

Abstract
Multiphoton microscopy (MPM) has emerged as one of the most powerful and widespread technologies to monitor the activity of neuronal networks in awake, behaving animals over long periods of time. MPM development spanned across decades and crucially depended on the concurrent improvement of calcium indicators that report neuronal activity as well as surgical protocols, head fixation approaches, and innovations in optics and microscopy technology. Here we review the last decade of MPM development and highlight how in vivo imaging has matured and diversified, making it now possible to concurrently monitor thousands of neurons across connected brain areas or, alternatively, small local networks with sampling rates in the kilohertz range. This review includes different laser scanning approaches, such as multibeam technologies as well as recent developments to image deeper into neuronal tissues using new, long-wavelength laser sources. As future development will critically depend on our ability to resolve and discriminate individual neuronal spikes, we will also describe a simple framework that allows performing quantitative comparisons between the reviewed MPM instruments. Finally, we provide our own opinion on how the most recent MPM developments can be leveraged at scale to enable the next generation of discoveries in brain function.
Keywords: imaging, in vivo, multiphoton, two-photon
Introduction
When Adrian, Morison, and others performed the first in vivo recordings in anesthetized animals in the late 1930s, they were not able to record from more than a handful of neurons at a time (Adrian, 1936; Renshaw et al., 1940). Today, novel optical in vivo imaging methods enable routine high-resolution recordings of thousands of genetically identified individual neurons in the brain of awake, behaving animals over weeks and months (Trachtenberg et al., 2002; Holtmaat et al., 2009; Andermann et al., 2010; Attardo et al., 2015). Such capabilities illustrate the enormous advances that neurotechnologies have made over the last decades and how modern imaging tools open new avenues for neuroscience research. Compared with classical single-photon wide-field fluorescence microscopy, multiphoton microscopy (MPM) offers optical sectioning as well as depth penetration capabilities, two factors that greatly facilitate in vivo neural recordings deep in the intact living brain. Indeed, it was MPM that led the way for many important neuroscience discoveries revealing, among others, how new spines form during learning (Trachtenberg et al., 2002), how local spatial distribution of neuronal activity patterns represents sensory information (Ohki et al., 2005), and the role of other types of brain cells, such as microglia (Nimmerjahn et al., 2005).
Since the first demonstration of a working two-photon (2P) microscope in 1990 (Denk et al., 1990), MPM technology has made remarkable progress, including the development of large FOV (Sofroniew et al., 2016; Stirman et al., 2016) and multibeam optical designs (Lecoq et al., 2014; Voigt et al., 2015), as well as high-speed laser scanning (Duemani Reddy et al., 2008; Grewe et al., 2010) and spatiotemporal multiplexing approaches (Cheng et al., 2011) (Fig. 1). Miniaturized, head-mounted versions of 2P microscopes were also developed (Helmchen et al., 2001; Piyawattanametha et al., 2006) and have recently reemerged (Zong et al., 2017).
Figure 1.

Historical timeline highlighting major milestones of MPM development and its applications. Adapted with permission from Kaiser and Garrett (1961), Denk et al., (1990), Svoboda et al. (1997), Helmchen et al. (2001), Stosiek et al. (2003), Grewe et al. (2010), Sofroniew et al. (2016), Kobat et al. (2011), Chavarha et al. (2018).
Although standard 2P microscopes are widely used in neuroscience, MPM still has some drawbacks and limitations in terms of the temporal resolution, large-scale network monitoring, long-term applicability, and its ability to record neural activity during natural behaviors in freely moving animals. In this review, we will mainly focus on the technological advances made to the initial MPM microscopy setup (for a basic schematic, see Fig. 2) over the past 10 years that, in our opinion, have the potential to substantially improve neuroscience research in the near future. Our goal is not to convey all relevant technical details but rather to make neuroscience researchers aware of recent technological developments in MPM as well as their basic strengths and limitations. Therefore, we organized this review to focus on developments for recording from a larger number of neurons, recording neurons at higher speed, and recording deeper in the brain; we also provide a comparison of MPM techniques. Due to space limitations, we do not cover any mobile and/or miniaturized MPM approaches that are described previously (Zong et al., 2017).
Figure 2.
