

Abstract
LTP, a fundamental mechanism of learning and memory, is a highly regulated process. One form of regulation is metaplasticity (i.e., the activity-dependent and long-lasting changes in neuronal state that orchestrate the direction, magnitude, and persistence of future synaptic plasticity). We have previously described a heterodendritic metaplasticity effect, whereby strong high-frequency priming stimulation in stratum oriens inhibits subsequent LTP in the stratum radiatum of hippocampal area CA1, potentially by engagement of the enmeshed astrocytic network. This effect may occur due to neuron-glia interactions in response to priming stimulation that leads to the release of gliotransmitters. Here we found in male rats that TNFα and associated signal transduction enzymes, but not interleukin-1β (IL-1β), were responsible for mediating the metaplasticity effect. Replacing priming stimulation with TNFα incubation reproduced these effects. As TNFα levels are elevated in Alzheimer's disease, we examined whether heterodendritic metaplasticity is dysregulated in a transgenic mouse model of the disease, either before or after amyloid plaque formation. We showed that TNFα and IL-1β levels were significantly increased in aged but not young transgenic mice. Although control LTP was impaired in the young transgenic mice, it was not TNFα-dependent. In the older transgenic mice, however, LTP was impaired in a way that occluded further reduction by heterosynaptic metaplasticity, whereas LTP was entirely rescued by incubation with a TNFα antibody, but not an IL-1β antibody. Thus, TNFα mediates a heterodendritic metaplasticity in healthy rodents that becomes constitutively and selectively engaged in a mouse model of Alzheimer's disease.
SIGNIFICANCE STATEMENT The proinflammatory cytokine TNFα is known to be capable of inhibiting LTP and is upregulated several-fold in brain tissue, serum, and CSF of Alzheimer's disease (AD) patients. However, the mechanistic roles played by TNFα in plasticity and AD remain poorly understood. Here we show that TNFα and its downstream signaling molecules p38 MAPK, ERK, and JNK contribute fundamentally to a long-range metaplastic inhibition of LTP in rats. Moreover, the impaired LTP in aged APP/PS1 mice is rescued by incubation with a TNFα antibody. Thus, there is an endogenous engagement of the metaplasticity mechanism in this mouse model of AD, supporting the idea that blocking TNFα might be of therapeutic benefit in the disease.
Keywords: Alzheimer's disease, glia, LTP, metaplasticity, neuroinflammation, TNFα
Introduction
Metaplasticity mechanisms have been proposed to modify the threshold for learning ability (Abraham, 2008), help maintain synaptic transmission within a dynamic range, and potentially prevent excitotoxicity (Abraham and Bear, 1996). In most experimental studies of metaplasticity, the effect of a prior bout of neural activity or “priming” on subsequent LTP or LTD is investigated. In one unusual form of metaplasticity, high-frequency priming stimulation in the stratum oriens (SO) of hippocampal area CA1 inhibits subsequent LTP elicited in the stratum radiatum (SR) of the same area (Hulme et al., 2012). Evidence to date indicates that this form of heterodendritic metaplasticity is independent of postsynaptic depolarization, action potential firing, and LTP generation in SO (Hulme et al., 2012), but requires the activation of muscarinic acetylcholine receptors and the release of Ca2+ from intracellular stores via activation of IP3 receptors (Hulme et al., 2012). This pattern of results suggests that non-neuronal cells, such as astrocytes, may play a role in this long-range communication as priming stimulation induces calcium elevations not only in SO astrocytes but also in SR astrocytes (Hulme et al., 2014). Moreover, the effect requires the opening of connexin-43 channels located on astrocytes, and the conversion of extracellular ATP to adenosine followed by activation of adenosine A2 receptors (Jones et al., 2013).
What are the glial mechanisms that could mediate the heterodendritic metaplasticity? Gliotransmitters that can affect synaptic plasticity include glutamate (Kang et al., 1998; Araque et al., 1999), d-serine (Mothet et al., 2005; Panatier et al., 2006; Henneberger et al., 2010), cytokines (Beattie et al., 2002; Schroeter and Jander, 2005; Stellwagen and Malenka, 2006), and ATP and adenosine (Fields and Burnstock, 2006; Haydon and Carmignoto, 2006; North and Verkhratsky, 2006). One proinflammatory cytokine known to inhibit LTP prominently is TNFα (Tancredi et al., 1992; Butler et al., 2004). Exogenously applied TNFα inhibits LTP in both CA1 (Tancredi et al., 1992) and the dentate gyrus (Cunningham et al., 1996) in vitro. Conversely, TNFα KO mice show increased performance in spatial memory and learning tasks compared with WT animals (Golan et al., 2004). TNFα inhibits the early phase of LTP by activation of TNFα receptor 1 (TNFR1, p55) (Butler et al., 2004), and the downstream activation of p38 mitogen-activated protein kinase (p38 MAPK) (Pickering et al., 2005), and c-Jun N-terminal kinase (JNK) (Liu et al., 2007). In contrast, another target of TNFR1, extracellular signal-related kinase (ERK), is usually required for LTP in CA1 (English and Sweatt, 1996; Jin and Feig, 2010) and is crucial for transcription-dependent long-term synaptic plasticity.
Given the regulation by p38 MAPK, JNK, and ERK of LTP and their activation by TNFα, we tested the involvement of these molecules in mediating the heterodendritic metaplasticity effect. For comparison, we investigated another candidate cytokine, IL-1β, which also causes inhibition of LTP in CA1 and CA3 (Katsuki et al., 1990; Bellinger et al., 1993; Hoshino et al., 2017), and in the dentate gyrus (Cunningham et al., 1996), and may contribute to aging- and stress-related impairments of LTP (Murray and Lynch, 1998).
As both TNFα and IL-1β levels are elevated in Alzheimer's disease (Griffin et al., 1989; Brosseron et al., 2014), we also tested the hypothesis that heterodendritic metaplasticity is constitutively engaged in a double-transgenic (Tg, APP/PS1) mouse model of the disease, accounting for the impairment of LTP in this model. Our data revealed that TNFα, but not IL-1β, mediates heterodendritic metaplasticity in area CA1 of both rat and mouse hippocampus, and that this mechanism is endogenously active in aged, but not young, APP/PS1 mice.
Materials and Methods
Animals.
Adult Sprague Dawley male rats (6–8 weeks old; 200–320 g), male WT B6C3 mice, and male Tg APPswe/PS1ΔE9 (The Jackson Laboratory; https://www.jax.org/strain/004462) littermates (4 or 14 months old) were used. All animals were bred in colonies maintained by the University of Otago, Dunedin. Rats were group-housed (3–5 animals per cage), while the mice were singly housed after weaning in individually ventilated cages. Ambient temperature and the light/dark cycle were maintained at 23 ± 2°C and 12:12 h, respectively, with lights on at 6:00 A.M. All methods of animal handling and manipulation were performed with approval by the University of Otago Animal Ethics Committee and in accordance with New Zealand animal welfare legislation.
Slice preparation.
Rats (6–8 weeks old, 180–300 g) were anesthetized with ketamine (100 mg/kg, i.p.) and decapitated using a guillotine. Brains were rapidly dissected out and submerged into ice-cold sucrose cutting solution (in mm as follows: 210 sucrose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 3 MgCl2, 20 d-glucose) continuously bubbled with carbogen (95% O2, 5% CO2). The hippocampus was then dissected out, and the most ventral hippocampus removed. CA3 was subsequently removed from the remaining dorsal hippocampus by a manual knife cut and slices (3–6 slices from each hemisphere) of 400 μm thickness prepared using a VT1000 S vibroslicer (Leica Microsystems). Prepared slices were incubated at 32°C in a humidified incubation chamber containing ACSF (in mm as follows: 124 NaCl, 3.2 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.5 CaCl2, 1.3 MgCl2, 10 d-glucose) and bubbled with carbogen. The slices remained thinly submerged for 30 min at 32°C and then at room temperature for 90 min. The slices were then submerged in a recording chamber containing recirculating ACSF at 32.5°C, and fEPSPs were recorded.
WT and Tg mice were deeply anesthetized with pentobarbital (200 mg/kg, i.p.) and transcardially perfused with ice-cold sucrose cutting solution. Immediate perfusion with cold cutting solution minimized compression of brain tissue while cutting slices using the VT1000 S vibroslicer (Leica Microsystems). After decapitation, one hemisphere was snap-frozen with dry ice for later ELISA and the other for preparation of 400 μm coronal slices of the whole hemisphere that included the dorsal hippocampus for electrophysiology. The mouse slices were treated similarly to the rat slices thereafter, except that the CA3 region was intact.
Electrophysiology.
Teflon-coated tungsten monopolar stimulating electrodes (50 μm) were placed centrally in both SR and SO of CA1 (see Fig. 1a) to elicit fEPSPs. The evoked fEPSPs were recorded using glass micropipettes (A-M Systems, 1.0 mm × 0.58 mm, 4 inch; catalog #601000) filled with ACSF (1.7–2.7 mΩ). Recording electrodes were placed 400 μm from their respective stimulating electrodes and connected to P511 AC amplifiers via high impedance probes (Grass Instruments). Signals were amplified (×1000) with half-amplitude filter cutoffs of 0.3 Hz and 3 kHz.
Figure 1.

Stimulation pattern and the heterodendritic metaplasticity effect in area CA1. a, Photograph of a hippocampal (CA3 removed) slice placed between the top and bottom nets within a recording chamber when viewed through a dissection microscope. Placement of the stimulating and recording electrodes for extracellular recording is shown. S1 and S2, Stimulating electrodes in SO and SR, respectively; R1 and R2, recording electrodes in SO and SR, respectively. The pyramidal neuron image shows its processes extending from stratum pyramidale (SP) toward SO (basal dendrites) and SR (apical dendrites). b, The stimulation pattern for priming consisted of 2 sets of 3 HFS (3 × 1 s at 100 Hz, spaced by 20 s) with 5 min between sets delivered in SO. LTP stimulation consisted of 2 trains of TBS (each train consists of 10 bursts and each burst consists of five pulses at 100 Hz with a 200 ms interburst interval) delivered in SR. *Interburst interval in the priming HFS. **Interpulse interval in the LTP-inducing TBS. ***Interburst interval in the TBS. c, Two trains of TBS successfully induced LTP in SR of nonprimed rat hippocampal slices. SO recordings were not affected by the TBS in SR. d, Priming HFS in SO induced LTP in SO; however, SR recordings were unaffected. e, Priming stimulation in SO inhibited LTP in SR, that is, priming stimulation induced heterodendritic inhibition of LTP compared with control, nonprimed slices. *p < 0.05, significant difference between the control and primed groups. Data are mean ± SEM. n = 8 slices per group. Inset, Representative waveforms for the figures are averages of the final 10 sweeps taken before (pre) and 50–60 min after TBS (post) for SR and 80–90 min after HFS (post) for SO graphs, respectively.
