

Abstract
Mitochondria and the endoplasmic reticulum (ER) have an intimate functional relationship due to tethering proteins that bring their membranes in close (~30 nm) apposition. One function of this inter-organellar junction is to increase the efficiency of Ca2+ transfer into mitochondria, thus stimulating mitochondrial respiration. Here we show that the ER cation-permeant channel polycystin 2 (PC2) functions to reduce mitochondria-ER contacts. In cell culture models, PC2 knockdown led to a 50% increase in mitofusin-2 (MFN2) expression, an outer mitochondrial membrane GTPase. Live-cell super-resolution and electron microscopy analyses revealed enhanced MFN2-dependent tethering between the ER and mitochondria in PC2 knockdown cells. PC2 knockdown also led to increased ER-mediated mitochondrial Ca2+ signaling, bioenergetic activation, and mitochondrial density. Mutation or deletion of the gene encoding for PC2 results in autosomal dominant polycystic kidney disease (ADPKD), a condition characterized by numerous fluid-filled cysts. In cell culture models and mice with kidney-specific PC2 knockout, knockdown of MFN2 rescued defective mitochondrial Ca2+ transfer, and markedly diminished cell proliferation in kidney cysts. Consistent with these results, cyst-lining epithelial cells from human ADPKD kidneys had a 2-fold increase in mitochondria and MFN2 expression. Our data suggest that PC2 normally serves to limit key mitochondrial proteins at the ER-mitochondrial interface, and acts as a checkpoint for mitochondrial biogenesis and bioenergetics through a transcriptional mechanism. Loss of this regulation may contribute to the increased oxidative metabolism and aberrant cell proliferation typical of kidney cysts in ADPKD.
One-sentence summary:
Polycystin 2 regulates the ion channel and tethering protein composition at the mitochondria-ER interface, and loss of polycystin 2 promotes increased ER-mitochondrial Ca2+ transfer that initiates changes in biogenetics and contributes to cystogenesis.
Introduction
Mitochondrial energy production requires a continuous supply of Ca2+ to maintain oxidative metabolism through regulation of Ca2+-dependent enzymes in the tricarboxylic acid (TCA) cycle1,2. However, the amount of Ca2+ entering mitochondria must be precisely regulated, as mitochondrial Ca2+ overload can lead to apoptosis through increased mitochondrial membrane permeabilization, depolarization, and opening of the permeability transition pore3-6.
Under non-pathological conditions, Ca2+ flux occurs between the endoplasmic reticulum (ER) and mitochondria at a region that forms close physical contacts (~30 nm) between these organelles, called the mitochondria-associated ER membrane (MAM)3,7,8. The MAM effectively integrates signal transduction with metabolic pathways to regulate communication between the ER and mitochondria. Regulated transfer of Ca2+ from the ER to mitochondria occurs through opening of the inositol 1,4,5-trisphosphate receptor (InsP3R) on the ER membrane and the mitochondrial Ca2+ uniporter (MCU) complex on the inner mitochondrial membrane3,9,10. Specifically, the type 3 InsP3R (InsP3R3), but not InsP3R111, is reported to localize primarily at the MAM to coordinate Ca2+ transfer to mitochondria.
In many disease states, including Alzheimer’s Disease12 and diabetes13, the contacts between the ER and mitochondria, along with Ca2+ transfer between these organelles, are altered, ultimately affecting cellular metabolism. The polycystin proteins, particularly Polycystin 1 (PC1), have emerged as modulators of mitochondrial metabolism. Loss-of-function mutations to the polycystin genes PKD1 and PKD2 result in Autosomal Dominant Polycystic Kidney Disease (ADPKD), a disease for which there is no cure. There is a striking change in the metabolism of cystic epithelial cells14,15, and parallels between cystogenesis and cancer development have been noted14. Indeed, loss of PC1 results in a shift to glycolytic metabolism, and treatment with 2-deoxyglucose, an inhibitor of glycolysis, was shown to reduce renal cysts in PC1-deficient mice 15-17. In addition, the polycystin complex (comprised of PC1 and polycystin 2 [PC2]) has been shown to be sensitive to oxygen concentrations, demonstrating its ability to sense environmental conditions and regulate cellular metabolism18. However, studies investigating whether PC2 depletion specifically alters mitochondrial function or Ca2+ signaling have been limited19. The vast majority of the protein product of PKD2, PC2, is found on the ER of epithelial cells, with the remainder localized to the primary cilium20,21. In contrast to the largely plasma membrane-localized PC1, PC2 can act as a Ca2+-dependent Ca2+ release channel in the ER membrane, and can also interact with and regulate the release of ER Ca2+ via the InsP3R22-24. Loss-of-function mutations in PC2 disrupt downstream Ca2+ signaling, leading to decreased intracellular Ca2+ release and increased cAMP signaling, a consequence of ADPKD25.
We hypothesized that a reduction of PC2 would diminish mitochondrial Ca2+ signals. However, in this study we found that loss of PC2 increased mitochondrial Ca2+ transfer. We also observed decreased mitochondrial movement due to increased expression of the ER-mitochondria tethering protein, mitofusin 2 (MFN2)26-28. Specific knockdown of MFN2 in the tubules of a PC2-deficient cystic mouse model restored mitochondrial Ca2+ signaling, morphological alterations, and reduced proliferation in cystic cells. Collectively, our results suggest that PC2 inhibits mitochondrial Ca2+ entry, and that its absence alters the complement of Ca2+ transfer molecules at the mitochondrial-ER interface, ultimately affecting Ca2+-mediated mitochondrial bioenergetics that contribute to cell proliferation.
Results
PC2 knockdown results in enhanced mitochondrial Ca2+ transients
As one of its main functions, PC2 modulates ER Ca2+ release. In light of work demonstrating metabolic alterations of ADPKD cystic cells, we examined mitochondrial Ca2+ signaling in an in vitro system comparing cells with PC2 knocked down (PC2 KD) to control (scrambled “SCR”) kidney cells (LLC-PK1) (Supplementary Fig. 1A). We used ATP as an agonist of G-protein coupled receptors that activate phospholipase C (PLC) to produce inositol trisphosphate (InsP3) to activate the InsP3R. PC2 KD cells had a significant decrease in the cytoplasmic Ca2+ transient in response to ATP stimulation, as analyzed by the area under the curve (Fig. 1A,B), consistent with previous reports22,29. We predicted that reduced cytoplasmic Ca2+ transients would be correlated with a reduction in mitochondrial Ca2+ uptake. To test this hypothesis, we used the mitochondrial-targeted ratiometric Ca2+ probe, pericam30,31, to interrogate mitochondrial Ca2+ signals (Supplementary Fig. 1B,C). Counter to our expectation given the decreased cytoplasmic Ca2+ release after stimulation with ATP, the mitochondrial Ca2+ transient was significantly larger in PC2 KD cells compared to SCR cells (Supplementary Fig. 1D). There was no alteration in baseline cytoplasmic Ca2+ or Ca2+ released from the ER stores following addition of ionomycin, an ionophore causing release of all ER Ca2+ stores (Supplementary Fig. 1E).
Figure 1: PC2 knockdown results in decreased cytoplasmic but increased mitochondrial Ca2+ signals.

A). Measurements of the cytoplasmic Ca2+ signal in response to 5 μM ATP (measured using GCaMP6F) in PC2 KD cells compared to SCR cells. B). Quantification of area under the curve of the GCaMP6F cytoplasmic Ca2+ amplitude. p < 0.0001 determined by Unpaired t test. n = 30-94 cells from at least 3 independent experiments. C). Example traces depicting mitochondrial Ca2+ measurements using the FRET sensor 4mitD3cpV in SCR cells (left) and PC2 KD cells (right) in response to 5 μM ATP. D). Averaged data of the baseline mitochondrial Ca2+ levels measured with 4mitD3cpV. Data determined not significant by Unpaired t test. E). Averaged mitochondrial maximum Ca2+ response after 5 μM ATP stimulus measured with 4mitD3cpV. p < 0.05 determined by Unpaired t test. SCR: n=3 individual experiments with 12 cells per experiment; PC2 KD: n=6 individual experiments with 27 cells per experiment.
Because ratiometric pericam has been shown to be sensitive to changes in pH32,33, these mitochondrial Ca2+ findings were corroborated using the pH-insensitive mitochondrial Ca2+ FRET sensor, 4mitD3cpV34,35 (Fig. 1C). There was no significant difference in the baseline mitochondrial Ca2+ (Fig. 1D). However, consistent with the pericam measurements, mitochondrial Ca2+ influx in response to ATP was significantly greater in PC2 KD cells than in SCR cells (Fig. 1E).
To confirm that our findings remained consistent in other cell lines, we utilized stable mouse kidney epithelial (mIMCD3) cells that were either transfected with and selected for clones expressing mutant Cas9 (IMCD3) or functional Cas9 and gRNA targeting PKD2 to genetically delete PKD2 (PC2 KO; Supplementary Fig. 1F). Cytoplasmic (gCaMP6F) and mitochondrial (mito-gCaMP6F) Ca2+ responses to ATP were measured in these cells and it was seen that, similar to the LLC-PK1 cells, PC2 KO cells had decreased cytoplasmic and increased mitochondrial Ca2+ responses compared to WT IMCD3 cells (Supplementary Fig. 1G,H).
