

Abstract
Development of brain circuitry requires precise regulation and timing of proliferation and differentiation of neural progenitor cells. The p75 neurotrophin receptor (p75NTR) is highly expressed in the proliferating granule cell precursors (GCPs) during development of the cerebellum. In a previous paper, we showed that proNT3 promoted GCP cell cycle exit via p75NTR. Here we used genetically modified rats and mice of both sexes to show that p75NTR regulates the duration of the GCP cell cycle, requiring activation of RhoA. Rats and mice lacking p75NTR have dysregulated GCP proliferation, with deleterious effects on cerebellar circuit development and behavioral consequences persisting into adulthood. In the absence of p75NTR, the GCP cell cycle is accelerated, leading to delayed cell cycle exit, prolonged GCP proliferation, increased glutamatergic input to Purkinje cells, and a deficit in delay eyeblink conditioning, a cerebellum-dependent form of learning. These results demonstrate the necessity of appropriate developmental timing of the cell cycle for establishment of proper connectivity and associated behavior.
SIGNIFICANCE STATEMENT The cerebellum has been shown to be involved in numerous behaviors in addition to its classic association with motor function. Cerebellar function is disrupted in a variety of psychiatric disorders, including those on the autism spectrum. Here we show that the p75 neurotrophin receptor, which is abundantly expressed in the proliferating cerebellar granule cell progenitors, regulates the cell cycle of these progenitors. In the absence of this receptor, the cell cycle is dysregulated, leading to excessive progenitor proliferation, which alters the balance of inputs to Purkinje cells, disrupting the circuitry and leading to functional deficits that persist into adulthood.
Keywords: cerebellum, eyeblink conditioning, p75NTR, proliferation, RhoA
Introduction
Cerebellar granule cells comprise over half the neurons of the human and rodent brain. Granule cells receive a wide range of input from all over the brain, including sensory, motor, reward, cognitive, and social information. This diversity of input matches the growing appreciation of the cerebellum as not just a motor structure, but also a structure for supporting cognitive functions (Leiner et al., 1986; Fiez et al., 1992; Kim et al., 1994), reward conditioning (Wagner et al., 2017; Carta et al., 2019), emotion (Turner et al., 2007; Strata et al., 2011), fear conditioning (Sacchetti et al., 2004), and flexible learning and social behavior (Badura et al., 2018; Carta et al., 2019). The cerebellum is implicated in several psychiatric disorders, such as anxiety (Phillips et al., 2015; Moreno-Rius, 2018), schizophenia (Martin and Albers, 1995; Andreasen and Pierson, 2008; Phillips et al., 2015), and autism spectrum disorder (Fatemi et al., 2012; Becker and Stoodley, 2013; Wang et al., 2014; D'Mello and Stoodley, 2015). Indeed, deficits in cerebellum-dependent learning have been identified in multiple mouse models of autism (Kloth et al., 2015), and cerebellar disruption leads to autism-like symptoms in neonates (Stoodley and Limperopoulos, 2016) and in postnatally developing mice (Badura et al., 2018). Disruptions to granule cell processing would thus be expected to have a wide range of motor, cognitive, and social consequences.
During cerebellar development, granule cell progenitors (GCPs) undergo a massive clonal expansion in the external granule layer (EGL) and eventually differentiate into cerebellar granule neurons (CGNs), the most abundant cell type in the brain. Proliferation of the GCPs in the EGL is dependent on the presence of sonic hedgehog (Shh) secreted from Purkinje cells (PCs). However, mechanisms that regulate the duration of the cell cycle are not well characterized. Previously, we showed that the p75 neurotrophin receptor (p75NTR) is highly expressed in proliferating GCPs, and exposure to the proNT3 ligand elicits cell cycle exit. The absence of p75NTR in the EGL during cerebellar development was sufficient to induce an increase in GCP proliferation, with an enlarged cerebellum and deficits in motor function (Zanin et al., 2016).
CGNs exert their functional influence via the mossy fiber/granule cell pathway, which passes though PCs. After terminal postmitotic differentiation, CGNs migrate toward the internal part of the cerebellar cortex, leaving behind ascending axons, which form a T-like branch to form the parallel fibers, the principal source of excitation to PC dendrites and interneurons. Plasticity at these excitatory synapses contributes to cerebellum-dependent learning mechanisms, including sensory associative conditioning. PCs provide the sole output of information from the cerebellar cortex to the rest of the nervous system, projecting to the deep cerebellar nuclei (DCN), which transmit information to the rest of the brain. CGNs also receive a wide variety of converging sensory, motor efference, and other information from all over the brain. In this way, CGNs, acting though PCs play a central role in cerebellar information processing, as well as adaptive learning from changes in the sensory, motor, and cognitive environment.
Many autism-related genes are expressed in the cerebellum, and disruption of autism-associated genes can impair cerebellum-dependent associative learning, as measured using delay eyeblink conditioning (Kloth et al., 2015). Although p75NTR itself is not an autism susceptibility gene, neurotrophin signaling is necessary for normal circuit development and function, and levels of p75NTR mRNA as well as neurotrophin ligands, specifically NT3, were altered in autism spectrum disorder patients compared with normal controls (Sajdel-Sulkowska et al., 2009; Segura et al., 2015).
In the present study, we demonstrate that unliganded p75NTR regulates the duration of the cell cycle, requiring activation of RhoA. Deletion of this receptor from proliferating GCPs alters the timing of their cell cycle, affecting the development of cerebellar circuitry with increased excitatory drive of PCs. To test adult cerebellar function, we performed delay conditioning behavior and found deficits in this behavior as a consequence of p75 deletion from the EGL.
Materials and Methods
Animals.
All animal studies were conducted using the National Institutes of Health guidelines for the ethical treatment of animals with the approval of the Rutgers Animal Care and Facilities Committee or the Princeton Animal Care and Use Committee. Rats lacking p75NTR were obtained from SAGE/Horizon Laboratories and confirmed by us using PCR, Western blot, and immunostaining (see Fig. 1B), to be lacking p75NTR.
Figure 1.

Increase in cell cycle speed in GCPs lacking p75NTR. A, Granule cell cultures from WT and p75NTR−/− rats labeled with EdU (red) and BrdU (green). Edu was added for 1 h and washed out; BrdU was added for 2 h window between 12 and 24 h after the EdU. Cells were fixed and stained for both markers. EdU- and BrdU-positive cells represent cells that undergo a second round of S phase in the indicated time after the EdU pulse. The proliferation fraction represented as the ratio of double-labeled cells over the total EdU-positive cells. p = 0.0003 (Sidak's post hoc test following a two-way ANOVA test, F(6,56) = 5.063). B, Validation of the p75 KO rat by genotyping (primer sequence, forward: CAGCGCAGTTCAGCTCAG and reverse: CGCTCGCTCTGAAAGTCCTA). Absence of p75 protein demonstrated by Western blot and immunostaining. C, p75NTR is expressed in proliferating GCPs of P7 WT (top) mouse cerebellum and is deleted from the EGL of p75NTRfl/fl; Atoh1Cre (bottom) mice. Ca–Ch, High magnification of lobe 9, immunostained for Ki67 (Ca, Cb, green), p75NTR (Cc, Cd, magenta), calbindin (Ce, Cf white), and merged images with DAPI (blue) (Cg, Ch). Note the deletion of p75NTR specifically from EGL (Cg, Ch), while it remains expressed in PCs colocalized with calbindin. D, Sagittal sections of lobes 5, 6, and 9 from WT and p75NTRfl/fl; Atoh1Cre. Animals were injected with EdU at P7, killed 24 h later, and stained for Ki67 (red) and EdU (green). EdU-positive/Ki6-positive cells represent proliferating cells; EdU-positive/Ki67-negative cells represent cells that exited the cell cycle during those 24 h. E, Quantification of the quitting fraction experiments shown in D. The quitting fraction is represented as the ratio of EdU-positive and Ki67-negative cells over the total number of EdU-positive cells. p < 0.0001 (Sidak's post hoc test following a two-way ANOVA test, F(1,38) = 26.73). F, Quantification of the quitting fraction experiments with EdU injected at P10 and analyzed 24 h later. p < 0.0001 (Sidak's post hoc test following a two-way ANOVA test, F(1,21) = 29.77). Error bars indicate mean ± SEM. *Significant at the indicated p value.