Baseline stimulation (0.1 ms half-wave duration) was set at 40% of maximum fEPSP slope elicited at a stimulation strength of ∼0.2 mA. Baseline stimulation was delivered every 30 s, alternating between stimulating electrodes placed in SR and SO, and was given for at least 30 min and until recordings were stable (<10% difference in synaptic responses from the initial 30 min of baseline recording). Control experiments consisted of 80 min of baseline stimulation followed by LTP induction in SR and a further 60 min of stimulation. Priming experiments consisted of 45 min of baseline stimulation followed by two sets of priming stimulation (separated by 5 min) in SO. A further 30 min of baseline stimulation was delivered after priming, followed by LTP induction in SR and further baseline stimulation for 60 min. Priming stimulation consisted of 2 sets of 3 high-frequency stimulation (HFS) trains delivered in SO (each train consisted of 100 Hz, 1 s, delivered 20 s apart; see Fig. 1b) (Wang and Wagner, 1999; Hulme et al., 2012). For LTP induction, 2 trains of theta burst stimulation (TBS, each train consisted of 10 bursts of 5 pulses at 100 Hz, 200 ms between bursts) were delivered in SR, 30 s apart. The stimulation intensities for HFS priming stimulation in SO and TBS stimulation in SR were the same as for their respective baseline test pulses.
Anti-rat TNFα Ab (catalog #MAB510), anti-rat IL-1β Ab (catalog #MAB501), anti-mouse TNFα Ab (catalog #MAB4101), and anti-mouse IL-1β Ab (catalog #MAB401) were purchased from R&D Systems. Thalidomide, SB203580, PD98059, and SP600125 were purchased from Tocris Bioscience and dissolved in DMSO (Sigma-Aldrich) and further diluted to 1:1000 in ACSF to give the working concentration. Other chemicals were purchased from Merck.
Data analysis.
The initial slope values of the fEPSPs were analyzed offline as a measure of synaptic efficacy. The baseline response value was calculated by averaging the responses over the last 10 min before LTP induction in SR, and all the synaptic responses throughout the experiment were then normalized to the baseline value and expressed as a percentage of baseline. LTP in SO due to priming stimulation was also recorded, and the baseline for SO was calculated by averaging the baseline responses 10 min before the first priming stimulation in SO. Finally, LTP was calculated in SR and SO by averaging the responses generated in the final 10 min of the experiment and normalized to their respective baseline values.
Western blotting.
Hippocampal slices were made and treated similarly to those in electrophysiology slice preparation. Field recordings were made using the same protocol as for the electrophysiology experiments, except slices were collected at the time the TBS would have been delivered to induce LTP. Slices were then micro-dissected to isolate SR from other regions of the hippocampus. The groups used were the control group that received test pulses only and the SO primed group that received test pulses plus SO priming stimulation identical to that used in the electrophysiology experiments. All the groups used for Western blotting experiments consisted of 12 samples, and each sample consisted of 5 micro-dissected SR slices from 1 animal pooled together. Slices were stored at −80°C before being thawed at room temperature for protein extraction and Western blot.
Proteins were first extracted from thawed slices into a detergent-containing buffer with both phosphatase and protease inhibitors: 50 mm Tris buffer, pH 7.6, 10 mm EDTA, 10 mm EGTA, 1.0 mm PMSF (Sigma-Aldrich, catalog #10837091001), 50 mm sodium fluoride, 80 μm sodium molybdate, 5 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 4 mm p-nitrophenyl phosphate, 1:50 phosphatase inhibitor III (Abcam, catalog #201114), 1:2000 phosphatase inhibitor II (Sigma-Aldrich, catalog #P5726), 0.01% (v/v) Triton-X, and a protease inhibitor (Complete Ultra Mini Tablet, Roche Diagnostics) used as per the manufacturer's instructions. Slices were homogenized by 30× pestle, and proteins were solubilized by ice bath sonication (5 min) to produce a lysate mixture. This solution was then spun in a centrifuge (Heraeus Biofuge Pico) at 13,000 rpm at 4°C for 20 min. The supernatant was taken and placed in a separate tube. These sonication and spinning steps were repeated once, and the supernatant was added to the first fraction. This combined lysate was used as the sample. A detergent-compatible protein assay (Bio-Rad) was used to quantify protein concentrations using BSA (Sigma Aldrich) as a standard.
Protein samples were separated on 12% (w/v) bis-acrylamide (Bio-Rad) gels before transferring to a nitrocellulose membrane (GE Healthcare). Blots were incubated in Odyssey blocking buffer (LI-COR) at room temperature for 1 h. The primary Ab solution was prepared using the following antibodies and concentrations in 0.1% NGS and 0.1% BSA in PBS/Tween. From Abcam: mouse anti-total p38 MAPK (1:1000; catalog #ab31828); from Novus Biologicals: rabbit anti-phospho-p38 MAPK (1:1000; catalog #NB500-138), rabbit anti-total ERK (1:1000; catalog #9102), mouse anti-phospho-ERK (1:1000; catalog #9106), rabbit anti-total JNK (1:1000; catalog #9252), mouse anti-phospho-JNK (1:1000; catalog #9255), and rabbit anti-α-tubulin (1:10,000; catalog #2144); and from Thermo Fisher Scientific: rabbit anti-GAPDH Ab (1:10,000; catalog #TAB1001).
The secondary Ab solution was composed of IRDye goat anti-rabbit 680 (1:10,000, LI-COR) and IRDye goat anti-mouse 800 (1:15,000, LI-COR) in PBS/Tween. The blots were incubated in this solution for 1 h at room temperature. Blots were imaged on a LI-COR Odyssey imaging system, quantified using Image Studio Lite 5.2 (LI-COR) after normalizing to the loading control protein (α-tubulin or GAPDH). We used the gray channel for the analysis of bands because it minimizes the background noise and provides better contrast. The results were normalized to a loading control protein (α-tubulin or GAPDH) and calculated as a proportion of all the samples on the blot. The relative level of phosphorylation was measured by the ratio of phosphorylated to the total protein.
ELISA.
The ELISA assay used an immobilized Ab specific for mouse TNFα or IL-1β coated on a 96-well plate. ELISA kits specific to mouse TNFα (catalog #BE69212), and IL-1β (catalog #IB49700) were purchased from IBL America. The tissue homogenate group used for the ELISA consisted of 10 samples each containing the hippocampus of a single hemisphere from each animal (the other hemisphere was used for electrophysiology experiments). Once dissected, these samples were flash frozen on dry ice and stored at −80°C. On the day of experiments, samples were thawed at room temperature, and each sample was homogenized in the solubilization buffer containing 1 mm EGTA, 1 mm EDTA, complete protease inhibitor, 1% (v/v) Triton-X, 1 mm PMSF, PBS, and 0.1% (w/v) SDS. The samples were dissolved in homogenization buffer by gentle 20× piston up and down followed by probe sonication for 10 s at room temperature. Tissue homogenate was centrifuged at ∼5000 rpm for 5 min, and detergent-compatible protein assay (Bio-Rad DC Protein Assay) was used to find the concentration of protein. Thereafter, the ELISA procedure was performed according to the manufacturer's instructions.
Statistical analysis.
Data are mean ± SEM unless stated otherwise, and n indicates the number of slices for electrophysiology (n = 7–10 slices per group), numbers of samples for Western blots (n = 12/group), and ELISAs (n = 10/group). To assess statistically the differences between groups, Student's t tests and ANOVA were conducted using SPSS Statistics software (version 24, IBM). Post hoc tests using Fisher's least significant difference tests were used to identify between-group differences, as appropriate. Statistical significance was determined as p < 0.05.
Results
Heterodendritic inhibition of LTP in CA1 is mediated by TNFα
TBS delivered to SR successfully induced LTP in SR of nonprimed rat hippocampal slices (148 ± 4%, n = 8), without affecting responses in SO (96 ± 3%, n = 8; Fig. 1c). HFS in SO, used for priming, produced significant LTP in that pathway (240 ± 12%, n = 8), also without directly affecting responses in SR (98 ± 3%, n = 8; Fig. 1d). Consistent with previous findings (Wang and Wagner, 1999; Hulme et al., 2012); however, the SO priming stimulation induced a strong heterodendritic inhibition of SR LTP induced 30 min later (SR control LTP = 148 ± 4%, n = 8; SR Primed LTP = 119 ± 3%, n = 8; t(15) = 11.78, p = 0.01; Fig. 1e). Although the priming stimulation, in this case, did produce rapid heterosynaptic depression at SR synapses, the responses quickly recovered fully before the LTP stimulation (Fig. 1e).
Bath application of a TNFα Ab (25 μg/50 ml) was conducted for 30 min before and during priming stimulation before washing out for 30 min before SR TBS. There was a significant main effect of group for SR LTP (F(2,22) = 22.38, p = 0.01). Post hoc analysis revealed that there was significant inhibition of LTP by the priming stimulation (Control ACSF: 146 ± 6%, n = 7; Primed ACSF: 120 ± 3%, n = 8; p = 0.001), whereas the TNFα Ab significantly reduced the extent of this priming effect on SR LTP (Primed ACSF: 120 ± 3%, n = 8; Primed TNFα Ab: 132 ± 4%, n = 8; p = 0.043; Fig. 2a). The TNFα Ab had no effect on control SR LTP by itself (t(12) = 0.38, p = 0.42). The SO LTP that occurred due to the priming stimulation in SO was also unaltered by the TNFα Ab treatment (Primed ACSF: 220 ± 9%, n = 9; Primed TNFα Ab: 222 ± 11%, n = 8, t(15) = 0.48, p = 0.96; Fig. 2b).
Figure 2.

Blocking TNFα activity prevented the heterodendritic metaplasticity effect at SR synapses. a, Priming-mediated inhibition of LTP was significantly reduced by the TNFα Ab treatment (25 μg/50 ml), although not completely. b, In contrast, priming stimulation elicited homosynaptic LTP in SO, which was unaltered by the TNFα Ab treatment. c, Preincubating slices with the TNFα Ab (25 μg/ml) completely prevented the inhibition of LTP in SR due to priming in SO. d, However, SO LTP remained unaltered by the TNFα Ab preincubation treatment. e, Application of thalidomide throughout the experiment prevented the inhibition of LTP in SR due to priming in SO. f, LTP in SO was unaltered by thalidomide treatment. *p < 0.05. Data are mean ± SEM. n = 7–9 slices per group. Inset, Representative waveforms for the figures are averages of the final 10 sweeps taken before (pre) and 50–60 min after TBS (post) for SR and 80–90 min after HFS (post) for SO graphs, respectively. The numbers above the waveforms correspond to the treatment group, as indicated in the figure legend.