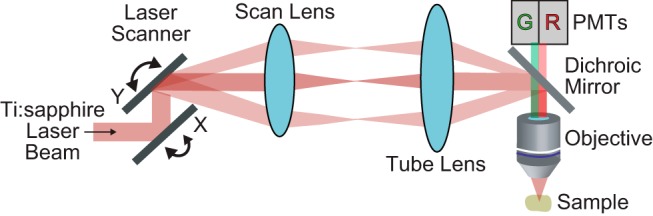
Schematic of a basic multiphoton microscope. An ultra-fast pulsed, near-infrared laser beam is directed onto a pair of X/Y scanning mirrors, which deflect the laser beam in two dimensions, depending on the voltage applied to their galvanometric elements. The beam then passes through the scan and tube lenses. The lens combination determines two fundamental parameters of scanning microscopy: the expansion of the beam entering the objective lens, which controls the size of the beam focus; and the scan angle of the laser beam into the objective, which is responsible for the size of FOV. Next, the laser beam is directed into the objective by a dichroic mirror that reflects near-infrared but passes light in the visible range. The objective lens focuses the short laser pulses into a tiny spot at the sample, resulting in multiphoton excitation of fluorescent indicator molecules. The emitted photons pass the dichroic mirror before being split into two channels (here, red and green) and are detected by two dedicated photomultiplier tubes (PMTs).
Meso-scale multiarea MPM approaches allow simultaneous in vivo recordings of thousands of neurons
Complex behavior often involves large brain networks that have both local and long-range connections and can span several millimeters in the mouse brain (Oh et al., 2014). To relate neuronal activity to behavior, it is crucial to simultaneously monitor the activity of such integrated long-range networks across many individual behavioral trials (Zhang et al., 2014). This experimental scientific need has fueled the development of new technologies that are capable of in vivo recordings from very large and distributed neuronal populations.
Interestingly, some of the biggest challenges in large-FOV optical systems were not optical. For example, only after the development of improved genetically encoded calcium indicators (GECIs) and related transgenic mouse lines (Dana et al., 2014; Madisen et al., 2015; Wekselblatt et al., 2016) did it become possible to label large populations of neuronal networks across multiple brain regions. In addition, early surgical protocols only provided limited optical access to a single isolated, superficial brain region either by thinning of the animal's skull (Stosiek et al., 2003) or by implanting a small cover glass as skull replacement (Trachtenberg et al., 2002). Such limitations were overcome by protocols using large optical windows (i.e., 5 mm diameter) implanted over a large portion of the mouse dorsal cortex (Goldey et al., 2014). Recently, a novel glass implant preparation known as the “crystal skull” allowed imaging of the entire dorsal cortex by implanting a single, curved glass window that mimicked the bone plate curvature (Kim et al., 2016). Thanks to these innovations, imaging of large populations of neurons is no longer limited by methods of sample preparation and cell labeling; however, we still lack the imaging technology to sample all these neurons efficiently. To address this bottleneck, several optical MPM design approaches have been developed over the last decade. Broadly, these fall into two categories: single-beam scanning techniques, which sequentially traverse large fields of neural tissue at high speed; and multibeam scanning techniques, which can simultaneously interrogate different locations of the neuronal tissue.
Single-beam MPM for large-scale interrogation of brain tissue
Initial attempts to scale up neuronal recordings focused on optimizing single-beam scanning and extending MPM to the third dimension. By axially moving the microscope objective with a fast piezo actuator, smart 3D line-scanning approaches (Göbel et al., 2007) allowed recording from ∼200 neurons at 10 Hz in the brain of live anesthetized rodents. An optimal use of the single laser-beam scanning time can be achieved by so-called random-access scanning (i.e., the laser beam is rapidly scanned over arbitrary selected points across the whole FOV). True random-access scanning can be realized with acousto-optic deflectors (AODs). These diffractive elements “deflect” a laser beam depending on the frequency of the sound wave generated by a piezo element that travels across a crystal positioned in the beam path. Two orthogonal AODs combine into a 2D scanner (Duemani Reddy et al., 2008; Grewe et al., 2010). If a pair of 2D-AODs are aligned in series with opposite sound propagation, 3D random-access imaging can be achieved with fast point-to-point transition times in the 10 μs range (Katona et al., 2012; Nadella et al., 2016). This allows spending the entire laser beam scan time on neuronal somata while avoiding scanning any unlabeled neuropil. However, all line- and point-scanning techniques are sensitive to sample motion, which is typically present when imaging in awake animals (Dombeck et al., 2007; Nimmerjahn et al., 2009). Various approaches were proposed to mitigate this issue, for example, by implementing a feedback loop for on-the-fly motion measurement and correction (Nadella et al., 2016) or by scanning local volumes around each area of interest (Szalay et al., 2016).
Despite these recent developments, fast raster scanning (i.e., sampling the FOV line by line) was favored for a long time, as motion artifacts could easily be resolved with custom postprocessing motion-correction algorithms (Kerr et al., 2005; Dombeck et al., 2007). This encouraged widespread adoption of galvanometric scanners operating at their resonance frequency, enabling fast, 2D scanning rates at ∼30 Hz (Fan et al., 1999). By combining such resonant scanners with an electrically tunable lens that shifts the axial focus of the laser beam by rapidly changing its shape, it became possible to scan two imaging planes at different depths at rates of up to 15 Hz, doubling the amount of recorded neurons (Grewe et al., 2011).
