

Abstract
Although several genes have been identified to promote axon regeneration in the CNS, our understanding of the molecular mechanisms by which mammalian axon regeneration is regulated is still limited and fragmented. Here by using female mouse sensory axon and optic nerve regeneration as model systems, we reveal an unexpected role of telomerase reverse transcriptase (TERT) in regulation of axon regeneration. We also provide evidence that TERT and p53 act downstream of c-Myc to control sensory axon regeneration. More importantly, overexpression of p53 in sensory neurons and retinal ganglion cells is sufficient to promote sensory axon and optic never regeneration, respectively. The study reveals a novel c-Myc-TERT-p53 signaling pathway, expanding horizons for novel approaches promoting CNS axon regeneration.
SIGNIFICANCE STATEMENT Despite significant progress during the past decade, our understanding of the molecular mechanisms by which mammalian CNS axon regeneration is regulated is still fragmented. By using sensory axon and optic nerve regeneration as model systems, the study revealed an unexpected role of telomerase reverse transcriptase (TERT) in regulation of axon regeneration. The results also delineated a c-Myc-TERT-p53 pathway in controlling axon growth. Last, our results demonstrated that p53 alone was sufficient to promote sensory axon and optic nerve regeneration in vivo. Collectively, the study not only revealed a new mechanisms underlying mammalian axon regeneration, but also expanded the pool of potential targets that can be manipulated to enhance CNS axon regeneration.
Keywords: axon regeneration, c-Myc, optic nerve regeneration, p53, telomerase reverse transcriptase
Introduction
One major reason for the failed axon regeneration in the adult mammalian CNS is the diminished intrinsic axon regeneration ability in neurons during maturation and aging (Sun and He, 2010). During the past decade, genes regulating the intrinsic axon regeneration ability have been identified, including Pten (Park et al., 2008; Liu et al., 2010), KLF4 (Moore et al., 2009), SOCS3 (Smith et al., 2009), SOX11 (Wang et al., 2015), Lin28 (Wang et al., 2018), and c-Myc (Belin et al., 2015). Despite significant progress, our understanding of the molecular mechanisms by which mammalian axon regeneration is regulated is still fragmented. In contrast, neurons from the peripheral nervous system (PNS) can regenerate their axons by reactivating the intrinsic axon growth abilities in response to peripheral nerve injury (Michaelevski et al., 2010; Chandran et al., 2016) via a transcription-dependent process (Smith and Skene, 1997; Saijilafu et al., 2013). Several transcription factors (TFs) have been identified to orchestrate such process, such as c-Jun (Raivich et al., 2004; Zhou et al., 2004), Smad1 (Parikh et al., 2011; Saijilafu et al., 2013), and ATF3, etc. (Seijffers et al., 2007). Many of these TFs functioned similarly in the CNS to regulate axon regeneration (Bareyre et al., 2011; Parikh et al., 2011; Finelli et al., 2013; Qin et al., 2013; Fagoe et al., 2015). Conversely, most of the aforementioned genes regulating the intrinsic axon regeneration ability of CNS neurons also acted to control PNS axon regeneration (Fischer et al., 2004; Jankowski et al., 2009; Jing et al., 2012; Saijilafu et al., 2013; Wang et al., 2018). Thus, PNS axon regeneration provides a useful model system to study the molecular mechanisms underlying mammalian axon regeneration.
A recent study indicated that overexpression of c-Myc in retinal ganglion cells (RGCs) could promote optic nerve regeneration (Belin et al., 2015). However, the underlying mechanisms by which c-Myc regulated axon regeneration remained unclear. Previous studies in non-neuronal systems have identified many downstream target genes regulated by c-Myc, such as p53 (Reisman et al., 1993) and TERT (Wu et al., 1999; Khattar and Tergaonkar, 2017). Thus, it is likely that c-Myc might control axon regeneration through these downstream targets in neurons. Indeed, it has been shown that p53 signaling was necessary for both PNS and optic nerve regeneration (Tedeschi et al., 2009; Gaub et al., 2010, 2011; Di Giovanni and Rathore, 2012; Floriddia et al., 2012; Joshi et al., 2015). TERT is the catalytic subunit of the enzyme telomerase, which is well known for its role in regulation of telomere extension, cellular aging, and cancer (Maciejowski and de Lange, 2017). In postmitotic neurons, TERT has been shown to protect neurons from oxidative damages and degenerative changes during aging (Spilsbury et al., 2015; Miwa and Saretzki, 2017; Liu et al., 2018). Inhibition of TERT during iPSC-induced neuronal differentiation could produce aged neurons suitable for studying late-onset neurodegenerative diseases (Vera et al., 2016). Whether TERT plays any role in regulation of axon regeneration has not been investigated.
In this study, we demonstrated that TERT level was markedly increased in sensory neurons upon peripheral nerve injury, and was functionally involved in regulation of sensory axon regeneration. Moreover, we showed that TERT acted downstream of c-Myc to regulate sensory axon regeneration. Last, we found that p53 acted downstream of c-Myc and TERT to control sensory axon regeneration. Importantly, overexpression of p53 in sensory neurons and RGCs was sufficient to promote sensory axon regeneration in vivo and optic nerve regeneration, respectively. Collectively, our data not only revealed an unexpected function of TERT in regulation of axon regeneration, but also suggested that c-Myc, TERT, p53 signaling might act coordinately to regulate both PNS and CNS axon regeneration.
Materials and Methods
Animals and surgical procedures.
Adult female mice, 10-week-old (weighing 25 g–30 g) were used. All animals were handled according to the protocols of the Institutional Animal Care and Use Committee of the Soochow University. For surgical procedures, mice were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg) via intraperitoneal injection. The cornea was protected with eye ointment containing atropine sulfate during the surgery.
Reagents and antibodies.
10058-F4, BIBR1532, PFTα, and Tenovin-6 were from Selleck Chemicals, and CAG was from Sigma-Aldrich. Antibody against the neuron-specific class III β-tubulin mouse mAb (Tuj1; 1:1000) was from Sigma-Aldrich. The antibody against c-Myc rabbit mAb (1:1000) was from GeneTex. Antibodies against TERT rabbit mAb (1:1000) and p53 mouse mAb (1:1000) were from Abcam. All fluorescence secondary antibodies were purchased from Invitrogen. The Adeno-associated virus-p53 viral vector was purchased from Cyagen Biosciences. pEX4-c-Myc and pEX3-p53 plasmids were from Gene Pharma. The small interfering RNA (siRNA) against TERT and c-Myc were from Gene Pharma. The sequence of the sic-Myc is as follows: 5′-AACGUUAGCUUCACCAACAUU-3′; The sequence of the first siRNA against TERT (siTERT1) is as follows: 5′-CAGAUCAAGAGCAGUAGUCTT-3′, and sequence of the second siRNA against TERT (siTERT2) is as follows: 5′-GCAUCAAUAUAUACAAGAUTT-3′.
Cell cultures and axon length quantification.
Dissection and culture of adult sensory neurons were performed as described in our previous protocol (Saijilafu et al., 2013). Briefly, dorsal root ganglia (DRGs) were dissected out from 10-week old mice and incubated with 1 mg/ml collagenase A (Roche) for 90 min and then with 1× TrypLE (Life Technologies) for 20 min at 37°C. Then, DRGs were dissociated in culture medium, which was minimum essential media containing 5% fetal bovine serum (FBS), 20 μm uridine, 20 μm 5-fluoro-2-deoxyuridine, and penicillin/streptomycin. The isolated neurons were cultured onto glass coverslips, which were coated with a mixture of 100 μg/ml poly-d-lysine (Sigma-Aldrich) and 10 μg/ml laminin (Sigma-Aldrich). For chondroitin sulfate proteoglycans (CSPGs) or myelin experiments, the coverslips were coated with 100 μl of CSPGs (5 μg/ml) or purified CNS myelin. Cortical and hippocampal neurons were isolated from embryonic day (E)15 or E18 mouse embryos and treated with TrypLE for 5 min at 37°C, then the supernatants were cultured in neurobasal medium supplemented with penicillin/streptomycin, 1× GlutaMAX, and B27 for 3 d.
All images were analyzed with the AxioVision 4.7 software (Carl Zeiss MicroImaging). In each experiment, at least 100 neurons per condition were selected randomly, and the longest axon of each neuron was measured manually using the “measure/curve” application. The average axon length was quantified from three independent experiments.
RNA interference.
The dissociated neurons were suspended in 100 μl of Amaxa electroporation buffer (Lonza Cologne) containing with a mixture of 0.2 nmol siRNA or plasmids (5 μg per transfection). Then, the cells were transferred to a 2.0 mm cuvette, and electroporated with the Amaxa Nucleofector apparatus. After that, cells were mixed with the 500 μl prewarmed culture medium and plated onto coverslips. After the neurons were attached to the coverslips (∼4 h), the culture medium containing the electroporation buffer was changed. Three days later, the cells were fixed with 4% paraformaldehyde (PFA) and then subjected to further analysis.
Myelin extract preparation.
Myelin fractions were isolated as described by Norton and Poduslo (1973). In brief, three carefully cleaned adult rat brains were homogenized in 100 ml of 0.32 m sucrose. Each 33 ml volume of homogenate was layered over 25 ml of 0.85 m sucrose, and then it was centrifuged at 25,000 rpm for 30 min. The crude myelin layers formed between the interfaces of the two sucrose solutions was collected. The collected myelin layers were then suspended in 180 ml pure water by homogenization. Then, it was centrifuged again at 25,000 rpm for 15 min. The supernatant fluid was carefully discarded, and the crude myelin pellets were dispersed in 180 ml water and centrifuged at 10,000 rpm for 10 min. These procedures were repeated twice. All of the myelin pellets were mixed and resuspended in 100 ml of 0.32 m sucrose. Then, 33 ml of this suspension was layered over 0.85 m sucrose again and centrifuged at 25,000 rpm for 30 min. The purified myelin was finally carefully collected from the interface of the two sucrose solutions, and stored at −80°C.