Primary cerebellum cell cultures.
Cerebella were removed under sterile conditions from WT or p75NTR−/− P7 rat pups of either sex after death by CO2. Meninges and small blood vessels were removed under a dissecting microscope. Tissue was minced and dissociated using the papain dissociation kit (Worthington, LK003150). Dissociated cells were plated onto 24-well plates (1 × 105 cells in 300 μl of serum-free media) or 6-well plates (1.5 × 106 cells per well in 1 ml of serum-free media) coated with poly-d-lysine (0.1 mg/ml). Serum-free medium consisted of 1:1 MEM and F12, with glucose (6 mg/ml), insulin (2.5 mg/ml), putrescine (60 μm), progesterone (20 nm), transferrin (100 μg/ml), selenium (30 nm), penicillin (0.5 U/ml), and streptomycin (0.5 μg/ml). Cells cultured under these conditions were >95% granule cells identified by immunostaining for Pax6.
Generation of EGL-specific deletion of p75NTR.
Mice with homozygous floxed alleles of p75NTR (p75NTRfl/fl) (Bogenmann et al., 2011) were mated with Atoh1-cre mice (The Jackson Laboratory, #011104), and pups were obtained postnatally at different developmental ages. The genotype of p75NTRfl/fl; Atoh1-Cre animals was confirmed by PCR, and the absence of p75NTR in the EGL was confirmed by immunostaining.
Immunohistochemistry.
Animals of either sex were deeply anesthetized with ketamine/xylazine and perfused with 4% PFA in PBS. Brains were removed and postfixed in 4% PFA/PBS overnight at 4°C and then cryopreserved with 30% sucrose. Sections (20 μm) were cut using a Leica Microsystems cryostat and mounted onto charged slides. Sections were permeabilized with 0.5% Triton in PBS for 20 min and blocked with 1% BSA and 5% donkey serum in PBS for 1 h at room temperature. Primary and secondary antibodies were prepared in 1% BSA. Sections were incubated with primary antibodies overnight at 4°C in a humidified chamber. Antibodies used were as follows: Ki67 (Abcam, 15580, RRID:AB_443209, 1:500), anti-p75 (R&D Systems, AF367, RRID:AB_2152638, 1:500), anti-p75 (Millipore MAB365, RRID:AB_2152788, 1:1000), and anti-BrdU (Millipore BU-1, RRID:AB_11212826, 1:50). All secondary antibodies were diluted 1:500 and incubated for 1 h at room temperature. Nuclei were labeled with 1 mm Draq5 (BioStatus, DR-50200) for 30 min or using Prolong Gold with DAPI mounting media (Invitrogen, P36931). Cultured cells were mounted using Prolong Gold (Invitrogen, P36931) or Prolong Gold + DAPI (Invitrogen, P36934). Controls for immunostaining included incubation with secondary antibodies in the absence of primary antibodies.
EdU/BrdU double-labeling cell assay.
Cells were obtained from WT and p75NTR−/− rats of either sex and cultured as described above in the presence of 0.1 μg/ml of Shh (R&D Systems, 1845-SH) for the entire experiment; 10 μm of EdU (Invitrogen, C10337) was added to the culture for 1 h and washed with serum-free media at 37°C. After different time points from 12 to 24 h, BrdU 20 μm (Sigma-Aldrich, B5002) was added for 2 h, after which cells were fixed with ice-cold 4% PFA/PBS for 15 min, washed with PBS 2 times, and permeabilized with 0.5% Triton ×100 for 20 min. Cells were treated with 2N HCl at room temperature for 30 min to increase BrdU detection, coverslips were washed 3 × 5 min with PBS. EdU was developed following the manufacturer's protocol (Invitrogen, C10337). For BrdU detection, after completing the EdU development, cells were washed 2 × 10 min with PBS and blocked with 1% BSA, 5% donkey serum in PBS for 1 h at room temperature. Cells were incubated with primary antibody overnight at 4°C. Anti-BrdU (Millipore, BU-1, RRID:AB_11212826) was diluted 1:20 according to the manufacturer's specification. The cells were washed with PBS 3 × 10 min and incubated with secondary antibody diluted 1:500 for 1 h at room temperature. Cells were then washed 3 × 15 min in PBS. Coverslips were mounted using Prolong Gold + DAPI (Invitrogen P36931) for nuclear labeling. Fifteen images per coverslip were taken with a Nikon Eclipse TE200 microscope. The number of EdU, BrdU, and EdU/BrdU-positive cells was quantified using ImageJ version 1.51s.
Quitting fraction analysis.
The analysis was performed according to Legué et al. (2016). Briefly, P7 WT and p75fl/fl; Atoh1Cre mouse pups were injected intraperitoneally with 10 mg/kg of EdU; 24 h after the injection, animals were perfused as described above; 20 μm cryostat sagittal sections at the vermis of the cerebellum were stained for EdU and Ki67. EdU was developed following the manufacturer's protocol (Invitrogen, C10337). Immediately after EdU staining, tissue was blocked with 1% BSA and 5% donkey serum for 1 h at room temperature. Tissue was incubated with anti-Ki67 (Abcam, 15580, RRID:AB_443209, 1:500) overnight at 4°C. At least 4 different sections separated by at least 100 μm from each animal were used for the analysis, and at least 2 pictures from the external portion of each lobe were taken. Pictures were taken with an LSM 510 Meta confocal microscope. The number of EdU+/ Ki67− cells, and EdU+/Ki67+ cells was quantified using ImageJ.
GTPase activation analysis.
Cells were obtained from P7 pups and cultured as described above. Cells were incubated for 24 h with and without 0.1 μg/ml Shh ± 10 ng/ml of proNT-3. Cells were processed for G-Lisa analysis according to the manufacturer's instructions. G-Lisa for RhoA (Cytoskeleton, #BK124), Cdc42 (Cytoskeleton, #BK127), and Rac1 (Cytoskeleton, #BK128).
RhoA inhibition experiments.
Cells were obtained from P7 pups and cultured as described above. Cells were incubated for 48 h with and without 0.1 μg/ml Shh. Two different RhoA pathway inhibitors were added to the cells, Rhosin (Tocris Bioscience, 5003) to a final concentration of 0.1, 0.35, and 3.5 μm, and Y27632 (Calbiochem, 688000) a Rho kinase inhibitor, to a final concentration of 1, 10, and 50 μm. All the inhibitors were added from the time of plating the cells and maintained for 48 h. Cells were processed for Western blot or immunocytochemistry analysis as described. In Western blots, proliferation levels were quantified using PCNA (BD 610664, RRID:AB_397991, 1:2000). For immunocytochemistry, 10 μm of EdU was added 2 h before fixing the cells, to identify proliferating cells.
Western blot and coimmunoprecipitation analysis.
Tissue or cultured cells were washed with ice-cooled PBS and homogenized using 1% NP40, 1% Triton, 10% glycerol in TBS buffer (50 mm Tris, pH 7.6, 150 mm NaCl) with protease inhibitor mixture (Roche, 11836153001). Proteins were quantified using the Bradford assay (Bio-Rad, 500-0006), and equal amounts of protein were run on SDS gels and transferred to nitrocellulose membrane. To ensure equal protein levels, blots were stained with Ponceau before incubation with antibody. For coimmunoprecipitation analysis, lysates were immunoprecipitated with anti-Rho-GDI (Sigma-Aldrich, R3025), pulled down with protein G magnetic beads (New England Biolabs, S1430S), and analyzed by Western blot with anti-p75NTR (Millipore, 07-476, 1:1000). The blots were then rinsed and blocked in 5% nonfat dried skim milk in TBS-T for 2 h at room temperature. Blots were incubated with primary antibodies diluted 1:1000 in 1% BSA in TBS buffer overnight at 4°C. The blots were washed with TBS-T 3 × 10 min each and incubated with Licor secondary antibody for 1 h at room temperature. All secondary antibodies were diluted 1:10,000. Membranes were washed 3 × 10 min each in TBS-T. The membranes were analyzed using Licor Odyssey infrared imaging system (LI-COR Biosciences). To confirm equal protein levels, blots were reprobed for actin. All analyses were performed at least three times in independent experiments. Bands were quantified using ImageJ version 1.51s.
