

Abstract
Triple-negative breast cancer (TNBC) commonly develops resistance to chemotherapy, yet markers predictive of chemoresistance in this disease are lacking. Here we define WNT10B-dependent biomarkers for β-CATENIN/HMGA2/EZH2 signaling predictive of reduced relapse-free-survival. Concordant expression of HMGA2 and EZH2 proteins is observed in MMTV-Wnt10bLacZ transgenic mice during metastasis and Hmga2 haploinsufficiency decreased EZH2 protein expression, repressing lung metastasis. A novel auto-regulatory-loop interdependent on HMGA2 and EZH2 expression is essential for β-CATENIN/TCF-4/LEF-1 transcription. Mechanistically, both HMGA2 and EZH2 displaced Groucho/TLE1 from TCF-4 and served as gatekeepers for K49 acetylation on β-CATENIN, which is essential for transcription. Additionally, we discover that HMGA2-EZH2 interact with the PRC2 complex. Absence of HMGA2 or EZH2 expression or chemical inhibition of Wnt signaling in a chemoresistant patient-derived xenograft (PDX) model of TNBC abolished visceral metastasis, repressing AXIN2, MYC, EZH2, and HMGA2 expression in vivo. Combinatorial therapy of a WNT inhibitor with doxorubicin synergistically activated apoptosis in vitro, re-sensitized PDX-derived cells to doxorubicin, and repressed lung metastasis in vivo. We propose that targeting the WNT10B biomarker network will provide improved outcomes for TNBC.
Introduction:
Triple-negative breast cancer (TNBC; estrogen receptor-negative [ER−], progesterone receptor-negative [PR−], and HER2-unamplified), including basal-like (BL) and mesenchymal (M) subtypes, is an aggressive and frequently occurs in BRCA1 mutation carriers and in young women of African descent. African-American (AA) women are ~3 times more likely than European American (EA) women to be diagnosed with TNBC (1, 2). TNBC adenocarcinomas, which are highly metastatic and have a poor prognosis, are treated with a combination of surgery, radiation and/or chemotherapy; however, unlike other breast cancer subtypes, there are no FDA-approved targeted agents (3). TNBCs preferentially metastasize to the brain or the lung (70%), rather than bone or liver (30%) (4) and the process regulating tropism is poorly understood.
WNT/β-catenin canonical-signaling activates co-receptors (i.e. LRP5/6/FZD), leading to the stabilization of β-catenin. Stabilized β-catenin translocates to the nucleus, interacts with TCF/LEF and induces specific cellular response transcriptional programs including, but not limited to, cellular proliferation, development, differentiation, neoplasia and stem cell maintenance (5). We previously showed that WNT10B expression is elevated in the majority of TNBCs from AA women (90%) relative to EA women (65%), and is associated with poor survival (6). The WNT10B direct target HMGA2 predicts survival outcome TNBC (6). Furthermore, HMGA2 expression alone predicts metastasis in TNBC (6).
Enhancer of Zeste 2 (EZH2) is a methyltransferase component of the polycomb repressive complex 2 (PRC2) essential for the epigenetic maintenance of histone 3 lysine 27 trimethylation (H3K27me3), a repressive chromatin mark (7). EZH2 is over-expressed in a variety of cancers including basal-like and TNBC (8), in which EZH2 expression correlates with inverse levels of H3K27me3, not observed in the ER+ or HER2 cancer subtypes (9), suggesting that EZH2/PRC2 activity is lacking in TNBCs. A high frequency of elevated EZH2 expression is observed in basal-like invasive carcinomas from AA women with West African or Ghanian heritage (10). EZH2 is known to physically interact with β-catenin and to enhance Wnt-ligand mediated transactivation of β-catenin independent of EZH2’s PRC2 methyltransferase activity (11),(12). EZH2 over-expression in mammary epithelial cells initiated hyperplasia but not neoplasia (13), which suggests that EZH2 requires other factor(s) for neoplasia of the mammary gland.
HMGA2 and EZH2 are master regulators of EMT. HMGA2/EZH2 mediate DNA methylation activity, repress expression of E-Cadherin (CDH1), induce transcription factors that repress CDH1 (i.e. Slug, Snail, Twist and Zeb1/2), and oppose genomic-stability mediated by BRCA1 (14–19). Importantly, both HMGA2 and EZH2 are linked to oncogenic β-catenin activity during initation and metastasis of TNBC (6, 11, 12). Mechanistically, the overlapping functions of HMGA2 and EZH2 and how they each coordinate oncogenic β-catenin signaling are unknown. Herein, our preclinical MMTV-Wnt10b/β-catenin tumor model with haplotype insufficiency expression of HMGA2 is linked to loss of EZH2 expression and metastasis. Futhermore, in vitro we link the activity of both HMGA2 and EZH2 as necessary gatekeepers of K49 acetylation of oncogenic β−catenin and prevention of repression mediated by Groucho/TLE1. The WNT10B-biomaker network we identified, composed of WNT10B, β−CATENIN, HMGA2, and EZH2, is predictive of poor survival in patients. We suggest that this WNT10B-biomaker network is clinically relevant and useful for development of novel precision therapies against metastatic, chemoresistant TNBC.
Materials and Methods
Human Breast Cancer Tissues:
Studies were based on formalin-fixed paraffin-embedded or frozen-fixed methanol-acetone sections. Details about breast tissue collection and preparation can be found in the Supplemental Experimental Procedures. Written informed consent was obtained from all subjects and experiments conformed to the principles set out in the WMA Declaration of Helsinki and the NIH Belmont Report. Studies were approved by the Institutional Review Boards of the collaborating pathologists at UCLA, UTHSC, Emory University School of Medicine, Duke University School of Medicine and City of Hope.
Mice:
MMTV-Wnt10bTG and MMTV-Wnt10b-IRES-LacZ mouse models were described (6, 20). Hmga2+/− mice were provided from the Lowry lab (21) that contained p53flx/flx alleles from a highly mixed mouse background and were backcrossed into the MMTV-Wnt10b-IRES-LacZ FVB/NJ strain for ten generations. Animal protocols were approved by the Office of Animal Research Oversight at UCLA and the Institutional Animal Care and Use Committee (IACUC) at UTHSC.
RNA Scope:
Advanced Cell Diagnostics, Inc. (ACD) designed the probes. In situ hybridization protocols were conducted according to the manufacturer’s instructions.
Cell Culture Assays:
All conventional cell lines were maintained in a humidified atmosphere with 5% CO2 in DMEM plus 1% Pen/Strep and 10% FCS. All cells were purchased from ATCC, and authenticated by Genetica prior to use in experiments.
Knockdown experiments:
MDA-MB-231 cells silenced for HMGA2 utilizing two independent lentivirus constructs has been described (6). EZH2 CRISPR/Cas9 clones were generated by purchasing independent sgRNAs from GeneCopoeia Corp. and following the manufacturer’s protocols.
Tumor Biology, Metastasis Experiments and PDX Tumor Modeling:
Transplantation, dilution and xenograft experiments were carried out using standard procedures. Therapy with ICG-001 was used at 100 mg/kg and/or 200 mg/kg every two days, administered by IP injections and mice were euthanized at nine weeks after surgical transplantation. When used in combination with doxorubicin (1.4 mg/kg), the ICG-001 dose was reduced to 50 mg/kg. Details can be found in the Supplemental Experimental Procedures.