PC2 is located at the MAM and interacts with VDAC, an outer mitochondrial membrane channel
The change in mitochondrial Ca2+ could be explained if PC2 KD cells exhibited increased physical tethering between the ER and mitochondria. To understand if tethering was influenced by the loss of PC2, we first sought to see if PC2 was present in crude mitochondrial fractions, which contain resident MAM proteins. Co-immunoprecipitation experiments demonstrated that endogenous PC2 was enriched in the crude mitochondrial fraction and was associated with the voltage-dependent anion channel (VDAC; Fig. 2A), the outer mitochondrial membrane protein responsible for Ca2+ uptake into the intermembrane space. These data agree with a previous study which identified VDAC as an interacting protein of PC2 by immunoprecipitation mass spectrometry of PC2 in the vasculature36.
Figure 2: PC2 resides in ER membranes close to mitochondria.
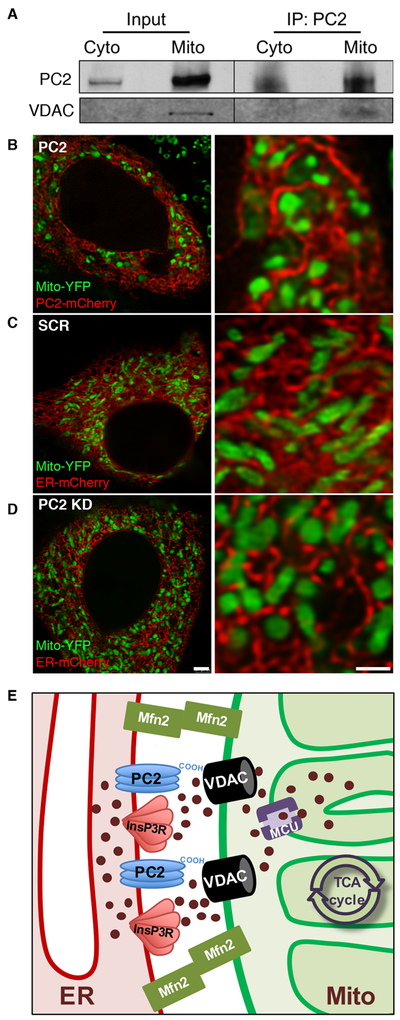
A). PC2 immunoprecipitates from cytoplasmic and MAM-containing crude mitochondrial fractions were immunoblotted for VDAC. B-D). Example images of live cells transfected with Mito-YFP and PC2-mCherry (B) or Mito-YFP and ER-mCherry in SCR (C) or PC2 KD cells (D) taken with STED super-resolution microscopy. Scale bar, 2.5 μm (left), 1 μm (right). n = 12-21 cells from at least 3 independent experiments. E). Proposed schematic of Ca2+-handling and tethering proteins at the MAM. PC2 and InsP3R reside on the ER membrane in close apposition to mitochondria to regulate ER Ca2+ release. The cytoplasmic C-terminal tail of PC2 spans the distance between the ER and mitochondria to interact with VDAC on the outer mitochondrial membrane. The MCU complex resides on the inner mitochondrial membrane, and MFN2 is a tethering protein that physically links the ER and mitochondrial membranes at the MAM.
We then determined if PC2 is present within mitochondria along with its localization in the MAM. Conventional confocal microscopy revealed areas of apparent co-localization between the ER and mitochondria as detected by mitochondrial-targeted yellow fluorescence protein (Mito-YFP) and ER-targeted mCherry (ER-mCherry) and quantified using Mandar’s Coefficient analyses (Supplementary Fig. 1A,B). However, with super-resolution STED microscopy, in which we obtained ~80 nm resolution with live cell imaging, a distinct reticular expression pattern was observed with mCherry-tagged PC2 (PC2-mCherry), consistent with PC2 adopting ER tubule localization (Fig. 2B). There was no indication that PC2 co-localized with Mito-YFP, and there was no loss of ER patterning with PC2 KD (Fig. 2B,C,D, and Supplementary Fig. 2C). These data suggest that intracellular PC2 is located on the ER membrane in close proximity to mitochondria, but not within mitochondria themselves. This interpretation is consistent with the MitoCarta 2.0 database, which does not list PC2 as a resident mitochondrial protein37. Together, these results present a picture of how these proteins of interest at the MAM coordinate to regulate Ca2+ transfer from the ER to mitochondria (Fig. 2E).
PC2 KD alters distribution of ER Ca2+ release channels in close proximity to mitochondria and increases mitochondrial Ca2+ uptake capacity
In addition to altering mitochondrial-ER tethering, redistribution of the Ca2+ channels at the MAM could also contribute to the differences between cytoplasmic and mitochondrial Ca2+. We therefore examined the MAM-containing mitochondria-enriched fraction for InsP3R isoform expression. The abundance of InsP3R3 in the mitochondrial fraction was significantly lower in PC2 KD cells than in SCR cells when normalized to VDAC, suggesting that PC2 modulates the composition of the Ca2+ release channels located at the MAM (Fig. 3A,B). In contrast, there was no change in InsP3R1 (Fig. 3C). Previous characterization of InsP3R isoform levels in whole cell lysate indicated no change in the overall amounts of InsP3R in PC2 KD cells38. The specific subtype of InsP3R is an important contributor to Ca2+ release dynamics, because InsP3R1 is more sensitive than InsP3R3 to modulation by both InsP3 and ATP levels39,40. As a result, more Ca2+ will be released for a lower concentration of stimulus when InsP3R1 is present.
Figure 3: PC2 knockdown modifies expression of ER Ca2+ release channels associated with the mitochondria.

A). Crude MAM-containing mitochondrial fractions from PC2 KD and SCR cells were immunoblotted for InsP3R3, InsP3R1, and VDAC. Two different example preparations are shown B). Quantification of InsP3R3 levels in crude mitochondrial fractions from SCR AND PC2 KD cells normalized to VDAC. p < 0.05 determined by Mann-Whitney U test. n = 4 independent experiments. C). Quantification of InsP3R1 levels in crude mitochondrial fractions from SCR and PC2 KD cells normalized to VDAC. Data determined not significant by Mann-Whitney U test. n = 4 independent experiments. D). MCU protein abundance in SCR and PC2 KD cells. Three different example preparations are shown. E). mRNA expression of MiCU2 and MCUb in SCR and PC2 KD cells. p < 0.05 determined by Unpaired t test. n = 4 independent experiments. F). Representative trace of Ca2+ uptake into mitochondria measured with Calcium Green 5N. 0.02% digitonin was added ~1000 seconds after the start of the assay to permeabilize the cell membrane. Each spike is the addition of 200 nmol calcium. G). Quantification of mitochondrial Ca2+ uptake in permeabilized SCR and PC2 KD cells. p < 0.0001 determined by Unpaired t-test. n = quadruplicate 96 wells averaged from 3 separate experiments.
We then measured the expression levels of the mitochondrial Ca2+ uniporter (MCU)9,10 because changes in MCU can affect mitochondrial Ca2+ uptake due to its role as the channel responsible for Ca2+ uptake into the mitochondrial matrix41. Whereas MCU expression was unchanged in PC2 KD cells (Fig. 3D, Supplementary Fig. 3A), the expression of mRNAs encoding the MCU inhibitors MiCU2 and MCUb was significantly lower in PC2 KD cells than in SCR cells (Fig. 3E). The expression of the mRNAs encoding MiCU1, MiCU3 and EMRE were unchanged (Supplementary Fig. 3B). This expression pattern is consistent with an increase in mitochondrial Ca2+ uptake capacity.
Mitochondrial Ca2+ uptake experiments in permeabilized cells (Fig. 3F) confirmed that mitochondria from PC2 KD cells took up more Ca2+ (Fig. 3G), consistent with a loss of inhibition due to decreased MiCU2 and MCUb. Together, the combined effects of the changes in the MCU complex coupled with the alterations in InsP3R distribution at the MAM help to explain the increased mitochondrial Ca2+ uptake observed in PC2 KD cells.
ER-mitochondrial tethering is increased with PC2 KD
Because our experiments indicated changes in the MCU complex and MAM-associated Ca2+ channels, we examined if these changes were accompanied by structural changes in mitochondria. Transmission Electron Microscopy (TEM) analysis revealed a significant increase in mitochondrial-ER tethering and mitochondrial morphological rearrangements in PC2 KD cells. Specifically, we found that the distance between the ER and mitochondria was decreased compared to SCR cells (Fig. 4A,B). In addition, mitochondria in PC2 KD cells had a larger area and a more circular morphology (Fig. 4C,D).
Figure 4: Ultrastructural analysis of PC2 KD cells reveals closer ER-mitochondrial distances and increased mitochondrial fragmentation.

A). Example TEM images from SCR and PC2 KD cells. Arrows denote example mitochondrial (m) distance to the ER. “n” denotes cell nucleus; ER denotes endoplasmic reticulum (ER). Area outlined in black is shown at higher magnification (bottom). Scale bars, 500 nm. B). Analysis of distance between mitochondria and their nearest ER, with each symbol representing a single calculated ER-mitochondrial distance. p < 0.0001 determined by Unpaired t test. C). Histogram of individual mitochondrial area as assessed by TEM images. D). Histogram of mitochondrial circularity as assessed by TEM images, where 1 represents a circle. n = 20 cells for each condition with 4 different regions imaged per cell.