Slice electrophysiology.
Acute parasagittal cerebellar slices (300 μm) of the vermis were obtained from 6- to 8-week-old p75fl/fl or p75fl/fl; Atoh1Cre littermate mice and maintained in ice-cold sucrose artificial cerebral spinal solution (sucrose aCSF, 85 mm NaCl, 75 mm sucrose, 24 mm NaHCO3, 25 mm glucose, 4 mm MgCl2, 2.5 mm KCl, 1.25 mm NaH2PO4, and 0.5 mm CaCl2) using a Leica Microsystems VT1200S vibratome. Slices were subsequently transferred to an incubation chamber containing an equal mixture of sucrose-aCSF and recording aCSF at 34°C for 30 min and then maintained at room temperature. The recording aCSF contained the following: 126 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 10 mm d-glucose. All solutions were saturated with 95% O2/5% CO2 and maintained at a pH of 7.4 for 1–6 h. Recordings were obtained using a patch pipet containing 126 K-gluconate, 4 KCl, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 phospho-creatinine, and biocytin (0.2%, Sigma-Aldrich) for post hoc cell identification.
Patch-clamp recording was done under infrared differential interference contrast techniques with a Nikon Eclipse FN-1 microscope, using a 40×, 0.80 NA Nikon water-immersion objective. Whole-cell voltage-clamp and current-clamp recordings from PCs were performed using patch pipettes with 4–6 mΩ resistance and recorded using MultiClamp 700B (Molecular Devices). Data were low-pass filtered at 3 kHz, digitized using Digidata 1440A, and acquired using pClamp10 at 10 kHz sampling frequency. For firing rate and input resistance determination, cells were held in current clamp at −70 mV with small current injections, and measurements were done using 1 s test pulses of sizes ranging from −120 to 200 pA with an increment of 40 pA. Glutamatergic synaptic currents were recorded in voltage clamp at −70 mV. In some recordings, TTX (1 μm, Sigma-Aldrich) was added to aCSF to monitor miniature, action potential independent, EPSCs (mEPSCs), and kynurenic acid (3 mm) was used to confirm the glutamatergic nature of the events. Recordings were rejected if the cell had more than a 20% change in series resistance over time. Clampfit 10 and SigmaPlot 12.3 were used to analyze the data. Following physiological recordings, slices were fixed in 0.1 m phosphate buffer containing 4% PFA at 4°C for 2 d. Biocytin staining was revealed with AlexaFluor-594-conjugated streptavidin (1:1000) overnight at room temperature. Sections were visualized and imaged with a Nikon A1R laser confocal microscope with a 60× water objective. sEPSCs and mEPSCs were analyzed using event detection Clampfit (Molecular Devices).
Spine density analysis: Golgi staining.
Animals were killed using CO2, and the cerebellum was immediately removed. The vermis was dissected out, cut in half, and incubated for Golgi staining using FD rapid Golgi staining kit (FD Neuro Technologies, PK401A) following the manufacturer's instructions; 100 μm cryostat sagittal sections of the cerebellar vermis were stained. At least 25 neurons were analyzed per cerebellum. Pictures were taken from the distal portion of PC located in lobes 6 and 7. Pictures were taken using a Nikon Eclipse TE200 connected to a motorized stage ASI MFC-2000. A z stack array covering the entire cell with a 1 μm interval was obtained. For each cell, at least 20 in-focus segments of at least 10 μm length were used for analysis. Each segment was processed according to Orlowski and Bjarkam (2012) for quantification. Spines were counted with ImageJ version 1.52 and using a custom-made macro to ensure homogeneity in the counting.
Behavioral analysis: delay eyeblink conditioning.
p75NTRfl/fl and p75NTRfl/fl; Atoh1Cre of both sexes were tested for classical delay eyeblink conditioning (Heiney et al., 2014; Kloth et al., 2015; Badura et al., 2018). The surgical procedure was based on Heiney et al. (2014). Briefly, mice were anesthetized with isoflurane and placed in a stereotaxic apparatus. At the midline, a small piece of the skin was removed, and a metal head plate was attached directly onto the skull using dental cement. During eyeblink conditioning, mice were head-fixed to a rotating wheel to allow free walking. The light flash (conditional stimulus [CS]) was presented using a blue LED for 280 ms. A 30 ms air puff (unconditional stimulus [US]) was delivered directly to the cornea using a 27-gauge needle and timed to end at the same time as the CS. The eyelid deflection (conditioned response [CR]) was detected using a modified playstation camera at 60 fps and analyzed using custom MATLAB software. The analysis of the CR was based on Heiney et al. (2014). The average of all responses was used to calculate the CR response.
Mice were habituated to the eyeblink conditioning apparatus for 2 d for 15, 30, and 60 min each day. After habituation, 14 d of training started (1 session/d, 7 d/week). Each session lasted 20 min, and the mice received 200 CS-US parings/session. Successive CS-US trials were separated by an interval of 6 s. Trials were automatically triggered using a custom-made python interface.
Statistical analysis.
Each experiment was designed to answer a particular question, and the proper statistical test was used accordingly, as indicated below.
In Figure 1A, the experiments were designed to test whether the absence of p75NTR can alter cell cycle speed of GCP. We used a two-way ANOVA to separate the two factors of the experiments: (1) genotype of the cells and (2) the time between EdU and BrdU addition. This experiment was repeated at least 4 times using pups from 4 different litters (n = 4). Two coverslips per condition were counted. For each coverslip, 15 pictures were randomly taken with 20× magnification using a Nikon Eclipse T1000 microscope. Cells were counted with ImageJ version 1.52 and using a custom-made macro to ensure homogeneity in the counting.
For Figure 1E, F, the experiments were designed to test whether the absence of p75NTR altered the proportion of cells that exit the cell cycle in vivo. We used a two-way ANOVA to separate the factors “genotype” and the “age” of the animals. Since the source of variability is the animals, in Figure 1E, n = 8 WT and n = 7 p75fl/fl; Atoh1Cre mice. Male and female animals were used from three different litters for both genotypes. In Figure 1F, n = 5 WT and n = 4 p75fl/fl; Atoh1Cre mice. Male and female animals from two different litters for both genotypes were used. For the analysis, at least four sections per brain, separated by at least 100 μm, were included in the quantification. Pictures were taken in the coronal part of the indicated lobe using a Carl Zeiss LSM 510 meta confocal microscope with a 63× magnification. Cells were counted with ImageJ version 1.52 and using a custom-made macro to ensure homogeneity in the counting.
For Figure 2, coimmunoprecipitation analysis of cultured GCPs were from male and female pups of three different litters in three independent experiments (n = 3). Statistical analysis was performed using a one-way ANOVA. The effect of Shh and Shh + proNT-3 on GTPase activation was tested. Statistical analysis was performed using a one-way ANOVA. This experiment was repeated n = 4 times for RhoA and n = 3 times for Rac1 and Cdc42. Each experiment used cells obtained from male and female pups of different litters. The experiments were run in duplicate per condition following the manufacturer's recommendations.
Figure 2.

Analysis of Rho family GTPases in GCPs. Activation of the Rho GTPases was analyzed in cultured GCPs treated with Shh ± proNT-3 for 48 h. A, Coimmunoprecipitation showing increased interaction between p75NTR and Rho-GDI with Shh treatment, which was decreased with proNT3 addition. Graph represents level of p75NTR pulled down by Rho-GDI immunoprecipitation relative to total Rho-GDI. *p = 0.0020 (one-way ANOVA, F(3,8) = 12.93). B, RhoA activation was increased by Shh and restored to basal levels with Shh + proNT3. **p = 0.0047 (one-way ANOVA with Tukey's post hoc test, F(2,9) = 10.28). C, Levels of active Cdc42 were not affected by any of the treatments. p = 0.9089 (one-way ANOVA followed by Tukey's post hoc test, F(2,6) = 0.09709). D, Active Rac1 in GCPs were unchanged in the presence of Shh or Shh + proNT3. p = 0.3004 (Tukey's post hoc test following a one-way ANOVA test, F(2,6) = 1.479). Error bars indicate mean ± SEM.
