (2014) T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther 22(1):102–111. 10.1038/mt.2013.240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nounamo B, Liem J, Cannon M et al. (2017) Myxoma virus optimizes cisplatin for the treatment of ovarian cancer in vitro and in a syngeneic murine dissemination model. Mol Ther Oncolytics 6:90–99. 10.1016/j.omto.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilkinson MJ, Smith HG, McEntee G et al. (2016) Oncolytic vaccinia virus combined with radiotherapy induces apoptotic cell death in sarcoma cells by down-regulating the inhibitors of apoptosis. Oncotarget 7 (49):81208–81222. 10.18632/oncotarget.12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cripe TP, Ngo MC, Geller JI et al. (2015) Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol Ther 23(3):602–608. 10.1038/mt.2014.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park BH, Hwang T, Liu TC et al. (2008) Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol 9 (6):533–542 [DOI] [PubMed] [Google Scholar]
- 31.Merrick AE, Ilett EJ, Melcher AA (2009) JX-594, a targeted oncolytic poxvirus for the treatment of cancer. Curr Opin Investig Drugs 10(12):1372–1382 [PubMed] [Google Scholar]
- 32.Gentschev I, Muller M, Adelfinger M et al. (2011) Efficient colonization and therapy of human hepatocellular carcinoma (HCC) using the oncolytic vaccinia virus strain GLV-1h68. PLoS One 6(7):e22069. 10.1371/journal.pone.0022069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Wennier S, McFadden G (2010) The immunoregulatory properties of oncolytic myxoma virus and their implications in therapeutics. Microbes Infect 12 (14–15):1144–1152. 10.1016/j.micinf.2010.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McFadden G (2015) The curious road from basic pathogen research to clinical translation. PLoS Pathog 11(6):e1004997. 10.1371/journal.ppat.1004997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McFadden G (2005) Poxvirus tropism. Nat Rev Microbiol 3(3):201–213. 10.1038/nrmicro1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lun XQ, Yang WQ, Alain T et al. (2005) Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res 65 (21):9982–9990. 10.1158/0008-5472.Can-05-1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stanford MM, Barrett JW, Nazarian SH et al. (2007) Oncolytic virotherapy synergism with signaling inhibitors: Rapamycin increases myxoma virus tropism for human tumor cells. J Virol 81(3):1251–1260. 10.1128/JVI.01408-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanford MM, Shaban M, Barrett JW et al. (2008) Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther 16(1):52–59. 10.1038/sj.mt.6300348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tosic V, Thomas DL, Kranz DM et al. (2014) Myxoma virus expressing a fusion protein of interleukin-15 (IL15) and IL15 receptor alpha has enhanced antitumor activity. PLoS One 9(10):e109801. 10.1371/journal.pone.0109801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urbasic AS, Hynes S, Somrak A et al. (2012) Oncolysis of canine tumor cells by myxoma virus lacking the serp2 gene. Am J Vet Res 73 (8):1252–1261. 10.2460/ajvr.73.8.1252 [DOI] [PubMed] [Google Scholar]
- 41.MacNeill AL, Moldenhauer T, Doty R et al. (2012) Myxoma virus induces apoptosis in cultured feline carcinoma cells. Res Vet Sci 93(2):1036–1038. 10.1016/j.rvsc.2011.10.016 [DOI] [PubMed] [Google Scholar]
- 42.Lun XQ, Zhou H, Alain T et al. (2007) Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res 67 (18):8818–8827. 10.1158/0008-5472.Can-07-1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartee MY, Dunlap KM, Bartee E (2016) Myxoma virus induces ligand independent extrinsic apoptosis in human myeloma cells. Clin Lymphoma Myeloma Leuk 16 (4):203–212. 10.1016/j.clml.2015.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bartee E, Chan WM, Moreb JS et al. (2012) Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol Blood Marrow Tr 18(10):1540–1551. 10.1016/j.bbmt.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lilly CL, Villa NY, Lemos de Matos A et al. (2017) Ex vivo oncolytic virotherapy with myxoma virus arms multiple allogeneic bone marrow transplant leukocytes to enhance graft versus tumor. Mol Ther Oncolytics 4:31–40. 10.1016/j.omto.2016.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gammon DB, Evans DH (2009) The 3′-to-5′ exonuclease activity of vaccinia virus DNA polymerase is essential and plays a role in promoting virus genetic recombination. J Virol 83(9):4236–4250. 10.1128/JVI.02255-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shuman S (1991) Site-specific interaction of vaccinia virus topoisomerase I with duplex DNA. Minimal DNA substrate for strand cleavage in vitro. J Biol Chem 266 (17):11372–11379 [PubMed] [Google Scholar]