Transient transfections:
Plasmids were obtained as follows: GFP (Lonza), pCMV/HA-EZH2 (#24230, Addgene) and pMIG-HMGA2-FLAG (#25409 Addgene). EZH2-HA and HMGA2-FLAG were transfected in 293T cells with Lipofectamine 3000 (Invitrogen) per the manufacturer’s protocol. pcDNA-WNT10B and constitutive active (ca) β-Catenin were transfected into MDA-MB-231cells as previously described (6, 22).
Statistical Analysis:
Two-sided Fisher-Boschloo (23) unconditional exact tests were used to compare the incidence of tumors and LM between different transplanted cell populations. To compare HMGA2 and EZH2 gene expression between TNBC and non-TNBC patients, published datasets were analyzed (24). For metastasis-free survival analysis of EZH2 in breast cancer patients, published datasets were analyzed (25).
RNA and Real-Time PCR:
Standard procedures were used as described (6). Details can be found in the Supplemental Material and Methods. Primers sequences for qPCR are listed in supplementary Table S1.
Chromatin immunoprecipitation (ChIP):
Standard procedures were used as described (26). Details can be found in the Supplemental Material and Methods. The primers sequences for ChIP are listed in supplementary Table S2
IHC for of HMGA2 and EZH2 in Hmag2+/+ or Hmga2+/−Wnt10bLacZ tumors:
To increase IHC sensitivity, we decreased the HMGA2 antibody dilution by 10X and increased the EZH2 antibody dilution by 10X relative to conditions used to generate Fig. 2Aii, and we demonstrated the relative decreases in both HMGA2 and EZH2 protein levels in the Hmga2+/− primary tumors.
Figure 2. Haplo-insufficiency of HMGA2 in Wnt10bLacZ mice decreases EZH2 protein expression and represses primary tumor growth and metastasis.

A–B H&E analysis and IHC conducted for HMGA2 and EZH2 at 3 and 6 months in Wnt10bLacZ female mammary glands (Ai), primary tumors from 9-month old mice (Aii) and lung metastases from 11-month old mice (B).
C–D Kaplan-Meier Survival graph comparing Wnt10bHmga2+/+ (n=23) and Wnt10bHmga2+/− (n=14) mice survival (p=0.004) (C). Comparison of tumor volumes in Wnt10bHmga2+/+ and Wnt10bHmga2+/− mice (p=0.0011; D).
E–F H&E analysis of a primary (E) or LM tumor (F) in Wnt10bHmga2+/+ and/or Wnt10bHmga2+/− female mouse. Two independent tumors are shown per genotype. Arrows and arrowheads depict macro-metastases and micro-metastases, respectively.
G IHC for HMGA2 and EZH2 on primary tumors from Hmga2+/+ and heterozygous Hmga2+/− Wnt10bLacZ female mice.
Results:
WNT10B/β-CATENIN/HMGA2/EZH2 is predictive of poor overall survival in basal-like and TNBC subtypes
The KM-Plotter (http://kmplot.com/analysis/index.php?p=service&cancer=breast) tool recently added ~3,000 new datasets; a new analysis with HMGA2 and WNT10B reveals worse recurrence-free survival (RFS) when both WNT10B and HMGA2 expression are combined (HR= 1.99, p=0.0083; Fig. 1Ai). The combination of WNT10B, β-CATENIN, HMGA2 and EZH2 increased the hazard ratio for RFS outcomes by ~2.4 times compared to patients lacking expression of the WNT10B-network (HR=2.39 and p=0.0112, Fig. 1Aii). In another publicly available dataset stratified by women diagnosed with TNBC versus other subtypes, which included specimens from 178 women (58 TNBC and 120 non-TNBC), combined HMGA2 and EZH2 expression levels were higher in TNBC vs. non-TNBC subtypes (Fig. 1Bi–ii), providing additional evidence that combined HMGA2 and EZH2 expression is associated only in TNBC. We analyzed the correlation between WNT10B, HMGA2 and EZH2 mRNA expression using the TCGA database (Supplementary Figure S1A–B). We show that the gene expression of WNT10B significantly correlates with HMGA2and EZH2 in TNBC (p=0.028 and p<0.001, respectively). Moreover, we did not find a correlation of WNT10B and HMGA2 in ER+ or HER2+ subtypes, but we did find a correlation between WNT10B and EZH2 ( Supplementary figure S1C–D)
Figure 1. WNT10B/β-CATENIN/HMGA2/EZH2 is predictive of poor survival in basal-like and TNBC subtypes.

A i-ii Kaplan-Meier survival analysis showing significantly improved recurrence-free survival of women with basal-like and TNBC associated with lower mRNA expression of WNT10B/HMGA2 (i) and WNT10B/β-CATENIN/HMGA2/EZH2 (ii).
B HMGA2 (i) and EZH2 (ii) gene expression compared between TNBC and non-TNBC patients (178 women total, 58 TNBC and 120 non-TNBC) (24); =p value was calculated by Mann-Whitney U test using the SPSS 17.0 software.
C IHC for WNT10B, HMGA2 and EZH2 in normal breast tissue from a low-risk AA woman
D IHC for WNT10B, HMGA2 and EZH2 in serial breast biopsies from two high-risk women undergoing breast MRI screening who subsequently developed an interval cancer. Normal (NL) biopsy (Bx) (arrow) designates biopsy site.
E Subcellular localization and co-expression patterns of HMGA2, EZH2, ABC and WNT10B analyzed in two lung metastases of TNBC patients, LM EA and LM AA.
We hypothesized that the WNT10B-signaling network might be useful in evaluating case samples of women at increased risk for TNBC. First, in normal breast tissue control cases, WNT10B, HMGA2 and EZH2 were rarely detectable by IHC (Fig. 1C). Second, we performed pilot retrospective immunohistochemistry analysis of WNT10B, HMGA2 and EZH2 expression in two high-risk women who developed interval cancers while undergoing breast Magnetic Resonance Imaging (MRI) screening (Fig. 1D). Selected Case 1 is a 27-year-old Caucasian woman with the BRCA1 mutation S868X. Selected Case 2 is a 54-year-old AA female with the BRCA1 mutation Y1563X. Two additional retrospectively selected high-risk cases (Selected Cases 3 and 4) are presented in Supplementary Fig. S2A. In each case, expression of WNT10B, HMGA2 and EZH2 is observed in non-cancerous breast tissue. These results provide preliminary evidence that WNT10B-network signaling associated in “early” tissue in women at high risk for TNBC.
Next, we pilot tested for WNT10B-network expression by IHC in two women with metastatic TNBC in the lung. The first metastasis, obtained from a woman of EA descent and the second metastasis was obtained from an AA woman, in both cases, we observed expression of HMGA2, EZH2, non-phosphorylated (Ser31Ser47Thr41) active β-CATENIN (referred to as activated β-catenin, ABC) and WNT10B (Fig. 1E). EZH2 expression was observed in a BRCA1-mutation carrier in primary TNBC, but not in “adjacent normal” tissue (Supplementary Fig. S2B).
These data provide evidence that our WNT10B-network signature is predictive of poorer survival outcomes in basal-like and TNBC patients. Furthermore, in preliminary investigations of “early tissue” sample biopsies from high-risk women, the WNT10B network is both associated with increased likelihood to develop interval TNBC and metastatic basal-like/TNBC.
Loss of a single HMGA2 allele decreases EZH2 protein expression with a concordant repression of tumor growth and lung metastasis.