The experiments shown in Figure 3 were designed to test the role of RhoA in GCP proliferation. A one-way ANOVA was used for all analyses in this figure. The experiments in Figure 3B were repeated n = 3 times using male and female pups from different litters. Two coverslips per condition were counted in each experiment. For each coverslip, 15 pictures were randomly taken with a 20× magnification using a Nikon Eclipse T1000 microscope. Cells were counted with ImageJ version 1.52 and using a custom-made macro to ensure homogeneity in the counting. n = 5 replicates for p75NTR and n = 4 replicates for PCNA quantification with the Rock inhibitor (see Fig. 3D,E) and n = 3 for p75NTR and PCNA quantification with the Rhosin inhibitor (see Fig. 3G,H). Each experiment used cells obtained from male and female pups from different litters. The Western blots were analyzed with ImageJ version 1.52 and using the Gel analysis plug in.
Figure 3.

Effects of RhoA inhibition on GCP proliferation. A, B, Granule cells were cultured in the presence or absence of Shh and exposed to different concentrations of the ROCK inhibitor Y27632. A, Cells stained for EdU (green) and DAPI (blue). B, Quantification of proliferation levels expressed as the number of EdU-positive cells divided by the number of DAPI-positive cells. Three independent experiments were analyzed. p < 0.0001 (Tukey's post hoc analysis following a one-way ANOVA test, F(7,16) = 39.89). C–E, Western blot analysis of GCP proliferation levels exposed to ROCK inhibitor in the absence or presence of Shh. C, Representative blot showing expression of p75NTR, PCNA, and actin. D, Quantification of PCNA levels normalized to actin levels. p < 0.0001 (Tukey's post hoc analysis following a one-way ANOVA test, F(5,18) = 35.46). E, Quantification of p75NTR expression levels normalized to actin. p < 0.0001 (Tukey's post hoc analysis following a one-way ANOVA test, F(5,24) = 27.43). At least four independent experiments were analyzed. F–H, Western blot analysis of GCP proliferation levels exposed to Rhosin, a RhoA inhibitor. F, Representative blot showing expression of p75NTR, PCNA, and actin. G, PCNA expression levels normalized with actin expression levels. p < 0.0001 (Tukey's post hoc analysis following a one-way ANOVA test, F(7,16) = 17.21). H, p75NTR expression levels normalized with actin expression levels. p < 0.0001 (Tukey's post hoc analysis following a one-way ANOVA test, F(7,16) = 11.19). Three independent experiments were analyzed. Error bars indicate mean ± SEM. *indicates different from untreated control, #indicates different from Shh alone.
Experiments in Figures 4 and 5 were designed to test the effect of the absence of p75NTR on intrinsic physiological parameters and synaptic currents. p75fl/fl without Cre and p75fl/fl; Atoh1Cre mice were compared (n = 3 male mice per group for physiological recordings). One cell was recorded and filled in each slice. Intrinsic physiological parameters between groups were compared using Student's t test in SigmaPlot 12.3. Cumulative probability plots of sEPSC and mEPSC parameters were constructed by pooling an equal number of sEPSCs and mEPSCs from each cell and compared using Kolmogorov–Smirnov test (SigmaPlot 12.3).
Figure 4.

Increase in spontaneous excitatory synaptic currents in cerebellar PCs of p75NTRfl/fl; Atoh1Cre mice compared with p75NTRfl/fl littermates. A, Example confocal image of biocytin-filled PCs recorded from a p75NTRfl/fl; Atoh1Cre mice. Scale bar, 40 μm. B, C, Representative current-voltage plots recorded in PCs in response to −200 and 80 pA current injections in p75NTRfl/fl (indicated as WT) (B) and p75NTRfl/fl; Atoh1Cre mice (C). D–F, Summary histograms compare resting membrane potential (D), input resistance measured in response to a −200 pA current injection (E), and average firing frequency in response to an 80 pA (F) between p75NTRfl/fl and p75NTRfl/fl; Atoh1Cre mice. G, H, Sample PC sEPSC traces in p75NTRfl/fl (WT) mice (G) and p75NTRfl/fl; Atoh1Cre mice (H). I, J, Summary cumulative probability plots of sEPSCs interevent interval (I) and amplitude (J) from 9 cells each from 3 WT mice and 3 p75NTRfl/fl; Atoh1Cre mice. p < 0.05 (Kolmogorov–Smirnov test). Error bars indicate mean ± SEM.
Figure 5.

PC mEPSC frequency is enhanced in p75NTRfl/fl; Atoh1Cre mice. A–C, Representative current traces of action potential independent mEPSCs in PCs from p75NTRfl/fl (indicated as WT) (A) and p75NTRfl/fl; Atoh1Cre mice (B) recorded in 1 μm TTX. All synaptic events were abolished in the presence of the glutamate receptor antagonist kynurenic acid (C). D, Cumulative probability plots of mEPSC interevent interval from 8 cells each from 3 WT mice and 3 p75NTRfl/fl; Atoh1Cre mice. p < 0.05 (Kolmogorov–Smirnov test). Inset, Histogram of mEPSC interevent interval averaged within each cell. *p < 0.05 (Student's t test). Error bars indicate mean ± SEM.
The experiments in Figure 6 were designed to test the effect of the absence of p75NTR on spine density of PCs. n = 6 mice per genotype; both male and female mice were used. Animals were from two different litters for the WT, and three different litters for p75fl/fl; Atoh1Cre mice. For the Golgi staining analysis, the full cerebellum in the area of the vermis was sectioned at 150 μm thickness. All sections were collected and stained; at least 25 cells per animal were counted. A z stack for each cell in the distal portion of the PC dendritic tree was generated using a Nikon Eclipse Ti 1000 connected to a motorized stage. Cells were obtained from lobes 6 and 7 only. Cells were counted with ImageJ version 1.52 and using a custom-made macro to ensure homogeneity in the counting, and statistical analysis was performed with an unpaired Student's t test.
Figure 6.
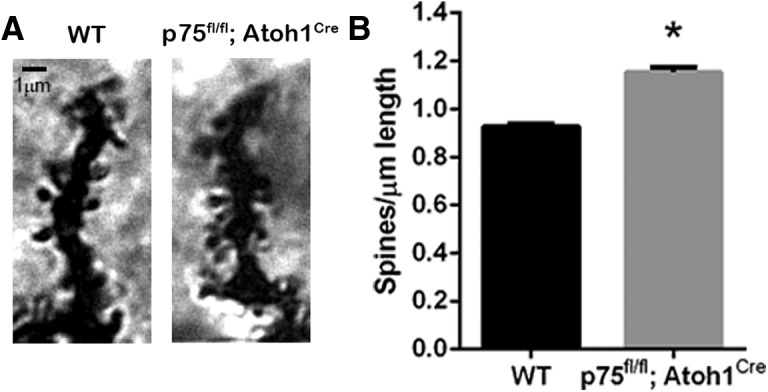
Dendritic spine analysis of PCs. A, Golgi staining of a distal dendritic branch of a PC in adult WT or p75fl/fl; Atoh1Cre mice. B, Dendritic spine analysis represented as the number of spines per length of the segment analyzed. Six animals per genotype were analyzed. *p < 0.0001 (unpaired Student's t test; Cohen's d effect size = 5.69). Error bars indicate mean ± SEM.
The experiments in Figure 7 tested the differences in eyeblink conditioning between p75NTRfl/fl and p75NTRfl/fl; Atoh1Cre. Since the same animals needed to be tested several times, we used a repeated-measures ANOVA. For the average of the last 4 d of training, we used an unpaired Student's t test. Animals from three different litters were tested, and each litter contained p75NTRfl/fl with or without the Atoh1-cre. n = 7 p75fl/fl and n = 11 p75fl/fl; Atoh1Cre mice. Both male and female mice were used.
Figure 7.