PC2 KD cells have increased MFN2 expression and altered mitochondrial metabolism
Consistent with increased mitochondrial-ER tethering, we found increased abundance of MFN2, a mitochondrial-ER tethering protein, and increased overall mitochondrial content in PC2 KD cells (Fig. 5A,B, Supplementary Fig. 4A). This increase in mitochondrial density was observed in in 3D cultured cysts as well (Fig. 5C,D). We hypothesized that this increase in MFN2 contributed to the altered mitochondrial Ca2+ dynamics seen in PC2 KD cells, as modulation of MFN2 levels directly influences mitochondrial Ca2+ uptake42. To test this, we knocked down MFN2 in PC2 KD cells (Supplementary Fig. 4B). Mitochondrial Ca2+ was measured in response to ATP using mitochondrial-targeted gCaMP6F, and, consistent with our hypothesis, knockdown of MFN2 in PC2 KD cells rescued the mitochondrial Ca2+ response to levels similar to SCR cells (Fig. 5E,F). In addition, knockdown of MFN2 in PC2 KD cells restored the cytoplasmic Ca2+ response to ATP (Supplementary Fig. 4C).
Figure 5: PC2 KD increases mitofusin 2 expression and mitochondrial metabolism through altered Ca2+ dynamics.

A). Western blot of MFN2 protein abundance in SCR and PC2 KD cells. Four different preparations are shown. B). Quantification of MFN2 protein abundance in SCR and PC2 KD cells, normalized to Actin. p < 0.05 determined by Mann-Whitney U test. n = 4 biological replicates. C). 3D cultured SCR and PC2 KD cysts were stained for Phalloidin (red), DAPI (blue) and TOM20 (green), an outer mitochondrial membrane protein. D). TOM20 intensity normalized to cell area was quantified. Scale bar, 5 μm. p < 0.0001 determined by Unpaired t-test. E). Example average trace of mitochondrial Ca2+ responses to 5 μM ATP using mito-gCaMP6F in SCR, PC2 KD, and PC2 KD MFN2 KD cells. F). Quantification of maximum mitochondrial Ca2+ responses in SCR, PC2 KD, and PC2 KD MFN2 KD cells. p < 0.0001 determined by Unpaired t test. n = 30 or more cells from 3 independent experiments. G). Oxygen consumption rates (OCR) and maximal respiration capacity as revealed by oxygen flux mitochondrial stress test analyses in SCR and PC2 KD cells. p < 0.05 determined by Unpaired t-test. n = 3 independent experiments with 24 replicates in each experiment. H,I). Simultaneous measurements of mitochondrial Ca2+ using mito-GCaMP6F (green line) and mitochondrial membrane potential (red line) in SCR (H) or PC2 KD cells (I) in response to 5 μM ATP, 2 μM oligomycin and 2 μM FCCP. Data are averaged from 3-6 independent experiments representing 12-27 individual cells.
In 2D culture, PC2 KD cells turned the growth medium acidic more quickly than SCR cells (Supplementary Fig, 4D), suggesting an effect of PC2 KD on the cells’ metabolic status. To test the metabolic capacity of PC2 KD versus SCR cells, we measured the oxygen consumption rate (OCR) of cells subjected to a mitochondrial stress test and found a significantly higher OCR in PC2 KD cells under basal conditions, but lower OCR following mitochondrial uncoupling with the ionophore carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP, Fig. 5G). Similar results for basal conditions were obtained using an assay to measure ATP/ADP (Supplementary Fig. 4E) and a Clarke electrode to measure OCR (Supplementary Fig. 4F). Collectively, these data suggested that following PC2 KD, cells have an increased basal metabolic profile, but an overall loss of maximal respiratory capacity. These results are consistent with our data demonstrating increased mitochondrial Ca2+ transients with PC2 KD cells, because increased mitochondrial Ca2+ uptake is associated with enhanced metabolism through Ca2+-dependent regulation of matrix dehydrogenases43.
To confirm that altered mitochondrial Ca2+ and not mitochondrial membrane potential was responsible for these metabolic changes, we measured mitochondrial Ca2+ dynamics (using mitochondrial-targeted GCaMP6F [mito-gCaMP6F]) and inner mitochondrial membrane potential (using TMRE) in PC2 KD and SCR cells under the same treatment conditions as the mitochondrial stress test. In agreement with the hypothesis that mitochondrial Ca2+ is responsible for the observed changes in OCR, we determined that PC2 KD cells had enhanced mitochondrial Ca2+ uptake following the addition of ATP and oligomycin, but the change in mitochondrial Ca2+ following FCCP addition was less pronounced in PC2 KD cells than in SCR cells (Fig. 5H,I). Conversely, TMRE measurements showed no difference between SCR and PC2 KD cells.
Super-resolution microscopy analysis of mitochondrial dynamics reveals decreased mitochondrial movement with PC2 KD
Changes in mitochondrial-ER tethering contacts result in altered mitochondrial movement dynamics, which is closely correlated with changes in metabolism44. Although TEM can obtain a snapshot of the ultrastructure, it does not provide information as to how stable these inter-organellar contacts are. Using time-lapse super-resolution microscopy under conditions that did not induce differential mitochondrial ROS production (Supplementary Fig. 5), we tracked mitochondrial dynamics for 5 minutes (Fig. 6A, Supplementary Video 1) and found that the although mitochondria in PC2 KD and SCR cells traveled similar distances at equivalent velocities (Fig. 6B,C), PC2 KD mitochondria showed a significant decrease in vectorial displacement (Fig. 6D), suggesting reduced linearity in their movement. Examination of the PC2 KD cells revealed that many mitochondria adopted dynamic ring-like structures, which transitioned between a linear to circular conformation without directed movement along the cellular cytoskeleton (Fig. 6E, arrow). These data suggest that PC2 KD mitochondria, which spend more time in contact with particular MAM regions of the ER, enable privileged and prolonged Ca2+ communication between the ER and mitochondria.
Figure 6: PC2 expression alters mitochondrial dynamics.

A). Single time point images of SCR (top left) or PC2 KD (bottom left) cells transfected with ER-mCherry and Mito-YFP. Scale bar, 2.5 μm. Tracks of individual mitochondrial movement from frame to frame are depicted, with each color representing a different individual mitochondrion over time. B). Averaged total distance traveled by mitochondria over a 5-minute period. C). Averaged velocity of mitochondria over a 5-minute period. Data in B and C are representative of at least 12 mitochondria per cell. Each symbol is representative of an individual cell from at least 3 different preparations. D). Absolute direction in x- and y-axes traveled by each mitochondrion over a 5-minute period. Each symbol is representative of 5-10 cells, with 87-267 mitochondria per cell. E). Time-lapse example of static circular mitochondria found exclusively in PC2 KD cells (white arrow).
The PGC1α-CREB axis is activated in PC2 KD cells
We wanted to further explore the mechanism by which MFN2 expression is increased to affect these observed mitochondrial changes. We tested the levels of PGC1α, an established upstream regulator of MFN2 expression, and found a >40-fold increase in the transcription factor in PC2 KD cells (Fig. 7A). As PGC1α is a known activator of mitochondrial biogenesis, its upregulation is also consistent with both the increased mitochondrial density observed in PC2 KD 3D and 2D cultures (Fig. 5C,D, Supplementary Fig. 4A), and with the increased expression of MFN2 (Fig. 5A,B). Increased expression of PGC1α is stimulated through CREB activation45, and in ADPKD kidneys, cAMP levels are elevated due to increased vasopressin receptor signaling and cAMP production46,47,48. Consistent with this literature, phosphorylated CREB (pCREB), which is activated through cAMP signaling, was significantly increased in the PC2 KD samples (Fig. 7B).
Figure 7: PC2 KD increases mitochondrial biogenesis through the PGC1α-CREB axis.

A). mRNA expression of PGC1α in SCR and PC2 KD cells. p < 0.05 determined by Unpaired t test. n = 4 independent experiments. B). Immunoblot for phosphorylation of the transcription factor CREB in SCR and PC2 KD cells. Four different example preparations are shown. Right: quantification of pCREB normalized to CREB in SCR and PC2 KD cells. p < 0.05 determined by Mann-Whitney U test. n = 4 independent experiments.
Although PGC1α was upregulated, there was no change in the expression of mRNAs encoding alternate mitochondrial fusion or other tethering proteins, OPA1 or Mitofusin 1 (MFN1, Supplementary Fig. 6A). Similarly, Sigma1 Receptor (Sigma1R), whose abundance is increased under conditions of ER stress and which can act as a tethering protein between the mitochondria and the ER49, showed no significant change in protein expression (Supplementary Fig. 6B). These results suggest that the increased abundance of MFN2 facilitates increased tethering between the ER and mitochondria in PC2 KD cells. In addition, dysregulated levels of PGC1α have been shown to render cells susceptible to hyper-fragmentation of mitochondria and altered mitochondrial dynamics50, as seen in PC2 KD cells (Fig. 4D, Fig. 6A,D,E).