Next, we hypothesized that HMGA2 and EZH2 protein expression levels would be co-expressed in response to Wnt10b/β-catenin signaling in normal epithelial cells of the mammary gland. Female Wnt10bLacZ transgenic females at 3-, 6-, 9- and 11-months of age were tracked for the number of pregnancies, tumor formation and associated lung metastases (Fig. 2A–B). A single pregnancy in a 3-month-old Wnt10bLacZ female did not induce expression of either HMGA2 or EZH2. In contrast, strong expression for both HMGA2 and EZH2 was observed in multiparous females at 6-months (n=3; Fig. 2Ai) and 9- months (n=4; Fig. 2Aii) of age, and in metastatic lung lesions harvested from 11-month females (n=6; Fig. 2B). Overlapping expression for HMGA2 and EZH2 is detected the hyperplasia stage in the 6-month old females.
We previously identified HMGA2 to be the most abundant biomarker in Wnt10b-driven tumors (6). To genetically address the requirement for HMGA2 expression for tumor growth and metastasis, we used Hmga2+/−;p53flx/flx mice (21). First, male Hmga2+/− mice were backcrossed to the females of FVB/NJ Wnt10bLacZ mice for ten generations to generate a syngeneic model. Wnt10bLacZ-Hmga2+/+ mice (n=23) were then compared to heterozygous Wnt10bLacZ-Hmga2+/− mice (n=14). Despite loss of a single copy of Hmga2, there was a significant increase in survival (Fig. 2C) and a reduction in tumor volume (Fig. 2D). Hmga2−/− female mice are sterile, and the Wnt10b-preclinical model rarely gives rise to nulliparous spontaneous tumors (latency > 1 year); therefore, we compared Hmga2−/− female mice to the virgin female Wnt10bLacZ mice (n=5, Supplementary Fig. S3A). The homozygous, Hmga2−/− null female mice showed suppression of Wnt10b-driven oncogenesis up to 20 months. Primary tumors from two Wnt10bLacZ-Hmga2+/+ (Fig. 2Ei–ii) mice were analyzed by H&E and compared to two Wnt10bLacZ-Hmga2+/− tumors (Fig. 2Eiii–iv); adenocarcinomas are smaller in Hmga2+/− mice. Corresponding lungs from the same mice (Fig.2F) plus one additional mouse pair were histomorphometricly quantified for metastases (Supplementary Fig. S3B, n=3), revealing fewer lesions in Hmga2+/− mice. HMGA2 and EZH2 protein expression levels were also decreased in Hmga2+/− and Hmga2+/+ primary tumors (Fig. 2G and Supplementary Fig. S3C–E).
In summary, the co-expression of both HMGA2 and EZH2 protein occurs throughout the metastatic cascade in Wnt10b/β−catenin-driven mammary tumors. More importantly, haplo-insufficient expression of HMGA2 decreased EZH2 protein expression and repressed lung metastasis in vivo in tumors driven by Wnt10b/β-catenin signaling.
A novel HMGA2-EZH2 protein-protein interaction controls regulation of nuclear core β-catenin/TCF-4/LEF-1 complexes and is a necessary gatekeeper for K49 Acetylation of β-catenin.
Next, we confirmed that the TOPFLASH Wnt reporter is activated by constitutively active ß-catenin or Wnt10b and is repressed by treatment with the WNT reporter ICG-001. In addition, EZH2, like HMGA2 (6), is a direct target of WNT10B and WNT10b-dependent EZH2 expression is down-regulated by ICG-001(6), which disrupts CBP-β-catenin protein interactions (Supplementary Fig. S4A–B). More importantly, HMGA2 is recruited at the promoters of AXIN2 and MYC; but, in the absence of HMGA2, β-CATENIN cannot be recruited to the promoters of AXIN2 and MYC, resulting in loss of gene expression and demonstrating that the absence of HMGA2 functionally represses Wnt transcriptional activity (Supplementary Fig. S4C–E).
EZH2 and β-CATENIN physically interact (11) and EZH2 cooperates to regulate Wnt-ligand mediate gene expression (12). Therefore, we hypothesized that an auto-regulatory loop between HMGA2 and EZH2 must be responsible for the loss of β-catenin recruitment to Wnt responsive elements. This was tested by 1) immunoprecipitating (IP) EZH2 in the absence of HMGA2 protein expression and 2) in the presence of ICG-001 (@ 10μM for 48hours [h]) followed by immunoblotting with non-phosphorylated β-catenin protein [amino acids Ser31/Ser37, Thr41], commonly referred to as transcriptionally active β-catenin (ABC) (Fig. 3A–B). A decrease of EZH2 protein expression and a loss of EZH2 interacting with ABC in the absence of HMGA2 expression is consistent with pharmacologic repression using ICG-001.
Figure 3. HMGA2-EZH2 protein-protein interactions are necessary for maintaining K49Ac of β-catenin and for displacing the Groucho/TLE1 repressor from TCF-4/LEF-1 transcriptional complexes.
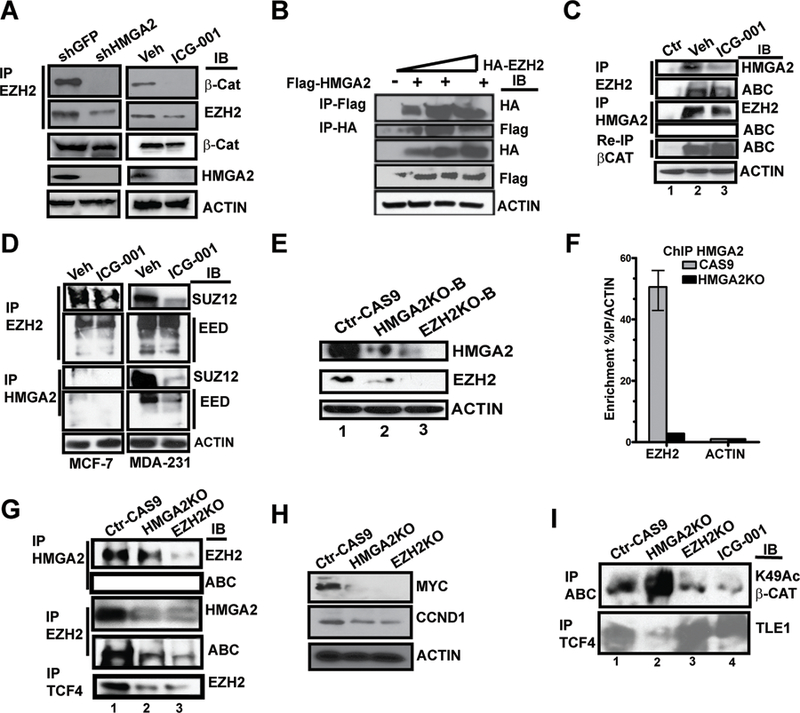
A Immunoblot for HMGA2 and EZH2 and the immunoprecipitation (IP) of EZH2 and immunoblot for both β-CATENIN and EZH2 in MDA-MB-231 cells, comparing shRNA-mediated knockdown of HMGA2 to the Sh-GFP vector only in the presence or absence of ICG-001 (@10μM for 48h). ACTIN serve as loading controls.
B IP of epitope-tagged FLAG-HMGA2 and HA-EZH2, followed by immunoblot for HA and/or FLAG. Immunoblot for FLAG, HA and ACTIN served as controls for the 293T cells transfected with increasing input of the HA-EZH2 vector in the presence of constant input of FLAG-HMGA2.
C IP of both endogenous EZH2 (immunoblot for HMGA2 and ABC) and HMGA2 (immunoblot for EZH2 and ABC). Re-IP for β-CAT and immunoblot (IB) ABC was from the original IP HMGA2 supernatant.
D IP of EHZ2 and or HMGA2, immunoblot for SUZ12 and EED in both MCF-7 and MDA-MD-231 cells.