p75NTR deletion from GCPs in the EGL-induced deficits in eyeblink conditioning in adult mice. A, Training sessions for eyeblink conditioning. Average of total CR amplitude (normalized to the amplitude of the unconditioned response to air puff) was reduced in the p75fl/fl; Atoh1Cre compared with control littermates. *p = 0.0086 (repeated-measures ANOVA; F(1,15) = 9.110; n = 7 WT; n = 10 p75fl/fl; Atoh1Cre). B, Average CR in the last 3 d of training. *p < 0.0233 (unpaired Student's t test; Cohen's d effect size = 1.35). Error bars indicate mean ± SEM.
Results
p75NTR regulates GCP cell cycle progression in vitro and in vivo
The p75NTR is highly expressed in all the proliferating cells in the EGL, and it is downregulated before cells differentiate and migrate (Fig. 1C), leading us to surmise that p75NTR might be involved in cell cycle progression of the GCPs. To measure the length of the cell cycle, we performed an EdU/BrdU double-labeling experiment. Cells were cultured from WT and p75NTR−/− rats (Fig. 1A) in the presence of Shh to promote proliferation. Cells were treated with EdU for 1 h to identify the cells in S phase, and the EdU was washed out with fresh media. To determine when the cells reentered S phase, the cells were exposed to BrdU for 2 h between 12 and 24 h after the EdU treatment before fixing. Double-labeled EdU+/BrdU+ cells represent the cells that went through a second S phase during that 2 h window of BrdU exposure. The proportion of EdU+/BrdU+ cells over the total EdU+ population provides an estimate of the duration of the cell cycle. Our results showed a peak of double-labeled cells obtained from WT animals 16–18 h after the initial EdU incorporation (Fig. 1A). Surprisingly, cells obtained from p75NTR−/− animals showed an earlier peak of BrdU incorporation, between 12 and 14 h and a second peak at 24–26 h after EdU incorporation, suggesting that the p75NTR−/− cells enter a second S phase more rapidly than the WT cells, indicating a shorter cell cycle (Fig. 1A).
Since p75NTR is expressed in PCs as well as GCPs, we analyzed effects of p75NTR on the GCP cell cycle in vivo using p75NTRfl/fl mice crossed with Atoh1-cre mice to specifically delete p75NTR from GCPs (Fig. 1C–F). To determine the length of the cell cycle in vivo, we injected EdU into WT and p75fl/fl; Atoh1Cre pups at postnatal day (P) 7 and waited for 24 h before killing the animal. After GCP proliferation, the daughter cells can either exit the cell cycle and begin to migrate to the IGL or stay in the cell cycle for another round of cell division. Cerebellar tissue sections were labeled for EdU incorporation and immunostained for Ki67 to identify cells remaining in the cell cycle. Cells positive for EdU and negative for Ki67 represent the quitting fraction (e.g., the cells that have exited the cell cycle in the 24 h since the EdU labeling) (Legué et al., 2016). We observed a significant reduction in the quitting fraction in the p75fl/fl; Atoh1Cre compared with the WT animals, both in lobe 5 and lobe 9 (Fig. 1D,E), suggesting that the absence of p75NTR in the EGL maintains cells in a proliferative state longer than WT cells. Interestingly, no significant difference was observed in lobe 6 at this age (Fig. 1D,E); but when EdU was injected in P10 pups and the brains analyzed 24 h later, a significant difference was seen in lobes 6 and 9, but no longer in lobe 5 (Fig. 1F). These observations are in agreement with Legué et al. (2016) who identified a differential rate of proliferation/differentiation among lobes at different developmental ages. However, a delay in GCPs exiting the cell cycle was demonstrated in the p75NTRfl/fl; Atoh1Cre mice in all lobes.
RhoA is required for GCP proliferation
Small GTPases are critical regulators of the cytoskeleton, which makes them important regulators of the cell cycle as well as cell migration (Olson et al., 1995; Ridley, 2001, 2015; Fukata et al., 2003; Raftopoulou and Hall, 2004; Villalonga and Ridley, 2006). Unliganded p75NTR can constitutively activate RhoA in granule cells, and this maintenance of RhoA activity is reduced upon ligand binding to the receptor (Yamashita et al., 1999; Yamashita and Tohyama, 2003). We confirmed the association of p75NTR with Rho-GDI in GCPs by coimmunoprecipitation analysis, showing that Shh maintains a high level of p75NTR:Rho-GDI interaction, which was decreased by the addition of proNT3 (Fig. 2A), To determine the role of Rho GTPases in GCP proliferation, cultured GCPs were treated for 48 h with Shh to induce proliferation and proNT-3 to promote cell cycle withdrawal, and the amount of active RhoA, Rac1, and Cdc42 was quantified (Fig. 2). Shh induced a 76% increase in active RhoA compared with control cultures without Shh, and treatment with proNT-3 reduced the level of active RhoA induced by Shh to control levels (Fig. 2B), consistent with the antiproliferative effect of proNT-3 we observed previously (Zanin et al., 2016). No changes in active Cdc42 (Fig. 2C) or Rac1 (Fig. 2D) were observed in the proliferating GCPs with either Shh or proNT3 treatment.
To assess the importance of RhoA in regulating the cell cycle of GCPs, we evaluated the effects of blocking RhoA signaling on GCP proliferation. Rho Kinase I and II (ROCK I and II) are downstream targets of RhoA and can affect cell cycle via Cyclin A and D1 as well as p27 (Croft and Olson, 2006). GCPs were cultured with Shh to induce proliferation, and different concentrations of the ROCK inhibitor Y27632 were added. EdU was provided in the last 2 h before fixing the cells to assess the level of proliferation. We observed a dose-dependent inhibition of EdU incorporation in the cells treated with Y27632, even in the presence of Shh (Fig. 3A,B). To further confirm this result, Western blot analysis showed a decrease in PCNA, a proliferation marker, with ROCK inhibition in a dose-dependent manner (Fig. 3C,D). Additionally, directly blocking RhoA with Rhosin (Fig. 3F,G) yielded a decrease in PCNA expression similar to the effect observed with ROCK inhibition. Moreover, in association with the decrease in PCNA, there was a reduction in the level of p75NTR expression as the cells exited the cell cycle (Fig. 3C,E,F,H). These results confirm the importance of RhoA signaling for the cell cycle progression of GCPs.
Increase in excitatory synaptic events in cerebellar PCs of p75fl/fl; Atoh1Cre mice
Since GCPs provide excitatory synaptic input to PCs, the differences observed in GCP proliferation in the p75fl/fl; Atoh1Cre mice suggested that the cerebellar circuitry of these animals might be affected. Whole-cell current and voltage recordings were obtained from cerebellar PCs in slices from p75fl/fl mice and p75fl/fl; Atoh1Cre mice. The PC identity was confirmed by post hoc biocytin immunostaining (Fig. 4A). The effect of altered GCP proliferation due to p75NTR deletion from the EGL on PC passive and active properties was examined by measuring membrane voltage responses evoked by a series of current injections. The resting membrane potential was measured when 0 current was injected under current clamp. There was no significant difference in the resting membrane potential between p75fl/fl without Cre (which were identical to WT) and p75fl/fl; Atoh1Cre mice (Fig. 4D; p75fl/fl: −63.54 ± 1.37, median −64.36, p75fl/fl; Atoh1Cre: 62.69 ± 1.26, median 62.93, n = 6 cells each, p = 0.657 by unpaired Student's t test). PC input resistance, measured in response to a −120 pA current injection, was not different between genotypes (Fig. 4E; input resistance in mΩ, p75fl/fl: 89.50 ± 7.32; p75fl/fl; Atoh1Cre: 83.17 ± 5.20, n = 6 cells each, p = 0.497 by unpaired Student's t test). Similarly, the average firing frequency was also not different between p75fl/fl and p75fl/fl; Atoh1Cre mice (Fig. 4F; firing frequency in response to 120 PA in Hz, p75fl/fl: 50.16 ± 2.95; p75fl/fl; Atoh1Cre: 48.50 ± 3.38, n = 6 cells each, p = 0.718 by unpaired Student's t test). These data indicate that basic intrinsic physiology of PCs was not altered by deletion of p75NTR in GCPs.