A second set of experiments aimed to obtain a more complete block of the priming effect. After the initial incubation for 30 min at 32°C in ACSF, hippocampal slices were transferred to a closed incubation chamber kept at room temperature continuously gassed with carbogen where they were incubated with the TNFα Ab (25 μg/ml ACSF) for at least 90 min at room temperature. After incubation with the TNFα Ab, slices were transferred to the recording chamber and maintained at 32.5°C in standard ACSF without further Ab treatment. Under these conditions, there was again an overall significant effect of the treatment group on SR LTP (F(2,18) = 13.05, p = 0.001). Post hoc analysis revealed that there was significant inhibition of LTP by priming (Control ACSF: 142 ± 4%, n = 7; Primed ACSF: 126 ± 3%, n = 7; p = 0.004) and, in this case, the TNFα Ab fully blocked the priming effect in SR (Control TNFα Ab: 145 ± 7%, n = 7; Primed TNFα Ab: 141 ± 4%, n = 7; p = 0.94; Fig. 2c). The TNFα Ab had no effect on control SR LTP by itself (t(12) = 0.81, p = 0.73). The SO LTP due to priming was also unaltered by preincubation with the TNFα Ab (Primed ACSF: 219 ± 12%, n = 8; Primed TNFα Ab: 197 ± 13%, n = 7, t(13) = 1.04, p = 0.86; Fig. 2d).
The TNFα mRNA synthesis inhibitor thalidomide (Moreira et al., 1993; Tramontana et al., 1995; Tavares et al., 1997) was also investigated to test whether it could abolish the heterodendritic metaplasticity effect. Thalidomide was bath-applied at 5 μm dissolved in DMSO throughout the experiments, as this concentration was found to be effective in downregulating TNFα mRNA synthesis (Enomoto et al., 2004). As for the TNFα Ab, there was an overall significant treatment effect on SR LTP (F(2,18) = 10.21, p = 0.001), such that there was significant inhibition of LTP by priming (Control DMSO: 147 ± 6%, n = 7; Primed DMSO: 127 ± 6%, n = 7; p = 0.002). Thalidomide completely blocked the priming effect in SR (Control Thalidomide: 150 ± 4%, n = 7; Primed Thalidomide: 153 ± 7%, n = 7; p = 0.910; Fig. 2e), whereas thalidomide by itself had no effect on control SR LTP compared with Control DMSO group (t(14) = 0.15, p = 0.42). The final level of SO LTP due to priming stimulation was unaltered by thalidomide (Primed DMSO: 176 ± 11%, n = 8; Primed Thalidomide: 191 ± 12%, n = 7, t(13) = 0.41, p = 0.76). However, significant enhancement in the early SO LTP induction phase (first 5 min after TBS) was observed in the primed thalidomide group compared with the primed DMSO control group (Primed DMSO: 189 ± 17%, n = 8; Primed Thalidomide: 283 ± 12%, n = 8, t(14) = 4.11, p = 0.035; Fig. 2f).
IL-1β Ab treatment does not affect heterodendritic metaplasticity in area CA1
To explore whether a cytokine other than TNFα was also recruited by priming stimulation to inhibit LTP, the role of IL-1β was investigated (Bellinger et al., 1993; Cunningham et al., 1996; Li et al., 1997). The hippocampal slices were preincubated with an IL-1β Ab (25 μg/ml ACSF) and treated thereafter as described for the second TNFα Ab experiment. For this experiment, there was no main group effect but an overall significant ANOVA interaction (F(2,20) = 6.54, p = 0.0417). Post hoc analysis revealed that, while there was significant inhibition of SR LTP by priming (Control ACSF: 140 ± 4%, n = 8; Primed ACSF: 120 ± 3%, n = 7; p = 0.005), the IL-1β Ab had no effect on priming mediated inhibition of SR LTP (Control IL-1β Ab: 143 ± 4%, n = 9; Primed IL-1β Ab: 122 ± 3%, n = 7; p = 0.008; Fig. 3a). The IL-1β Ab by itself had no effect on either the control SR LTP (t(15) = 0.81, p = 0.73) or the SO LTP due to priming (Primed ACSF: 179 ± 10%, n = 8; Primed IL-1β Ab: 168 ± 14%, n = 7, t(13) = 0.22, p = 0.139; Fig. 3b). These data suggest a selective involvement of TNFα over IL-1β in generating the inhibition of LTP by priming stimulation.
Figure 3.

IL-1β Ab pretreatment did not affect inhibition of LTP due to priming. a, Preincubating slices with the IL-1β Ab (25 μg/ml) treatment did not affect inhibition of LTP in SR due to priming in SO. b, Priming stimulation elicited homosynaptic LTP in SO, which was not altered by the IL-1β Ab preincubation. *p < 0.05. Data are mean ± SEM. n = 7–9 slices per group. Inset, Representative waveforms for the figures are averages of the final 10 sweeps taken before (pre) and 50–60 min after TBS (post) for SR and 80–90 min after HFS (post) for SO graphs, respectively. The numbers above the waveforms correspond to the treatment group, as indicated in the figure legend.
Pharmacological priming by TNFα and IL-1β
We next investigated whether incubation with the TNFα protein could reproduce the metaplastic inhibition of LTP in the absence of electrical priming stimulation. As predicted, bath application of TNFα protein (1.18 nm, 10 min) followed by a 30 min washout period inhibited LTP in SR to a similar extent as the electrical priming in SO reported above (Control ACSF: 149.5 ± 6%, n = 7; 1.18 nm TNFα: 127.8 ± 8%, n = 7; t(14) = −7.48, p = 0.003; Fig. 4a). In contrast, neither the same concentration of IL-1β (1.18 nm) nor a higher concentration (1.5 nm) affected SR LTP (F(2,19) = 0.031, p = 0.970; Fig. 4b). On the other hand, a much higher concentration of IL-1β (5 nm) did produce an inhibition of SR LTP (Control ACSF: 155.1 ± 12%, n = 7; 1.5 nm IL-1β: 132.1 ± 8%, n = 7; p = 0.007; Fig. 4c).
Figure 4.
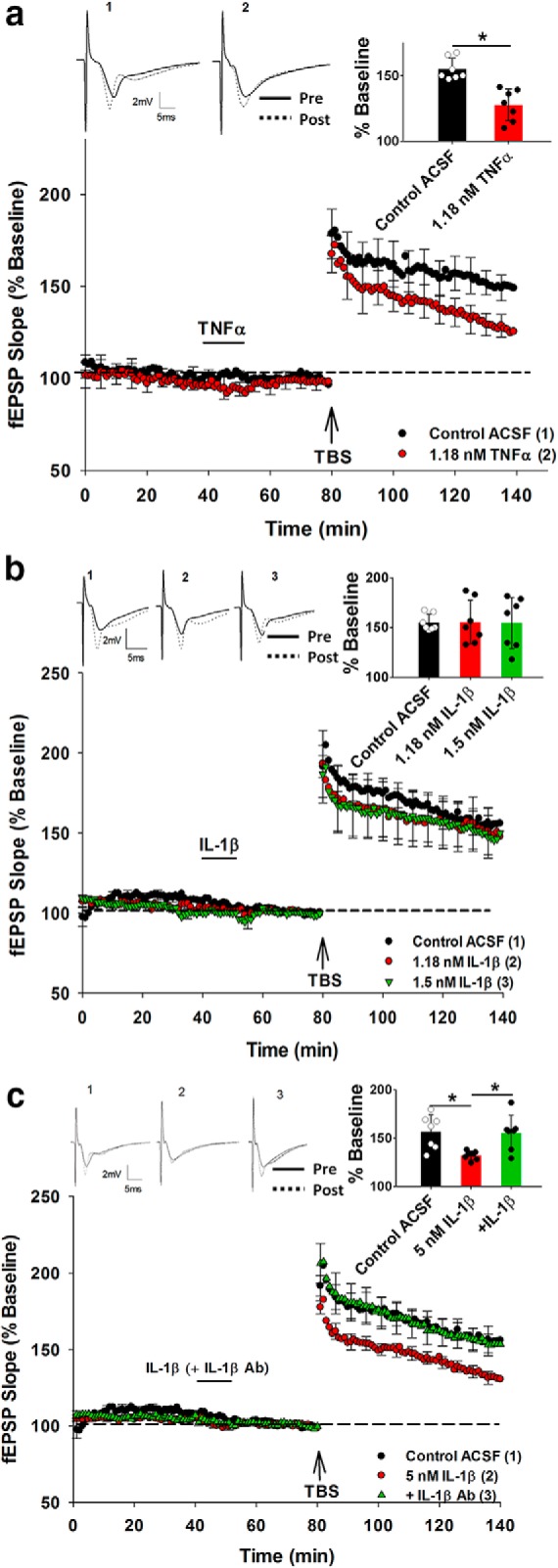
Pharmacological priming with TNFα and IL-1β protein. a, The priming effect was established by 1.18 nm TNFα in the acute rat slices. b, In contrast, IL-1β failed to produce priming effect at either the 1.18 or 1.5 nm concentration. c, IL-1β did produce a priming effect at the higher concentration of 5 nm, and this was abolished by 25 μg/ml IL-1β Ab preincubation. *p < 0.05. Data are mean ± SEM. n = 7–9 slices per group. Inset, Representative waveforms for the figures are averages of the final 10 sweeps taken before (pre) and 50–60 min after TBS (post) for SR. The numbers above the waveforms correspond to the treatment group, as indicated in the figure legend.
Given that IL-1β at 5 nm could generate the priming effect, we needed to assess whether the IL-1β Ab that failed to block the electrical priming effect actually had function-blocking capability. Therefore, to achieve this, a mixture of IL-1β (5 nm) and IL-1β Ab (25 μg/ml) was prepared at least 1 h before the perfusion of the mixture commenced, 25 min before first priming activity. Analysis revealed an overall treatment effect on SR LTP (F(2,18) = 5.971, p = 0.01), such that the mixture of IL-1β and its Ab blocked the priming effect, leaving LTP intact (IL-1β: 132.1 ± 8%, n = 7; mixture: 152.4 ± 13%, n = 7; p = 0.009; Fig. 4c). Thus, the failure of the IL-1β Ab to prevent the electrical priming of LTP (Fig. 3a) was not due to an inability to inhibit IL-1β activity.