- 48.Shuman S (1992) Two classes of DNA end-joining reactions catalyzed by vaccinia topoisomerase I. J Biol Chem 267 (24):16755–16758 [PubMed] [Google Scholar]
- 49.Petersen BO, Shuman S (1997) DNA strand transfer reactions catalyzed by vaccinia topoisomerase: hydrolysis and glycerololysis of the covalent protein-DNA intermediate. Nucleic Acids Res 25(11):2091–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia AD, Otero J, Lebowitz J et al. (2006) Quaternary structure and cleavage specificity of a poxvirus holliday junction resolvase. J Biol Chem 281(17):11618–11626. 10.1074/jbc.M600182200 [DOI] [PubMed] [Google Scholar]
- 51.Scheiflinger F, Dorner F, Falkner FG (1992) Construction of chimeric vaccinia viruses by molecular cloning and packaging. Proc Natl Acad Sci U S A 89(21):9977–9981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Panicali D, Paoletti E (1982) Construction of poxviruses as cloning vectors-insertion of the thymidine kinase gene from herpes-simplex virus into the DNA of infectious vaccinia virus. Proc Natl Acad Sci-Biol 79 (16):4927–4931. 10.1073/pnas.79.16.4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mackett M, Smith GL, Moss B (1982) Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proc Natl Acad Sci U S A 79(23):7415–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moss B (1996) Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc Natl Acad Sci U S A 93(21):11341–11348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J, Wennier S, Reinhard M et al. (2009) Myxoma virus expressing interleukin-15 fails to cause lethal myxomatosis in European rabbits. J Virol 83(11):5933–5938. 10.1128/JVI.00204-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barrett JW, Shun Chang C, Wang G et al. (2007) Myxoma virus M063R is a host range gene essential for virus replication in rabbit cells. Virology 361(1):123–132. 10.1016/j.virol.2006.11.015 [DOI] [PubMed] [Google Scholar]
- 57.Bratke KA, McLysaght A, Rothenburg S(2013) A survey of host range genes in poxvirus genomes. Infect Genet Evol 14:406–425. 10.1016/j.meegid.2012.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barrett JW, Sypula J, Wang F et al. (2007) M135R is a novel cell surface virulence factor of myxoma virus. J Virol 81(1):106–114. 10.1128/JVI.01633-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogbomo H, Zemp FJ, Lun X et al. (2013) Myxoma virus infection promotes NK lysis of malignant gliomas in vitro and in vivo. PLoS One 8(6):e66825. 10.1371/journal.pone.0066825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu J, McFadden G (2015) SAMD9 is an innate antiviral host factor with stress response properties that can be antagonized by poxviruses. J Virol 89(3):1925–1931. 10.1128/JVI.02262-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stewart L, Burgin AB (2005) Whole gene synthesis: a gene-O-matic future. Front Drug Des Discov 1(1):297–341. 10.2174/1574088054583318 [DOI] [Google Scholar]
- 62.Eindhoven T (2015) The Cloning Guide.80 [Google Scholar]
- 63.Katzen F (2007) Gateway((R)) recombinational cloning: a biological operating system. Expert Opin Drug Discov 2(4):571–589. 10.1517/17460441.2.4.571 [DOI] [PubMed] [Google Scholar]
- 64.Liang X, Peng L, Baek CH et al. (2013) Single step BP/LR combined Gateway reactions. BioTechniques 55(5):265–268. 10.2144/000114101. [DOI] [PubMed] [Google Scholar]
- 65.Mohamed MR, Rahman MM, Lanchbury JS et al. (2009) Proteomic screening of variola virus reveals a unique NF-kappaB inhibitor that is highly conserved among pathogenic orthopoxviruses. Proc Natl Acad Sci U S A 106(22):9045–9050. 10.1073/pnas.0900452106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu J, Wennier S, Zhang L et al. (2011) M062 is a host range factor essential for myxoma virus pathogenesis and functions as an antagonist of host SAMD9 in human cells. J Virol 85(7):3270–3282. 10.1128/JVI.02243-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smallwood SE, Rahman MM, Werden SJ et al. (2011) Production of Myxoma virus gateway entry and expression libraries and validation of viral protein expression. Curr Protoc Microbiol Chapter 14:Unit 14A 12 10.1002/9780471729259.mc14a02s21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Broyles SS (2003) Vaccinia virus transcription. J Gen Virol 84(Pt 9):2293–2303. 10.1099/vir.0.18942-0 [DOI] [PubMed] [Google Scholar]