To test whether the altered GCP proliferation resulted in altered input to the PCs, sEPSCs were recorded in PCs from a holding potential of −70 mV. Consistent with an increase in excitatory input, the sEPSC frequency in PCs from p75fl/fl; Atoh1Cre mice was significantly increased compared with p75fl/fl (Fig. 4I,J), as demonstrated by the reduction in sEPSC interevent interval (reciprocal of frequency) in p75fl/fl; Atoh1Cre mice (sEPSC interevent interval in seconds, p75fl/fl: 0.89 ± 0.05, median 0.51 s, interquartile range [IQR] 0.26 to1.12; p75fl/fl; Atoh1Cre: 0.18 ± 0.004, median: 0.17, IQR 0.12–0.23; n = 9 cells each, k − s (greatest y difference between plots) = 61%, p = 0.000001 by Kolmogorov–Smirnov test). Additionally, the amplitude of sEPSC in PCs from p75fl/fl; Atoh1Cre mice was significantly increased (Fig. 4J; sEPSC amplitude in pA, p75fl/fl: 24.23 ± 0.57, median 22.33, IQR 15.21–31.11; p75fl/fl; Atoh1Cre: 29.94 ± 0.91, median 24.79, IQR 16.26–38.10, x-y; n = 9 cells each, k − s = 13.78%, p = 0.0039 by Kolmogorov–Smirnov test).
mEPSCs were recorded in the presence of the sodium channel antagonist TTX (1 μm) to isolate action potential independent synaptic events (Fig. 5). As with sEPSCs, there was a significant increase in mEPSC frequency in p75fl/fl; Atoh1Cre mice (mEPSC interevent interval in seconds p75fl/fl: 1.14 ± 0.07, median 0.63 s, IQR, 0.20–1.52; p75fl/fl; Atoh1Cre: 0.22 ± 0.01, median 0.15, IQR: 0.09–0.25; n = 8 cells each, k − s = 53%, p = 0.00001 by Kolmogorov–Smirnov test; Fig. 5D). However, mEPSC amplitude was not different between the genotypes (mEPSC amplitude in pA, p75fl/fl: 16.90 ± 0.31, median 15.51, IQR 12.21–20.60; p75fl/fl; Atoh1Cre: 16.97 ± 0.34, median 15.79, IQR 11.99–20.32; n = 8 cells each, k − s = 9%, p = 0.81 by Kolmogorov–Smirnov test). Together, these data demonstrate an increase in excitatory input to PCs in p75fl/fl; Atoh1Cre mice.
p75fl/fl; Atoh1Cre mice show an increase in PC dendritic spines
The altered proliferation of GCPs, which give rise to the parallel fibers that synapse on the distal part of the PC dendritic tree, and the increased excitatory drive of the PCs in the p75fl/fl; Atoh1Cre compared with WT animals suggested an increase in excitatory synapses onto the PCs. Golgi staining was used to study the spine density in the distal part of PC dendritic tree where the parallel fibers are known to synapse (Fig. 6A). The distal portion of PC neurons was analyzed in semiautomatic spine counting (Orlowski and Bjarkam, 2012). In p75fl/fl; Atoh1Cre mice, the spine density in the distal dendrites was increased 26% compared with the WT animals (Fig. 6B). These results suggest that the altered PC firing frequency might be due to increased excitatory input from the granule cell parallel fibers.
Deletion of p75NTR from the developing EGL results in deficient cerebellum-dependent learning
p75fl/fl and p75fl/fl; Atoh1Cre mice were assessed on delay eyeblink conditioning, a behavioral paradigm that depends on intact cerebellar function. For conditioning, mice were presented with a light flash (CS), which coterminated with an air puff to the cornea of the animal (US). Repeated CS-US pairing eventually produced a learned anticipatory blink in response to the CS, the CR. p75fl/fl mice learned to anticipate the air puff and generated a larger average CR than p75fl/fl; Atoh1Cre mice (ANOVA repeated measurements, p = 0.0086, F(1,15) = 11.15). p75fl/fl; Atoh1Cre showed a significant difference in the last days of training compared with p75fl/fl mice (p75fl/fl mean = 0.3316 ± 0.03; p75fl/fl; Atoh1Cre mean = 0.19 ± 0.05; unpaired t test, p = 0.02, effect size Cohen's d = 1.23) (Fig. 7).
Discussion
In the present work, we demonstrated how a developmental dysregulation of progenitor proliferation can affect the connectivity and physiology of the cerebellum and have a major impact on adult behavior. p75NTR is expressed in multiple areas of the developing CNS, and it influences many aspects of brain development. p75NTR is abundantly expressed in the external EGL of the developing cerebellum, and it colocalizes with proliferation markers, such as Ki67 (Fig. 1C). In previous work, we demonstrated that proNT-3, via p75NTR, promotes cell cycle withdrawal of GCPs, even in the presence of Shh, a potent mitogen for this cell population (Zanin et al., 2016). However, the presence of this receptor in every proliferating cell in the EGL suggested that p75NTR might be controlling other aspects of cell cycle progression. In the present work, we demonstrated that p75NTR is required to maintain proper timing of cell cycle progression of GCPs during development of the cerebellum, and this role requires the RhoA pathway to maintain cells in a proliferative state. We also demonstrated that the absence of p75NTR specifically from the GCPs was enough to induce an increase in PC firing frequency, likely due to an excess of glutamatergic CGN inputs onto these cells, leading to cerebellar circuitry dysregulation and consequent behavioral deficits.
p75NTR regulates cell cycle length
The length of each phase in the cell cycle is crucial for proper cell cycle progression and dictates the transition from proliferation to differentiation. In particular, a longer G1 phase is often associated with the accumulation of factors as the cells prepare to leave the cell cycle and differentiate (Hardwick and Philpott, 2014; Hardwick et al., 2015), whereas a shorter G1 phase maintains the cell in a proliferative state (Calder et al., 2013; Roccio et al., 2013). Our double-labeling experiments demonstrated the importance of p75NTR in proper cell cycle control. The absence of p75NTR led to a 25% reduction of the time for reentry into the second S phase compared with WT cells. This acceleration would be in favor of maintaining the GCPs in a proliferative cycle. Moreover, the in vivo quitting fraction experiments support the maintenance of the GCPs in the cell cycle, since we observed a decrease in the quitting fraction of GCPs in the p75fl/fl; Atoh1Cre mice (i.e., fewer cells exiting the cell cycle in the absence of p75NTR). Together, these results suggest that the absence of p75NTR can cause an increase in the cell cycle speed, favoring continued GCP proliferation, which will eventually increase the final CGN numbers leading to the increase in cerebellar size that we observed previously (Zanin et al., 2016). Interestingly, since the different lobes of the cerebellum develop at different times, and lobe 6 is the last lobe in which the GCPs stop proliferating, we observed the differences in the quitting fraction at a later age in lobe 6 than in lobes 5 and 9. However, in all cases, the p75NTRfl/fl; Atoh1Cre mice showed delayed cell cycle exit compared with WT.
The role of RhoA in cell cycle regulation
The Rho GTPases are important cytoskeletal regulators that can control numerous cellular functions, including different aspects of the cell cycle, such as the appropriate transition between each phase. The Rho subfamily of GTPases can promote G1 to S phase transition via cyclin D1 and cyclin E activation (Villalonga and Ridley, 2006; David et al., 2012). RhoA and Rac1 are important regulators of cerebellar development (Mulherkar et al., 2014). p75NTR can regulate RhoA (Yamashita et al., 1999; Yamashita and Tohyama, 2003) and rac1 (Harrington et al., 2002; Zeinieh et al., 2015) activity in different contexts. p75NTR is expressed in all the proliferating GCPs in the external EGL, making this receptor a potential candidate to directly regulate the GCP cell cycle. p75NTR was previously shown to constitutively activate RhoA in an unliganded fashion, which was reduced upon ligand binding to the receptor (Yamashita et al., 1999; Yamashita and Tohyama, 2003). Shh, which is a mitogen for GCPs and maintains high levels of p75NTR expression, induced a significant increase in active RhoA. proNT-3, which we showed previously induces cell cycle exit (Zanin et al., 2016), reduced RhoA activity to control levels. Moreover, we observed a dose-dependent inhibition of proliferation after blocking RhoA signaling, which might indicate that a proper threshold level of RhoA activity is required to maintain a proliferative state. The proper balance between the level of active RhoA and other members of the GTPase family, in particular Cdc42, is critical for appropriate development. A recent study demonstrated the importance of Cdc42 for GCP cell polarity and migration, demonstrating a reduction in the number of migratory cells when Cdc42 was inhibited, suggesting that altered regulation of these GTPases may change the balance of proliferation and migration (Govek et al., 2018).