An alternative to inertia-limited axial scanners, such as piezo actuators or electrically tunable lenses, are spatial light modulators (SLMs) that are conjugated to the X/Y scanner and the Fourier plane of the objective (Liu et al., 2018). SLMs apply a variety of diffractive illumination patterns to the laser beam and thereby can shift MPM excitation laterally or axially. Contrary to inertia-limited electrically tunable lenses or piezo actuators, SLMs can provide fast (i.e., 5 ms) switching of the beam focus across the sample.
Another powerful concept for 3D imaging is remote focusing, which uses a second objective lens that focuses the beam on a mirror conjugated to the sample (Botcherby et al., 2012). Remote focusing not only corrects for optical aberrations introduced by the primary objective lens, but also provides a path for fast axial scanning as a lightweight mirror at the focus of the secondary objective can be moved by a fast actuator (e.g., a voice coil). Similarly, ultrasound lenses (Kong et al., 2015) refocus light by radially changing their refractive index. If used in a remote focusing setup, ultrasound lenses provide rapid axial scanning by changing the focus of the secondary objective.
An alternative approach to increase the number of recorded neurons is to enlarge the effective FOV by adjusting the microscope objective design. Commercially available objectives typically support scanning with a lateral extension of 500–800 × 500–800 μm2, but they do not allow large scanning angles without deteriorating the optical resolution of the whole imaging system (Stosiek et al., 2003; Ji et al., 2016). This issue was resolved through tailored in vivo 2P imaging objectives for very large FOVs (Tsai et al., 2015; Sofroniew et al., 2016; Stirman et al., 2016). Combined with more sophisticated single-beam scanner arrangements, 1000–2000 neurons were recorded within a 5 × 5 mm2 FOV (Sofroniew et al., 2016) at a sampling rate of 10 Hz (Fig. 3A,B). However, large-FOV objectives with sufficient NA require large and heavyweight optics that cannot be moved quickly for fast axial scanning. Therefore, next-generation large-FOV systems rely on remote focusing, SLMs, or electrically tunable lenses (Sofroniew et al., 2016; Stirman et al., 2016). To further extend the FOV, custom postobjective scanners, which position the scanning beam in the sample space using pairs of micro-mirrors moved by a rotating stage, were recently introduced (Terada et al., 2018).
Figure 3.

Large-area recording techniques. A, Schematic representation of a mesoscale (large FOV) imaging system known as 2P random-access microscope (2P-RAM). This imaging system supports slow scanning of a 5 mm2 FOV or high-resolution imaging of multiple 500 × 500 μm2 subregions with lateral separation of up to 4.5 mm. B, Low-magnification image acquired with the 2P-RAM system at 4.3 Hz frame rate, with an FOV of 5 mm2 (top). Four separate areas of 500 × 500 μm2 were sampled sequentially at 9.6 Hz (bottom). All data were acquired from a mouse expressing the calcium indicator GCaMP6f under a thy-1 promoter. C, Schematic of the dual optical axis MPM system. Two excitation arms are fully independent and have separate optical paths. To solve geometrical constraints, one of the objectives is coupled with micro-optical elements positioned onto the mouse's head. D, Two distal areas (S1 and M1) were imaged with the dual-axis imaging system in a GCaMP6s mouse. Scale bar, 300 μm. E, Schematic of a multibeam system with two simultaneous beams that scan two nearby areas. The scan areas were positioned within the FOV of a commercial objective and were laterally separated by <1 mm. F, Sequentially (top and middle) and simultaneously recorded images (bottom). Red boxes represent the area of FOV overlap. Computational algorithms and structural priors were used instead of excitation-emission encoding to separate the fluorescence signals of the two different planes. Scale bar, 50 μm. Adapted with permission from Lecoq et al. (2014), Sofroniew et al. (2016), and Yang et al. (2016).
Despite the possibility that new in vivo preparations, such as the crystal skull (Kim et al., 2016), would now even allow recording from ∼1 million cortical neurons, most of these single-beam scanning methods can only adequately detect calcium events across a very small subset of neurons simultaneously.
Multibeam MPM for sampling large neuronal population distributed across multiple distant sites
Perhaps one of the simplest multibeam imaging approaches is to use two independent pathways (Fig. 3C,D) by using micro-optics that provide the freedom of choosing two distant imaging sites (Lecoq et al., 2014). While this approach works well for distant recording sites (>1–2 mm apart), for adjacent areas, positioning multiple beams through a single objective can be more practical (Fig. 3E,F). For instance, with independent pathways, choosing new imaging areas requires a new optical alignment.