Tissue-specific targeting of MFN2 in cystic mice restores Ca2+ signaling and decreases proliferation
As we had observed that diminishing the tethering between the ER and mitochondria reversed the increased mitochondrial Ca2+ signaling in PC2 KD cells (Fig. 5E,F, Supplementary Fig. 4B,C), we asked whether this manipulation would also be effective in cystogenic signaling pathways present in animal models. We therefore created MFN2-specific siRNA expressed under a Cre promoter (pSico lenti-viral vector51). The addition of two siRNA sequences against MFN2 was found to be effective in knocking down MFN2 in collecting duct cells from Pkd2F/F mice with adenoviral Cre added ex-vivo, (Fig. 8A, Supplementary Fig. 7A). Pkd2F/FPkhd1Cre mice (which develop cysts from birth due to PC2 knockout in the collecting ducts, Supplementary Fig. 7B) were transduced with the lentivirus (LV) through retro-orbital injection at 3 weeks of age. MFN2 abundance was relatively uniform across tubules in Pkd2F/F control mice (Fig. 8B, left). MFN2 abundance was increased in the cystic kidneys of Pkd2F/FPkhd1Cre mice (Fig. 8B, middle), and was reduced by LV-siMFN2 transduction (Fig. 8B, right).
Figure 8: Reduction of MFN2 expression in murine cystic cells restores mitochondrial Ca2+ signaling and hyper-proliferation.

A). PC2 and MFN2 protein abundance in collecting duct Pkd2F/F cells after the addition of adenoviral Cre ex vivo and lentiviral transduction of two different siRNAs against MFN2. Actin is used as a loading control. Representative of 2 independent experiments (see also Supplementary Fig. 7A). B). Expression of MFN2 (green) in the tubules of Pkd2F/F mice (left), Pkd2F/FPkhd1Cre cystic tubules (middle), and Pkd2F/FPkhd1Cre cystic tubules 4 weeks after injection of siMFN2 (right). Representative of 4 different mice per group. Scale bar, 10 μm. C). Mitochondrial Ca2+ changes upon addition of 5 μM ATP. Traces from a single coverslip as a representation of the data is shown. Data were collected from 4 separate coverslips with at least 20 cells per coverslip, representing 2-3 different mice per genotype. D). Ki67 staining (green) for proliferating cells in kidney sections from Pkd2F/FPkhd1Cre mice (middle) and siMFN2-transfected Pkd2F/FPkhd1Cre mice (right) (see also Supplementary Fig. 8C,D) compared to Pkd2F/F mice (left). Sections from additional mice are presented in Supplementary Fig. 8D. Representative of 2-3 different mice per group. Scale bar, 10 μm.
The cystic collecting duct cells (Supplementary Fig. 8A) from Pkd2F/FPkhd1Cre recapitulated several of the features observed in the LLC-PK1 PC2 KD cells. In cells transfected with Mito-YFP and ER-mCherry, the mitochondria were more circular in cystic cells, a phenotype that was corrected by LV-siMFN2 treatment (Supplementary Fig. 8B). Compared to Pkd2F/F control cells (Fig. 8C, left), mitochondrial Ca2+ signals following addition of ATP were enhanced in cystic cells (Fig. 8C, middle) and restored to control values by LV-siMFN2 transduction (Fig. 8C, right).
We also examined cystic parameters of cell proliferation and apoptosis after siMFN2 KD. Three weeks of siMFN2 KD resulted in a marked reduction in cell proliferation in the LV-siMFN2 treated mice that was comparable to Pkd2F/F mice, as measured by Ki67 staining (Fig. 8D, Supplementary Fig. 8C,D). However, apoptosis measured through caspase-3 activation did not differ between cystic and non-cystic cells (Supplementary Fig. 8E).
Cystic kidneys from ADPKD patients have increased mitochondrial density and MFN2
We examined kidney sections from non-cystic (normal) and ADPKD diagnosed patients. In kidney sections from normal individuals, MFN2 and the mitochondrial protein TOM20 were increased in the distal convoluted tubule and collecting ducts (stained by the lectin Dolichos biflorus agglutinin, DBA) compared to other more proximal nephron regions (Fig. 9A,B, gray compared to red bars, Supplementary Fig 9A). Moreover, MFN2 and TOM20 staining localized to the basolateral membrane in DBA-positive tubules (Fig. 9A, bottom, 9B, Ap. compared to BL gray bars), consistent with previous findings52. In ADPKD patient samples, extensive fibrosis was found, and cysts were generally DBA positive (Fig. 9C). MFN2 and TOM20 staining were increased in cyst-lining cells (Fig. 9D, blue bars; Supplementary Fig. 9B). Note that non-cystic tubules in ADPKD patients had MFN2 and TOM20 levels comparable to the WT control samples (Fig. 9A, bottom, compared to Fig. 9C, bottom left; ratio of MFN2 to TOM20 is presented in Supplementary Figs. 9C,D). These data indicate that the mitochondrial density and the MAM-tethering protein MFN2 are increased in cysts from human subjects with ADPKD.
Figure 9: Renal cyst-lining cells from human ADPKD patients have increased MFN2 and mitochondria.

A). Human kidney samples from normal patients were stained with antibodies against TOM20 (green) and MFN2 (red), and counterstained with the lectin DBA (blue) as a marker for collecting duct tubules. Scale bar, 50 μm (top), 5 μm (bottom). B). Quantification of MFN2 staining normalized to area in normal human kidney, separated into DBA lectin positive tubules (gray bars) and DBA lectin negative tubules (red bars). Within each group, MFN2 staining was further separated by its intensity in the Whole Tubule (W Tb.), Apical localization (Ap.), and Basolateral localization (BL). p < 0.05 determined by Mann-Whitney U test. C). Human kidney samples from ADPKD patients were stained with antibodies against TOM20 (green) and MFN2 (red), and counterstained with lectin DBA (blue). Scale bar, 50 μm (top), 5 μm (bottom left, bottom right) D). Quantification of MFN2 staining in DBA positive tubules (gray bars), DBA negative tubules (red bars) and cyst lining epithelium (blue bars) from ADPKD patients. p < 0.05 determined by Mann-Whitney U test. Quantification is of 5 images per group (5 lectin positive, 5 lectin negative, 5 cystic samples) and 2 tissues of each (2 normal human kidney and 2 ADPKD). W Tb: Whole tubule; AP: apical side; BL: basolateral side; W Cy.: Whole cyst.
Discussion
Here we establish that PC2 serves to limit Ca2+ signaling to mitochondria by modulating proteins located at the MAM and by increasing mitochondrial biogenesis and mitochondrial-ER tethering through PGC1α. Loss of PC2 resulted in enhanced mitochondrial Ca2+ uptake, mitochondrial bioenergetics, and increased mitochondrial-ER tethering through increased MFN2 abundance. These findings were also present in cysts from human ADPKD patients and kidney tubule cells from cystic mice. Knockdown of MFN2 restored mitochondrial Ca2+ signaling, morphological changes, and hyperproliferation in cystic cells. Collectively, these findings demonstrate that PC2 is an important regulator of mitochondrial function, which is relevant in pathological settings.
We propose that PC2 modulates the ER protein composition associated with mitochondria and can conduct this function by directly interacting with VDAC. The InsP3R is the main intracellular release channel that provides Ca2+ to the mitochondria, and our data do not point to PC2 being another intracellular release channel for the mitochondria, but rather a regulator of Ca2+ entering the mitochondria. This can be achieved two ways: (i) by directly linking mitochondria to the ER, and (ii) by modulating InsP3R subtype expression in the MAM. Indeed, the extensive C-terminal tail of PC2 is long enough to span the 10–20 nm distance between the ER and mitochondria30,53 to interact with VDAC, and, in addition, increased MFN2 via CREB-mediated upregulation helps to facilitate more sustained ER-mitochondrial contacts. Our finding that the expression of InsP3R3 is decreased in the PC2 KD MAM is notable, as InsP3R3 is considered the dominant InsP3R at the mitochondrial-ER junction11. Because PC2 is not only found in the kidney but in many other organs including the vasculature36 and cardiomyocytes54, our data suggest that PC2 expression in other cell types may well affect cytoplasmic and mitochondrial Ca2+ signals independent of and unrelated to cyst formation.
A second way by which PC2 loss modulates mitochondrial signaling is by increasing PGC1α expression through pCREB activation, which leads to enhanced mitochondrial biogenesis and increased abundance of the ER-mitochondrial tethering protein MFN2. The functional role of MFN2 is complex, as loss of MFN2 has been associated with loss of ER-mitochondrial contacts26-28,55-60, whereas other reports suggest that loss of MFN2 results in increased ER-mitochondrial contacts41,61. Although static EM studies have been previously used to assess ER-mitochondrial contacts, here we combined EM data with super-resolution live-cell imaging of organelles to demonstrate a reduction of vectorial movement of mitochondria upon PC2 knockdown, indicating enhanced long-term contacts between mitochondria and ER. Our data suggest that changes in MAM proteins, along with altered mitochondrial morphology and movement, enable permissive uptake of Ca2+ into mitochondria, which leads to a heightened metabolic state (Fig. 5G, Supplementary Fig. 4D,E,F), as would be expected in cystic tissue. Consistent with this, human cysts also had increased MFN2 expression and, in our cell culture and murine cystic models, decreasing MFN2 expression restored both mitochondrial and cytoplasmic Ca2+ signaling. These data suggest that mitochondrial-ER tethering through MFN2 is critical for balancing the amount of ER Ca2+ released into the cytoplasm versus into mitochondria. Because mitochondrial signaling was restored through MFN2 KD alone, the expression of MiCU2 and MCUb are likely reciprocally regulated by the altered intracellular Ca2+ dynamics that can then influence transcriptional regulation. As such, cAMP-mediated pCREB/PGC1α activation in PC2 KD cells may ultimately impact mitochondrial function via increased abundance of MFN2, altering the path of ER Ca2+ release to preferentially be taken up into mitochondria, which is then exacerbated by transcriptional increases of MiCU2 and MCUb. Nevertheless, despite these corrections and the reduction in proliferation with MFN2 knockdown, no gross reduction in cysts was observed. However, as the nascent renal cysts had already developed by the time the LV-siMFN2 was introduced to the mice at 3 weeks of age, pre-treatment may have a larger impact on cyst formation.