E CRISPR/Cas9 HMGA2-KO and EZH2-KO in MDA-MB-231 cell lines were immunoblotted for HMGA2 and EZH2.
F HMGA2-ChIP experiments conducted on the EZH2 promoter in either parental CAS9 only MDA-MB-231 cells or HMGA2-KO cells, enrichment is expressed relative to the ACTB promoter.
G IP of both endogenous HMGA2 (immunoblot for EZH2 and ABC) and EZH2 (immunoblot for HMGA2 and ABC) and IP of TCF4 and immunoblot for EZH2 in Control CAS9 only cells and in HMGA2-KO and EZH2-KO cell lines.
H Immunoblot for MYC, CCND1, VIMENTIN and ACTIN in cells shown in panel J.
I IP for both ABC and TCF-4 followed by immunoblot with K49Ac-β-CAT and/or Groucho/TLE1 in the HMGA2-KO and EZH2-KO cell lines relative to Ctr-CAS9 cells. ICG-001 treated cells (10 μM for 48 hours), are included as a control for repression of K49Ac β-CATENIN.
These results prompted us to determine if HMGA2 mediates EZH2-ABC protein-protein interactions through direct interactions with EZH2. To this end, 293T cells were transfected with a constant input of FLAG-HMGA2 (2.5 μg) and increasing amounts of HA-EZH2 plasmid (1.0, 2.5 or 5.0 μg; Fig. 3B). Complexes were IP’ed with anti-FLAG/HA-epitope antibodies, and blotted with anti-HA or anti-FLAG antibodies. We observed that HMGA2 and EZH2 physically interact to form a novel protein-protein complex and confirmed that endogenous EZH2 and HMGA2 protein-protein interactions occur, disruption of EZH2 interactions with either ABC or HMGA2 are lost in the presence of ICG-001 (Fig. 3C). HMGA2 does not physically interact with ABC as shown by re-IP for pan-total β-CATENIN and blotting for transcriptionally ABC. Similar mechanisms were demonstrated by IP in two additional TNBC cell lines, MDA-MB-468 and SUM159PT (Supplementary Fig. S4F i–ii). In contrast, neither ER+ cells (MCF-7) nor two “normal” breast cancer cell lines, MCF-10A and HUMEC, exposed to ICG-001 for 48 hours had effects on the protein expression of either EZH2 or HMGA2 (Supplementary Fig. S4G); therefore this network is unique to TNBC cancer cells.
We then tested whether the HMGA2-EZH2 protein-protein complex could associate with other complexes, for example: 1) WNT-core components, TCF-4/LEF-1, and 2) PRC2-complex member SUZ12 and or EED. Like ABC, both TCF-4/LEF-1 physically interact with EZH2, but that HMGA2 does not interact with either of the two (Supplementary Fig. S4H). Next, we conducted IP assays with both HMGA2 and EZH2 ± ICG-001 in both MCF-7 cells and in MDA-MB231 cells, and blotted for either SUZ12 or EED (Fig. 3D). We confirmed that EZH2 interacts with either SUZ12 or EED in MCF-7 cells and the interactions are not disrupted by ICG-001. In contrast, the interaction of EZH2 with SUZ12, but not with EED, is disrupted in the presence of ICG-001 in MDA-MB-231 cells. Moreover, HMGA2 physically interacts with both SUZ12 and EED and exposure to ICG-001 inhibits either interactions; similar results were observed in MDA-MB-468 cells (Supplementary Fig. S4I).
We generated gene knockouts using CRISPR technology for both HMGA2 and EZH2 in MDA-MB-231 cells to determine if they participate in an auto-regulatory loop. We generated two independent clonal sub-lines for each gene (i.e. Clones A and B) and loss of mRNA and protein expression was verified (Fig. 3E and Supplementary Fig. S5A–i-B). In each gene’s clonal KO line, mRNA expression for WNT-direct targets and for proliferation/EMT-markers were down-regulated (Supplementary Fig. S5Ci–ii and S5Di–ii).
To gain mechanistic insights on how HMGA2 regulates EZH2, we performed ChIP with an antibody to HMGA2 for the EZH2 proximal promoter in cells silenced for HMGA2 expression (Fig. 3F), demonstrating decreased recruitment of HMGA2. An IP with EZH2 in the HMGA2 KO cells verified disruptions of EZH2 interact with endogenous HMGA2, ABC and TCF-4 (Fig. 3G). In both EZH2 and HMGA2 KO cells, MYC expression was lost and CCND1 expression was repressed (Fig. 3H).
The acetylation of lysine 49 (K49Ac) of β−CATENIN, mediated by CBP, can be substituted by trimethylation (beta-catMe3) on K49 by EZH2 in embryonic stem cells (ESC), causing β−CATENIN to function as a transcriptional co-repressor (27). We hypothesized that in the absence of either HMGA2 and or EZH2, the K49Ac on ABC mediated by CBP would be lost. We IP ABC and immunoblotted for β-CATENIN K49Ac, demonstrating loss of acetylation activity in both cells and in ICG-001-treated cells (Fig. 3I). In contrast, K49Ac of ABC increased in HGMA2KO cells. We next investigated the status of transducin-like enhancer family member Groucho/TLE, which physically interacts with TCF-4/LEF-1, recruiting histone deacetylases (HDAC) to counter CBP/P300 HAT activity (28, 29). We immunoprecipitated TCF-4 and immunoblotted for TLE1 to demonstrate that in the absence of EZH2 protein expression, and or exposure to ICG-001, TLE1 protein interactions with TCF-4 are increased compared to basal-levels (Fig. 3I). Paradoxically, in the absence of HMGA2, we demonstrate a decreased in the basal-level interaction between TCF-4 and TLE1. These results strongly suggest that enzymatic HAT activity, mediated by CBP, which maintains K49Ac of oncogenic, ABC is interdependent on HMGA2 and EZH2 protein-protein interactions.
HMGA2 and EZH2 are each necessary for tumor growth, visceral metastasis and repression of E-CADHERIN in vivo.
Both HMGA2 and EZH2 are master regulators of EMT (30–32), directly targeting E-CADHERIN repression and coinciding with the promotion of VIMENTIN expression (16, 33, 34). In KO clones for HMGA2 and EZH2, protein loss of VIMENTIN was associated with an inverse increase of E-CADHERIN (Fig. 4A), suggesting that loss of either gene reverts mesenchymal MDA-MB-231 cells to a more “normal” like breast epithelial cell expressing E-CADHERIN. Moreover, several EMT markers were downregulated, including VIMENTIN mRNA expression (Supplementary Fig. S5Di–ii).
Figure 4. HMGA2 and EZH2 are each essential for both tumor growth and lung metastasis in vivo.