Phosphorylation of p38 MAPK, ERK, and JNK following priming stimulation
We next investigated possible downstream targets of TNFα activated by priming stimulation in SO, using a Western blot approach. Each slice used received electrical stimulation just like electrophysiology experiments until just before the induction of LTP. The control group slices received test pulses only and were allowed to incubate in the recording chamber for 80 min. However, in the primed group, along with test pulses, the slices were electrically primed using two sets of high-frequency electrical stimulation and were allowed to incubate in the recording chamber for 30 min after priming (i.e., until just before LTP-inducing TBS would have been delivered). Since we were interested in finding the effect on SR due to priming in SO, we microdissected the slices to isolate SR from other regions of the hippocampus for Western blot samples.
Western blot analysis showed an increase in p38 MAPK phosphorylation relative to total p38 MAPK due to priming (Control: 1.07 ± 0.03, n = 12; Primed: 2.14 ± 0.087, n = 12; t(22) = 2.19, p = 0.018; Fig. 5a), whereas there was no significant effect of priming on the total p38 MAPK levels (Control total p38 MAPK: 0.108 ± 0.02, n = 12; Primed total p38 MAPK: 0.141 ± 0.03; t(22) = 0.33, p = 0.19; Fig. 5a). Similarly, there was an increase in phosphorylated JNK (JNK1/2 combined) relative to total JNK levels in primed relative to control groups (Control: 1.09 ± 0.09, n = 12; Primed: 2.08 ± 0.06, n = 12, t(22) = 2.18, p = 0.004; Fig. 5b), without a significant effect on total JNK levels (Control total JNK: 0.180 ± 0.01, n = 12; Primed total JNK: 0.17 ± 0.01; t(22) = 0.21, p = 0.64; Fig. 5b). Similar to p38 MAPK and JNK, there was an increase in phosphorylated ERK (ERK 1/2 combined) relative to total ERK levels in primed versus control groups (Control: 0.79 ± 0.02, n = 12; Primed: 1.37 ± 0.09, n = 12, t(22) = 2.89, p = 0.007; Fig. 5c), whereas there was no significant effect of priming on total ERK levels (Control total ERK: 0.06 ± 0.01, n = 12; Primed total ERK: 0.05 ± 0.006; t(22) = 0.49, p = 0.68; Fig. 5c).
Figure 5.
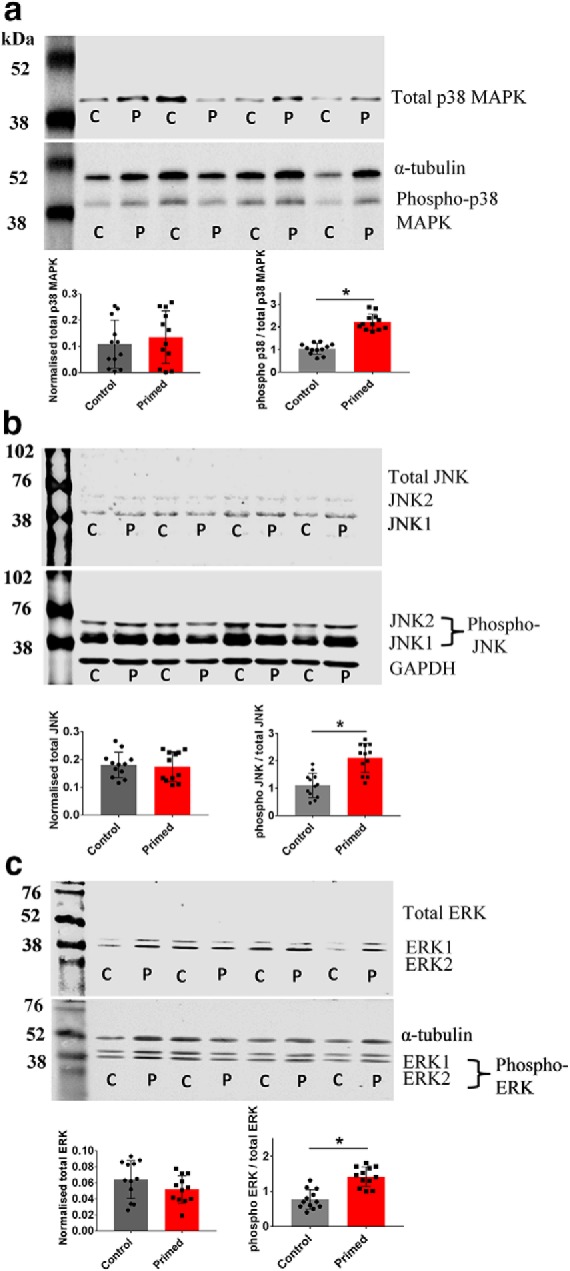
Priming-induced phosphorylation of p38 MAP, JNK, and ERK. a, A representative Western blot image of total p38 MAPK (41 kDa) and phosphorylated p38 MAPK and α-tubulin (50 kDa) in control (C) and primed (P) slices. The summary histogram for total p38 MAPK shows no difference in the control versus primed group. However, the ratio of phosphorylated p38 MAPK to total p38 MAPK was significantly increased in the primed group compared with the control group. b, A representative Western blot image of total JNK1 (46 kDa) and JNK2 (54 kDa) and phosphorylated JNK1, JNK2, and GAPDH (35 kDa) in control (C) and primed (P) slices. The summary histogram for total JNK exhibited no difference in the control versus primed group. However, the ratio of phosphorylated JNK to total JNK was significantly increased in the primed group compared with the control group. c, A representative Western blot image of total ERK1 (44 kDa) and ERK2 (42 kDa) and phosphorylated ERK1, ERK2, and α-tubulin (50 kDa) in control (C) and primed (P) slices. The summary histogram for total ERK exhibited no difference in the control versus primed group. However, the ratio of phosphorylated ERK to total ERK was significantly increased in the primed group than for the control group. For JNK and ERK, the bands for their two isoforms were treated as a single band for analysis. *p < 0.05. Data are mean ± SEM. n = 12 samples (each sample consisted of 5 micro-dissected SR slices).
Inhibition of TNFα cascade kinases abolishes heterodendritic metaplasticity
We next determined which of p38 MAPK, JNK, and ERK contributes to the inhibition of LTP in SR due to priming in SO. To test the contribution of p38 MAPK, the inhibitor SB203580 (10 μm) was bath-applied before and during priming stimulation before being washed out before SR TBS. There was a significant overall treatment effect (F(2,20) = 16.87, p = 0.0006) such that there was a significant inhibition in LTP by priming (Control DMSO: 150 ± 8%, n = 7; Primed DMSO: 127 ± 6%, n = 7; p = 0.006). Application of SB203580 blocked the priming effect in SR (Control SB203580: 156 ± 6%, n = 7; Primed SB203580: 151 ± 4%, n = 9; p = 0.355; Fig. 6a), whereas SB203580 by itself had no effect on control SR LTP (t(12) = 0.77, p = 0.92). The SO LTP was also unaltered by SB203580 (Primed DMSO: 170 ± 8%, n = 8; Primed SB203580: 157 ± 6%, n = 9, t(15) = 1.54, p = 0.68; Fig. 6b).
Figure 6.

Electrical priming-mediated inhibition of LTP is mediated by p38 MAPK, JNK, and ERK. a, SB203580 (p38 MAPK inhibitor; 10 μm) treatment before and during priming prevented the inhibition of LTP in SR due to priming in SO. b, Priming stimulation elicited homosynaptic LTP in SO, which was unaltered by SB203580 treatment. c, SP600125 (JNK 1/2 inhibitor; 10 μm) treatment before and during priming prevented the inhibition of LTP in SR due to priming in SO. d, Priming stimulation elicited homosynaptic LTP in SO that was unaltered by SP600125. e, Similar to SB203580 and SP600125, PD98059 (ERK 1/2 inhibitor; 10 μm) treatment before and during priming prevented the inhibition of LTP in SR due to priming in SO. f, However, priming stimulation elicited homosynaptic LTP in SO that was unaltered by PD98059. *p < 0.05. Data are mean ± SEM. n = 7–9 slices per group. Inset, Representative waveforms for the figures are averages of the final 10 sweeps taken before (pre) and 50–60 min after TBS (post) for SR and 80–90 min after HFS (post) for SO graphs, respectively. The numbers above the waveforms correspond to the treatment group, as indicated in the figure legend.
To investigate the role of JNK1/2 in heterodendritic metaplasticity, SP600125 (10 μm), a selective inhibitor of JNK that competes with ATP to inhibit the phosphorylation of c-Jun (Wei et al., 2011), was bath-applied to the slices. There was an overall significant treatment effect on SR LTP (F(2,18) = 20.62, p = 0.0003). Post hoc analysis revealed that there was significant inhibition of LTP by priming (Control DMSO: 150 ± 3%, n = 7; Primed DMSO: 124 ± 4%, n = 7; p = 0.009) that was blocked by SP600125 (Control SP600125: 154 ± 3%, n = 7; Primed SP600125: 163 ± 5%, n = 7; p = 0.0007; Fig. 6c). SP600125 had no effect on control SR LTP by itself (t(15) = 0.19, p = 1.11), nor on SO LTP (Primed ACSF: 157 ± 12%, n = 8; Primed SP600125: 163 ± 8%, n = 7, t(13) = 0.39, p = 0.86; Fig. 6d).
To investigate the role of ERK1/2 in heterodendritic metaplasticity, PD98059 (10 μm), which specifically inhibits MEK-mediated activation of ERK1/2, was bath-applied to the slices (Dudley et al., 1995). There was again a significant effect of treatment on SR LTP (F(2,20) = 6.622, p = 0.006) whereby there was significant inhibition of LTP by priming (Control DMSO: 153 ± 7%, n = 8; Primed DMSO: 127 ± 4%, n = 7; p = 0.002) that was blocked by PD98059 (Control PD98059: 149 ± 3%, n = 8; Primed PD98059: 142 ± 4%, n = 8; p = 0.31; Fig. 6e). In the control experiments, after washout, PD98059 had no effect on SR LTP by itself (t(14) = 0.39, p = 1.12). The final level of SO LTP due to the priming was also not significantly affected by PD98059 (Primed DMSO: 205 ± 12%, n = 8; Primed PD98059: 185 ± 8%, n = 8, t(14) = 0.29, p = 0.94; Fig. 6f), although there was a significant attenuation of SO LTP induction in the first 5 min after TBS (Primed DMSO: 279 ± 16%, n = 8; Primed PD98059: 216 ± 15%, n = 8, t(14) = 10.92, p = 0.033; Fig. 6f).
Blockade of TNFα priming by P38 MAPK, ERK, and JNK inhibitor
To confirm that p38 MAPK, ERK, and JNK were contributing to the TNFα-mediated heterodendritic metaplasticity effect, we tested whether inhibitors of these kinases would also block the pharmacological priming effect by TNFα. The inhibitors used in the study below were allowed to perfuse for 10 min before TNFα (1.18 nm) was delivered to the slices and then washed out for 30 min before the SR TBS to induce LTP.