Although we have focused on PC2, further studies are needed to examine if mitochondrial Ca2+ and dynamics are likewise disrupted in PKD1-associated cysts. Unlike PC2, PC1 does not appear to be present on the ER membrane, and thus a direct perturbation of mitochondrial Ca2+ uptake via ER Ca2+ release would only be likely to occur if the ER-mitochondrial membrane trafficking of PC2 and/or InsP3R distribution is altered by PC1. Because PC2 regulates the abundance and trafficking of PC162-65, it is likely that mutations to PC1 can also reciprocally alter expression and function of PC2 at mitochondrial-ER contacts, as previously reported in the cilia66. Indeed, it has been reported that a cleavage product of the PC1 C-terminal tail can enhance mitochondrial respiration and alter morphology when over-expressed in heterologous systems67. However, how this cleavage product is implicated in cyst formation or mitochondrial dysfunction in ADPKD, when the polycystins are absent or mutated, remains to be discovered. Morphological differences seen in mitochondria from kidneys of ADPKD patients and renal epithelial cells isolated from Pkd1ko/ko mice fit well with our finding that i) PC2 KD cyst-modeling cells exhibit altered metabolic capacity, and ii) that mitochondria from these cells, along with ADPKD kidneys, show a high degree of fragmentation. This fragmentation and decreased mitochondrial network formation has been noted in cystic tissue derived from PC1-deficient rats as well68. Our finding that mitochondria from PC2 KD cells have a decreased overall metabolic capacity accords well with this phenotype of hyper-fragmentation and circularity, as mitochondria that are unable to form elongated intracellular networks become dysfunctional and consequent targets for mitophagy, thus necessitating increased mitochondrial biogenesis69. However, although morphological differences appear to be a common phenotype between both PKD1- and PKD2-caused ADPKD development, PGC1α-mediated mitochondrial density differs depending on whether cystic cells derive from PKD1 or PKD2 loss-of-function. Whereas kidney tissue from PC1-deficient mice show a decrease in PGC1α expression levels68, our data demonstrate a significant increase in PGC1α in PC2 KD cells. These different phenotypes may be a result of the metabolic differences between PC1- and PC2-mediated cyst development, as, in contrast to our results in PC2 KD cells, Pkd1−/− cells have been shown to have decreased basal metabolic rates18, which may also be influenced by the lack of the PC1 cleavage product to enhance mitochondrial function. In addition, our human data demonstrate altered ER-mitochondria contacts and mitochondrial content in cystic tissue. Because PKD2 mutations contribute to a lower frequency of ADPKD (~20%) than PKD1 mutations, the majority of the human tissue samples are presumed to have PKD1 mutations. Therefore, our findings accord well with the idea that mitochondria-ER communication is a common mechanism contributing to altered metabolism observed in ADPKD 15.
Our finding that PC2 levels alter mitochondrial function has implications for cyst-promoting cell proliferation pathways associated with loss of polycystins, as evidenced by the decreased Ki67 staining in the MFN2 knockdown cyst samples. Cystic cell proliferation is due to a combination of elevated cAMP and decreased Ca2+ signaling, which collectively initiate a growth-promoting BRAF signaling pathway 70,71. Enhanced mitochondrial function could also aid in cellular proliferation when combined with BRAF pathway activation, because cell proliferation requires high ATP production, which, under the hypoxic conditions of cysts, would require a shift to anaerobic glycolytic pathways 15. Increased ATP production can inhibit AMPK, which may activate the cell survival-associated mTOR pathway. A component of the mTOR signaling pathway, Mammalian TOR Complex 2 (mTORC2), has also been shown to be physically present within the MAM 72,73. These pathways, along with a cAMP signaling pathway, are enhanced in ADPKD, making them possible targetable therapeutic pathways73-76. Here we found that in a cystic mouse model, knockdown of MFN2 markedly reduces cell proliferation, uncovering this linker protein as a potential therapeutic target for patients with PKD2 mutations to restore homeostatic calcium and the resulting altered signaling pathways in these cells.
In conclusion, we describe a new functional role for PC2 in the regulation of physical contacts between the mitochondria and the ER. Moreover, these contacts are perturbed in ADPKD, similar to several other disease states77,78. The contribution of PC2 to mitochondrial function implicates a pathway involved in cystogenesis in ADPKD. Finally, the wide tissue distribution of PC2 raises the possibility that PC2 may contribute to disease states in other tissues where mitochondria and cellular metabolism are disrupted.
Materials and methods
Cell culture and creation of stable knockdown cells
shRNA against PC2 and MFN2 were designed to be specific to the pig sequence. The PC2 shRNA constructs were previously described and characterized38,79 These were put into a psiRNA vector (InvivoGen, San Diego, CA), and LLC-PK1 cells stably transfected with the selection agent G418. G418 was maintained in the media at 2.5 mg/ml, and removed 2 days prior to experimentation, as previously described 38,79. All cells were maintained in DMEM supplemented with 10% fetal bovine serum and kept at 37°C in 5% CO2. mIMCD3 control and PC2 KO cells were obtained from the laboratory of Dr. S. Somlo (Yale University) and maintained in DMEM/F12 supplemented with 10% fetal bovine serum and kept at 37°C in 5% CO2.
Extracellular flux analysis
The mitochondrial respiration capabilities of LLC-PK1 SCR and PC2 KD cells were tested using a Seahorse XF96 Extracellular Flux Analyzer (Agilent Technologies). Cells were plated in 96-well plates at a density of 8×10^4 cells/well 24 hours prior to the assay. Two hours before analysis, cells were washed twice with, and then changed to Seahorse Assay Medium (Agilent Technologies) containing 4.5g/L D-glucose, 2mM L-glutamine and 1mM sodium pyruvate before being placed in a CO2-free incubator at 37˚C. Basal OCR measurements were taken before the sequential addition of 2μM Oligomycin-A, 2μM FCCP, and 1μM Rotenone to measure ATP production and proton leak, maximal mitochondrial respiration, and non-mitochondrial respiration, respectively. Following completion of Seahorse analysis, OCR results were normalized to cell number by lysing cells and quantifying the dsDNA in each well using a Quant-iT PicoGreen dsDNA Assay Kit (ThermoFisher) according to the manufacturer’s instructions. Experiments were performed in quadruplicates or greater.
Clarke electrode:
1× 10^6 cells were trypsinized and resuspended in respiration buffer (in mM: 120 KCl, 5 KH2PO4, 10 Tris-HCl , 3 MgCl2, 1 EDTA, pH = 7, modified from80) and permeabilized with 0.03% wt/vol digitonin. Cells were placed in a Clarke electrode oxygraph and allowed to equilibrate at 37˚C. The following reagents were added sequentially with Hamilton syringes: ADP (2–8 mM), pyruvate (5 mM) and malate (10 mM), and oligomycin (6 μM, used to block ATP Synthase).
ATP/ADP assay
Cells were plated in 96-well plates and ATP/ADP ratios measured according to manufacturer’s instructions (ADP/ATP Ratio Assay Kit, Sigma-Aldrich). Experiments were performed in quadruplicates.
Mitochondrial enrichment, Western Blot and immunoprecipitation
Cells from two T75 cm flasks were combined, and the cells spun down. Crude mitochondrial extracts were obtained by using a mitochondrial extraction kit (ThermoFisher, Waltham, MA). The final spin was conducted at 1000 rpm for 15 min at 4°C to obtain a cleaner mitochondrial fraction with reduced lysosome contamination. Mitochondria were lysed in 2% Chaps with Tris buffer and the protein concentration measured via BCA Assay (ThermoFisher).
For immunoprecipitation experiments, 100 μg of protein was added to pre-cleared A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) and allowed to bind with 1–2 μg of antibody overnight at 4°C. Antibodies used were: VDAC (Abcam, Cambridge, UK) and PC2 (gift from Dr. Y. Cai, Yale University). The adhered protein was removed from the beads by boiling in loading buffer and reducing agent for 10 min after extensive washing of the beads (4 times) with lysis buffer.
Samples for all Western Blot analyses were loaded on 4–12% gradient gels (NuPage gels, Life Technologies) and run with either MES or MOPS buffers. Protein was transferred to PVDF membranes by wet transfer. After blocking in 5% milk, primary antibodies were applied overnight.
Antibodies
Antibodies for Western Blot and immunohistochemistry were as follows: InsP3R1, InsP3R3 (BD Bioscience), PC2 (gift of Dr. Y. Cai, Yale University), VDAC (Abcam), mitochondrial Ca2+ uniporter (MCU, Cell Signaling Technology, Danvers, MA), MFN2 (Cell Signaling Technology), TOM20 (Santa Cruz Biotechnology), phospho-CREB and CREB (Cell Signaling Technology), Tubulin (Abcam). VDAC and TOM20 were used to assess mitochondrial quality and purity. β-Actin (Abcam) was used as a loading control and to assess cytoplasmic contamination of mitochondrial samples. For Western Blot analyses, HRP-conjugated biotin-labeled secondary antibodies were used and detected with West Dura Extended Duration Substrate (ThermoFisher).