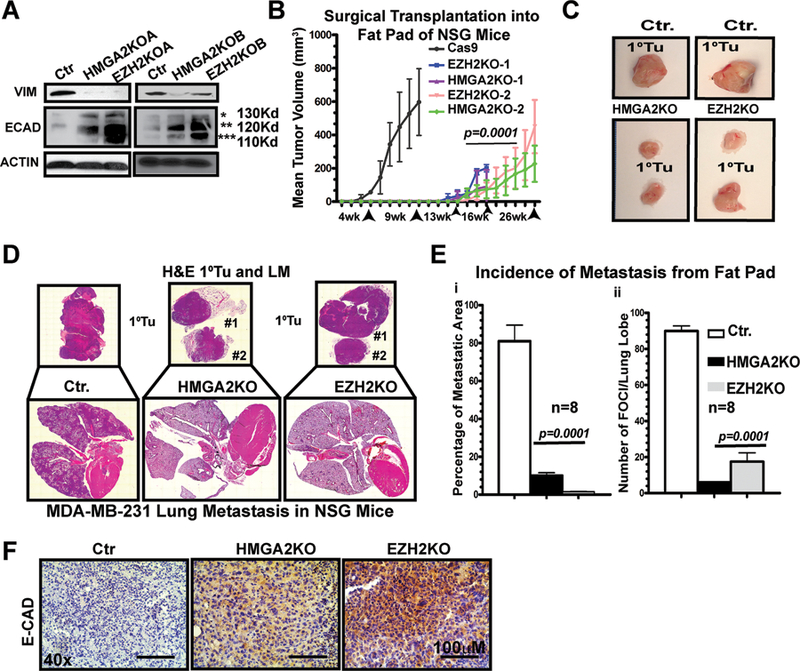
A HMGA2-KO and EZH2-KO MDA-MB-231 cell lines were immunoblotted for VIMENTIN, E-CADHERIN and ACTIN.
B CAS9, HMGA2-KO and EZH2-KO MDA-MB-231cells (1.25×106 cells; n=4 mice; bilaterally injected,) were transplanted into the inguinal mammary fat pad of (NSG) female mice and tumor growth tracked by caliper measurements.
C Images of end-stage primary tumors from panel A.
D H&E staining of the primary tumors and lung metastatic lesions from mice shown in panel A.
E Histomorphometric quantification of the lungs by H&E staining; metastases were derived from the mammary fat pad surgically transplanted cells shown in panel B. The percentage of metastatic area (i) per total lung area and the number of foci per lung lobe (ii) are compared.
F IHC for E-CADHERIN in primary tumors from mice shown in panel A; p-values for C–E by 1-WAY ANOVA (p=0.0001; p=0.0002, respectively), Averages are presented as +/− SEM
Next, we compared the effects of CRISPR KO on MDA-MB-231 tumor growth in vivo (n=8 mice; Fig. 4B). Control cells (Cas9 only) cells gave rise to 16/16 tumors by ~3–4 weeks post-transplant; all mice in this cohort were euthanized by ~6–9 weeks. In contrast, only 8/16 of possible tumors grew in either the HMGA2KO or EZH2KO cell lines by ~12–14 weeks, although tumors were detected by ~16–18 weeks. The experiment was terminated at ~26 weeks post-transplantation. Deletion of either HMGA2 or EZH2 significantly reduced tumor volume (Fig. 4B–C; p=0.001) and greatly decreased frequency of metastases in corresponding lung sections (Fig. 4D–Ei–ii) and Supplementary Fig. S5Ei–ii). Both The percentage of metastatic area and the number of metastatic foci per lung lobe are significantly reduced in the KO clones relative to the control cells (i.e. p=0.0001). Tail-vein injection (“experimental metastasis”) assays using these cells confirmed the loss of visceral metastasis (Supplementary Fig. S5F). More importantly, we confirm the restoration of E-CADHERIN expression in primary tumors derived from both CRISPR KO models (Fig. 4F).
Wnt10b/β-catenin direct targets AXIN2, EZH2, CCND1, MYC, CD44 and HMGA2 are lost in drug-resistant TNBC PDX tumors treated with ICG-001, inhibiting both primary and metastatic tumor growth.
We next posited that in mice bearing a highly chemoresistant TNBC PDX, exposure to ICG-001 would inhibit metastasis and block Wnt10b/β-catenin signaling to its downstream targets. First, we profiled a series of previously characterized PDX breast cancer lines obtained from the Huntsman Cancer Institute (35) for expression of β-CATENIN, ABC and HMGA2, confirming active Wnt/β-catenin signaling in several PDX models (Supplementary Fig.S6A). We noted that the HCI-2 (“treatment-naïve”, i.e. never received chemotherapy) and HCI-10 (“chemo-resistant”) TNBC PDX models expressed high levels of all markers. Primary cell lines were derived from these models. We exposed cHCI-2 (10 μM) and cHCI-10 (30 μM) PDX cells to ~IC50 doses of ICG-001 as previously described (22). AXIN2, EHZ2 and HMGA2 mRNA are decreased in cHCl-10 cells in response to ICG-001 therapy (Supplementary Fig. S6B). Therefore, the HCI-10 PDX model, derived from a patient who failed multiple frontline therapies, and who died from chemo-refractory disease, was chosen for in vivo therapeutic testing since it is metastatic to axillary lymph nodes and lungs. When passaged in mice, this PDX retains a basal-like/stem-cell signature, expressing elevated CD44+ (36). A subline of HCI-10Luc2 PDX tumors was derived to express luciferase to permit bioluminescent tracking of tumor growth and metastasis and this derivative was first characterized for timing of primary tumor onset and visceral metastasis to identify an optimal therapeutic window for ICG-001 therapy (HCI-10Luc2; Fig. S6C–D).
H&E staining of primary HCI-10Luc2 tumors revealed a proliferation front (dashes) and a posterior necrotic region (N) posterior and IHC with an anti-human mitochondria (i.e. Hu-Mito) antibody (Fig. 5Ai–ii) distinguished human cells from normal mammary gland (MG) stroma, including tumor cells invading into stroma (white arrowheads). Lymph node (LN) metastases were co-stained for ABC, which overlapped with the Hu-Mito pattern (Fig. 5A iii–iv). WNT10B, ABC, AXIN2 and HMGA2 were immunostained in serial sections of primary tumors (Fig. 5B), demonstrating all four WNT10B-axis proteins are highly expressed.
Figure 5. TNBC PDX tumors exposed to ICG-001 show repressed metastases by exerting an epigenetic repression of the β-Catenin/HMGA2/EZH2 signaling axis in chemoresistant CD44+ cells.

A H&E (i) staining and anti-human mitochondria antibody (Hu-MITO) IHC on primary tumors (ii), LN metastases (iii) and ABC (iv), demonstrating histological confirmation of metastatic lesions in the LN.
B IHC for WNT10B, ABC, AXIN2 and HMGA2 in primary tumors from C, demonstrating expression of the axis in the HCI-10-Luc2 PDX.
C Phase images of tumors harvested at end-stage and tumor volume from mice treated with vehicle (Veh. #1), or intraperitoneally (IP) injected with ICG-001: 100 mg/kg (Group #2) or 200 mg/kg (Group#3).
D Total light flux was compared in primary tumors (i) and in LN-Mets (ii); p-values generated by one-way ANOVA followed by pairwise t-tests (**>p=0.01, *** >p=0.001).
E Immunoblot for pan (total)-β-CATENIN (β-CATT), Act-β-CATENIN (ΑΒΓ), AXIN2, HMGA2, EZH2, PCNA, and MYC; TUBULIN serves as a loading control.
F IP for either endogenous LEF-1 or TCF-4, followed by immunoblot for EZH2 in tumors from each cohort shown in F.
G RNAscope in situ RNA hybridization detection of CD44 (PE) and HMGA2/AXIN2 (Cy5) in tumors shown in E, counterstained with DAPI. p-values for C by 1-WAY ANOVA (p=0.001), Averages are presented as +/− SEM.
To test whether ICG-001 would prevent primary tumor growth and/or inhibit metastasis, we began therapy three weeks post-transplantation (Fig. 5). Vehicle (Group #1) or ICG-001 was given intraperitoneally every other day for two weeks at a low (Group #2; 100 mg/kg) or high dosage (Group#3; 200 mg/kg). Tumor growth was monitored by calipers (Fig. 5C) and by luciferase flux (Fig. 5Di); LN metastases were monitored by luciferase flux (Fig. 5D ii). Overall, ICG-001 therapy at the highest dose significantly reduced both primary tumor and LN metastases burden.