Immunohistochemistry.
Under deep anesthesia, mice were perfused with 4% PFA. The lumbar L4–L5 DRGs were dissect out and postfixed with 4% PFA at 4°C overnight, followed by dehydration in 10, 20, and 30% sucrose solution (w/v) respectively. They were then cryosectioned into slices of 12 μm in thickness. For immunostaining, the sections were washed in PBS solution containing 0.5% Triton X-100, then, followed by blocking in 5% FBS and 0.3% Triton X-100 for 1 h at room temperature. Sections were incubated with the indicated primary antibodies overnight at 4°C, and then incubated with the corresponding secondary antibodies for 2 h at room temperature. Finally, the sections were mounted with mounting medium (Vector Laboratories, H-1400).
RNA isolation and RT-PCR.
The total RNA was extracted using the TRIzol reagent (Invitrogen), and the RNA was reverse transcribed into cDNA using Maxima H Minus Reverse Transcriptase according to the manufacturer's instructions (ThermoFisher Scientific). Quantitative PCR (qPCR) was performed with SYBR-Green Real-Time PCR Master Mix (Toyobo). Standard curves (cycle threshold values vs template concentration) were prepared for each target gene and the endogenous reference (18S) in each sample. The primer sequences were as follows; c-Myc: forward, 5′-ATCACAGCCCTCACTCAC-3′ and reverse, 5′-ACAGATTCC ACAAGGTGC-3′; TERT: forward, 5′-TGGTGGAGGTTGCCAA-3′ and reverse, 5′-CCACTGCATACTGGCGGATAC-3′; p53: forward, 5′-CGACGACATTCGGAT AAG-3′ and reverse, 5′-TTGCCAGATGAGGGACTA-3′. The amplification step involved an initial denaturation at 95°C for 10 s followed by 40 cycles of denaturation at 95°C for 15 s and annealing at 55°C for 30 s and extension at 72°C for 30 s. Reactions were performed in triplicate and the mRNA expression was normalized against the endogenous reference (18S). Data were quantified by CFX96TM real-time PCR detection system (Bio-Rad).
Western blot analysis.
Tissues or dissociated neurons were lysed with a RIPA buffer for 30 min. Equal amount of extracted proteins were separated with 10% SDS-PAGE, and transferred to the polyvinylidene difluoride (PVDF) membrane (Millipore). Then, PVDF membranes were blocked with 5% nonfat milk in tris-buffered saline buffer for 1 h at room temperature, and reacted with primary antibody overnight at 4°C. The membrane was incubated with horseradish peroxidase-conjugated secondary antibody (dilution of 1:500) at room temperature for 1 h. Last, the membrane was developed using an ECL Prime Western Blotting Detection Reagent (GE Healthcare). The protein bands density was quantified using the ImageJ software (National Institutes of Health) from three independent experiments.
In vivo electroporation of adult sensory neurons.
The in vivo electroporation of adult DRG neurons was performed as described previously (Saijilafu et al., 2011). In brief, under anesthesia, the left L4–L5 DRGs of 10-week-old mice were surgically exposed. A 1 μl solution containing indicated siRNAs or c-Myc plasmid, together with the plasmid encoding EGFP, were microinjected into the DRG using a glass needle pipette by the Picospritzer III (Parker). The scrambled siRNA or the EGFP encoding plasmid was used in the control groups. After microinjection, the electroporation was performed using a tweezer-like electrode (Ø1.2 mm) and an ECM830 Electro Square Porator BTX (five 15 ms pulses at 35 V with 950 ms interval). After the incision sites were closed, the animals were then allowed to recover. Two days later, the crush injury was made on the ipsilateral sciatic nerve with fine forceps and an 11-0 nylon epineural suture were place on the crush site. Another 3 d after the nerve crush, the animals were transcardially perfused with ice-cold 4% PFA, and the entire sciatic nerve segment was dissected out. The lengths of all EGFP-labeled regenerating axons were measured from the epineural suture point to the distal tips. The number of EGFP-positive axons was determined by the transfection efficiency of the plasmid encoding EGFP, which was ∼10% based on our previous study (Saijilafu et al., 2011). Only nerves containing at least 15 quantifiable axons were included in the results.
Sciatic nerve axotomy.
Under anesthesia, a 5-mm-long skin incision was cut aseptically on the legs. The overlaying muscles were then separated, and the sciatic nerve was transected at the sciatic notch with ophthalmic scissors. After a specific period of time, the L4 and L5 DRGs were isolated for further analysis, such as cell culture, tissue section, qPCR, or Western blotting. Mice who received sham surgery were used as “no-injury” controls.
Intravitreal injection and optic nerve crush.
After mice were anesthetized, a glass micropipette was inserted into the vitreous body of 4-week-old C57BL/6 mice avoiding damaging the lens, and a 1.0 μl of AAV2-EGFP or AAV2-p53 (titer 1.0 × 1012) viral vector suspension was carefully injected with a Picospritzer III (Parker; pressure: 9 psi; duration: 5 ms). Two weeks after the virus injection, the optic nerve was exposed intra-orbitally by blunt dissection, and a crush injury was performed ∼1.0 mm behind the optic disc using fine forceps for 2–3 s.
RGC axon anterograde labeling and quantification of optic nerve regeneration.
To label regenerating optic nerve axons, 12 d after the optic nerve crush injury, 2 μl of fluorescent AlexaFluor 488-conjugated cholera toxin β subunit (CTB; 2.0 μg/μl, Invitrogen;) was microinjected into the vitreous body using a Picospritzer III (Parker; pressure: 9 psi; duration: 5 ms). Two days later, animals were perfused with 4% PFA via the left ventricle. The entire optic nerve was then harvested. Regenerating RGC axons were calculated as previously described (Kurimoto et al., 2010). Using 12-μm-thick longitudinal sections of the optic nerves, the numbers of CTB-labeled axons were counted in each of the five sections (every 4 sections) at a different distance from the lesion site. Only CTB-labeled green fibers longer than 5 μm were included as axon fragments. The width of cross-section was measured at the counting point and used to calculate the axons number per millimeter of nerve width. Then, the average axon number per millimeter on all parts was measured. The total number of axons in the nerve with radius “r” was estimated by summing all the parts with thickness t(12 μm): Σad = πr2 × [average axons/mm]/t.
Whole-mount retina staining.
After perfusion, whole retinas were harvested and incubated in PBS containing 10% FBS and 0.3% Triton X-100 for 1 h. Then, they were followed by application of primary antibody Tuj1 (1:500) overnight at 4°C. After rinsing the retinas with blocking buffer three times for 30 min, the tissues were incubated with Alexa Fluor 594-conjugated secondary antibody for 2 h at room temperature. Cell survival rate was reported as the number of Tuj1-positive RGCs per square millimeter averaged over the eight random fields per retina, and then averaged again across in each experimental condition.
Statistics.
Statistical analyses were presented as the mean ± SEM. No statistical methods were used to predetermine sample sizes. Comparisons between two groups were performed using two tailed Student's t test. Multiple group comparisons were performed with one-way ANOVA followed by Bonferroni's post-test as post hoc test. GraphPad Prism software was used for all statistical analysis. Statistical significance was considered when the p value was < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
c-Myc is necessary and sufficient for supporting sensory axon regeneration in vitro and in vivo
A recent study demonstrated that peripheral axotomy could upregulate c-Myc expression in adult DRG sensory neurons (Belin et al., 2015). However, the functional roles of c-Myc in peripheral axon regeneration remain unknown. Similarly, our immunostaining results showed that c-Myc expression was significantly upregulated in L4 and L5 DRG neurons after sciatic nerve injury (Fig. 1A,B). Western blot analysis showed that peripheral nerve injury-induced upregulation of c-Myc expression in sensory neurons reached a peak at ∼3 d post-injury, and then decreased 7 d post-injury (Fig. 1C,D). The mRNA levels of c-Myc were also increased after peripheral axotomy, indicating enhanced gene transcription or reduced mRNA degradation (Fig. 1E). To investigate the functional role of c-Myc in regulation of sensory axon regeneration, we applied the specific c-Myc inhibitor, 10058-F4 (10 μm) to cultured adult DRG neurons for 3 d. The result showed that 10058-F4 significantly impaired the regenerative axon growth (Fig. 1F,G). Similarly, downregulation of c-Myc expression via a siRNA against c-Myc (Chen et al., 2010) also reduced axon growth of cultured DRG neurons (Fig. 1H,I), confirming the pharmacological results.
Figure 1.