Cytosolic Ca2+, mitochondrial membrane potential and mitochondrial ROS measurements
Cells were transfected with GCaMP6F (Addgene81) and imaged the next day. The mitochondrial membrane potential (Δψm) was assessed with 10 nM of the mitochondrial voltage-sensitive dye tetramethylrhodamine, methyl ester (TMRE, Life Technologies)82. After loading for 15 min at 37°C, the media was exchanged to imaging buffer without TMRE, and cells left to equilibrate at RT for 15 min. The imaging buffer consisted of: 1.25 mM CaCl2, 19.7 mM Hepes, 4.7 mM KCl, 1.2 mM KH2PO4, 1 mM MgSO4, 130 mM NaCl, 5 mM dextrose, pH = 7.2 at RT. Cells were imaged using the excitation wavelengths 488 nm (GCaMP6F) and 561 nm (TMRE), and average florescence intensities over 30 min acquired. As a positive control for loss of Δψm, cells were treated with 50 μM FCCP (Life Technologies) to depolarize mitochondria at the end of the experiment. Cells were imaged at RT on a Leica SP5 microscope with a 63x 1.4 NA oil objective and 2.5x zoom. Fluorescent intensities were quantified by a combination of Leica software TCS5 software and ImageJ (NIH).
For measurement of superoxides generated by mitochondria, cells were incubated with MitoSOX (Life Technologies) according to manufacturer’s recommendations. Cells were then washed and imaged.
Mitochondrial Ca2+ imaging experiments
Cells were transiently transfected with the ratiometric indicator, pericam, directed to the mitochondria. Cells were imaged using dual excitation at both 380 nm and 480 nm (Lambda DG-4, Sutter Instruments, Novato, CA), and images acquired with an Orca camera (Hamamatsu Photonics, Middlesex, NJ) with Metamorph software (Axon Instruments, Sunnyvale CA). Mitochondrial Ca2+ transients were elicited by the addition of 1 μM ATP. Data was analyzed using PRISM software (GraphPad Software, La Jolla, CA).
For FRET imaging experiments of matrix Ca2+, cells were transiently transfected with pcDNA-4mitD3cpV (a gift from Amy Palmer and Roger Tsien; Addgene plasmid #3632435). FRET emission ratios (535 nm/485 nm; 440 nm excitation) were acquired every 5 seconds using fluorescence ratio imaging systems running MetaFluor software (Molecular Devices). Cells were stimulated with 5 μM ATP followed by 5 μM ionomycin and imaged using Nikon TE200 or TE2000-U inverted fluorescence microscopes equipped with a QuantEM 512 camera (Photometrics, Tuscon, AZ) or an ORCA ER camera (Hamamatsu Photonics), respectively. Data were analyzed using MetaFluor software.
For mito-GCaMP6F experiments (a gift from Diego Stefani), cells were transfected with mito-GCaMP6F. Cells were imaged using sequential excitation at 488 nm, and images acquired with emission bandwidth of 501–555nm. Images were acquired every second on a Leica SP5 confocal microscope with a 40x objective lens. 5μM ATP was added to stimulate mitochondrial Ca2+ signals, followed by oligomycin and FCCP.
Super-resolution microscopy
Cells were plated in 8-well, 1.5 mm-thick cover glass chambers (LabTek, Rochester, NY) and transfected the day before the experiment. Cells were imaged on a Leica SP8 equipped with a STED depletion laser (Heidelberg, Germany). Cells were imaged with a white light laser at 488 and 568 nm in sequential line mode, and a 740 nm STED depletion laser used with 100x oil objective and 2–4x zoom. For time-lapse imaging, cells were imaged using bi-directional scanning and each frame acquired approximately 45 seconds apart to minimize photobleaching. Images were then subjected to deconvolution using Huygens software (SVI, Hilversum, Netherlands).
mRNA analysis
mRNA was extracted from cells grown to confluency using a RNeasy kit and QiaShredder (Qiagen, Hilden, Germany), and reverse transcribed to cDNA using Multiscribe (Life Technologies) and random oligomers as primers (Applied Biosystems, Foster City, CA). For real time RT-PCR, 20 ng of cDNA was used as transcript in a reaction with POWER SYBR green MasterMix (Life Technologies) on a 7500 Fast machine (Applied Biosystems). Fold change in mRNA transcript levels was determined by using the 2-ΔΔ CT method. Actin was used as control.
Measurement of mitochondrial DNA
DNA was isolated from cultured cells using a DNeasy blood and tissue kit according to manufacturer’s instructions (Qiagen). 20 ng of DNA was used as transcript with POWER SYBR green MasterMix (Life Technologies) on a 7500 Fast machine (Applied Biosystems). Primers against the porcine sequences of mitochondrial genes mtATP6 (F 5’: CTACCTATTGTCACCTTA; R 5’: GAGATTGTGCGGTTATTAATG) and mtATP8 (F 5’: ATCTACATGATTCATTACAAT; R 5’: CTATGTTTTTGAGTTTTGAGTTCA) were used. Primers against the DNA sequence of myosin were used to measure genomic DNA (F 5’: TTGTGCAAATCCTGAGACTCAT; R 5’: ATACCAGTCCCTGGGTTCAT).
Image analysis
For the calculation of TOM20 or VDAC staining, ImageJ was used for image processing. The ImageJ plugin MTrackJ was used to calculate mitochondrial movement 83. For analysis of time-lapse images, a different investigator coded the image sequences so that the analysis was done in a blinded manner. For circularity measurements, a mask of the mitochondria was created in ImageJ (NIH). Huygens software package analysis was used to analyze for co-localization.
Transmission Electron Microscopy
SCR and PC2 KD cells were plated on glass coverslips and allowed to grow to 90% confluency before fixing with 4% glutaraldehyde. Cells were incubated in 1% osmium tetroxide and dehydrated in increasing ethanol concentrations, then embedded in Durcupan resin. Ultrathin sections were cut on a Leica Ultra-Microtome, collected on Formvar-coated single-slot grids, and analyzed with a Tecnai 12 Biotwin electron microscope (FEI, Hillsboro, OR). The investigator collecting the TEM images was blinded to the identity of the samples.
Mitochondrial Ca2+ uptake assay
Cells were plated in a 96-well plate and grown to confluency. The protocol broadly followed previously published studies 84. Briefly, the media was changed to a potassium buffer (125 mM KCl, 2 mM K2HPO4, 1 mM MgCl2, 20 mM Hepes, 5 mM glutamate and 5 mM malate, pH = 7.0) with 0.1 μM Calcium Green 5N. After baseline measurements, digitonin (0.02%) was added to permeabilize cells, followed by the serial addition of 200 nM CaCl2 solution in the same potassium buffer. Measurements were conducted on a BioTek spectrophotometer, and experiments were conducted in quadruplicate. Data was collected using Gen 5 software.
Human tissue
ADPKD and normal human kidney tissues were obtained by the PKD Biomaterials Core at the University of Kansas Medical Center. The use of these tissues for research has been approved by the IRB. Tissues were fixed with 4% paraformaldehyde at 4° C overnight, embedded in paraffin and 5 μm sections were cut for immunofluorescence. Following deparaffinization and rehydration, antigen retrieval was performed by incubating the sections in a steamer for 20 min in sodium citrate buffer (10 mM Tri-sodium citrate, 0.05% Tween-20, pH = 6.0). Sections were then quenched of autofluorescence, blocked and permeabilized, and incubated with anti-TOM20 (Cell Signaling) and anti-MFN2 (Cell Signaling) antibodies. Tissues were mounted in ProLong Diamond Antifade Mountant (Life Technologies), counterstained with Dolichos biflorus agglutinin, (DBA) and imaged with 100× oil-immersion lenses on a Leica SP8 STED Microscope using Leica LAS X software (Heidelberg, Germany). To enable comparison between samples, the same laser power, gain and acquisition settings were used.
Animal studies
Pkd2fl/flPkhd1Cre mice were obtained from the laboratory of Dr. S. Somlo (Yale University). Animals were housed under a 12-h light/dark cycle and genotyped at 3 weeks of age with primers described previously 85,86. Pkd2fl/fl littermates were used as controls. All animal experiments were performed in a blinded manner. The Yale University Animal Ethics committee (IACUC) approved the animal housing conditions and experimental procedures conducted in this study.
Primary culture of collecting duct cells
Kidneys were isolated from isoflurane-anesthetized mice after perfusion with 0.9% NaCl. The kidney capsule was removed, and the tissue cut into small pieces. The tissue was incubated at 37°C under continuous agitation (220 rpm) for 1.5–2h with a mixture of hyaluronidase (1 mg/mL) and collagenase (2.2 mg/mL) for protease digestion. The cell suspension was washed 3 times with PBS and incubated at RT for 30 min with magnetic beads (Dynabeads 11047 – Life Technologies) previously coated with Biotinylated Dolichos Biflorus Agglutinin (DBA). The cells were washed 3 times with Isolation buffer (DPBS supplemented with 0.1% BSA and 2 mM EDTA, pH = 7.4), suspended and plated in DMEM supplemented with 10% FBS and 1% Pen/Strep.