To determine if proteins downstream of WNT10B/β-CATENIN were affected by ICG-001, we immunoblotted for 1) pan-β-CATENIN and ABC, 2) WNT direct targets: AXIN2, HMGA2 and EZH2 and 3) WNT direct targets of proliferation: CCND1, PCNA and MYC (Fig. 5E), confirming that Wnt-dependent direct target genes and proliferation markers are down-regulated in vivo. We observed by IP with either LEF-1 and/or TCF-4 that EZH2 is present in a complex, but after ICG-001 exposure, it is lost (Fig. 5F, i.e. #1, #2 and #3 refers to above dosages in Fig. 5C). The results reveal that the prevailing Wnt-mechanism demonstrated in MDA-MB-231 cells (Fig. 3–4), is active in the TNBC PDX model to initiate tumor growth and metastatic events. Moreover, IHC staining of primary tumors and lung sections confirmed loss of AXIN2 and the human mitochondria marker, respectively (Supplementary Fig. S6E–F). Next, we hypothesized that in HCI-10 PDX tumors chemoresistant CD44+(R) (37) cells would express the Wnt-direct targets HMGA2 and AXIN2. To co-localize HMGA2 and/or AXIN2 with CD44+(R) cells we conducted multiplex fluorescence RNAscope analysis, an in situ RNA-hybridization assay, in vehicle and ICG-001 treated tumors (Fig. 5G). Single RNA molecules for CD44 (Cy3), HMGA2 (Cy5) and AXIN2 (Cy5) were abundantly detected and in untreated tumors, but were greatly reduced in response to ICG-001 (Supplementary Fig. S6G).
Therefore, ICG-001 therapy targets the core WNT10B/β-CATENIN/HMGA2/EZH2 signaling axis in a chemo-refractory TNBC PDX model also characterized by enrichment of CD44+ stem-like cells to inhibit both primary tumors and visceral metastases.
Wnt inhibitor ICG-001 sensitizes doxorubicin to block metastasis of chemoresistant TNBC PDX tumors.
Treating stage IV metastatic breast cancer is an overarching clinical challenge, as the case for the patient from whom the HCI-10 PDX tumor was derived (35). The patient underwent systemic chemotherapy, which included multiple rounds of various anthracycline-based treatments, for over 21/2 years; her disease became highly chemorefractory and she died. We hypothesized that inhibition of Wnt/β-catenin pathways would re-sensitize cells derived from this patient to DOX to inhibit in vivo visceral metastasis.
Sensitivity of the cHCI-10 PDX cells to therapy was tested in vitro via WST-1 assays. ICG-001 (0.1 μM to 30 μM range: 0.1, 1, 5, 10, 15, 20 and 30 μM) or DOX (1 μM to 20 μΜ range: 1–20 0.02, 0.04, 0.2, 1, 5, 10 and 20μM) were added for 48 hours, resulting in calculated IC50 value of ~32 μM for ICG-001 (Fig. 6A). At the highest dose of DOX, ~75% of the cells were proliferating relative to the control. To address potential synergy, a sub-IC50 concentration of ICG-001 (10 μM; constant, CONST dose) was tested with varying doses of DOX. Vice versa, cells were exposed to a CONST dose of DOX (5μM) with varying ICG-001 doses and WST1 activity measured up to 96 hours. By 48 hours, the lowest ICG-001 dose with a CONST dose of DOX decreased proliferation ~40% as compared to either ICG-001 or DOX alone in the same dose range. Similarly, the lowest dose of DOX with CONST ICG-001 decreased proliferation by ~35% relative to single therapy. Moreover, by 96 hours, the lowest ICG-001 dose (0.1 μM) with a combination of DOX at 0.2 μM decreased proliferation by more than 56% (Supplementary Fig.S7A).
Figure 6. ICG-001 sensitizes chemoresistant TNBC PDX tumor cells to doxorubicin, preventing visceral lung metastasis.
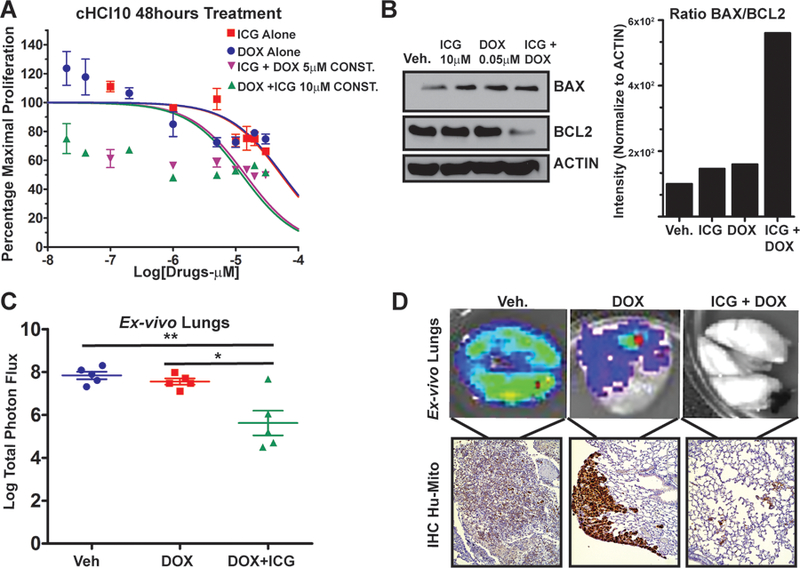
A cHCI-10 cells were exposed to various dosages of ICG-001 and doxorubicin (DOX) for up to 96h; data normalized to the maximal proliferation of vehicle treated cells; results are shown at 48h are representative of biological triplicates, expressed as the mean ± SEM.
B cHCI-10 cells exposed to ICG-001 (10 μM), DOX (0.05 μM) or in combination for 48h. Immunoblot for BCL-2, BAX and ACTIN. The signal intensity ratio for BCL-2/BAX was first normalized to ACTIN, conducted in biological triplicates.
C cHCI-10 cells (1.25×106) isolated from a primary PDX tumor were tail vein injected into NSG females and treated with DOX alone (1.4 mg/kg, IP) or in combination with ICG-001 (50 mg/kg, IP). Total Flux (p/s) was quantified by ex vivo bioluminescence imaging of lungs; p-values generated by one-way ANOVA followed by pairwise t-test (**>p=0.01).
D (Top) Ex vivo images of luciferase activity from metastatic lesions in the lung. (Bottom) To confirm that luciferase signal corresponds with metastases, IHC using an anti-human mitochondria antibody (Hu-MITO) was performed using the same lungs.
To test whether effects observed with ICG-001 and DOX are additive or synergistic, we calculated the IC30, IC50 and IC70 curves for each drug and then plotted isobole curves as in (35) (Supplementary Fig. S7B) to calculate the combination index (CI; Supplementary Table S3), revealing synergistic interactions. To understand the mechanism of synergy, we exposed cHCl-10 cells with a sub-IC50 dose of ICG-001 alone (10 μM) or DOX alone (0.5 μM), or in combination, for 48 hours and conducted IB analysis for BCL-2 and BAX (Fig. 6B); only in response to combination therapy does BCL-2 decrease. In contrast, ICG-001 or DOX alone is sufficient to increase BAX levels versus vehicle control, and drug combination did not further increase BAX expression. Quantification of the BCL2/BAX death “intensity” ratio (38) demonstrated the highest ratio when drugs were combined, thereby enhancing apoptosis (Fig. 6B, representative of three biological replicates).