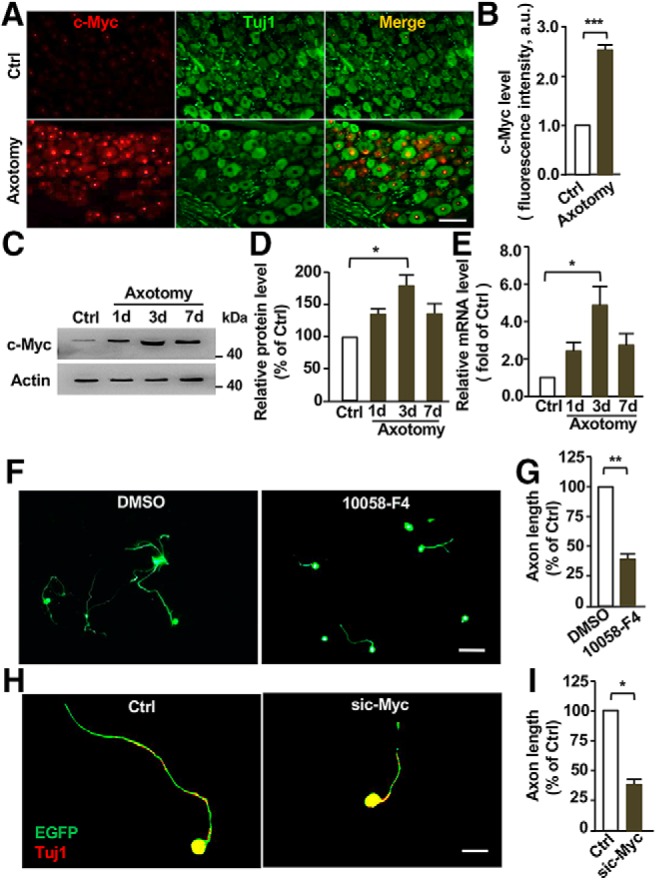
c-Myc is upregulated in adult sensory neurons upon peripheral nerve injury and functionally required for sensory axon regeneration in vitro. A, Representative immunostaining of DRG sections showing nucleus localization of c-Myc in dorsal root ganglion (DRG) neurons and upregulation of c-Myc levels after sciatic nerve axotomy. Scale bar, 50 μm. B, Quantification of nucleus fluorescence intensity of c-Myc (n = 3 independent experiments; p = 0.0001). C, Representative Western blot images showing significantly increased levels of c-Myc in adult DRG neurons in vivo 3 d after sciatic nerve axotomy. D, Quantification of Western blot results from three independent experiments. p = 0.021. E, qPCR results showing significantly increased mRNA levels of c-Myc in adult DRG neurons in vivo 3 d after sciatic nerve axotomy (n = 3 independent experiments; p = 0.029). F, Representative images showing that inhibition of c-Myc in cultured DRG neurons with the pharmacological agent, 10058-F4, for 3 d dramatically reduced DRG axon growth. Scale bar, 200 μm. G, Quantification of average axon lengths of control neurons treated with DMSO and neurons treated with the c-Myc inhibitor from three independent experiments. p = 0.0083. H, Representative images showing that knocking down c-Myc with a specific siRNA in cultured DRG neurons dramatically reduced axon growth after 3 d in culture. Scale bar, 50 μm. I, Quantification of average axon lengths of control neurons expressing a scrambled siRNA and neurons expressing sic-Myc from three independent experiments. p = 0.018. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Overexpression of c-Myc in RGCs has been shown to promote optic nerve regeneration (Belin et al., 2015) in the inhibitory CNS environment. We therefore examined whether overexpression of c-Myc in sensory neurons could promote axon regeneration over inhibitory substrates. When c-Myc was overexpressed in cultured DRG neurons in vitro, we found that sensory axon growth was significantly enhanced on supportive substrates laminin (Fig. 2A,B). CSPGs and myelin are two well known major inhibitory substrates of axon regeneration (Lee and Zheng, 2012; Ohtake and Li, 2015). We thus investigated whether overexpression of c-Myc was able to overcome the inhibitory effects of these substrates. Dissociated sensory neurons transfected with c-Myc plasmid were cultured on CSPGs or myelin-coated cell culture plates for 3 d. The results showed that either CSPGs or myelin substrates significantly inhibited the axon growth of DRG neurons, and overexpression of c-Myc markedly enhanced their axon growth on either CSPGs (Fig. 2C,D) or myelin (Fig. 2E,F) substrates.
Figure 2.
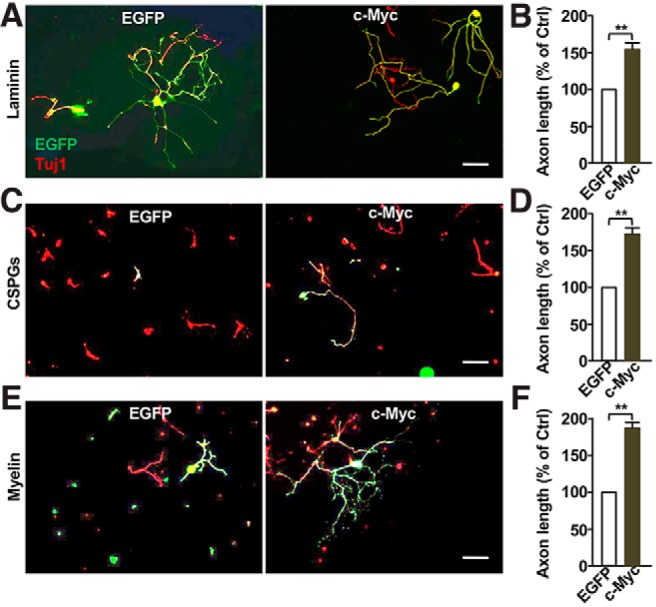
Overexpression of c-Myc in sensory neurons is sufficient to promote axon growth over both supportive and inhibitory substrates. A, Representative images showing that overexpression of c-Myc significantly enhanced regenerative axon growth of sensory neurons cultured on laminin. Scale bar, 200 μm. B, Quantification of A from three independent experiments. p = 0.00986. C, Representative images showing that overexpression of c-Myc significantly enhanced regenerative axon growth of sensory neurons cultured on the inhibitory substrates, chondroitin sulfate proteoglycans. Scale bar, 200 μm. D, Quantification of C from three independent experiments. p = 0.0094. E, Representative images showing that overexpression of c-Myc significantly enhanced regenerative axon growth of sensory neurons cultured on myelin inhibitory substrates. Scale bar, 200 μm. F, Quantification of E from three independent experiments. p = 0.0091.Data are represented as mean ± SEM. **p < 0.01.
We next examined whether c-Myc was necessary for sensory axon regeneration in vivo using the DRG electroporation technique (Saijilafu et al., 2011, 2014). The specific siRNA against c-Myc, together with the plasmid encoding enhanced green fluorescent protein (EGFP), were co-electroporated into adult mouse L4–L5 DRGs in vivo. The control group was electroporated with a scrambled siRNA and EGFP plasmid. Two days later, a sciatic nerve crush was performed using fine forceps and the injured site was labeled with 11-0 nylon epineural sutures as previously described (Saijilafu et al., 2014). After 3 more days, the animals were perfused with PFA and the entire sciatic nerve was dissected out. The resulting EGFP labeled sensory axons were imaged, manually traced, and measured from the injury site to the distal axon tips to quantify axon regeneration by a different person not directly involved in the experiments. The results showed that the siRNA against c-Myc significantly reduced sensory axon regeneration compared with that of control neurons (Fig. 3A–C). Western blot analysis showed that the siRNA against c-Myc successfully reduced the protein level of c-Myc in DRGs (Fig. 3D,E). To determine whether c-Myc was sufficient to promote PNS axon regeneration in vivo, we transfected L4 and L5 DRGs of adult mouse with the c-Myc plasmid via in vivo DRG electroporation, and performed sciatic nerve crush injury 2 d later. After 3 d, we found that overexpression of c-Myc significantly promoted sensory axon regeneration in vivo (Fig. 3F–H).
Figure 3.

c-Myc is both necessary and sufficient for sensory axon regeneration in vivo. A, Top, Time line of the experiments. Bottom, Representative images showing that knocking down c-Myc with siRNA (sic-Myc) in sensory neurons in vivo significantly impaired sensory axon regeneration. The red line indicates the nerve crush sites, and the red arrows indicate the distal ends of regenerating axons. Left, Images in the white boxes were enlarged and presented on the right. Scale bars: left, 500 μm; right, 250 μm. B, Quantification of A showing that knocking down c-Myc significantly impaired sensory axon regeneration in vivo (n = 6 nerves from 6 mice for each condition; p = 0.0001). C, Cumulative distribution curves showing that knocking down c-Myc in sensory neurons significantly inhibited sensory axon regeneration in vivo. D, Representative Western blot image showing reduced protein level of c-Myc in DRGs after knocking down c-Myc in vivo. E, Quantification of Western blot results from three independent experiments. p = 0.0289. F, Top, Time line of the experiments. Bottom, Representative images showing that c-Myc overexpression promoted sensory axon regeneration in vivo 3 d after sciatic nerve crush injury. The red line indicates the nerve crush sites and the red arrows indicate distal ends of selected regenerating axons. Left, Images in the white boxes were enlarged and presented on the right. Scale bars: left, 500 μm; right, 250 μm. G, Quantification of F showing that overexpression of c-Myc significantly promoted sensory axon regeneration in vivo (n = 6 nerves from 6 mice for each condition; p = 0.0001). H, Cumulative distribution curves showing that overexpression of c-Myc in sensory neurons significantly promoted sensory axon regeneration in vivo. Please note that the control group in A–C is the same as that in F–H. Data are represented as mean ± SEM. *p < 0.05, ***p < 0.001.