- 69.Yang Z, Maruri-Avidal L, Sisler J et al. (2013) Cascade regulation of vaccinia virus gene expression is modulated by multistage promoters. Virology 447(1–2):213–220. 10.1016/j.virol.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keck JG, Baldick CJ, Moss B (1990) Role of DNA replication in vaccinia virus gene expression: a naked template is required for transcription of three late trans-activator genes. Cell 61(5):801–809 [DOI] [PubMed] [Google Scholar]
- 71.Davison AJ, Moss B (1989) Structure of vaccinia virus early promoters. J Mol Biol 210 (4):749–769 [DOI] [PubMed] [Google Scholar]
- 72.Davison AJ, Moss B (1989) Structure of vaccinia virus late promoters. J Mol Biol 210 (4):771–784 [DOI] [PubMed] [Google Scholar]
- 73.Skinner MA, Laidlaw SM, Eldaghayes I et al. (2005) Fowlpox virus as a recombinant vaccine vector for use in mammals and poultry. Expert Rev Vaccines 4(1):63–76. 10.1586/14760584.4.1.63 [DOI] [PubMed] [Google Scholar]
- 74.Chakrabarti S, Sisler JR, Moss B (1997) Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques 23(6):1094–1097 [DOI] [PubMed] [Google Scholar]
- 75.Sanchez-Sampedro L, Perdiguero B, Mejias-Perez E et al. (2015) The evolution of poxvirus vaccines. Viruses 7(4):1726–1803. 10.3390/v7041726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cochran MA, Puckett C, Moss B (1985) Invitro mutagenesis of the promoter region for a vaccinia virus gene: evidence for tandem early and late regulatory signals. J Virol 54 (1):30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thomas DL, Doty R, Tosic V et al. (2011) Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol Immunother 60(10):1461–1472. 10.1007/s00262-011-1045-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weir JP, Moss B (1987) Determination of the promoter region of an early vaccinia virus gene encoding thymidine kinase. Virology 158(1):206–210. 10.1016/0042-6822(87)90254-6 [DOI] [PubMed] [Google Scholar]
- 79.Baldick CJ Jr, Moss B (1993) Characterization and temporal regulation of mRNAs encoded by vaccinia virus intermediate-stage genes. J Virol 67(6):3515–3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hagen CJ, Titong A, Sarnoski EA et al. (2014) Antibiotic-dependent expression of early transcription factor subunits leads to stringent control of vaccinia virus replication. Virus Res 181:43–52. 10.1016/j.virusres.2013.12.033 [DOI] [PubMed] [Google Scholar]
- 81.Senkevich TG, Ward BM, Moss B (2004) Vaccinia virus A28L gene encodes an essential protein component of the virion membrane with intramolecular disulfide bonds formed by the viral cytoplasmic redox pathway. J Virol 78(5):2348–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hammond JM, Oke PG, Coupar BE (1997) A synthetic vaccinia virus promoter with enhanced early and late activity. J Virol Methods 66(1):135–138 [DOI] [PubMed] [Google Scholar]
- 83.Rice AD, Gray SA, Li Y et al. (2011) An efficient method for generating poxvirus recombinants in the absence of selection. Viruses 3 (3):217–232. 10.3390/v3030217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yao XD, Evans DH (2003) High-frequency genetic recombination and reactivation of orthopoxviruses from DNA fragments transfected into leporipoxvirus-infected cells. J Virol 77(13):7281–7290. 10.1128/Jvi.77.13.7281-7290.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Di Lullo G, Soprana E, Panigada M et al. (2010) The combination of marker gene swapping and fluorescence-activated cell sorting improves the efficiency of recombinant modified vaccinia virus Ankara vaccine production for human use. J Virol Methods 163 (2):195–204. 10.1016/j.jviromet.2009.09.016 [DOI] [PubMed] [Google Scholar]
- 86.Franke CA, Rice CM, Strauss JH et al. (1985) Neomycin resistance as a dominant selectable marker for selection and isolation of vaccinia virus recombinants. Mol Cell Biol 5 (8):1918–1924. 10.1128/Mcb.5.8.1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhou J, Crawford L, Sun XY et al. (1991) The hygromycin-resistance-encoding gene as a selection marker for vaccinia virus recombinants. Gene 107(2):307–312 [DOI] [PubMed] [Google Scholar]