There was an overall significant treatment effect of SB203580 on SR LTP (F(2,18) = 6.071, p = 0.01), such that LTP was significantly inhibited by TNFα (Control ACSF: 149.5 ± 6%, n = 7; TNFα: 127.8 ± 8%, n = 7; p = 0.008). This effect was blocked by treatment with SB203580 (10 μm) (TNFα: 127.8 ± 8%, n = 7; TNFα + SB203580: 153.6 ± 9%, n = 7; p = 0.007; Fig. 7a), confirming a role for p38 MAPK in the inhibition of LTP by TNFα. Similar results were obtained for the JNK1,2 inhibitor SP600125 (10 μm), for which there was an overall treatment effect on SR LTP (F(2,18) = 5.621, p = 0.013), with SP600125 blocking the TNFα priming effect (TNFα: 127.8 ± 8%, n = 7; TNFα + SP600125: 154.3 ± 10%, n = 7; p = 0.008; Fig. 7b). In experiments testing the effects of the ERK inhibitor PD98059 (10 μm), there was again an overall significant treatment effect on SR LTP (F(2,18) = 6.335, p = 0.008), with PD98059 blocking the TNFα mediated priming effect in SR (TNFα: 127.8 ± 8%, n = 7; TNFα + PD98059: 143.7 ± 8%, n = 7; p = 0.039; Fig. 7c).
Figure 7.
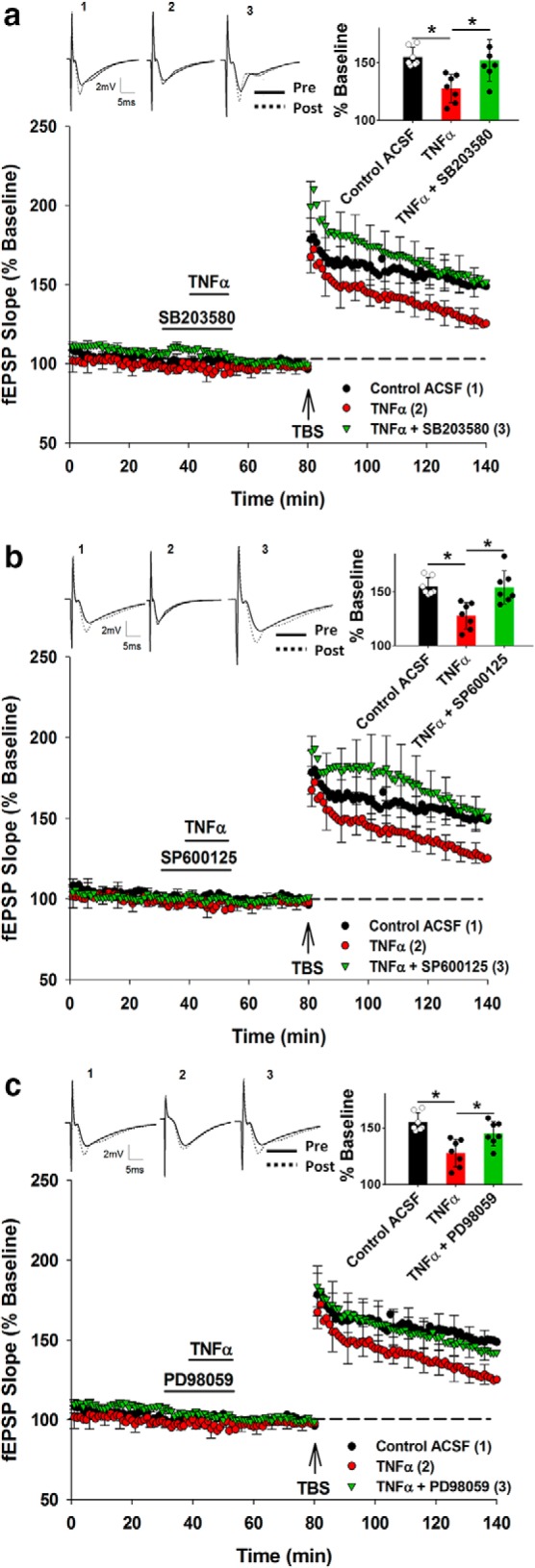
Pharmacological priming with TNFα and its block by inhibitors of p38 MAPK, JNK, and ERK. a, TNFα produced a priming effect that was blocked by application of p38 MAPK inhibitor SB203580. b, The JNK inhibitor SP600125 also blocked the priming effect produced by TNFα. c, Similarly, application of ERK inhibitor PD98059 blocked the priming effect produced by TNFα. *p < 0.05. Data are mean ± SEM. n = 7–9 slices per group. Inset, Representative waveforms for the figures are averages of the final 10 sweeps taken before (pre) and 50–60 min after TBS (post) for SR. The numbers above the waveforms correspond to the treatment group, as indicated in the figure legend.
Together, these experiments confirm that TNFα plays a key role in mediating the heterodendritic metaplasticity, as TNFα by itself can reproduce the priming effect, through activating the same signaling cascades as electrical priming.
Role of TNFα in APP/PS1 mice
Role of TNFα in mediating LTP impairment in APP/PS1 mice
We next addressed whether TNFα has a role to play in the impairment of LTP in a mouse model of the AD. We focused on two different mouse ages, 4 and 14 months, as Congo Red staining (Ryan et al., 2018) has demonstrated a negligible amount of Aβ plaque deposition in this APP/PS1 model at 4 months of age whereas Aβ plaque deposition was considerable in Tg mice 14–15 months of age.
We first performed ELISAs to quantify the amount of TNFα and IL-1β in WT and Tg brain tissue to correlate with the impairment of LTP as previously reported in this mouse model of AD. A one-way ANOVA revealed an overall group difference in TNFα (F(3,36) = 64.8, p = 0.0012). Although there was no significant difference in the concentration of TNFα between young WT versus Tg mice (young WT: 44.2 ± 13.8 pg/mg total protein, n = 10; young Tg: 56.7 ± 14.4 pg/mg total protein, n = 10; p = 0.07), the TNFα concentration in aged mice was twofold higher in Tg mice compared with their WT littermates (aged WT: 49.6 ± 14.3 pg/mg total protein, n = 10; aged Tg: 121.5 ± 18.4 pg/mg total protein, n = 10; p = 0.0002; Fig. 8a).
Figure 8.
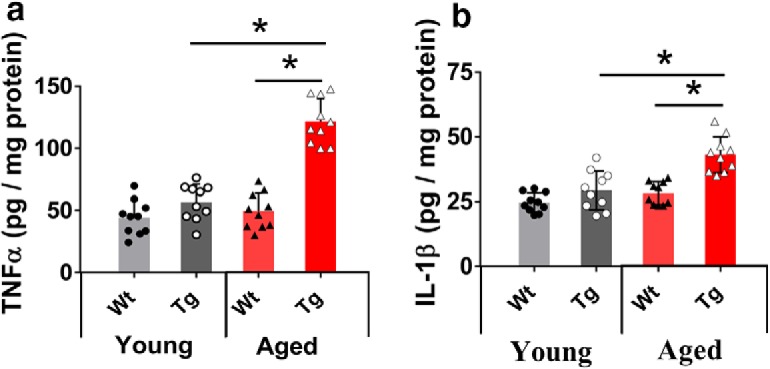
Quantification of TNFα and IL-1β concentrations in young and aged mouse hippocampus. Scatter plot with histogram represents the concentration of TNFα and IL-1β (pg/mg protein) analyzed by ELISA in the hippocampus of young (4 months) and aged mice (14 months). a, In the young mice, the concentration of TNFα was not different between genotypes. However, in aged Tg mice, the concentration of TNFα was elevated more than twofold compared with their WT littermates. The elevation in the concentration of TNFα in aged mice was also significantly different from the concentration of TNFα in young Tg mice. b, In the young mice, the concentration of IL-1β was not different between genotypes. In aged Tg mice, the concentration of IL-1β was elevated compared with their WT littermates. The elevation in the concentration of IL-1β in aged mice was also significantly different from the concentration of IL-1β in young Tg mice. *p < 0.05. Data are mean ± SEM. n = 10 samples, each sample containing the hippocampus from a single hemisphere.
One-way ANOVA also revealed a significant overall group difference in the concentration of IL-1β (F(3,36) = 21.92, p = 0.007). As for TNFα, there was no significant difference in IL-1β concentration between young WT and Tg mice (young WT: 24.7 ± 3.6 pg/mg total protein, n = 10; young Tg: 29.3 ± 7.4 pg/mg total protein, n = 10; p = 0.08), whereas the IL-1β concentration in aged Tg mice was significantly higher than in littermate WT mice, but only by ∼50% (aged WT: 28.2 ± 4.4 pg/mg total protein, n = 10; aged Tg: 43.1 ± 6.91 pg/mg total protein, n = 10; p = 0.003; Fig. 8b). Similar concentrations of TNFα and IL-1β in APP/PS1 brain slices have been reported previously (Patel et al., 2005).
TNFα mediates heterodendritic metaplasticity in young WT but not Tg mice
We next addressed whether TNFα-mediated heterodendritic metaplasticity also occurs in young WT and Tg mice. As for rats, a one-way ANOVA revealed that there was a significant treatment effect on SR LTP in young WT mice (F(3,28) = 4.6, p = 0.013), with a significant inhibition of SR LTP by priming (Control ACSF: 159 ± 8%, n = 7; Primed ACSF: 131 ± 5%, n = 7; p = 0.012), and its prevention by preincubation with the TNFα Ab (25 μg/ml) (Control TNFα Ab: 156 ± 7%, n = 8; Primed TNFα Ab: 155 ± 12%, n = 8; p = 0.24). The TNFα Ab had no effect on control SR LTP by itself (t(13) = 0.22, p = 0.39; Fig. 9a) nor on the priming-mediated LTP in SO (Fig. 10a).
Figure 9.

TNFα-dependent heterodendritic metaplasticity in WT and aged APP/PS1 but not young APP/PS1 mice. a, At SR synapses in young WT mice, priming-mediated inhibition of LTP was prevented by TNFα Ab preincubation. b, Control LTP in young Tg mice was impaired compared with WT mice and was not rescued by the TNFα Ab. However, treatment with the TNFα Ab in the primed group prevented the impairment caused by priming. c, At SR synapses in aged WT mice, priming-mediated inhibition of LTP was entirely prevented by the TNFα Ab preincubation. The control ACSF group LTP was not significantly different from the Control TNFα Ab group LTP, so only Control TNFα Ab data are shown in the graphs for clarity. d, Endogenous impairment of LTP in aged Tg mice was TNFα-dependent. At SR synapses, TNFα Ab rescued the endogenous inhibition of LTP in aged Tg mice. e, At SR synapses, IL-1β Ab did not affect priming-mediated inhibition of LTP in aged WT mice. The control ACSF group LTP was not significantly different from the Control IL-1β Ab group LTP, so only Control IL-1β Ab data are shown in the graphs for clarity. f, IL-1β Ab did not affect the endogenous inhibition of LTP in aged Tg mice. *p < 0.05. Data are mean ± SEM. n = 7–9 slices per group. Inset, Representative waveforms for the figures are averages of the final 10 sweeps taken before (pre) and 50–60 min after TBS (post) for SR. The numbers above the waveforms correspond to the treatment group, as indicated in the figure legend.