TERT functions to regulate sensory axon regeneration in vitro and in vivo
We first examined the protein levels of TERT in mouse DRGs during development and maturation. The results showed that the level of TERT gradually decreased from E14 to adult (Fig. 4A,C), suggesting that TERT function to support axon growth during development. To determine the role of TERT in regulation of sensory axon regeneration, we examined the level of TERT in sensory neurons upon peripheral nerve injury. Western blot analysis showed increased levels of TERT after the peripheral axotomy (Fig. 4B,D). Consistent with this observation, the immunostaining analysis showed that TERT expression in DRG neurons was significantly increased after peripheral axotomy (Fig. 4E,F). It should be noted that TERT was mostly localized in the cytoplasm of sensory neurons, which is consistent with previous studies of brain neurons (Spilsbury et al., 2015; Liu et al., 2018). One potential explanation is that in postmitotic neurons TERT is localized in mitochondria to reduce oxidative stress and protect neurons from apoptosis (Liu et al., 2018). Functionally, inhibition of TERT with a specific pharmacological inhibitor BIBR1532 (Lavanya et al., 2018) impaired regenerative axon growth of sensory neurons in a dose-dependent manner without any toxic effects on neuronal survival (Fig. 4G–I). To confirm the pharmacological results, we knocked down TERT with a specific siRNA (siTERT1; Wei et al., 2013) and found that it also effectively reduced sensory axon regeneration in vitro (Fig. 5A–C). Immunostaining analysis demonstrated that siTERT1 significantly reduced the level of TERT in sensory neurons (Fig. 5D,E). To examine whether TERT functioned to regulate sensory axon regeneration in vivo, we transfected L4 and L5 DRG neurons with the specific siTERT1 by using in vivo electroporation technique, and performed sciatic nerve crush injury 2 d later. Three days after the nerve crush, sensory axon regeneration was analyzed. We found that knocking down TERT significantly reduced sensory axon regeneration in vivo (Fig. 5F–H), indicating that TERT contributed to sensory axon regeneration. Western blot analysis verified that siTERT1 successfully reduced the protein level of TERT in vivo (Fig. 5I,J). These results revealed an unexpected role of telomere regulatory complex in regulating axon regeneration.
Figure 4.

TERT is upregulated in sensory neurons upon sciatic nerve axotomy. A, Representative Western blot images showing gradually reduced proteins levels of TERT in DRG tissues during developmental and maturation. B, Representative Western blot images showing increased protein levels of telomerase reverse transcriptase (TERT) in DRG tissues at different dates after sciatic nerve axotomy. C, Quantification of Western blot results shown in A from three independent experiments. p = 0.0073 for P7 vs E14, p = 0.0003 for adult vs E14. D, Quantification of Western blot results shown in B from three independent experiments. p = 0.0076. E, Representative immunostaining images of sectioned DRGs showing significantly elevated TERT levels in sensory neurons 3 d after sciatic nerve axotomy. Note that most TERT staining in adult sensory neurons was localized in the cytoplasm. Scale bar, 100 μm. F, Quantification of fluorescence intensity of TERT staining from three independent experiments. p = 0.0001. G, Representative images showing that inhibition of TERT activity with the pharmacological inhibitor, BIBR1532, at different concentrations, blocked regenerative axon growth of sensory neurons. Scale bar, 200 μm. H, Quantification of the average axon lengths of DMSO-treated control neurons and neurons treated with three different concentration of BIBR1532 from three independent experiments. p = 0.0041 for 25 μm vs 0 μm, p = 0.0007 for 50 μm vs 0 μm, p = 0.0002 for 100 μm vs 0 μm. I, Quantification of neuronal survival after treatment with DMSO or different concentration of BIBR1532; ns indicates no significant difference. Data are represented as mean ± SEM. **p < 0.01, ***p < 0.001.
Figure 5.

Knocking down TERT impairs sensory axon regeneration in vitro and in vivo. A, Representative Western blot images showing successful knockdown of TERT protein levels in cultured DRG neurons with the siRNA against TERT (siTERT1). B, Representative images showing that knocking down of TERT in cultured sensory neurons with a specific siRNA markedly decreased axon growth after 3 d. Scale bar, 50 μm. C, Quantification of average axon lengths of control neurons or neurons expressing siTERT1 in cultured sensory neurons from three independent experiments. p = 0.0004. D, Representative immunostaining images showing reduced level of TERT in cultured sensory neurons after transfection with siTERT1 compared with control neurons transfected with the scrambled siRNA. E, Quantification of D from three independent experiments. p = 0.0001. F, Top, Time line of the experiment. Bottom, Representative images of sensory axon regeneration in vivo following TERT knockdown 3 d after sciatic nerve crush injury. The red line indicates the crush sites, and the red arrows indicate the distal ends of selected regenerating axons. Left, Images in the white boxes were enlarged and presented on the right. Scale bars: left, 500 μm; right, 250 μm. G, Quantification of F showing that knocking down TERT in sensory neurons significantly inhibited axon regeneration in vivo (n = 6 nerves from 6 mice for each condition; p = 0.0002). H, Cumulative distribution curves showing that knocking down TERT in sensory neurons significantly reduced sensory axon regeneration in vivo. I, Representative Western blot images showing successful knockdown of TERT protein level in DRG tissues in vivo with the siTERT1. J, Quantification of Western blot results shown in I from three independent experiments. p = 0.0024. Please note the control group in F–J is the same as that shown in Figure 2. Data are represented as mean ± SEM. **p < 0.01, ***p < 0.001.
To determine whether TERT acted downstream of c-Myc in sensory neurons to regulate axon regeneration, we examined the expression level of TERT after knocking down c-Myc with siRNA. The results showed that downregulation of c-Myc resulted in significantly reduced level of TERT in cultured sensory neurons (Fig. 6A,B). Conversely, overexpression of c-Myc in sensory neurons markedly enhanced TERT expression (Fig. 6C,D). Functionally, when we treated sensory neurons with a specific pharmacological TERT activator CAG (Ip et al., 2014), we found that activation of TERT with CAG alone had no promoting effect on sensory axon growth cultured for 3 d, likely because of upregulation of endogenous TERT. However, CAG (25 μm) treatment significantly rescued regenerative axon growth inhibited by c-Myc knockdown (Fig. 6E,F). Collectively, these results indicated that TERT acted downstream of c-Myc to regulate axon regeneration.
Figure 6.
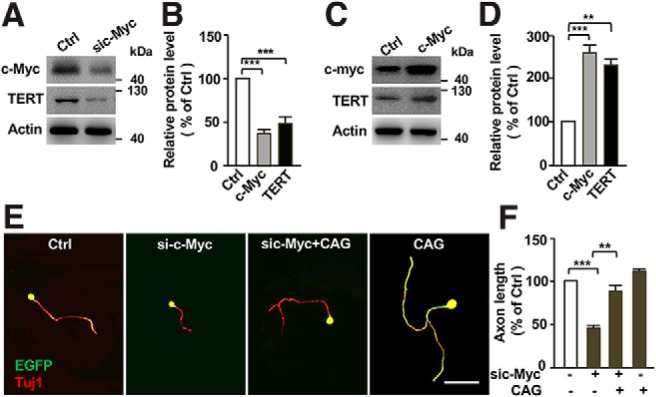
TERT acts downstream of c-Myc in sensory neuron to control axon regeneration. A, Representative Western blot images showing that knocking down c-Myc led to reduced TERT protein levels in sensory neurons. B, Quantification of Western blot results shown in A from three independent experiments. p = 0.0007 for c-Myc vs control, p = 0.0008 for TERT vs control. C, Representative Western blot images showing that overexpression of c-Myc led to increased TERT protein levels in sensory neurons. D, Quantification of Western blot results shown in C from three independent experiments. p = 0.0001 for c-Myc vs control, p = 0.0071 for TERT vs Control. E, Representative images showing that treatment of cultured sensory neurons with the specific TERT activator, CAG (25 μm), for 3 d, successfully rescued sensory axon growth inhibited by c-Myc knockdown. Scale bar, 50 μm. F, Quantification of average axon lengths of E in different conditions from three independent experiments. p = 0.0005 for sic-Myc vs control, p = 0.0032 for sic-Myc vs CAG+sic-Myc. Data are represented as mean ± SEM. **p < 0.01, ***p < 0.001.
p53 is necessary and sufficient for supporting sensory axon regeneration in vitro and in vivo
To investigate whether p53 acted in the c-Myc-TERT pathway to regulate axon regeneration, we examined the level of p53 in sensory neurons upon peripheral nerve injury. The results showed that p53 was upregulated in sensory neurons following sciatic nerve injury (Fig. 7A,C), suggesting that p53 act to support axon regeneration. In consistent, the protein levels of p53 in DRGs were gradually decreased when sensory axon growth ability subsides upon maturation (Fig. 7B,D). Functionally, the specific membrane-permeable p53 inhibitor, PFTα, markedly inhibited sensory axon growth in a dose-dependent manner without affecting neuronal survival (Fig. 7E–G), indicating that p53 was necessary for regenerative sensory axon growth. These results were consistent with previous studies (Tedeschi et al., 2009; Gaub et al., 2010, 2011; Di Giovanni and Rathore, 2012; Floriddia et al., 2012; Joshi et al., 2015), which showed that p53 was required for mammalian axon regeneration. Here we showed that overexpression of p53 in cultured sensory neurons alone significantly promoted axon growth (Fig. 7H–J). Moreover, p53 overexpression was able to restore axon growth suppressed by c-Myc knockdown (Fig. 7I,J), suggesting that p53 act downstream of c-Myc. Together, these results demonstrated that p53 was both necessary and sufficient to support sensory axon regeneration in vitro.
Figure 7.