We next tested whether combination therapy would repress visceral metastasis using cells derived from a freshly disassociated cHCI-10Luc2 PDX tumor introduced vial tail vein injection (Fig. 6B). To limit toxicity of combinatorial therapy, we used ICG-001 at 50 mg/kg, 4-fold less than shown in Fig. 5, with 1.4 mg/kg of DOX, which we found to be tolerated in NSG mice (IP) using a dosing regimen of once every two weeks. With DOX alone, there was no significant difference in lung metastases flux versus vehicle (Fig. 5B). In contrast, the DOX+ICG-001 combinatorial therapy significantly repressed metastatic flux compared to vehicle control or to DOX alone, as shown by ex vivo luciferase images and IHC analysis with Hu-MITO (Fig. 6C–D). Moreover, DOX alone did not prevent formation of large “clusters” of metastatic cells, which stained strongly with the Hu-MITO antibody; these larger metastatic lesions were not detectable following combinatorial therapy.
Discussion:
There is significant heterogeneity within the five breast cancer subtypes identified originally from microarray analysis (39), resulting in major therapeutic challenges. In order to develop targeted agents for TNBC, there is a great need for relevant pre-clinical mouse models. Our data support the conclusion that the Wnt10b/β-catenin/Hmga2/Ezh2 signaling axis is clinically relevant and that targeting this network is likely to prolong survival by repressing primary tumor growth and visceral metastasis.
In a preliminary retrospective study utilizing MRI-guided biopsies in women at high risk for developing TNBC we show that expression of WNT10B, HMGA2 and EZH2 proteins in biopsied material precedes the development of TNBC. Although limited by a small number of clinical cases (four), from profiling publicly available datasets, it is clear that survival is reduced by 2.4-fold in women with an elevated WNT10B-network, suggesting this network is clinically relevant.
Our preclinical Wnt10b model is distinct from the predominant MMTV-Wnt1-driven transgenic as Wnt1-tumors do not metastasize (40), therefore, we could not assess the impact of Hmga2 on metastasis (32). In contrast, lung metastasis is potently blocked in the haplotype insufficient Hmga2+/− mice in the context of Wnt10b/β-catenin-driven tumors,. More importantly, we link loss of HMGA2 expression in vivo with loss of EZH2 protein expression, decreasing visceral metastatic disease. EZH2 promotes expansion of breast tumor initiating cells (TICs) requiring β-catenin (11), but, on its own, EZH2 is incapable of transforming the mammary epithelium (13). We provide genetic evidence that HMGA2 expression is an essential cofactor for neoplasia and/or metastasis. The co-expression of both HMGA2 and EZH2 proteins in Wnt10b/β-catenin-driven early hyperplasia lesions after only two pregnancies at 6 months of age is maintained throughout the metastatic cascade providing additional evidence.
We provide compelling mechanistic evidence that both HMGA2 and EZH2 are necessary for the maintenance of K49 acetylation of β-catenin mediated by CBP (27). The Wnt nuclear complex composed of β-CATENIN/TCF-4/LEF-1(29) is necessary for Wnt-direct target gene expression, but was only maintained in the presence of both HMGA2 and EZH2. Transcription factors like TCF-4/LEF-1 regulate Wnt targets through a “transcriptional switch” that is “off” in the absence of Wnt ligands, or “on” in their presence. Co-repressors Groucho/TLE1 contribute by interacting physically with TCF/LEF-1 and disrupting Wnt signaling through β-catenin. We demonstrate that Groucho is displaced from TCF-4 only when EZH2 protein expression is present and, that in the absence of EZH2, a strong association between TLE1 and TCF-4 is observed. HMGA2 did not phenocopy this observation; in contrast, loss of HMGA2 expression increased K49 acetylation on β-catenin. We propose that HMGA2 guides nuclear architecture remodeling by partnering with CBP, or perhaps by disrupting HDACs recruitment to the Groucho proteins. HMGA2 is known to displace HDAC1 from pRB protein in pituitary tumors (41), so this hypothesis remains to be tested experimentally.
PRC2 is composed of the trimeric core proteins SUZ12, EED and EZH2 (42). EZH2-PRC2 is a multi-complex chromatin modifier that mediates enzymatic methylation of H3K27 to contribute to cellular identity. Basal-like and TNBC are cellularly distinct from other breast cancer subtypes, and our novel discovery that HMGA2 can physically interact with either of the PRC2-complex members SUZ12 and EED was observed only in TNBC cells, suggesting a unique network not found in ER+ or normal breast cancer cells that is a key player in TNBC aggressive biology. The functional consequences of these intriguing observations that the HMGA2-EZH2 complex is found within PRC2 requires further exploration. For example, EZH2-PRC2 specific inhibitors, such as DZNep (EZH2 degradation specific) or ZLD1029 (EZH2-blocking H3K27me3) enzymatic activity (43) tested in combination with ICG-001 on a variety of breast cancer subtypes would provide novel epigenetic insights into chemoresistance.
ICG-001 (PRI-724) is under clinical trials in combination with various FDA-approved agents for metastatic disease, but has not been tested against chemoresistant metastatic TNBC (https://clinicaltrials.gov/). We demonstrated that ICG-001 was effective at preventing both tumor growth and metastasis in a highly metastatic, chemoresistant TNBC PDX model derived from an EA woman, HCI-10 (44). Loss of Wnt target gene expression was concordant with the loss of CD44+ cells, known to contribute to chemoresistance (45, 46). The dose of ICG-001 currently tested in clinical trials ranges from 650 mg/m2 or 902 mg/m2 for a variety of solid tumors. Our mouse ICG-001 dosage (50 mg/kg) is equal to the human equivalent dosage (HED(47)) of 150 mg/m2, well below the phase II trial dosages. Importantly, combining doxorubicin (1.4 mg/kg; HED of 45.96 mg/m2) with ICG-001 prevented lung metastasis in this highly chemoresistant TNBC model demonstrating synergistic action in vivo. The NCCN guidelines recommend that doxorubicin be used at 60–75 mg/m2, or when used in combinatorial therapy to be lower than 50 mg/m2 once weekly in stage IV patients. DOX cytotoxicity is usually associated with its dose (48). The incidence of cardiomyopathy is about 4% with a dose of 500–550 mg/m2, 18% when the dose is 551–600 mg/m2 and 36% when the dose exceeds 600 mg/m2. We suggest that adding ICG-001 to a DOX regimen would be well-tolerated and efficacious in patients, without evidence of cardiac toxicity typically observed at a total dose of 400 mg/m2.We propose that WNT inhibitors could have broad clinical use in aggressive TNBC in combination with FDA-approved therapies. In conclusion, inhibition of the WNT10B-network signaling axis in chemoresistant TNBC patients could create new opportunities for treatment regimens.
Supplementary Material
Statement of Significance:
Findings reveal targeting the WNT signaling pathway as a potential therapeutic strategy in triple-negative breast cancer.
Acknowledgements
We thank K. Chada for his generosity in providing the Hmga2+/− mice to W. Lowry. UCLA Jonsson Comprehensive Cancer Center and UCLA CSTI grant (UL1TR000124) provided funding to GAMC. A. Welm (Huntsman Cancer Institute) provided flash frozen fragments of HCI PDX models for western blotting, and cryopreserved tissue fragments to passage the PDX models at UTHSC. Development of the Luc2-labeled HCI PDX subline was supported by NCI CA138488 (to TNS). This work was supported by a Susan G. Komen for the Cure Grant KG110317 to SAK. GAMC is currently supported by a R21 grant from NCI/Center to Reduce Cancer Health Disparities (CA179735), UTHSC/West Cancer Center grant and a NIH U01 grant (CA189283).