Lentiviral shRNA construction
Two different shRNA against mouse MFN2 was constructed using Broad Institute software, and cloned into pSICO (Addgene), one beginning at base pair 2009, the other at 3355. A scrambled sequence was also constructed. The lentivirus was then produced by co-transfecting the pSICO vector harboring the siRNA sequence with the following packing, Res and Pol plasmids: pRSV-Rev, pMDLg/pRRE, pMD2.G. Transfection was achieved using polyethylenimine, and the supernatant containing the virus harvested at 48 and 72 h afterwards. The effectiveness of the lentivirus was tested on HEK cells to determine titer.
The virus was injected in to 3-week-old mice via retro-orbital injection. Animals were sacrificed 4 weeks after injection, and the kidneys removed for histological analysis and the cells isolated for experiments.
Statistical analysis:
For cell-based experiments, data were calculated from individual cells and from multiple preparations. Where appropriate, one-way ANOVA with multiple comparisons or Students T-test were applied. For population-based experiments (e.g.: Western Blots), multiple experiments and samples were analyzed and subjected to nonparametric Mann-Whitney U tests. Data are presented as the averaged mean with standard error of the mean (SEM). In all experiments, p<0.05 was considered to be statistically significant. * represents p<0.05, ** represents p<0.01, *** represents p<0.001.
Supplementary Material
Fig. S1. PC2 KD and PC2 KO result in increased mitochondrial Ca2+ signals.
Fig. S2. PC2 resides in ER membranes close to mitochondria.
Fig. S3. PC2 KD does not alter the expression of MCU or multiple MCU complex components.
Fig. S4. MFN2 KD rescues Ca2+ signaling in PC2 KD cells and PC2 KD alters mitochondrial metabolism and density.
Fig. S5. Super-resolution STED microscopy does not induce differential mitochondrial ROS production.
Fig. S6. PC2 KD does not alter the expression of alternative ER-mitochondria linkers.
Fig. S7. LV-siMFN2 is effective at knocking down MFN2 in cells from Pkd2F/FPkhd1Cre mice, which develop kidney cysts from birth.
Fig. S8. LV-siMFN2 treatment rescues multiple effects of PC2 deficiency in cystic cells.
Fig. S9. Mitochondrial density is increased in cyst-lining cells from ADPKD kidneys.
Video S1. Super-resolution movie of organelle dynamics.
Acknowledgements:
We thank Dr. Michael Forte (Vollum Institute, OHSU) for the ratiometric pericam construct and Dr. Stefan Somlo and Ming Ma (Yale University) for the Pkd2F/F and Pkhd1Cre mice. We thank Dr. Diego Stefani (University of Padua) for the mito-GCaMP6F construct. Helpful discussions with Drs. Lawrence H. Young, Vlad G. Zaha, Marcos A. Carpio, Samuel G. Katz, John Hwa, Kisuk Min, Anton M. Bennett, Kathleen Martin and Gerald Shadel (Yale University) are acknowledged. We thank Lily Nguyen (Yale University) and Dr. Silvana Curci (VA, Harvard Medical School) for technical assistance and advice. Dr. Klara Szigeti-Buck (Yale University) is thanked for the TEM sample preparation and imaging and Nikki Mikush (Yale University) for technical expertise with echocardiograms. We thank Dr. Sue Kaech and Jun Low (Yale University) for use of the Seahorse Flux analyzer. We thank Dr. John Hwa and Zainab Jaji (Yale University) for use of the spectrometer, and Dr. Anton M. Bennett for the use of the Clark electrode.
Funding: We acknowledge use of the Yale Cell Biology Microscopy Core (NIH grants 5P30DK034989 and OD020142). Federal and NIH grant support is acknowledged: K99 DK101585 (IYK), 5P01DK057751 and P30DK090744 (BEE), 1F31EB018718 (EPK), VA-ORD 1 I01 BX000968–01 and UL1 TR001102 (AMH), and R24DK106743 (DLK). IMCD3 WT and PC2 KO cells were created by members of the George M. O’Brien Kidney Center at Yale (P30 DK079310). The PKD Biomaterials Core is part of the Kansas PKD Core Center, NIH P30 DK106912 (DPW).
Footnotes
Competing Interests:
The authors declare that they have no competing interests.
References
- 1.McCormack JG, Halestrap AP & Denton RM Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70, 391–425 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Cardenas C et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283, doi: 10.1016/j.cell.2010.06.007 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayashi T, Rizzuto R, Hajnoczky G & Su TP MAM: more than just a housekeeper. Trends Cell Biol 19, 81–88, doi: 10.1016/j.tcb.2008.12.002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giorgi C, De Stefani D, Bononi A, Rizzuto R & Pinton P Structural and functional link between the mitochondrial network and the endoplasmic reticulum. IJBCB 41, 1817–1827, doi:S1357-2725(09)00130-7 [pii] 10.1016/j.biocel.2009.04.010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinton P, Giorgi C, Siviero R, Zecchini E & Rizzuto R Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418, doi:onc2008308 [pii] 10.1038/onc.2008.308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Decuypere JP et al. The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta, doi:S0167-4889(10)00309-5 [pii] 10.1016/j.bbamcr.2010.11.023 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Rowland AA & Voeltz GK Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13, 607–625, doi: 10.1038/nrm3440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kornmann B The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol 25, 443–448, doi: 10.1016/j.ceb.2013.02.010 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Baughman JM et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345, doi: 10.1038/nature10234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Stefani D, Raffaello A, Teardo E, Szabo I & Rizzuto R A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340, doi: 10.1038/nature10230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendes CC et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem 280, 40892–40900, doi: 10.1074/jbc.M506623200 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Hedskog L et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proceedings of the National Academy of Sciences of the United States of America 110, 7916–7921, doi: 10.1073/pnas.1300677110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tubbs E et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63, 3279–3294, doi: 10.2337/db13-1751 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Seeger-Nukpezah T, Geynisman DM, Nikonova AS, Benzing T & Golemis EA The hallmarks of cancer: relevance to the pathogenesis of polycystic kidney disease. Nat Rev Nephrol, doi: 10.1038/nrneph.2015.46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowe I et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med 19, 488–493, doi: 10.1038/nm.3092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riwanto M et al. Inhibition of Aerobic Glycolysis Attenuates Disease Progression in Polycystic Kidney Disease. PloS one 11, e0146654, doi: 10.1371/journal.pone.0146654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiaravalli M et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J Am Soc Nephrol 27, 1958–1969, doi: 10.1681/ASN.2015030231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Padovano V et al. The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol Biol Cell 28, 261–269, doi: 10.1091/mbc.E16-08-0597 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wegierski T et al. TRPP2 channels regulate apoptosis through the Ca2+ concentration in the endoplasmic reticulum. EMBO J 28, 490–499, doi:emboj2008307 [pii] 10.1038/emboj.2008.307 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoder BK, Hou X & Guay-Woodford LM The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 13, 2508–2516 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Cai Y et al. Identification and characterization of polycystin-2, the PKD2 gene product. J Biol Chem 274, 28557–28565 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Koulen P et al. Polycystin-2 is an intracellular calcium release channel. Nature cell biology 4, 191–197, doi: 10.1038/ncb754 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Mekahli D et al. Polycystin-1 and polycystin-2 are both required to amplify inositol-trisphosphate-induced Ca2+ release. Cell Calcium 51, 452–458, doi: 10.1016/j.ceca.2012.03.002 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Sammels E et al. Polycystin-2 activation by inositol 1,4,5-trisphosphate-induced Ca2+ release requires its direct association with the inositol 1,4,5-trisphosphate receptor in a signaling microdomain. J Biol Chem 285, 18794–18805, doi: 10.1074/jbc.M109.090662 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres VE & Harris PC Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol 25, 18–32, doi: 10.1681/ASN.2013040398 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Brito OM & Scorrano L Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610, doi: 10.1038/nature07534 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Chen Y et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circulation research 111, 863–875, doi: 10.1161/circresaha.112.266585 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alford SC, Ding Y, Simmen T & Campbell RE Dimerization-dependent green and yellow fluorescent proteins. ACS synthetic biology 1, 569–575, doi: 10.1021/sb300050j (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian Q et al. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum Mol Genet 12, 1875–1880 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Barsukova A et al. Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur J Neurosci 33, 831–842, doi: 10.1111/j.1460-9568.2010.07576.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagai T, Sawano A, Park ES & Miyawaki A Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proceedings of the National Academy of Sciences of the United States of America 98, 3197–3202, doi: 10.1073/pnas.051636098051636098 [pii] (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuyama S, Llopis J, Deveraux QL, Tsien RY & Reed JC Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nature cell biology 2, 318–325, doi: 10.1038/35014006 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Rizzuto R, Simpson AW, Brini M & Pozzan T Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358, 325–327, doi: 10.1038/358325a0 (1992). [DOI] [PubMed] [Google Scholar]
- 34.Loro G, Ruberti C, Zottini M & Costa A The D3cpv Cameleon reports Ca(2)(+) dynamics in plant mitochondria with similar kinetics of the YC3.6 Cameleon, but with a lower sensitivity. J Microsc 249, 8–12, doi: 10.1111/j.1365-2818.2012.03683.x (2013). [DOI] [PubMed] [Google Scholar]