p53 is necessary and sufficient for supporting sensory axon regeneration in vitro. A, Representative Western blot images showing markedly increased p53 protein levels in DRG tissues at different dates after sciatic nerve axotomy. B, Representative Western blot images showing gradually reduced levels of p53 in DRG tissues during development and maturation. C, Quantification of the Western blot results shown in A from three independent experiments. p = 0.0006. D, Quantification of the Western blot results shown in B from three independent experiments. p = 0.0211 for P7 vs E14, p = 0.0026 for adult vs E14. E, Representative images showing that inhibition of p53 with PFTα in cultured sensory neurons for 3 d dramatically blocked regenerative axon growth. Scale bar, 200 μm. F, Quantification of the average axon lengths in E from three independent experiments. p = 0.0001 for 10 μm vs 0 μm, p = 0.0001 for 20 μm vs 0 μm. G, Quantification of neuronal survival after treatment with DMSO or different concentration of PFTα. ns, No significant difference. H, Representative Western blot images showing increased level of p53 after p53 overexpression (OE) in adult sensory neurons. I, Quantification of the average axon lengths in J from three independent experiments. p = 0.0021 for sic-Myc vs control, p = 0.0152 for sic-Myc vs sic-Myc+p53, p = 0.0034 for p53 vs control. J, Representative images showing that overexpression of p53 were sufficient to promote sensory axon regeneration by itself in vitro and restored axon regeneration inhibited by c-Myc knockdown. Scale bar, 50 μm. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Previous studies in non-neuronal cells have shown that p53 activity was controlled by c-Myc (Reisman et al., 1993) or TERT (Zhou et al., 2017). Therefore, we investigated whether p53 acted downstream of c-Myc and TERT in sensory neurons to control axon regeneration. Indeed, the specific TERT inhibitor BIBR1532, which has been shown to reduce the protein level of TERT (Lavanya et al., 2018), also led to reduced p53 level in sensory neurons (Fig. 8A,B), indicating that p53 was regulated by the c-Myc-TERT signaling in sensory neurons. To determine whether p53 acted functionally downstream of the c-Myc-TERT pathway, we used another siRNA against TERT, siTERT2 (Fujiki et al., 2010), which reduced the protein level of TERT in sensory neurons (Fig. 8C,D). The results showed that either siTERT1 or siTERT2 impaired sensory axon growth in vitro. In addition, the p53 activator Tenovin-6 alone was sufficient to enhance axon growth, consistent with the p53 overexpression result shown above. More importantly, p53 activation was able to restore sensory axon growth inhibited by knocking down c-Myc or TERT via siRNA (Fig. 8E,F), confirming that p53 acted downstream of c-Myc and TERT to regulate axon regeneration.
Figure 8.
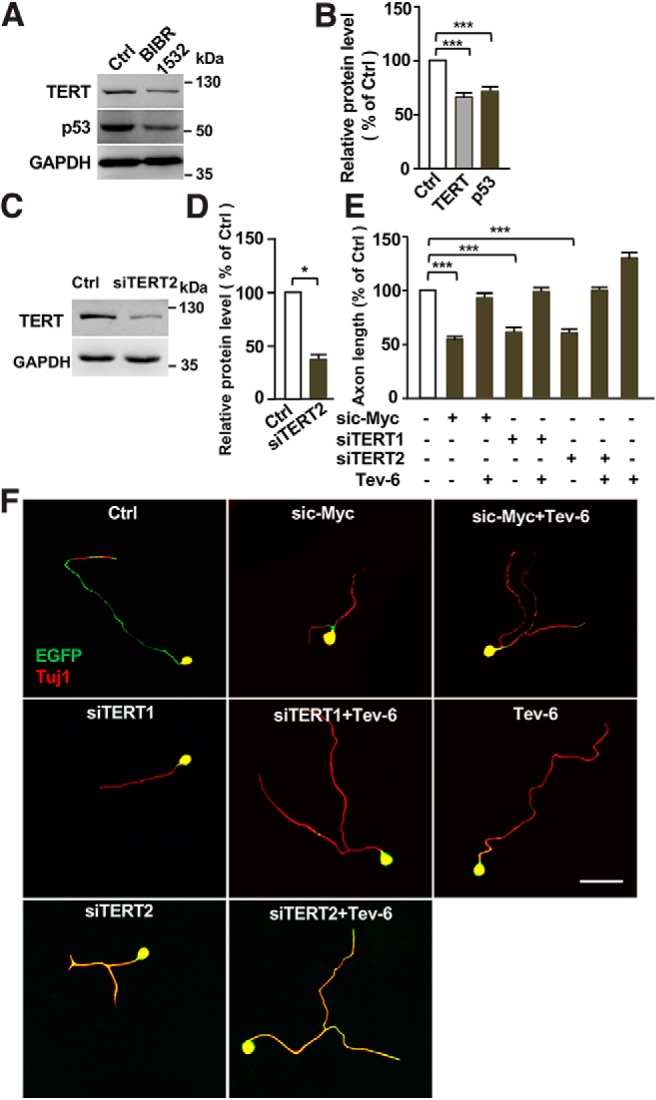
p53 acts downstream of c-Myc and TERT to regulate regenerative sensory axon growth. A, Representative Western blot images showing that inhibition of TERT activity with its specific inhibitor BIBR1532 led to reduced levels of TERT and p53 in sensory neurons. B, Quantification of the Western blot results shown in A from three independent experiments. p = 0.0001 for TERT vs control, p = 0.0002 for p53 vs control. C, Representative Western blot images showing that a different siRNA against TERT (siTERT2) resulted in reduced level of TERT in sensory neurons. D, Quantification of the Western blot results shown in C from three independent experiments. p = 0.0114. E, Quantification of the average axon lengths in F from three independent experiments. p = 0.0007 for sic-Myc vs control, p = 0.0009 for siTERT1 vs control, p = 0.0008 for siTERT2 vs control. F, Representative images showing that activation of p53 activity with tenovin-6 (0.5 μm) restored sensory axon regeneration defects induced by knocking down c-Myc with sic-Myc or TERT with two different siRNAs (siTERT1, siTERT2). Scale bar, 50 μm. Data are represented as mean ± SEM. *p < 0.05, ***p < 0.001.
Last, we examined the roles of p53 in regulation of sensory axon regeneration in vivo via the electroporation approach. The results showed that overexpression of p53 alone was able to enhance sensory axon regeneration in vivo (Fig. 9A–E). Moreover, overexpression of p53 significantly restored sensory axon regeneration impaired by c-Myc knockdown. Together, our results demonstrated that p53 functioned to regulate sensory axon regeneration in vitro and in vivo, likely downstream of the c-Myc-TERT pathway.
Figure 9.
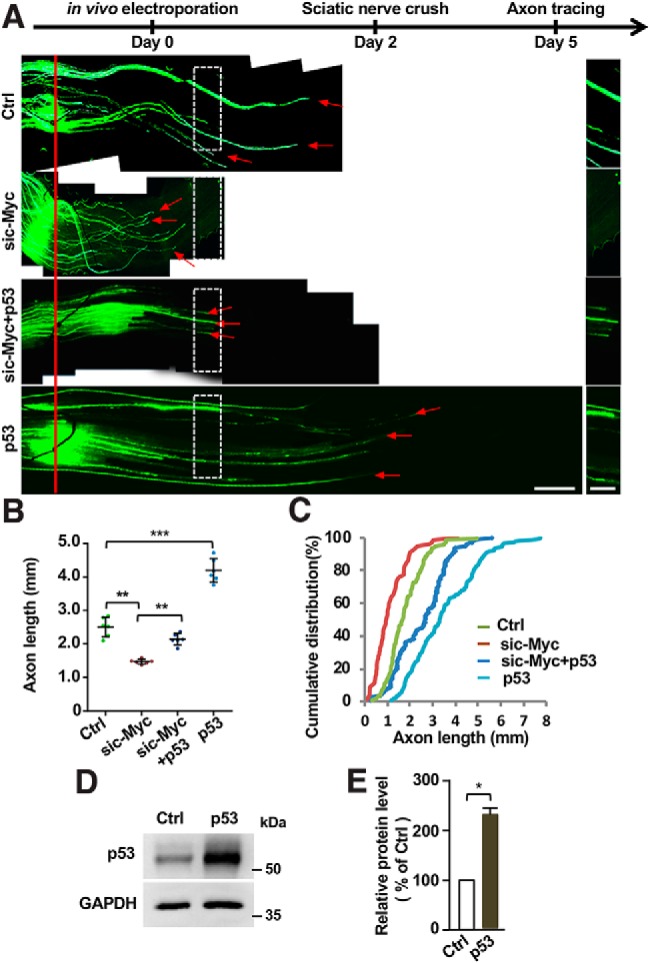
Overexpression of p53 in sensory neurons is sufficient to promote sensory axon regeneration and restore sensory axon regeneration impaired by c-Myc knockdown in vivo. A, Top, Time line of the experiment. Bottom, Representative images of sensory axon regeneration in vivo of control condition, c-Myc knockdown, overexpression of p53, or combined c-Myc knockdown and p53 overexpression. The red line indicates the nerve injury sites and the red arrows indicate the distal ends of selected regenerating axons. Left, Images in the white boxes were enlarged and presented on the right. Scale bars: left, 500 μm; right, 250 μm. B, Quantification of the results in A showing that overexpression of p53 alone could promote sensory axon regeneration in vivo and p53 overexpression significantly restored sensory axon regeneration impaired by c-Myc knockdown (n = 6 nerves from 6 mice for each condition; p = 0.0023 for sic-Myc vs control, p = 0.0031 for sic-Myc vs sic-Myc+p53, p = 0.0001 for p53 vs control). C, Cumulative distribution curves showing the same results as those in B. D, Representative Western blot images showing elevated p53 protein level in adult DRG tissues after overexpression of p53. E, Quantification of Western blot results shown in D from three independent experiments. p = 0.0264. Please note the results of the control and sic-Myc groups in this figure are the same as those shown in Figure 2. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
p53 regulates CNS axon growth and enhances optic nerve regeneration
Our results above showed that p53 was able to enhance mature sensory axon regeneration in vitro and in vivo. A previous study has shown that p53 was highly expressed in mammalian hippocampal neurons (Qin et al., 2009). Thus, we thought that p53 overexpression might enhance hippocampal axon growth as well. Therefore, we explored the functional roles of p53 in regulating axon growth of embryonic hippocampal or cortical neurons. E18 hippocampal neurons or E15 cortical neurons were treated with either the p53 inhibitor PFTα or activator tenovin-6. The results showed that inhibiting p53 with PFTα treatment markedly reduced axonal growth of cortical neurons (Fig. 10A,E), whereas activating p53 with tenovin-6 significantly enhanced cortical axon growth (Fig. 10B,F). Similar results were also observed using the hippocampal neuron axon growth assay (Fig. 10C,D,G,H). These results demonstrated that p53 was also an important regulator of axon growth from embryonic CNS neurons.