Figure 10.
Summary of TNFα Ab treatment effects on SO LTP in young and aged mice. a, In young Tg mice, SO LTP was not impaired, and not affected by preincubation with the TNFα Ab. In young Tg mice, LTP in SO was similar to that in WT age-matched littermates and also not affected by preincubation the TNFα Ab. Thus, TNFα did not affect SO LTP in young Tg mice or WT mice. b, In aged WT and Tg mice, priming stimulation in SO elicited homosynaptic LTP. The WT LTP was similar to young WT LTP (Fig. 4a), and preincubation with the TNFα Ab did not affect the SO LTP. In contrast, SO LTP in aged Tg mice was already inhibited compared with the LTP in the WT littermates. However, there was no significant effect of the TNFα Ab. *p < 0.05. Data are mean ± SEM. n = 7–9 slices per group.
The SR LTP for the young Tg mice was already inhibited, even without priming compared with their WT littermates (Control ACSF (WT) = 159 ± 8%, n = 7; Control ACSF (Tg): 133 ± 5%, n = 10; t(15) = 2.03, p = 0.042). However, for the Tg mice, there was still an overall significant treatment effect on SR LTP (F(3,33) = 10.05, p = 0.03). Post hoc analysis revealed that there was a small but significant inhibition of LTP by priming (Control ACSF: 133 ± 5%, n = 10; Primed ACSF: 122 ± 4%, n = 13; p = 0.042), that was prevented by the TNFα Ab (Control ACSF: 133 ± 5%, n = 10; Primed TNFα Ab: 146 ± 4%, n = 7; p = 0.03; Fig. 9b); however, the inhibited LTP in the Tg control group was not rescued to WT levels by the TNFα Ab (t(15) = 1.05, p = 0.24). In contrast to SR LTP, SO LTP in the Tg mice was not different from the SR LTP in their WT littermates. Preincubation with TNFα Ab also did not affect the SO LTP (Fig. 10a). These data indicate that, while there is an endogenous inhibition of LTP in the young Tg mice, it is TNFα-independent and only apparent in SR. This is in contrast to the additional inhibition of LTP by priming stimulation that was TNFα-dependent.
TNFα mediates heterodendritic metaplasticity in aged WT and endogenous inhibition of LTP in Tg mice
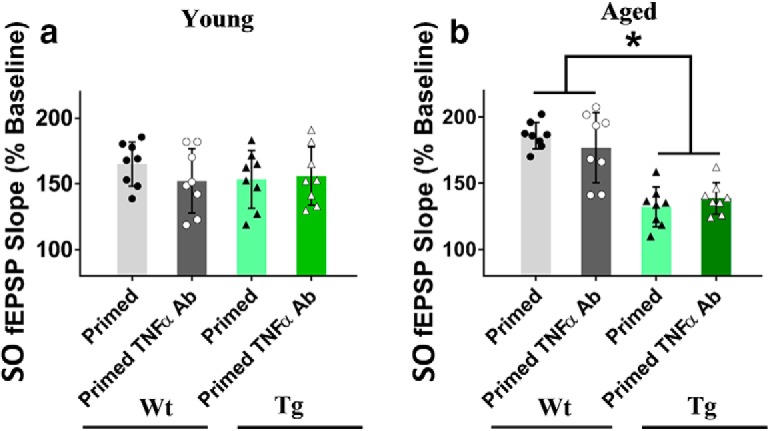
For the aged WT mice, there was a significant treatment effect on SR LTP (F(2,21) = 3.05, p = 0.027), such that priming impaired LTP in control mice (Control ACSF: 142 ± 5%, n = 9; Primed ACSF: 121 ± 3%, n = 9; p = 0.01), while the priming effect was blocked by preincubation with the TNFα Ab (25 μg/ml) (Control TNFα Ab: 146 ± 8%, n = 9; Primed TNFα Ab: 140 ± 8%, n = 9; p = 0.02; Fig. 9c). The TNFα Ab had no effect on control SR LTP by itself (t(16) = 0.93, p = 0.83), nor on the SO LTP elicited by priming stimulation (Fig. 10b). Thus, the heterodendritic metaplasticity effect was evident in both young and aged WT mice.
Compared with WT littermates, the SR LTP for Tg mice was significantly impaired (Control ACSF (WT): 142 ± 5%, n = 9; Control ACSF (Tg): 128 ± 5%, n = 9; t(16) = 3.77; p = 0.018). Moreover, there was an overall significant treatment effect on SR LTP for the Tg mice (F(2,24) = 5.01, p = 0.025) whereby, in contrast to the young Tg mice, there was no significant further inhibition of due to priming (Control ACSF: 128 ± 5%, n = 9; Primed ACSF: 123 ± 3%, n = 9; p = 0.22). Moreover, the endogenous inhibition of LTP was completely rescued by preincubation with the TNFα Ab in nonprimed Tg mice (Control ACSF: 128 ± 5%, n = 9; Control TNFα Ab: 142 ± 8%, n = 9; p = 0.017; Fig. 9d). Priming stimulation elicited homosynaptic LTP in SO, but this was not affected by the TNFα Ab treatment in Tg mice (Fig. 10b). Interestingly, two-way ANOVA demonstrated that there was a significant genotype effect present for the aged mice (i.e., less SO LTP in the aged mice albeit not as strongly impaired as in SR), but there was no TNFα Ab treatment effect for either genotype in the SO pathway (Fig. 10b).
IL-1β independent heterodendritic metaplasticity in aged WT mice
Although the TNFα Ab completely prevented the heterodendritic metaplasticity in aged WT mice, we nonetheless also investigated whether the IL-1β Ab would be equally effective. For the aged WT mice, there was a significant group effect for SR LTP (F(2,22) = 3.51, p = 0.041), with a significant inhibition of LTP by priming (Control ACSF: 146 ± 8%, n = 8; Primed ACSF: 119 ± 6%, n = 8; p = 0.002), but no effect of the IL-1β Ab (25 μg/ml) treatment (Control IL-1β Ab: 141 ± 8%, n = 8; Primed IL-1β Ab: 125 ± 11%, n = 9; p = 0.027; Fig. 9e). The IL-1β Ab also had no effect on either the control level of SR LTP (t(14) = 1.12, p = 0.97) or the SO LTP (data not shown).
For the aged Tg group, and as reported above for the TNFα experiment, SR LTP was significantly inhibited in the nonprimed control group compared with their WT littermates (Control ACSF (WT): 146 ± 8%, n = 8; Control ACSF (Tg): 128 ± 4%, n = 9; t(16) = 4.14, p = 0.02). However, unlike for the TNFα Ab, there was no overall group effect for the aged Tg mice (F(2,27) = 0.88, p = 0.098). While, as before, there was no inhibition of LTP due to priming (Control ACSF: 128 ± 4%, n = 9; Primed ACSF: 121 ± 5%, n = 9; p = 0.71), the endogenous inhibition of LTP in the nonprimed control group was not affected by the IL-1β Ab (Control ACSF: 128 ± 4%, n = 9; Control IL-1β Ab: 131 ± 4%, n = 9; t(16) = 0.57, p = 0.12; Fig. 9f), and thus there was no rescue of the LTP to WT levels. Two-way ANOVA of the SO LTP data confirmed an overall impairment in the Tg mice, but the IL-1β Ab (as for the TNFα Ab) had no effect on LTP for either genotype (data not shown).
Discussion
Synaptic strengthening in area CA1 of the rat hippocampus can be constrained by episodes of prior neural activity that affects subsequent plasticity thresholds (Wang and Wagner, 1999; Hulme et al., 2012) via metaplasticity mechanisms (Abraham and Bear, 1996). Since priming stimulation in the basal dendritic region impairs LTP in the apical dendritic region (Jones et al., 2013), it is possible that this long-range communication uses intercellular interactions between neurons and glia, as has been reported for spinal cord LTP (Kronschläger et al., 2016). Astrocytes are known to respond to M1-AChR activation by releasing gliotransmitters that regulate plasticity (Takata et al., 2011; Navarrete et al., 2012), and thus astrocytes activated in CA1 following priming stimulation (Kang et al., 1998; Hulme et al., 2014) might be the missing link in mediating the heterodendritic metaplasticity effect. Alternatively, electrical stimulation-activated astrocytic Ca2+ waves elevate microglial activity (Schipke et al., 2002), causing release of cytokines (Elkabes et al., 1996; Hanisch, 2002) as well as neurotransmitters (Hayashi et al., 2006; Pascual et al., 2012) from microglia. Therefore, we hypothesized that electrical priming stimulation in SO inhibits LTP in SR by eliciting the widespread release of cytokines from neuron-glia interactions that spread across CA1 strata.
In the present experiments, acute treatment of rat slices with TNFα antibodies as well as thalidomide, but not IL-1β antibodies, blocked the LTP inhibition due to priming stimulation. Moreover, treatment with exogenous 1.18 nm TNFα, but not IL-1β, at the same concentration was sufficient to metaplastically impair LTP to the same extent as produced by electrical priming. These findings extend prior studies that concurrently administered TNFα impairs LTP in the hippocampus (Liu et al., 2017; Samidurai et al., 2018). At a higher concentration, however, IL-1β (5 nm) was able to produce the priming effect. As the IL-1β Ab blocked this effect, the failure of the IL-1β Ab to block the electrical priming effect was thus not due to an inability to block the actions of IL-1β protein, confirming a selective contribution by TNFα to the metaplasticity effect.
We then addressed the intracellular signaling pathways activated by TNFα to generate the effect. TNFα induces phosphorylation of p38 MAPK (Barbin et al., 2001), ERK (McLeish et al., 1998), and JNK (Barbin et al., 2001). Our Western blot analyses revealed that priming caused substantial increases in phosphorylation of all three kinases at the time that the SR TBS would have been delivered. Correspondingly, we found that bath-applying inhibitors of each of the three kinases abolished the inhibition of LTP by both electrical priming and by exogenous TNFα. Although whether these kinases work in parallel or serial cascade fashion, and in the same cellular compartments, remains to be studied, the results indicate that TNFα signal transduction cascades are recruited by priming stimulation to generate the inhibition of LTP.