Footnotes
Conflicts of Interest: The authors declare that they have no conflict of interest.
REFERENCES:
- 1.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. Jama 2006;295:2492–502. [DOI] [PubMed] [Google Scholar]
- 2.Dietze EC, Sistrunk C, Miranda-Carboni G, O’Regan R, Seewaldt VL. Triple-negative breast cancer in African-American women: disparities versus biology. Nat Rev Cancer 2015;15:248–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pal SK, Childs BH, Pegram M. Triple negative breast cancer: unmet medical needs. Breast cancer research and treatment 2011;125:627–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010;363:1938–48. [DOI] [PubMed] [Google Scholar]
- 5.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell 2012;149:1192–205. [DOI] [PubMed] [Google Scholar]
- 6.Wend P, Runke S, Wend K, Anchondo B, Yesayan M, Jardon M, et al. WNT10B/beta-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol Med 2013;5:264–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoo KH, Hennighausen L. EZH2 methyltransferase and H3K27 methylation in breast cancer. Int J Biol Sci 2012;8:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hussein YR, Sood AK, Bandyopadhyay S, Albashiti B, Semaan A, Nahleh Z, et al. Clinical and biological relevance of enhancer of zeste homolog 2 in triple-negative breast cancer. Hum Pathol 2012;43:1638–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holm K, Grabau D, Lovgren K, Aradottir S, Gruvberger-Saal S, Howlin J, et al. Global H3K27 trimethylation and EZH2 abundance in breast tumor subtypes. Mol Oncol 2012;6:494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pang J, Toy KA, Griffith KA, Awuah B, Quayson S, Newman LA, et al. Invasive breast carcinomas in Ghana: high frequency of high grade, basal-like histology and high EZH2 expression. Breast cancer research and treatment 2012;135:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer cell 2011;19:86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung HY, Jun S, Lee M, Kim HC, Wang X, Ji H, et al. PAF and EZH2 induce Wnt/beta-catenin signaling hyperactivation. Molecular cell 2013;52:193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Gonzalez ME, Toy K, Filzen T, Merajver SD, Kleer CG. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am J Pathol 2009;175:1246–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006;439:871–4. [DOI] [PubMed] [Google Scholar]
- 15.Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, Gomes S, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci U S A 2013;110:9920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008;27:7274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez ME, DuPrie ML, Krueger H, Merajver SD, Ventura AC, Toy KA, et al. Histone methyltransferase EZH2 induces Akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res 2011;71:2360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Zeng X, Chen S, Ding L, Zhong J, Zhao JC, et al. BRCA1 is a negative modulator of the PRC2 complex. EMBO J 2013;32:1584–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmed KM, Tsai CY, Lee WH. Derepression of HMGA2 via removal of ZBRK1/BRCA1/CtIP complex enhances mammary tumorigenesis. J Biol Chem 2010;285:4464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miranda-Carboni GA, Krum SA, Yee K, Nava M, Deng QE, Pervin S, et al. A functional link between Wnt signaling and SKP2-independent p27 turnover in mammary tumors. Genes Dev 2008;22:3121–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White A, Flores A, Ong J, Lowry WE. Hmga2 is dispensable for cutaneous squamous cell carcinoma. Exp Dermatol 2016;25:409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fatima I, El-Ayachi I, Taotao L, Lillo MA, Krutilina R, Seagroves TN, et al. The natural compound Jatrophone interferes with Wnt/beta-catenin signaling and inhibits proliferation and EMT in human triple-negative breast cancer. PLoS One 2017;12:e0189864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lydersen S, Fagerland MW, Laake P. Recommended tests for association in 2 × 2 tables. Statistics in medicine 2009;28:1159–75. [DOI] [PubMed] [Google Scholar]
- 24.Tabchy A, Valero V, Vidaurre T, Lluch A, Gomez H, Martin M, et al. Evaluation of a 30-gene paclitaxel, fluorouracil, doxorubicin, and cyclophosphamide chemotherapy response predictor in a multicenter randomized trial in breast cancer. Clin Cancer Res 2010;16:5351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005;365:671–9. [DOI] [PubMed] [Google Scholar]
- 26.Krum SA, Miranda-Carboni GA, Lupien M, Eeckhoute J, Carroll JS, Brown M. Unique ERalpha cistromes control cell type-specific gene regulation. Molecular endocrinology 2008;22:2393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmeyer K, Junghans D, Kanzler B, Kemler R. Trimethylation and Acetylation of beta-Catenin at Lysine 49 Represent Key Elements in ESC Pluripotency. Cell Rep 2017;18:2815–24. [DOI] [PubMed] [Google Scholar]
- 28.Daniels DL, Weis WI. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat Struct Mol Biol 2005;12:364–71. [DOI] [PubMed] [Google Scholar]
- 29.Ramakrishnan AB, Sinha A, Fan VB, Cadigan KM. The Wnt Transcriptional Switch: TLE Removal or Inactivation? Bioessays 2017. [DOI] [PMC free article] [PubMed]
- 30.Chang CJ, Hung MC. The role of EZH2 in tumour progression. British journal of cancer 2012;106:243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fedele M, Palmieri D, Fusco A. HMGA2: A pituitary tumour subtype-specific oncogene? Mol Cell Endocrinol 2010;326:19–24. [DOI] [PubMed] [Google Scholar]
- 32.Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y, et al. HMGA2 is a driver of tumor metastasis. Cancer Res 2013;73:4289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, Moustakas A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem 2008;283:33437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan J, Zhang Y, Shi W, Ren C, Liu Y, Pan Y. The critical role of HMGA2 in regulation of EMT in epithelial ovarian carcinomas. Tumour Biol 2016;37:823–8. [DOI] [PubMed] [Google Scholar]
- 35.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 2011;17:1514–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015;526:131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008;100:672–9. [DOI] [PubMed] [Google Scholar]
- 38.Liu B, Han M, Sun RH, Wang JJ, Zhang YP, Zhang DQ, et al. ABL-N-induced apoptosis in human breast cancer cells is partially mediated by c-Jun NH2-terminal kinase activation. Breast Cancer Res 2010;12:R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A 2003;100:8418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Podsypanina K, Huang S, Mohsin SK, Chamness GC, Hatsell S, et al. Estrogen receptor positivity in mammary tumors of Wnt-1 transgenic mice is influenced by collaborating oncogenic mutations. Oncogene 2005;24:4220–31. [DOI] [PubMed] [Google Scholar]
- 41.Fedele M, Pierantoni GM, Visone R, Fusco A. E2F1 activation is responsible for pituitary adenomas induced by HMGA2 gene overexpression. Cell Div 2006;1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conway E, Healy E, Bracken AP. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol 2015;37:42–8. [DOI] [PubMed] [Google Scholar]
- 43.Song X, Gao T, Wang N, Feng Q, You X, Ye T, et al. Selective inhibition of EZH2 by ZLD1039 blocks H3K27 methylation and leads to potent anti-tumor activity in breast cancer. Sci Rep 2016;6:20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dietze EC, Sistrunk C, Miranda-Carboni G, O’Regan R, Seewaldt VL. Triple-negative breast cancer in African-American women: disparities versus biology. Nature reviews 2015. [DOI] [PMC free article] [PubMed]
- 45.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A 2009;106:13820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep 2014;6:514–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008;22:659–61. [DOI] [PubMed] [Google Scholar]
- 48.Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology 2010;115:155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.