Figure 10.

p53 regulates axon growth of developing cortical and hippocampal neurons. A, Representative images showing that inhibition of p53 activity with the pharmacological inhibitor, PFTα, blocked cortical neuron axon growth. Scale bar, 200 μm. B, Representative images showing that activation of p53 activity with the small molecule, tenovin-6, promoted cortical neuronal axon growth. Scale bar, 200 μm. C, Representative images showing that inhibition of p53 activity with the pharmacological inhibitor, PFTα, blocked hippocampal neuron axon growth. Scale bar, 200 μm. D, Representative images showing that activation of p53 activity with the small molecule tenovin-6, promoted hippocampal neuron axon growth. Scale bar, 200 μm. E, Quantification of the average axon lengths of cortical neurons shown in A from three independent experiments. p = 0.0006 for 5 μm vs 0 μm, p = 0.0002 for 10 μm vs 0 μm. F, Quantification of the average axon lengths of cortical neurons shown in B from three independent experiments. p = 0.0002 for 0.1 μm vs 0 μm, p = 0.0001 for 0.25 μm vs 0 μm. G, Quantification of the average axon lengths of hippocampal (Hippo) neurons shown in C from three independent experiments. p = 0.0261 for 5 μm vs 0 μm, p = 0.0007 for 10 μm vs 0 μm. H, Quantification of the average axon lengths of hippocampal neurons shown in D from three independent experiments. p = 0.0006 for 0.1 μm vs 0 μm, p = 0.0005 for 0.25 μm vs 0 μm. Data are represented as mean ± SEM. *p < 0.05, ***p < 0.001.
Next, we investigated whether p53 overexpression could enhance optic nerve regeneration in vivo. AAV2 viral vector encoding p53 (AAV2-p53) was injected into the vitreous body between the lens and the retina of the eye in a mouse model. The AAV2-EGFP viral vectors were used as the control. After 2 weeks of AAV2-p53 or AAV2-EGFP infection, we found that the expression levels of p53 were significantly enhanced in mouse retina tissue (Fig. 11A) and RGCs (Fig. 11B,C) infected with AAV2-p53 compared to those infected with AAV2-EGFP. To evaluate the functional role of p53 in optic nerve regeneration, optic nerve crush injury was performed 2 weeks after AAV2-p53 injection. After 12 more days, the fluorescently AlexaFluor 488-conjugated CTB was injected into the vitreous body to label regenerating axons. The results showed that the number of axons crossing the lesion site in the AAV2-EGFP mice 2 weeks after the optic nerve crush were minimum (Fig. 12A,B). In contrast, p53 overexpression induced marked CTB fluorescent labeled regenerating axons up to 2 mm from the crush site (Fig. 12A,B). Because p53 is a well known regulator of cell apoptosis, we assessed the cell viability of RGCs in whole-mount retina immunostained with the neuron-specific tubulin marker, Tuj1. By quantifying the number of cells positive for Tuj1, we found that compared with the AAV2-EGFP control group, the survival rates of RGCs were the same as that in the p53 overexpression experimental group, two weeks after the optic nerve crush (Fig. 12C,D). These results demonstrated that overexpression of p53 alone was sufficient to promote optic nerve regeneration without affecting RGC survival.
Figure 11.
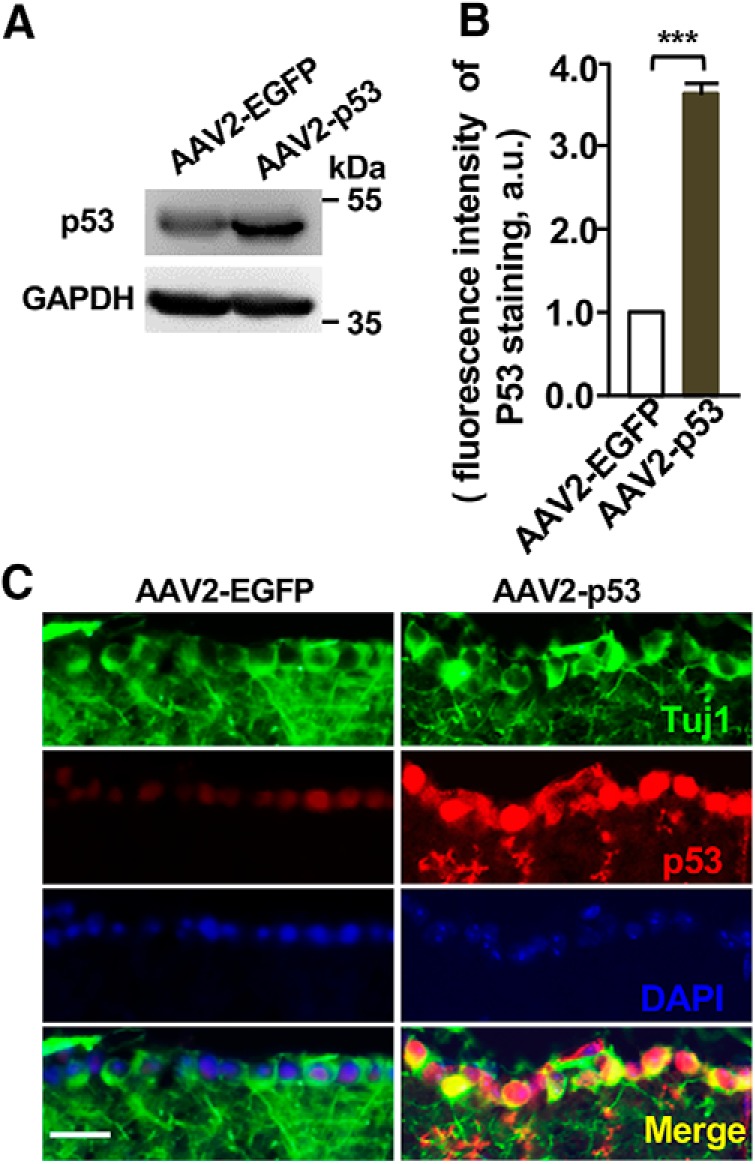
AAV2–p53-mediated overexpression of p53 in retina ganglion cells (RGCs). A, Representative Western blot images showing increased expression of the p53 protein in mouse retina tissues infected with AAV2–p53 compared with the control tissues infected with AAV2-EGFP. B, Quantification of fluorescence intensity of p53 in RGCs shown in C from three independent experiments. p = 0.0001. C, Representative images of immunostaining of sectioned retina with neuronal marker Tuj-1, p53, and nuclear DNA dye DAPI. Note the markedly increased p53 staining in RGCs 2 weeks after infected with AAV2–p53 compared to that infected with AAV2-EGFP. Scale bar, 50 μm. Data are represented as mean ± SEM. ***p < 0.001.
Figure 12.

Overexpression of p53 enhances optic nerve regeneration. A, Top, Time line of the experiment. Bottom, Representative images showing that p53 overexpression in RGCs induced drastic optic nerve regeneration 2 weeks after the optic nerve crush. The right eight columns show enlarged images of nerves at places marked by white boxes on the left. The red line indicates the crush sites. Scale bars, 50 μm. B, Quantification of regenerating optic nerve axons at different distances from the nerve crush site (n = 6 nerves from 6 mice for each condition; p = 0.0001 at 200 μm, p = 0.0001 at 500 μm). C, Representative images of whole-mount retina stained with the neuron-specific βIII tubulin antibody Tuj-1. Scale bar, 100 μm. D, Quantification of Tuj-1-positive cells in C showing that overexpression of p53 did not affect RGC survival 2 weeks after optic nerve crush. ns, No significant difference. Data are represented as mean ± SEM. ***p < 0.001.
Discussion
Identification of novel genes and pathways regulating axon regeneration would not only provide potential new target molecules for treating CNS injuries, but also help us better understand the molecular mechanisms by which mammalian axon regeneration is regulated. Here we provided evidence that c-Myc, previously identified to enhance optic nerve regeneration, was both necessary and sufficient for sensory axon regeneration in vitro and in vivo. These findings provide a great opportunity and model system to explore novel signaling pathways related to the c-Myc signaling that can be manipulated to regulate mammalian axon regeneration. In this study, we revealed an unexpected role of TERT, a key component of the telomere regulatory complex, in regulating mammalian axon regeneration downstream of c-Myc. Moreover, we provided evidence that p53 likely acted downstream of c-Myc and TERT to regulate sensory axon regeneration. More importantly, we showed that overexpression of p53 alone was sufficient to promote sensory axon and optic nerve regeneration in vivo.