It is also likely that additional proteins are necessary to interact with p75NTR for regulation of the transition from proliferation to migration. The structure of p75NTR allows constitutive interaction with Rho-GDI, leading to RhoA activation (Lin et al., 2015). Moreover, a previous study showed that the dual-specificity GEF, Kalirin9, can induce p75NTR-mediated RhoA activation in CGNs (Harrington et al., 2008). However, upon ligand binding, conformational changes in the receptor structure induce a displacement of Rho-GDI, favoring the interaction with Rip2 (Lin et al., 2015). Therefore, the activation of p75NTR might reduce the levels of RhoA, but at the same time recruit a second interactor protein, required to induce cell differentiation. In the absence of p75NTR, this recruitment would also be compromised, causing a failure to differentiate. While activation of RhoA appears to be permissive for proliferation throughout the cell cycle, it is possible that additional proteins must interact with p75NTR to regulate the cell cycle speed.
Neural circuitry deficits
PCs are the only cells that provide output information from the cerebellar cortex. PCs synapse onto the DCN; and from there, the cerebellum projects to the rest of the CNS. PCs receive excitatory inputs from the climbing fibers of the inferior olive and the mossy fibers via the parallel fibers of the granule cells. Our results indicate that the accelerated cell cycle of the GCPs in the absence of p75NTR caused increased proliferation, potentially providing an excess of CGNs, reflected in the increased size of the cerebellum in the adult p75fl/fl; Atoh1Cre mice (Zanin et al., 2016) and increased excitatory input to PCs.
PCs also receive inhibitory input from the stellate and basket cells, which are GABAergic interneurons located in the molecular layer, providing a strong inhibitory signal for PCs. Although these inhibitory interneurons may also receive increased excitation from the excess granule cells, they clearly do not balance out the increased excitatory drive the PCs receive from the granule cells, since the firing activity of PCs showed elevated excitatory drive in the p75fl/fl; Atoh1Cre mice compared with WT mice. This is supported by the significant increase in the density of spines observed in the PC dendrites of p75fl/fl; Atoh1Cre mice.
Behavioral effects
Eyeblink conditioning is a reflex motor learning task in which the cerebellum plays an essential role (Schneiderman et al., 1962; Heiney et al., 2014; Kloth et al., 2015; Badura et al., 2018). Similar to other motor learning behavior, it is acquired gradually though the course of multiple training sessions (Heiney et al., 2014). The integrity of parallel fiber-PC synapses is required for the performance of this task, as lack of Cbln1, a synaptic organizer necessary for the integrity of this pathway, caused impaired eyeblink conditioning as well as ataxic-gait and motor deficits (Matsuda et al., 2010; Emi et al., 2013).
In the present work, we demonstrated how defects in cerebellar circuitry can generate a deficit in eyeblink conditioning learning in adult mice. The increase in excitatory synaptic drive to PCs of the p75fl/fl; Atoh1Cre might therefore explain the cellular basis for the behavioral deficits observed. Ultimately, all the input information (sensory and motor) converges on the PCs, which synapse onto the DCN neurons; and from there, the cerebellum output to the rest of the CNS is executed. Altering the excitatory inputs to the PCs may affect information processing within the cerebellum as well as the cerebellum's interactions with other regions, which ultimately determine the behavioral response. Our results suggest that an excess of CGNs is responsible for the increased excitatory synaptic drive of PCs, which elicits a net reduction of the DCN to the rest of the CNS.
Together, these findings demonstrate that the p75 neurotrophin receptor regulates the GCP cell cycle, and lack of this receptor alters regulation of GCP proliferation during development. This elicits an alteration in the balance of excitatory/inhibitory inputs to PCs, leading to persistent changes in adult behavior. This study confirms the importance of appropriate cell cycle regulation in the development of circuitry that modulates associative learning behavior dependent on cerebellar function.
Footnotes
This work was supported by National Institutes of Health Grants R56NS094589 to W.J.F., R01NS045193 and R01MH115750 to S.S.-H.W., and R01NS069861 and R01NS097750 to V.S. We thank Aaron Cooper for assistance with experiments; and Ben Deverett for assistance with data analysis.
The authors declare no competing financial interests.
References
- Andreasen NC, Pierson R (2008) The role of the cerebellum in schizophrenia. Biol Psychiatry 64:81–88. 10.1016/j.biopsych.2008.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badura A, Verpeut JL, Metzger JW, Pereira TD, Pisano TJ, Deverett B, Bakshinskaya DE, Wang SS (2018) Normal cognitive and social development require posterior cerebellar activity. Elife 7:1–36. 10.7554/eLife.36401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker EB, Stoodley CJ (2013) Autism spectrum disorder and the cerebellum. Int Rev Neurobiol 113:1–34. 10.1016/B978-0-12-418700-9.00001-0 [DOI] [PubMed] [Google Scholar]
- Bogenmann E, Thomas PS, Li Q, Kim J, Yang LT, Pierchala B, Kaatrinen V (2011) Generation of mice with a conditional allele for the p75(NTR) neurotrophin receptor gene. Genesis 49:862–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder A, Roth-Albin I, Bhatia S, Pilquil C, Lee JH, Bhatia M, Levadoux-Martin M, McNicol J, Russell J, Collins T, Draper JS (2013) Lengthened G1 phase indicates differentiation status in human embryonic stem cells. Stem Cells Dev 22:279–295. 10.1089/scd.2012.0168 [DOI] [PubMed] [Google Scholar]
- Carta M, Chen C, Schott A, Dorizan S, Khodakhah K (2019) Cerebellar modulation of the reward circuitry and social behavior. Science 248:1–10. 10.1126/science.aav0581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft DR, Olson MF (2006) The rho GTPase effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by distinct mechanisms. Mol Cell Biol 26:4612–4627. 10.1128/MCB.02061-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M, Petit D, Bertoglio J (2012) Cell cycle regulation of ρ signaling pathways. Cell Cycle 11:3003–3010. 10.4161/cc.21088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello AM, Stoodley CJ (2015) Cerebro-cerebellar circuits in autism spectrum disorder. Front Neurosci 9:408. 10.3389/fnins.2015.00408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emi K, Kakegawa W, Miura E, Ito-Ishida A, Kohda K, Yuzaki M (2013) Reevaluation of the role of parallel fiber synapses in delay eyeblink conditioning in mice using Cbln1 as a tool. Front Neural Circuits 7:180. 10.3389/fncir.2013.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ, Chauhan A, Chauhan V, Dager SR, Dickson PE, Estes AM, Goldowitz D, Heck DH, Kemper TL, King BH, Martin LA, Millen KJ, Mittleman G, Mosconi MW, Persico AM, et al. (2012) Consensus paper: pathological role of the cerebellum in autism. Cerebellum 11:777–807. 10.1007/s12311-012-0355-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiez JA, Petersen SE, Cheney MK, Raichle ME (1992) Impaired non-motor learning and error detection associated with cerebellar damage: a single case study. Brain 115:155–178. 10.1093/brain/115.1.155 [DOI] [PubMed] [Google Scholar]
- Fukata M, Nakagawa M, Kaibuchi K (2003) Roles of ρ-family GTPases in cell polarisation and directional migration. Curr Opin Cell Biol 15:590–597. 10.1016/S0955-0674(03)00097-8 [DOI] [PubMed] [Google Scholar]