The importance of p38 MAP kinase and JNK activation for the inhibition of LTP is consistent with previous studies of these kinases (Saleshando and O'Connor, 2000; Butler et al., 2004; Costello and Herron, 2004), but the connection between ERK and LTP inhibition was surprising given that it has traditionally been associated with the activation of protein synthesis and late-phase LTP (English and Sweatt, 1996; Jin and Feig, 2010). However, protein synthesis is also essential for late-phase LTD and metabotropic glutamate receptor-dependent LTD (Gallagher et al., 2004), and any activation of these mechanisms or other ERK-mediated mechanisms by TNFα might act to suppress the LTP mechanisms. Another possibility is that the TNFα mechanisms are upstream of extrasynaptic GluN2B-containing NMDA receptor activation in area CA1. It has been demonstrated that TNFα promotes glutamate release (Santello et al., 2011) and the activation of GluN2B-containing receptors that leads to cognitive impairment (Hu et al., 2009). Previous studies have indicated that glutamate-mediated activation of GluN2B-containing receptors inhibits LTP and enhances LTD under specific conditions (Li et al., 2011; Yang et al., 2017). Moreover, GluN2B antagonists block the LTP impairment due to amyloid-β (Hu et al., 2009). These arguments, therefore, suggest a possible critical role of GluN2B receptors in mediating inhibition of LTP in both healthy and disease conditions.
Interestingly, the SO LTP was not changed by any of these antagonists, even though ERK antagonists have in particular been found to impair LTP in SR of CA1 (Winder et al., 1999). One possible explanation is that the requirements for plasticity induction in SR differ from those in SO due to the difference in their protein-synthesis dependency, suggesting distinct roles in mediating short- and long-term plasticity (Hagena and Manahan-Vaughan, 2013). Alternatively, HFS (for SO priming) and TBS (for SR LTP) can trigger synaptic plasticity by activating different intracellular pathways (Zhu et al., 2015), potentially accounting for why SO LTP was unaffected by the ERK inhibitor. The difference between SO and SR LTP is emphasized by the lack of effect of soluble amyloid-β on SO LTP (Zhao et al., 2018), and the relative lack of LTP impairment in SO in the Tg mice in the present study. Thus, LTP in SO and SR involves at least partially different mechanisms, likely due to their different morphologies, biophysical properties, and molecular mechanisms (Son et al., 1996; Kramár and Lynch, 2003), which impart different characteristics to the two compartments.
The other objective for this study was to test the hypothesis that the heterodendritic metaplasticity mechanism is aberrantly engaged in the APP/PS1 mouse model of AD, thus accounting for the impaired SR LTP in these mice. We first confirmed that TNFα Ab blocked the heterodendritic metaplasticity in young WT mice, similar to young adult rats. Young Tg mice already displayed an impairment of LTP under control conditions and only a small further impairment following SO priming. The TNFα Ab blocked the small priming effect but could not reverse the endogenous impairment of LTP, indicating that the endogenous impairment of LTP in young Tg mice was independent of TNFα. This was consistent with the lack of an increased level of TNFα in the tissue of young Tg mice. Thus, the constitutive release of TNFα in the young Tg (and WT) mouse brain is insufficient to inhibit LTP, and only by raising the level of TNFα released into the extracellular space by priming stimulation can the LTP impairment be revealed. At this point, the TNFα inhibitors become active and rescue the impaired LTP to normal levels. What then explains the moderate impairment of LTP in the young Tg mice? Since soluble Aβ aggregates are likely to account for this endogenous inhibition of LTP (Takeda et al., 2013), other non-TNFα mechanisms must be in play, such as internalization of NMDA receptors from the plasma membrane (Snyder et al., 2005).
The priming effect was also visible in aged WT mice, and the TNFα Ab again blocked the effect, indicating a ubiquitous mechanism across species and ages. However, Tg littermates already exhibited a strong endogenous inhibition of LTP, even without priming that completely occluded any further reduction in LTP by the priming stimulation. Importantly, the TNFα Ab completely rescued the endogenously inhibited LTP to WT levels, and this finding was consistent with the highly elevated levels of TNFα in the aged Tg brains detected by ELISA. We speculate that the presence of the Aβ plaques in the extracellular space at this age (Palop and Mucke, 2010) causes activation of astrocytes (Rojo et al., 2008), which has been linked with active forms of p38 MAPK, JNK, and ERK (Migheli et al., 1997; Mandell and VandenBerg, 1999; Roy Choudhury et al., 2014), or microglia (Yin et al., 2017), potentially mediating the constitutive release of TNFα into the extracellular space. This supports our hypothesis that constitutive release of TNFα in the disease condition could lead to aberrant endogenous engagement of the TNFα-mediated metaplasticity mechanisms, thereby causing the impairment of LTP. Unlike the TNFα Ab, the IL-1β Ab did not affect the heterodendritic metaplasticity effect in aged WT and the endogenous impairment in aged Tg mice. Therefore, this study provides evidence that IL-1β does not play a role in heterodendritic metaplasticity in WT brain, nor the Tg brain, even though the cytokine is at an elevated level in the latter. However, it remains possible that IL-1β becomes involved at a later age of Tg mice, or in a different model.
Overall, we propose that heterodendritic metaplasticity may be an important form of homeostatic synaptic plasticity regulation and potentially memory storage in the hippocampus that entails the release of the cytokine TNFα. However, when this mechanism is aberrantly engaged, it mediates the impairment of LTP in at least one model of AD. Although such metaplasticity could work to reduce excitotoxicity from the hyperexcitability seen in such amyloid-based mouse models of AD (Hall and Roberson, 2012), it could come at the cost of impairing memory and cognition. Nonetheless, given the potential involvement of TNFα in AD as suggested by preclinical experiments (He et al., 2007; McAlpine et al., 2009), it is worth exploring the optimal dosage and route of administration of TNFα antagonists/inhibitors in the treatment of this currently incurable disease (Clark and Vissel, 2016; MacPherson et al., 2017).
Footnotes
This work was supported by the Health Research Council of New Zealand to W.C.A., B.G.M., and O.D.J., Neurological Foundation of New Zealand to W.C.A. and O.D.J., and a University of Otago postgraduate scholarship to A.S.
The authors declare no competing financial interests.
References
- Abraham WC. (2008) Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci 9:387. 10.1038/nrn2356 [DOI] [PubMed] [Google Scholar]
- Abraham WC, Bear MF (1996) Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci 19:126–130. 10.1016/S0166-2236(96)80018-X [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215. 10.1016/S0166-2236(98)01349-6 [DOI] [PubMed] [Google Scholar]
- Barbin G, Roisin MP, Zalc B (2001) Tumor necrosis factor alpha activates the phosphorylation of ERK, SAPK/JNK, and P38 kinase in primary cultures of neurons. Neurochem Res 26:107–112. 10.1023/A:1011086426652 [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC (2002) Control of synaptic strength by glial TNFα. Science 295:2282–2285. 10.1126/science.1067859 [DOI] [PubMed] [Google Scholar]
- Bellinger FP, Madamba S, Siggins GR (1993) Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res 628:227–234. 10.1016/0006-8993(93)90959-Q [DOI] [PubMed] [Google Scholar]
- Brosseron F, Krauthausen M, Kummer M, Heneka MT (2014) Body fluid cytokine levels in mild cognitive impairment and Alzheimer's disease: a comparative overview. Mol Neurobiol 50:534–544. 10.1007/s12035-014-8657-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MP, O'Connor JJ, Moynagh PN (2004) Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience 124:319–326. 10.1016/j.neuroscience.2003.11.040 [DOI] [PubMed] [Google Scholar]
- Clark IA, Vissel B (2016) Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents. J Neuroinflammation 13:236. 10.1186/s12974-016-0708-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello DA, Herron CE (2004) The role of c-jun N-terminal kinase in the A beta-mediated impairment of LTP and regulation of synaptic transmission in the hippocampus. Neuropharmacology 46:655–662. 10.1016/j.neuropharm.2003.11.016 [DOI] [PubMed] [Google Scholar]
- Cunningham AJ, Murray CA, O'Neill LA, Lynch MA, O'Connor JJ (1996) Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett 203:17–20. 10.1016/0304-3940(95)12252-4 [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR (1995) A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A 92:7686–7689. 10.1073/pnas.92.17.7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabes S, DiCicco-Bloom EM, Black IB (1996) Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci 16:2508–2521. 10.1523/JNEUROSCI.16-08-02508.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD (1996) Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem 271:24329–24332. 10.1074/jbc.271.40.24329 [DOI] [PubMed] [Google Scholar]
- Enomoto N, Takei Y, Hirose M, Ikejima K, Kitamura T, Sato N (2004) Thalidomide prevents alcoholic liver injury in rats through inhibition of Kupffer cell sensitization. Comp Hepatol 3 [Suppl 1]:S37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Burnstock G (2006) Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci 7:423–436. 10.1038/nrn1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM (2004) Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci 24:4859–4864. 10.1523/JNEUROSCI.5407-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan H, Levav T, Mendelsohn A, Huleihel M (2004) Involvement of tumor necrosis factor alpha in hippocampal development and function. Cereb Cortex 14:97–105. 10.1093/cercor/bhg108 [DOI] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL 3rd, Araoz C (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A 86:7611–7615. 10.1073/pnas.86.19.7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagena H, Manahan-Vaughan D (2013) Differentiation in the protein synthesis-dependency of persistent synaptic plasticity in mossy fiber and associational/commissural CA3 synapses in vivo. Front Integr Neurosci 7:10. 10.3389/fnint.2013.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AM, Roberson ED (2012) Mouse models of Alzheimer's disease. Brain Res Bull 88:3–12. 10.1016/j.brainresbull.2011.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK. (2002) Microglia as a source and target of cytokines. Glia 40:140–155. 10.1002/glia.10161 [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Ishibashi H, Hashimoto K, Nakanishi H (2006) Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors. Glia 53:660–668. 10.1002/glia.20322 [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86:1009–1031. 10.1152/physrev.00049.2005 [DOI] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y (2007) Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol 178:829–841. 10.1083/jcb.200705042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA (2010) Long-term potentiation depends on release of d-serine from astrocytes. Nature 463:232–236. 10.1038/nature08673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Hasegawa K, Kamiya H, Morimoto Y (2017) Synapse-specific effects of IL-1beta on long-term potentiation in the mouse hippocampus. Biomed Res 38:183–188. 10.2220/biomedres.38.183 [DOI] [PubMed] [Google Scholar]