In addition to its well known function to maintain telomere length during cell division, TERT has been shown to be involved in many other biological functions independent of its telomere regulatory function, such as the regulation of cell metabolism, growth factor secretion, mitochondria function, energy balance, and apoptosis (Smith et al., 2003; Chung et al., 2005; Bagheri et al., 2006; Massard et al., 2006; Lin et al., 2007; Passos et al., 2007; Liu et al., 2012). In postmitotic neurons, previous studies (Spilsbury et al., 2015; Miwa and Saretzki, 2017; Liu et al., 2018) have shown that TERT was mainly localized in the cytoplasm and functioned to protect neurons from oxidative or degenerative stresses in aged neurons. It is likely that TERT acts in neurons to regulate mitochondria function by reducing reactive oxidative species (Spilsbury et al., 2015; Miwa and Saretzki, 2017; Liu et al., 2018). In this study, we observed that TERT was also mainly localized in the cytoplasm of sensory neurons, similar to that observed in previous studies, and the expression of TERT was significantly increased in sensory neurons after peripheral injury. Functionally, TERT was required for the sensory axon regeneration in vitro and in vivo. TERT has been shown to be regulated by multiple positive and negative regulators, such as c-Myc, Sp1, p53, Wilms tumor 1, E2F (Cukusić et al., 2008), and the nuclear factor κ B (Lingner et al., 1997; Nakamura et al., 1997; Yin et al., 2000). Here we showed that in adult sensory neurons downregulation of c-Myc significantly reduced TERT expression, whereas the upregulation of c-Myc significantly increased TERT expression. Functionally, pharmacological activation of TERT significantly rescued the axon regeneration impaired by c-Myc knockdown in adult sensory neurons. Thus, our data strongly suggest that TERT acts as a downstream target of c-Myc during sensory axon regeneration.
The p53 protein is well known for its tumor suppressor role, functioning primarily to promote cell cycle arrest, DNA repair, and apoptosis (Levine and Oren, 2009). In postmitotic neurons, p53 has been shown to be necessary for proper axon guidance, growth, and regeneration (Arakawa, 2005; Di Giovanni and Rathore, 2012; Tedeschi et al., 2009; Gaub et al., 2010, 2011; Floriddia et al., 2012; Joshi et al., 2015). However, whether p53 is sufficient to promote axon growth and regeneration is unclear. In this study, we showed that overexpression of p53 in adult sensory neurons was sufficient to promote sensory axon regeneration in vitro and in vivo. In addition, the results showed that the protein level of p53 was regulated by TERT, suggesting that p53 act downstream of c-Myc and TERT to regulate sensory axon regeneration. Indeed, pharmacological activation of p53 was able to significantly restore sensory axon regeneration in vitro impaired by either c-Myc or TERT knockdown. Moreover, overexpression of p53 could significantly restore sensory axon regeneration impaired by c-Myc knockdown both in vitro and in vivo, indicating that p53 acted downstream of c-Myc and TERT to regulate axon regeneration. In addition to peripheral sensory neurons, we also showed that p53 was an important regulator of axon growth of developing cortical or hippocampal neurons, indicating its similar function in controlling CNS axon growth. Most importantly, overexpression of p53 in RGCs was sufficient to promote optic nerve regeneration.
Although either c-Myc or p53 has been shown previously to be involved in regulation of axon regeneration, our study provided evidence that they acted in the same pathway. In addition, we showed directly that c-Myc was both necessary and sufficient for sensory axon regeneration in vivo, and overexpression of p53 alone was sufficient to enhance both PNS and CNS axon regeneration in vivo. We also demonstrated a new role of TERT in regulation of axon regeneration as a signaling mediator between c-Myc and p53. How c-Myc, TERT, and p53 regulate axon regeneration remains unclear. One potential mechanism is through regulation of cell metabolism. In support of this hypothesis, c-Myc is well known to enhance anabolic metabolism of glycolysis via reprogramming mitochondria (Ward and Thompson, 2012). A recent study (Kim et al., 2019) also showed that wild-type p53 can act in the mitochondria to suppress oxidative phosphorylation and thus increase glycolysis, similar to the function of c-Myc. As aforementioned, TERT has been shown to be localized in mitochondria of neurons and regulates mitochondria function. It would be interesting in the future to show whether TERT functions to enhance CNS axon regeneration in vivo, and explore whether other factors or pathways regulating neuronal metabolism could also promote axon regeneration.
Footnotes
This work was supported by Grants from the National Natural Science Foundation of China (Nos. 81772353 and 81571189), the National Key Research and Development Program (Nos. 2016YFC 1100203), The Priority Academic Program Development of Jiangsu Higher Education Institutions, and Innovation and Entrepreneurship Program of Jiangsu Province to Saijilafu, and by the NIH (R01NS064288, R01NS085176, R01GM111514, R01EY027347), the Craig H. Neilsen Foundation, and the BrightFocus Foundation to F.-Q.Z.
The authors declare no competing financial interests.
References
- Arakawa H. (2005) p53, apoptosis and axon-guidance molecules. Cell Death Differ 12:1057–1065. 10.1038/sj.cdd.4401601 [DOI] [PubMed] [Google Scholar]
- Bagheri S, Nosrati M, Li S, Fong S, Torabian S, Rangel J, Moore DH, Federman S, Laposa RR, Baehner FL, Sagebiel RW, Cleaver JE, Haqq C, Debs RJ, Blackburn EH, Kashani-Sabet M (2006) Genes and pathways downstream of telomerase in melanoma metastasis. Proc Natl Acad Sci U S A 103:11306–11311. 10.1073/pnas.0510085103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareyre FM, Garzorz N, Lang C, Misgeld T, Büning H, Kerschensteiner M (2011) In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc Natl Acad Sci U S A 108:6282–6287. 10.1073/pnas.1015239108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin S, Nawabi H, Wang C, Tang S, Latremoliere A, Warren P, Schorle H, Uncu C, Woolf CJ, He Z, Steen JA (2015) Injury-induced decline of intrinsic regenerative ability revealed by quantitative proteomics. Neuron 86:1000–1014. 10.1016/j.neuron.2015.03.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Coppola G, Nawabi H, Omura T, Versano R, Huebner EA, Zhang A, Costigan M, Yekkirala A, Barrett L, Blesch A, Michaelevski I, Davis-Turak J, Gao F, Langfelder P, Horvath S, He Z, Benowitz L, Fainzilber M, Tuszynski M, et al. (2016) A systems-level analysis of the peripheral nerve intrinsic axonal growth program. Neuron 89:956–970. 10.1016/j.neuron.2016.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bathula SR, Li J, Huang L (2010) Multifunctional nanoparticles delivering small interfering RNA and doxorubicin overcome drug resistance in cancer. J Biol Chem 285:22639–22650. 10.1074/jbc.M110.125906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Cheong C, Song J, Lee HW (2005) Extratelomeric functions of telomerase. Curr Mol Med 5:233–241. 10.2174/1566524053586635 [DOI] [PubMed] [Google Scholar]
- Cukusić A, Skrobot Vidacek N, Sopta M, Rubelj I (2008) Telomerase regulation at the crossroads of cell fate. Cytogenet Genome Res 122:263–272. 10.1159/000167812 [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Rathore K (2012) p53-dependent pathways in neurite outgrowth and axonal regeneration. Cell Tissue Res 349:87–95. 10.1007/s00441-011-1292-5 [DOI] [PubMed] [Google Scholar]
- Fagoe ND, Attwell CL, Kouwenhoven D, Verhaagen J, Mason MR (2015) Overexpression of ATF3 or the combination of ATF3, c-jun, STAT3 and Smad1 promotes regeneration of the central axon branch of sensory neurons but without synergistic effects. Hum Mol Genet 24:6788–6800. 10.1093/hmg/ddv383 [DOI] [PubMed] [Google Scholar]
- Finelli MJ, Wong JK, Zou H (2013) Epigenetic regulation of sensory axon regeneration after spinal cord injury. J Neurosci 33:19664–19676. 10.1523/JNEUROSCI.0589-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Petkova V, Thanos S, Benowitz LI (2004) Switching mature retinal ganglion cells to a robust growth state in vivo: gene expression and synergy with RhoA inactivation. J Neurosci 24:8726–8740. 10.1523/JNEUROSCI.2774-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floriddia EM, Rathore KI, Tedeschi A, Quadrato G, Wuttke A, Lueckmann JM, Kigerl KA, Popovich PG, Di Giovanni S (2012) p53 regulates the neuronal intrinsic and extrinsic responses affecting the recovery of motor function following spinal cord injury. J Neurosci 32:13956–13970. 10.1523/JNEUROSCI.1925-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki T, Udono M, Kotake Y, Yamashita M, Shirahata S, Katakura Y (2010) NFAT5 regulates transcription of the mouse telomerase reverse transcriptase gene. Exp Cell Res 316:3342–3350. 10.1016/j.yexcr.2010.10.001 [DOI] [PubMed] [Google Scholar]
- Gaub P, Tedeschi A, Puttagunta R, Nguyen T, Schmandke A, Di Giovanni S (2010) HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ 17:1392–1408. 10.1038/cdd.2009.216 [DOI] [PubMed] [Google Scholar]
- Gaub P, Joshi Y, Wuttke A, Naumann U, Schnichels S, Heiduschka P, Di Giovanni S (2011) The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain 134:2134–2148. 10.1093/brain/awr142 [DOI] [PubMed] [Google Scholar]
- Ip FC, Ng YP, An HJ, Dai Y, Pang HH, Hu YQ, Chin AC, Harley CB, Wong YH, Ip NY (2014) Cycloastragenol is a potent telomerase activator in neuronal cells: implications for depression management. Neurosignals 22:52–63. 10.1159/000365290 [DOI] [PubMed] [Google Scholar]