- Govek EE, Wu Z, Acehan D, Molina H, Rivera K, Zhu X, Fang Y, Tessier-Lavigne M, Hatten ME (2018) Cdc42 regulates neuronal polarity during cerebellar axon formation and glial-guided migration. iScience 1:35–48. 10.1016/j.isci.2018.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick LJ, Philpott A (2014) Nervous decision-making: to divide or differentiate. Trends Genet 30:254–261. 10.1016/j.tig.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick LJ, Ali FR, Azzarelli R, Philpott A (2015) Cell cycle regulation of proliferation versus differentiation in the central nervous system. Cell Tissue Res 359:187–200. 10.1007/s00441-014-1895-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AW, Kim JY, Yoon SO (2002) Activation of rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. J Neurosci 22:156–166. 10.1523/JNEUROSCI.22-01-00156.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AW, Li QM, Tep C, Park JB, He Z, Yoon SO (2008) The role of Kalirin9 in p75/Nogo receptor-mediated RhoA activation in cerebellar granule neurons. J Biol Chem 283:24690–24697. 10.1074/jbc.M802188200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiney SA, Wohl MP, Chettih SN, Ruffolo LI, Medina JF (2014) Cerebellar-dependent expression of motor learning during eyeblink conditioning in head-fixed mice. J Neurosci 34:14845–14853. 10.1523/JNEUROSCI.2820-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Ugurbil K, Strick PL (1994) Activation of cerebellar output nucleus during cognitive processing. Science 265:949–951. 10.1126/science.8052851 [DOI] [PubMed] [Google Scholar]
- Kloth AD, Badura A, Li A, Cherskov A, Connolly SG, Giovannucci A, Bangash MA, Grasselli G, Peñagarikano O, Piochon C, Tsai PT, Geschwind DH, Hansel C, Sahin M, Takumi T, Worley PF, Wang SS (2015) Cerebellar associative sensory learning defects in five mouse autism models. Elife 4:e06085. 10.7554/eLife.06085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legué E, Gottshall JL, Jaumouillé E, Roselló-Díez A, Shi W, Barraza LH, Washington S, Grant RL, Joyner AL (2016) Differential timing of granule cell production during cerebellum development underlies generation of the foliation pattern. Neural Dev 11:17. 10.1186/s13064-016-0072-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiner HC, Leiner AL, Dow RS (1986) Does the cerebellum contribute to mental skill? Behav Neurosci 100:443–454. 10.1037/0735-7044.100.4.443 [DOI] [PubMed] [Google Scholar]
- Lin Z, Tann JY, Goh ET, Kelly C, Lim KB, Gao JF, Ibanez CF (2015) Structural basis of death domain signaling in the p75 neurotrophin receptor. Elife 4:1–21. 10.7554/eLife.11692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Albers M (1995) Cerebellum and schizophrenia: a selective review. Schizophr Bull 21:241–250. 10.1093/schbul/21.2.241 [DOI] [PubMed] [Google Scholar]
- Matsuda K, Miura E, Miyazaki T, Kakegawa W, Emi K, Narumi S, Fukazawa Y, Ito-Ishida A, Kondo T, Shigemoto R, Watanabe M, Yuzaki M (2010) Cbln1 is a ligand for an orphan glutamate receptor d2, a bidirectional synapse organizer. Science 328:363–368. 10.1126/science.1185152 [DOI] [PubMed] [Google Scholar]
- Moreno-Rius J. (2018) The cerebellum in fear and anxiety-related disorders. Prog Neuropsychopharmacol Biol Psychiatry 85:23–32. 10.1016/j.pnpbp.2018.04.002 [DOI] [PubMed] [Google Scholar]
- Mulherkar S, Uddin MD, Couvillon AD, Sillitoe RV, Tolias KF (2014) The small GTPases RhoA and Rac1 regulate cerebellar development by controlling cell morphogenesis, migration and foliation. Dev Biol 394:39–53. 10.1016/j.ydbio.2014.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, Hall A (1995) An essential role for ρ, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269:1270–1272. 10.1126/science.7652575 [DOI] [PubMed] [Google Scholar]
- Orlowski D, Bjarkam CR (2012) A simple reproducible and time saving method of semi-automatic dendrite spine density estimation compared to manual spine counting. J Neurosci Methods 208:128–133. 10.1016/j.jneumeth.2012.05.009 [DOI] [PubMed] [Google Scholar]
- Phillips JR, Hewedi DH, Eissa AM, Moustafa AA (2015) The cerebellum and psychiatric disorders. Front Public Health 3:66. 10.3389/fpubh.2015.00066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A (2004) Cell migration: ρ GTPases lead the way. Dev Biol 265:23–32. 10.1016/j.ydbio.2003.06.003 [DOI] [PubMed] [Google Scholar]
- Ridley AJ. (2001) Rho GTPases and cell migration. J Cell Sci 114:2713–2722. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. (2015) ScienceDirect ρ GTPase signalling in cell migration. Curr Opin Cell Biol 36:103–112. 10.1016/j.ceb.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccio M, Schmitter D, Knobloch M, Okawa Y, Sage D, Lutolf MP (2013) Predicting stem cell fate changes by differential cell cycle progression patterns. Development 140:459–470. 10.1242/dev.086215 [DOI] [PubMed] [Google Scholar]
- Sacchetti B, Scelfo B, Tempia F, Strata P (2004) Long-term synaptic changes induced in the cerebellar cortex by fear conditioning. Neuron 42:973–982. 10.1016/j.neuron.2004.05.012 [DOI] [PubMed] [Google Scholar]
- Sajdel-Sulkowska EM, Xu M, Koibuchi N (2009) Increase in cerebellar neurotrophin-3 and oxidative stress markers in autism. Cerebellum 8:366–372. 10.1007/s12311-009-0105-9 [DOI] [PubMed] [Google Scholar]
- Schneiderman N, Fuentes I, Gormezano I (1962) Acquisition and extinction of the classically conditioned eyelid response in the albino rabbit. Science 136:650–652. 10.1126/science.136.3516.650 [DOI] [PubMed] [Google Scholar]
- Segura M, Pedreño C, Obiols J, Taurines R, Pàmias M, Grünblatt E, Gella A (2015) Neurotrophin blood-based gene expression and social cognition analysis in patients with autism spectrum disorder. Neurogenetics 16:123–131. 10.1007/s10048-014-0434-9 [DOI] [PubMed] [Google Scholar]
- Stoodley CJ, Limperopoulos C (2016) Structure–function relationships in the developing cerebellum: evidence from early-life cerebellar injury and neurodevelopmental disorders. Semin Fetal Neonatal Med 21:356–364. 10.1016/j.siny.2016.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strata P, Scelfo B, Sacchetti B (2011) Involvement of cerebellum in emotional behavior. Physiol Res 60:S38–S48. [DOI] [PubMed] [Google Scholar]
- Turner BM, Paradiso S, Marvel CL, Pierson R, Boles Ponto LL, Hichwa RD, Robinson RG (2007) The cerebellum and emotional experience. Neuropsychologia 45:1331–1341. 10.1016/j.neuropsychologia.2006.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalonga P, Ridley AJ (2006) Rho GTPases and cell cycle control. Growth Factors 24:159–164. 10.1080/08977190600560651 [DOI] [PubMed] [Google Scholar]
- Wagner MJ, Kim TH, Savall J, Schnitzer MJ, Luo L (2017) Cerebellar granule cells encode the expectation of reward. Nature 544:96–100. 10.1038/nature21726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Kloth AD, Badura A (2014) The cerebellum, sensitive periods, and autism. Neuron 83:518–532. 10.1016/j.neuron.2014.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Tohyama M (2003) The p75 receptor acts as a displacement factor that releases ρ from ρ-GDI. Nat Neurosci 6:461–467. 10.1038/nn1045 [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA (1999) Neurotrophin binding to the p75 receptor modulates ρ activity and axonal outgrowth. Neuron 24:585–593. 10.1016/S0896-6273(00)81114-9 [DOI] [PubMed] [Google Scholar]
- Zanin JP, Abercrombie E, Friedman WJ (2016) Proneurotrophin-3 promotes cell cycle withdrawal of developing cerebellar granule cell progenitors via the p75 neurotrophin receptor. Elife 5:e16654. 10.7554/eLife.16654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeinieh M, Salehi A, Rajkumar V, Barker PA (2015) p75NTR-dependent Rac1 activation requires receptor cleavage and activation of an NRAGE and NEDD9 signaling cascade. J Cell Sci 128:447–459. 10.1242/jcs.152173 [DOI] [PubMed] [Google Scholar]