- 88.Falkner FG, Moss B (1988) Escherichia coli gpt gene provides dominant selection for vaccinia virus open reading frame expression vectors. J Virol 62(6):1849–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Isaacs SN, Kotwal GJ, Moss B (1990) Reverse guanine phosphoribosyltransferase selection of recombinant vaccinia viruses. Virology 178(2):626–630 [DOI] [PubMed] [Google Scholar]
- 90.Lorenzo MM, Blasco R (1998) PCR-based method for the introduction of mutations in genes cloned and expressed in vaccinia virus. BioTechniques 24(2):308–313 [DOI] [PubMed] [Google Scholar]
- 91.Wong YC, Lin LCW, Melo-Silva CR et al. (2011) Engineering recombinant poxviruses using a compact GFP-blasticidin resistance fusion gene for selection. J Virol Methods 171(1):295–298. 10.1016/j.jviromet.2010.11.003 [DOI] [PubMed] [Google Scholar]
- 92.Guo ZS, Liu Z, Sathaiah M et al. (2017) Rapid generation of multiple loci-engineered marker-free poxvirus and characterization of a clinical-grade oncolytic vaccinia virus. Mol Ther Methods Clin Dev 7:112–122. 10.1016/j.omtm.2017.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dambach MJ, Trecki J, Martin N et al. (2006) Oncolytic viruses derived from the γ34. 5-deleted herpes simplex virus recombinant R3616 encode a truncated UL3 protein. Mol Ther 13(5):891–898 [DOI] [PubMed] [Google Scholar]
- 94.Rintoul JL, Wang J, Gammon DB et al. (2011) A selectable and excisable marker system for the rapid creation of recombinant poxviruses. PLoS One 6(9): e24643. 10.1371/journal.pone.0024643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Grosenbach DW, Jordan R, Hruby DE (2011) Development of the small-molecule antiviral ST-246 as a smallpox therapeutic. Futur Virol 6(5):653–671. 10.2217/fvl.11.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Olson VA, Smith SK, Foster S et al. (2014) In vitro efficacy of brincidofovir against variola virus. Antimicrob Agents Chemother 58 (9):5570–5571. 10.1128/AAC.02814-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trost LC, Rose ML, Khouri J et al. (2015) The efficacy and pharmacokinetics of brincidofovir for the treatment of lethal rabbitpox virus infection: a model of smallpox disease. Antivir Res 117:115–121. 10.1016/j.antiviral.2015.02.007 [DOI] [PubMed] [Google Scholar]
- 98.Guimaraes AP, de Souza FR, Oliveira AA et al. (2015) Design of inhibitors of thymidylate kinase from Variola virus as new selective drugs against smallpox. Eur J Med Chem 91:72–90. 10.1016/j.ejmech.2014.09.099 [DOI] [PubMed] [Google Scholar]
- 99.Lederman ER, Davidson W, Groff HL et al. (2012) Progressive vaccinia: case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J Infect Dis 206(9):1372–1385. 10.1093/infdis/jis510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vora S, Damon I, Fulginiti V et al. (2008) Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin Infect Dis 46(10):1555–1561. 10.1086/587668 [DOI] [PubMed] [Google Scholar]
- 101.Adelfinger M, Bessler S, Frentzen A et al. (2015) Preclinical testing oncolytic vaccinia virus strain GLV-5b451 expressing an antiVEGF single-chain antibody for canine cancer therapy. Viruses 7(7):4075–4092. 10.3390/v7072811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patil SS, Gentschev I, Nolte I et al. (2012) Oncolytic virotherapy in veterinary medicine: current status and future prospects for canine patients. J Transl Med 10:3 10.1186/1479-5876-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paszkowski P, Noyce RS, Evans DH (2016) Live-cell imaging of vaccinia virus recombination. PLoS Pathog 12(8):e1005824. 10.1371/journal.ppat.1005824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Villa NY, Bartee E, Mohamed MR et al. (2010) Myxoma and vaccinia viruses exploit different mechanisms to enter and infect human cancer cells. Virology 401 (2):266–279. 10.1016/j.virol.2010.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fang Q, Yang L, Zhu W et al. (2005) Host range, growth property, and virulence of the smallpox vaccine: vaccinia virus Tian Tan strain. Virology 335(2):242–251. 10.1016/j.virol.2005.02.014 [DOI] [PubMed] [Google Scholar]
- 106.Henderson B (1996) In: Ausubel F, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (eds) Short protocols in molecular biology, 3rd edn John Wiley and Sons, Chichester: xxxii+ 700 pages,£ 6.00 (1995). Wiley Online Library [Google Scholar]
- 107.Liu J, Wennier S, Moussatche N et al. (2012) Myxoma virus M064 is a novel member of the poxvirus C7L superfamily of host range factors that controls the kinetics of myxomatosis in European rabbits. J Virol 86 (9):5371–5375. 10.1128/jvi.06933-11 [DOI] [PMC free article] [PubMed] [Google Scholar]