- Jankowski MP, McIlwrath SL, Jing X, Cornuet PK, Salerno KM, Koerber HR, Albers KM (2009) Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Res 1256:43–54. 10.1016/j.brainres.2008.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing X, Wang T, Huang S, Glorioso JC, Albers KM (2012) The transcription factor Sox11 promotes nerve regeneration through activation of the regeneration-associated gene Sprr1a. Exp Neurol 233:221–232. 10.1016/j.expneurol.2011.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi Y, Sória MG, Quadrato G, Inak G, Zhou L, Hervera A, Rathore KI, Elnaggar M, Cucchiarini M, Marine JC, Magali C, Marine JC, Puttagunta R, Di Giovanni S (2015) The MDM4/MDM2–p53-IGF1 axis controls axonal regeneration, sprouting and functional recovery after CNS injury. Brain 138:1843–1862. 10.1093/brain/awv125 [DOI] [PubMed] [Google Scholar]
- Khattar E, Tergaonkar V (2017) Transcriptional regulation of telomerase reverse transcriptase (TERT) by MYC. Front Cell Dev Biol 5:1. 10.3389/fcell.2017.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Yu L, Chen W, Xu Y, Wu M, Todorova D, Tang Q, Feng B, Jiang L, He J, Chen G, Fu X, Xu Y (2019) Wild-type p53 promotes cancer metabolic switch by inducing PUMA-dependent suppression of oxidative phosphorylation. Cancer Cell 35:191–203.e8. 10.1016/j.ccell.2018.12.012 [DOI] [PubMed] [Google Scholar]
- Kurimoto T, Yin Y, Omura K, Gilbert HY, Kim D, Cen LP, Moko L, Kügler S, Benowitz LI (2010) Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. J Neurosci 30:15654–15663. 10.1523/JNEUROSCI.4340-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavanya C, Venkataswamy MM, Sibin MK, Srinivas Bharath MM, Chetan GK (2018) Down regulation of human telomerase reverse transcriptase (hTERT) expression by BIBR1532 in human glioblastoma LN18 cells. Cytotechnology 70:1143–1154. 10.1007/s10616-018-0205-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Zheng B (2012) Role of myelin-associated inhibitors in axonal repair after spinal cord injury. Exp Neurol 235:33–42. 10.1016/j.expneurol.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Oren M (2009) The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9:749–758. 10.1038/nrc2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Wu J, Holstein D, Upadhyay G, Rourk W, Muller E, Lechleiter JD (2007) Ca 2+ signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes. Neurobiol Aging 28:99–111. 10.1016/j.neurobiolaging.2005.11.004 [DOI] [PubMed] [Google Scholar]
- Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276:561–567. 10.1126/science.276.5312.561 [DOI] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Connolly L, Steward O, Zheng B, He Z (2010) PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 13:1075–1081. 10.1038/nn.2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MY, Nemes A, Zhou QG (2018) The emerging roles for telomerase in the central nervous system. Front Mol Neurosci 11:160. 10.3389/fnmol.2018.00160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hu Y, Zhu L, Chen C, Zhang Y, Sun W, Zhou Q (2012) Overexpression of the mTERT gene by adenoviral vectors promotes the proliferation of neuronal stem cells in vitro and stimulates neurogenesis in the hippocampus of mice. J Biomed Res 26:381–388. 10.7555/JBR.26.20110078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, de Lange T (2017) Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18:175–186. 10.1038/nrm.2016.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massard C, Zermati Y, Pauleau AL, Larochette N, Métivier D, Sabatier L, Kroemer G, Soria JC (2006) hTERT: a novel endogenous inhibitor of the mitochondrial cell death pathway. Oncogene 25:4505–4514. 10.1038/sj.onc.1209487 [DOI] [PubMed] [Google Scholar]
- Michaelevski I, Segal-Ruder Y, Rozenbaum M, Medzihradszky KF, Shalem O, Coppola G, Horn-Saban S, Ben-Yaakov K, Dagan SY, Rishal I, Geschwind DH, Pilpel Y, Burlingame AL, Fainzilber M (2010) Signaling to transcription networks in the neuronal retrograde injury response. Sci Signal 3:ra53. 10.1126/scisignal.2000952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa S, Saretzki G (2017) Telomerase and mTOR in the brain: the mitochondria connection. Neural Regen Res 12:358–361. 10.4103/1673-5374.202922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL (2009) KLF family members regulate intrinsic axon regeneration ability. Science 326:298–301. 10.1126/science.1175737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR (1997) Telomerase catalytic subunit homologs from fission yeast and human. Science 277:955–959. 10.1126/science.277.5328.955 [DOI] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE (1973) Myelination in rat brain: method of myelin isolation1. J Neurochem 21:749–757. 10.1111/j.1471-4159.1973.tb07519.x [DOI] [PubMed] [Google Scholar]
- Ohtake Y, Li S (2015) Molecular mechanisms of scar-sourced axon growth inhibitors. Brain Res 1619:22–35. 10.1016/j.brainres.2014.08.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh P, Hao Y, Hosseinkhani M, Patil SB, Huntley GW, Tessier-Lavigne M, Zou H (2011) Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc Natl Acad Sci U S A 108:E99–E107. 10.1073/pnas.1100426108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z (2008) Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322:963–966. 10.1126/science.1161566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, von Zglinicki TV (2007) DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res 35:7505–7513. 10.1093/nar/gkm893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Baudry M, Liao G, Noniyev A, Galeano J, Bi X (2009) A novel function for p53: regulation of growth cone motility through interaction with rho kinase. J Neurosci 29:5183–5192. 10.1523/JNEUROSCI.0420-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Zou Y, Zhang CL (2013) Cross-talk between KLF4 and STAT3 regulates axon regeneration. Nat Commun 4:2633. 10.1038/ncomms3633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L, Wolfer DP, Lipp HP, Aguzzi A, Wagner EF, Behrens A (2004) The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43:57–67. 10.1016/j.neuron.2004.06.005 [DOI] [PubMed] [Google Scholar]
- Reisman D, Elkind NB, Roy B, Beamon J, Rotter V (1993) C-myc trans-activates the P53 promoter through a required downstream CACGTG motif. Cell Growth Differ 4:57–65. [PubMed] [Google Scholar]
- Saijilafu, Hur EM, Zhou FQ (2011) Genetic dissection of axon regeneration via in vivo electroporation of adult mouse sensory neurons. Nat Commun 2:543–543. 10.1038/ncomms1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijilafu, Hur EM, Liu CM, Jiao Z, Xu WL, Zhou FQ (2013) PI3K-GSK3 signalling regulates mammalian axon regeneration by inducing the expression of Smad1. Nat Commun 4:2690. 10.1038/ncomms3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijilafu, Zhang BY, Zhou FQ (2014) In vivo electroporation of adult mouse sensory neurons for studying peripheral axon regeneration. Methods Mol Biol 1162:167–175. 10.1007/978-1-4939-0777-9_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seijffers R, Mills CD, Woolf CJ (2007) ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci 27:7911–7920. 10.1523/JNEUROSCI.5313-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DS, Skene JH (1997) A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci 17:646–658. 10.1523/JNEUROSCI.17-02-00646.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LL, Coller HA, Roberts JM (2003) Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat Cell Biol 5:474–479. 10.1038/ncb985 [DOI] [PubMed] [Google Scholar]
- Smith PD, Sun F, Park KK, Cai B, Wang C, Kuwako K, Martinez-Carrasco I, Connolly L, He Z (2009) SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron 64:617–623. 10.1016/j.neuron.2009.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilsbury A, Miwa S, Attems J, Saretzki G (2015) The role of telomerase protein TERT in Alzheimer's disease and in tau-related pathology in vitro. J Neurosci 35:1659–1674. 10.1523/JNEUROSCI.2925-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, He Z (2010) Neuronal intrinsic barriers for axon regeneration in the adult CNS. Curr Opin Neurobiol 20:510–518. 10.1016/j.conb.2010.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Nguyen T, Puttagunta R, Gaub P, Di Giovanni S (2009) A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ 16:543–554. 10.1038/cdd.2008.175 [DOI] [PubMed] [Google Scholar]
- Vera E, Bosco N, Studer L (2016) Generating late-onset human iPSC-based disease models by inducing neuronal age-related phenotypes through telomerase manipulation. Cell Rep 17:1184–1192. 10.1016/j.celrep.2016.09.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Reynolds A, Kirry A, Nienhaus C, Blackmore MG (2015) Overexpression of Sox11 promotes corticospinal tract regeneration after spinal injury while interfering with functional recovery. J Neurosci 35:3139–3145. 10.1523/JNEUROSCI.2832-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XW, Li Q, Liu CM, Hall PA, Jiang JJ, Katchis CD, Kang S, Dong BC, Li S, Zhou FQ (2018) Lin28 signaling supports mammalian PNS and CNS axon regeneration. Cell Rep 24:2540–2552.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21:297–308. 10.1016/j.ccr.2012.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Lv PP, Chen XM, Yue ZG, Fu Q, Liu SY, Yue H, Ma GH (2013) Codelivery of mTERT siRNA and paclitaxel by chitosan-based nanoparticles promoted synergistic tumor suppression. Biomaterials 34:3912–3923. 10.1016/j.biomaterials.2013.02.030 [DOI] [PubMed] [Google Scholar]
- Wu KJ, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J, Dalla-Favera R (1999) Direct activation of TERT transcription by c-MYC. Nat Genet 21:220–224. 10.1038/6010 [DOI] [PubMed] [Google Scholar]
- Yin L, Hubbard AK, Giardina C (2000) NF-κB regulates transcription of the mouse telomerase catalytic subunit. J Biol Chem 275:36671–36675. 10.1074/jbc.M007378200 [DOI] [PubMed] [Google Scholar]
- Zhou D, Liu P, Sun DW, Chen ZJ, Hu J, Peng SM, Liu YL (2017) USP22 downregulation facilitates human retinoblastoma cell aging and apoptosis via inhibiting TERT/P53 pathway. Eur Rev Med Pharmacol Sci 21:2785–2792. [PubMed] [Google Scholar]
- Zhou FQ, Walzer MA, Snider WD (2004) Turning on the machine: genetic control of axon regeneration by c-jun. Neuron 43:1–2. 10.1016/j.neuron.2004.06.020 [DOI] [PubMed] [Google Scholar]