- Hu NW, Klyubin I, Anwy R, Rowan MJ (2009) GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc Natl Acad Sci U S A 106:20504–20509. 10.1073/pnas.0908083106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme SR, Jones OD, Ireland DR, Abraham WC (2012) Calcium-dependent but action potential-independent BCM-like metaplasticity in the hippocampus. J Neurosci 32:6785–6794. 10.1523/JNEUROSCI.0634-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme SR, Jones OD, Raymond CR, Sah P, Abraham WC (2014) Mechanisms of heterosynaptic metaplasticity. Philos Trans R Soc Lond B Biol Sci 369:20130148. 10.1098/rstb.2013.0148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Feig LA (2010) Long-term potentiation in the CA1 hippocampus induced by NR2A subunit-containing NMDA glutamate receptors is mediated by ras-GRF2/Erk map kinase signaling. PLoS One 5:e11732. 10.1371/journal.pone.0011732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones OD, Hulme SR, Abraham WC (2013) Purinergic receptor- and gap junction-mediated intercellular signalling as a mechanism of heterosynaptic metaplasticity. Neurobiol Learn Mem 105:31–39. 10.1016/j.nlm.2013.05.010 [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M (1998) Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci 1:683–692. 10.1038/3684 [DOI] [PubMed] [Google Scholar]
- Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M (1990) Interleukin-1 beta inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol 181:323–326. 10.1016/0014-2999(90)90099-R [DOI] [PubMed] [Google Scholar]
- Kramár EA, Lynch G (2003) Developmental and regional differences in the consolidation of long-term potentiation. Neuroscience 118:387–398. 10.1016/S0306-4522(02)00916-8 [DOI] [PubMed] [Google Scholar]
- Kronschläger MT, Drdla-Schutting R, Gassner M, Honsek SD, Teuchmann HL, Sandkühler J (2016) Gliogenic LTP spreads widely in nociceptive pathways. Science 354:1144–1148. 10.1126/science.aah5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AJ, Katafuchi T, Oda S, Hori T, Oomura Y (1997) Interleukin-6 inhibits long-term potentiation in rat hippocampal slices. Brain Res 748:30–38. 10.1016/S0006-8993(96)01283-8 [DOI] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31:6627–6638. 10.1523/JNEUROSCI.0203-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, Wei X, Xu T, Xin WJ, Pang RP, Li YY, Qin ZH, Murugan M, Mattson MP, Wu LJ, Liu XG (2017) TNF-α differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J Neurosci 37:871–881. 10.1523/JNEUROSCI.2235-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YL, Zhou LJ, Hu NW, Xu JT, Wu CY, Zhang T, Li YY, Liu XG (2007) Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology 52:708–715. 10.1016/j.neuropharm.2006.09.011 [DOI] [PubMed] [Google Scholar]
- MacPherson KP, Sompol P, Kannarkat GT, Chang J, Sniffen L, Wildner ME, Norris CM, Tansey MG (2017) Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol Dis 102:81–95. 10.1016/j.nbd.2017.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell JW, VandenBerg SR (1999) ERK/MAP kinase is chronically activated in human reactive astrocytes. Neuroreport 10:3567–3572. 10.1097/00001756-199911260-00019 [DOI] [PubMed] [Google Scholar]
- McAlpine FE, Lee JK, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, Das P, Golde TE, LaFerla FM, Oddo S, Blesch A, Tansey MG (2009) Inhibition of soluble TNF signaling in a mouse model of Alzheimer's disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis 34:163–177. 10.1016/j.nbd.2009.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeish KR, Knall C, Ward RA, Gerwins P, Coxon PY, Klein JB, Johnson GL (1998) Activation of mitogen-activated protein kinase cascades during priming of human neutrophils by TNF-alpha and GM-CSF. J Leukoc Biol 64:537–545. 10.1002/jlb.64.4.537 [DOI] [PubMed] [Google Scholar]
- Migheli A, Piva R, Atzori C, Troost D, Schiffer D (1997) c-jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 56:1314–1322. 10.1097/00005072-199712000-00006 [DOI] [PubMed] [Google Scholar]
- Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G (1993) Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J Exp Med 177:1675–1680. 10.1084/jem.177.6.1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G (2005) Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter d-serine. Proc Natl Acad Sci U S A 102:5606–5611. 10.1073/pnas.0408483102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CA, Lynch MA (1998) Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci 18:2974–2981. 10.1523/JNEUROSCI.18-08-02974.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete M, Perea G, Fernandez de Sevilla D, Gómez-Gonzalo M, Núñez A, Martín ED, Araque A (2012) Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol 10:e1001259. 10.1371/journal.pbio.1001259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Verkhratsky A (2006) Purinergic transmission in the central nervous system. Pflugers Arch 452:479–485. 10.1007/s00424-006-0060-y [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2010) Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 13:812–818. 10.1038/nn.2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH (2006) Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell 125:775–784. 10.1016/j.cell.2006.02.051 [DOI] [PubMed] [Google Scholar]
- Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A (2012) Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A 109:E197–E205. 10.1073/pnas.1111098109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ (2005) Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer's disease. J Neuroinflammation 2:9. 10.1186/1742-2094-2-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O'Connor JJ (2005) Actions of TNF-α on glutamatergic synaptic transmission in the central nervous system. Exp Physiol 90:663–670. 10.1113/expphysiol.2005.030734 [DOI] [PubMed] [Google Scholar]
- Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB (2008) Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer's disease. Arch Med Res 39:1–16. 10.1016/j.arcmed.2007.10.001 [DOI] [PubMed] [Google Scholar]
- Roy Choudhury G, Ryou MG, Poteet E, Wen Y, He R, Sun F, Yuan F, Jin K, Yang SH (2014) Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res 1551:45–58. 10.1016/j.brainres.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MM, Guévremont D, Mockett BG, Abraham WC, Williams JM (2018) Circulating plasma microRNAs are altered with amyloidosis in a mouse model of Alzheimer's disease. J Alzheimers Dis 66:835–852. 10.3233/JAD-180385 [DOI] [PubMed] [Google Scholar]
- Saleshando G, O'Connor JJ (2000) SB203580, the p38 mitogen-activated protein kinase inhibitor blocks the inhibitory effect of beta-amyloid on long-term potentiation in the rat hippocampus. Neurosci Lett 288:119–122. 10.1016/S0304-3940(00)01210-6 [DOI] [PubMed] [Google Scholar]
- Samidurai M, Ramasamy VS, Jo J (2018) β-amyloid inhibits hippocampal LTP through TNFR/IKK/NF-κB pathway. Neurol Res 40:268–276. [DOI] [PubMed] [Google Scholar]
- Santello M, Bezzi P, Volterra A (2011) TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69:988–1001. 10.1016/j.neuron.2011.02.003 [DOI] [PubMed] [Google Scholar]
- Schipke CG, Boucsein C, Ohlemeyer C, Kirchhoff F, Kettenmann H (2002) Astrocyte Ca2+ waves trigger responses in microglial cells in brain slices. FASEB J 16:255–257. 10.1096/fj.01-0514fje [DOI] [PubMed] [Google Scholar]
- Schroeter M, Jander S (2005) T-cell cytokines in injury-induced neural damage and repair. Neuromolecular Med 7:183–195. 10.1385/NMM:7:3:183 [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8:1051–1058. 10.1038/nn1503 [DOI] [PubMed] [Google Scholar]
- Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER (1996) Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell 87:1015–1023. 10.1016/S0092-8674(00)81796-1 [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial. Nature 440:1054–1059. 10.1038/nature04671 [DOI] [PubMed] [Google Scholar]
- Takata N, Mishima T, Hisatsune C, Nagai T, Ebisui E, Mikoshiba K, Hirase H (2011) Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J Neurosci 31:18155–18165. 10.1523/JNEUROSCI.5289-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Hashimoto T, Roe AD, Hori Y, Spires-Jones TL, Hyman BT (2013) Brain interstitial oligomeric amyloid beta increases with age and is resistant to clearance from brain in a mouse model of Alzheimer's disease. FASEB J 27:3239–3248. 10.1096/fj.13-229666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tancredi V, D'Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F (1992) Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett 146:176–178. 10.1016/0304-3940(92)90071-E [DOI] [PubMed] [Google Scholar]
- Tavares JL, Wangoo A, Dilworth P, Marshall B, Kotecha S, Shaw RJ (1997) Thalidomide reduces tumour necrosis factor-alpha production by human alveolar macrophages. Respir Med 91:31–39. 10.1016/S0954-6111(97)90134-7 [DOI] [PubMed] [Google Scholar]
- Tramontana JM, Utaipat U, Molloy A, Akarasewi P, Burroughs M, Makonkawkeyoon S, Johnson B, Klausner JD, Rom W, Kaplan G (1995) Thalidomide treatment reduces tumor necrosis factor alpha production and enhances weight gain in patients with pulmonary tuberculosis. Mol Med 1:384–397. 10.1007/BF03401576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wagner JJ (1999) Priming-induced shift in synaptic plasticity in the rat hippocampus. J Neurophysiol 82:2024–2028. 10.1152/jn.1999.82.4.2024 [DOI] [PubMed] [Google Scholar]
- Wei J, Wang W, Chopra I, Li HF, Dougherty CJ, Adi J, Adi N, Wang H, Webster KA (2011) c-jun N-terminal kinase (JNK-1) confers protection against brief but not extended ischemia during acute myocardial infarction. J Biol Chem 286:13995–14006. 10.1074/jbc.M110.211334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER (1999) ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron 24:715–726. 10.1016/S0896-6273(00)81124-1 [DOI] [PubMed] [Google Scholar]
- Yang Q, Zhu G, Liu D, Ju JG, Liao ZH, Xiao YX, Zhang Y, Chao N, Wang J, Li W, Luo JH, Li ST (2017) Extrasynaptic NMDA receptor dependent long-term potentiation of hippocampal CA1 pyramidal neurons. Sci Rep 7:3045. 10.1038/s41598-017-03287-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Raj D, Saiepour N, Van Dam D, Brouwer N, Holtman IR, Eggen BJ, Möller T, Tamm JA, Abdourahman A, Hol EM, Kamphuis W, Bayer TA, De Deyn PP, Boddeke E (2017) Immune hyperreactivity of Abeta plaque-associated microglia in Alzheimer's disease. Neurobiol Aging 55:115–122. 10.1016/j.neurobiolaging.2017.03.021 [DOI] [PubMed] [Google Scholar]
- Zhao J, Li A, Rajsombath M, Dang Y, Selkoe DJ, Li S (2018) Soluble Abeta oligomers impair dipolar heterodendritic plasticity by activation of mGluR in the hippocampal CA1 region. iScience 6:138–150. 10.1016/j.isci.2018.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Liu Y, Wang Y, Bi X, Baudry M (2015) Different patterns of electrical activity lead to long-term potentiation by activating different intracellular pathways. J Neurosci 35:621–633. 10.1523/JNEUROSCI.2193-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]