- 35.Palmer AE et al. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol 13, 521–530, doi: 10.1016/j.chembiol.2006.03.007 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Sharif-Naeini R et al. Polycystin-1 and −2 dosage regulates pressure sensing. Cell 139, 587–596, doi:S0092–8674(09)01125–8 [pii] 10.1016/j.cell.2009.08.045 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Calvo SE, Clauser KR & Mootha VK MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–1257, doi: 10.1093/nar/gkv1003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuo IY et al. Cyst formation following disruption of intracellular calcium signaling. Proceedings of the National Academy of Sciences of the United States of America 111, 14283–14288, doi: 10.1073/pnas.1412323111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foskett JK, White C, Cheung KH & Mak DO Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593–658, doi: 10.1152/physrev.00035.2006 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagar RE & Ehrlich BE Regulation of the type III InsP(3) receptor by InsP(3) and ATP. Biophys J 79, 271–278, doi: 10.1016/S0006-3495(00)76289-8 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Filadi R et al. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proceedings of the National Academy of Sciences of the United States of America 112, E2174–2181, doi: 10.1073/pnas.1504880112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ainbinder A, Boncompagni S, Protasi F & Dirksen RT Role of Mitofusin-2 in Mitochondrial Localization and Calcium Uptake in Skeletal Muscle. Cell calcium 57, 14–24, doi: 10.1016/j.ceca.2014.11.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denton RM Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787, 1309–1316, doi: 10.1016/j.bbabio.2009.01.005 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Schrepfer E & Scorrano L Mitofusins, from Mitochondria to Metabolism. Molecular cell 61, 683–694, doi: 10.1016/j.molcel.2016.02.022 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Finck BN & Kelly DP PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 116, 615–622, doi: 10.1172/JCI27794 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Wu Y, Ward CJ, Harris PC & Torres VE Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 19, 102–108, doi: 10.1681/ASN.2007060688 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamaguchi T et al. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int 57, 1460–1471, doi: 10.1046/j.1523-1755.2000.00991.x (2000). [DOI] [PubMed] [Google Scholar]
- 48.Pinto CS, Reif GA, Nivens E, White C & Wallace DP Calmodulin-sensitive adenylyl cyclases mediate AVP-dependent cAMP production and Cl- secretion by human autosomal dominant polycystic kidney cells. Am J Physiol Renal Physiol 303, F1412–1424, doi: 10.1152/ajprenal.00692.2011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hayashi T & Su TP Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610, doi: 10.1016/j.cell.2007.08.036 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Dabrowska A et al. PGC-1alpha controls mitochondrial biogenesis and dynamics in lead-induced neurotoxicity. Aging (Albany NY) 7, 629–647, doi: 10.18632/aging.100790 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ventura A et al. Cre-lox-regulated conditional RNA interference from transgenes. Proceedings of the National Academy of Sciences of the United States of America 101, 10380–10385, doi: 10.1073/pnas.0403954101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dorup J Ultrastructure of distal nephron cells in rat renal cortex. J Ultrastruct Res 92, 101–118 (1985). [DOI] [PubMed] [Google Scholar]
- 53.Csordas G et al. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Molecular cell 39, 121–132, doi: 10.1016/j.molcel.2010.06.029 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuo IY et al. Decreased polycystin 2 expression alters calcium-contraction coupling and changes beta-adrenergic signaling pathways. Proceedings of the National Academy of Sciences of the United States of America 111, 16604–16609, doi: 10.1073/pnas.1415933111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schneeberger M et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155, 172–187, doi: 10.1016/j.cell.2013.09.003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sugiura A et al. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Molecular cell 51, 20–34, doi: 10.1016/j.molcel.2013.04.023 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Li D, Li X, Guan Y & Guo X Mitofusin-2-mediated tethering of mitochondria and endoplasmic reticulum promotes cell cycle arrest of vascular smooth muscle cells in G0/G1 phase. Acta biochimica et biophysica Sinica 47, 441–450, doi: 10.1093/abbs/gmv035 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Naon D et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proceedings of the National Academy of Sciences of the United States of America 113, 11249–11254, doi: 10.1073/pnas.1606786113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M & Snoeck HW Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529, 528–531, doi: 10.1038/nature16500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beikoghli Kalkhoran S et al. Assessing the effects of mitofusin 2 deficiency in the adult heart using 3D electron tomography. Physiological reports 5, doi: 10.14814/phy2.13437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cosson P, Marchetti A, Ravazzola M & Orci L Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PloS one 7, e46293, doi: 10.1371/journal.pone.0046293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cai Y et al. Altered trafficking and stability of polycystins underlie polycystic kidney disease. J Clin Invest 124, 5129–5144, doi: 10.1172/JCI67273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chapin HC, Rajendran V & Caplan MJ Polycystin-1 surface localization is stimulated by polycystin-2 and cleavage at the G protein-coupled receptor proteolytic site. Mol Biol Cell 21, 4338–4348, doi:E10-05-0407 [pii] 10.1091/mbc.E10-05-0407 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gainullin VG, Hopp K, Ward CJ, Hommerding CJ & Harris PC Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J Clin Invest, doi: 10.1172/JCI76972 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cebotaru V et al. Polycystin-1 negatively regulates Polycystin-2 expression via the aggresome/autophagosome pathway. J Biol Chem 289, 6404–6414, doi: 10.1074/jbc.M113.501205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Freedman BS et al. Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. J Am Soc Nephrol 24, 1571–1586, doi: 10.1681/ASN.2012111089 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin C-C et al. A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Scientific Reports 8, 2743, doi: 10.1038/s41598-018-20856-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ishimoto Y et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Molecular and Cellular Biology 37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suarez-Rivero JM et al. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases (Basel, Switzerland) 5, doi: 10.3390/diseases5010001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamaguchi T et al. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 279, 40419–40430, doi: 10.1074/jbc.M405079200 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Yamaguchi T, Hempson SJ, Reif GA, Hedge AM & Wallace DP Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol 17, 178–187, doi: 10.1681/ASN.2005060645 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Betz C et al. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proceedings of the National Academy of Sciences of the United States of America 110, 12526–12534, doi: 10.1073/pnas.1302455110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Torres VE et al. Prospects for mTOR inhibitor use in patients with polycystic kidney disease and hamartomatous diseases. Clin J Am Soc Nephrol 5, 1312–1329, doi: 10.2215/CJN.01360210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shillingford JM et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America 103, 5466–5471, doi: 10.1073/pnas.0509694103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boletta A Emerging evidence of a link between the polycystins and the mTOR pathways. Pathogenetics 2, 6, doi:1755–8417-2–6 [pii] 10.1186/1755-8417-2-6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Becker JU et al. The mTOR pathway is activated in human autosomal-recessive polycystic kidney disease. Kidney and Blood Pressure Research 33, 129–138, doi: 10.1159/000314380 (2010). [DOI] [PubMed] [Google Scholar]
- 77.Fu S et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531, doi: 10.1038/nature09968 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arruda AP et al. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med 20, 1427–1435, doi: 10.1038/nm.3735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.DesRochers TM, Kuo IY, Kimmerling EP, Ehrlich BE & Kaplan DL The Effects of Mycoplasma Contamination upon the Ability to Form Bioengineered 3D Kidney Cysts. PloS one 10, e0120097, doi: 10.1371/journal.pone.0120097 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Simonnet H, Vigneron A & Pouyssegur J Conventional techniques to monitor mitochondrial oxygen consumption. Methods Enzymol 542, 151–161, doi: 10.1016/B978-0-12-416618-9.00008-X (2014). [DOI] [PubMed] [Google Scholar]
- 81.Chen TW et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300, doi: 10.1038/nature12354 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roy SS & Hajnoczky G Calcium, mitochondria and apoptosis studied by fluorescence measurements. Methods 46, 213–223, doi: 10.1016/j.ymeth.2008.09.024 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meijering E, Dzyubachyk O & Smal I Methods for cell and particle tracking. Methods Enzymol 504, 183–200, doi: 10.1016/B978-0-12-391857-4.00009-4 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Murphy AN, Bredesen DE, Cortopassi G, Wang E & Fiskum G Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proceedings of the National Academy of Sciences of the United States of America 93, 9893–9898 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Patel V et al. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet 17, 1578–1590, doi: 10.1093/hmg/ddn045 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Traykova-Brauch M et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med 14, 979–984, doi: 10.1038/nm.1865 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. PC2 KD and PC2 KO result in increased mitochondrial Ca2+ signals.
Fig. S2. PC2 resides in ER membranes close to mitochondria.
Fig. S3. PC2 KD does not alter the expression of MCU or multiple MCU complex components.
Fig. S4. MFN2 KD rescues Ca2+ signaling in PC2 KD cells and PC2 KD alters mitochondrial metabolism and density.
Fig. S5. Super-resolution STED microscopy does not induce differential mitochondrial ROS production.
Fig. S6. PC2 KD does not alter the expression of alternative ER-mitochondria linkers.
Fig. S7. LV-siMFN2 is effective at knocking down MFN2 in cells from Pkd2F/FPkhd1Cre mice, which develop kidney cysts from birth.
Fig. S8. LV-siMFN2 treatment rescues multiple effects of PC2 deficiency in cystic cells.
Fig. S9. Mitochondrial density is increased in cyst-lining cells from ADPKD kidneys.
Video S1. Super-resolution movie of organelle dynamics.