When designing multibeam scanning techniques, special attention must be given to crosstalk between the two excitation beams. Indeed, the presence of scattered photons in neural tissue (Oheim et al., 2001) limits the ability to later disambiguate multiple fluorescence emitters because fluorescence photons from one imaging plane are often detected in other imaging planes. This issue can be addressed either by post hoc source separation methods (Lecoq et al., 2014; Yang et al., 2016) or by temporal multiplexing schemes that support attributing the fluorescence signal to each individual scanning site (Cheng et al., 2011; Voigt et al., 2015). Temporal multiplexing can be easily implemented because MPM relies on high-power laser oscillators emitting very short (e.g., ∼100–200 femtosecond laser pulses at 80 MHz repetition rate). These short laser pulses excite a fluorescence signal that lasts a couple of nanoseconds (GFP fluorescence lifetime ≃ 3 ns) (Striker et al., 1999). This allows the use of multiple laser excitation beams simultaneously but requires the pulses of each beamlet to be delayed in time. This delay is crucial because it later allows temporal separation of the individual fluorescence signals that have been excited by the different beams (Cheng et al., 2011; Voigt et al., 2015; Stirman et al., 2016).
Recently, reverberation optical loops that circulate (loop) the excitation beams several times through a single delay line were introduced to efficiently distribute laser pulses that differ in time and axial position (Beaulieu et al., 2018). Such temporal approaches typically allow multiplexing of up to 4 beams and thus increase the number of imaged neurons by a factor of 4 accordingly. However, introducing >4 beams would lead to an increased overlap of fluorescence decays in time and thus limit the ability to distinguish the signal sources. Another major drawback that might have limited the broad adoption of multiplexing MPM is the need for custom-built high-bandwidth electronics operating at least twice the rate of the multiplexed laser pulse rate. When multiplexing 4 beams from a standard 80 MHz laser, this results in a sampling frequency of > 640 MHz to comply with Nyquist sampling criterion.
An alternative to online demultiplexing are offline computational algorithms that remove multiplexing crosstalk (Mukamel et al., 2009; Pnevmatikakis et al., 2016), which, in combination with SLM-based multibeam microscope, allow post hoc source separation (Yang et al., 2016) (Fig. 3E,F). In this case, all simultaneously acquired planes are overlapping during the acquisition, and calibration data need to be collected during the experiment to estimate a (de)mixing matrix.
Custom beam engineering to match the excitation beam to fluorescently labeled neuronal populations
When dealing with sparsely labeled tissue, raster scanning techniques mostly sample nonlabeled pixels. One possible approach to minimize this issue is to elongate the point-spread-function (PSF) in the axial direction to more efficiently sample the tissue along this imaging axis. This so-called “Bessel beam scanning” projects a volume of tissue into a single plane (Lu et al., 2017) and thus enables volumetric 400 × 400 × 100 μm3 MPM scan rates of 30–45 Hz. Although Bessel beams lack axial resolution along the z dimension, high-resolution image priors that are acquired with standard raster scanning can be used to retrieve the longitudinal coordinates of all recorded cells. Interestingly, Bessel beams at diameters well above the diffraction limit have also been successfully used in the context of densely labeled tissue. Indeed, diffraction-limited PSFs are often unnecessary for in vivo imaging, and a loss in resolution can be traded off for higher imaging speeds (Lecoq et al., 2014; Stirman et al., 2016). In the extreme case, one can fully abandon the diffraction-limited PSF and very efficiently scan the tissue with a large PSF (e.g., ∼5 × 5 × 10 μm3) that approximately matches the neuronal soma size. Recent works from the Vaziri group (Prevedel et al., 2016; Weisenburger et al., 2019) explored this direction. By combining temporal focusing, beam engineering, and smart laser pulse energy control with fast laser scanning, they could record neuronal activity from up to ∼12,000 neurons contained in a volume of 1 × 1 × 1.2 mm3. This combination of methods, however, is technically challenging because it requires temporal focusing and a custom laser source to maintain sufficient signal-to-noise ratio (SNR) levels. In addition, larger PSFs effectively harm the ability to distinguish the activity of nearby cells, which has to be accounted for.
Fast MPM approaches to resolve neuronal activity at the millisecond time scale
Neuronal networks in the brain process incoming stimuli within a few tens of milliseconds. To understand such fast neuronal dynamics, it is of key importance to investigate neuronal activity patterns with an adequate temporal resolution. With the introduction of fast calcium indicators (Chen et al., 2013; Dana et al., 2019) and recent improvements of voltage indicators (Gong et al., 2015; Lin and Schnitzer, 2016; Kannan et al., 2018), high-speed imaging has become a major direction in MPM development (St Pierre, 2017; Chavarha et al., 2018). Strategies to realize fast imaging speeds in vivo share principles similar to those used for imaging large FOVs, but instead use the extra recording bandwidth to increase the sampling rate. Fast MPM approaches can be categorized into three classes that include the following: (1) single-beam line scanning methods or AOD-based scanning that allow fast random-access scanning of neuronal populations; (2) multibeam scanning techniques that divide the FOV into subregions; and (3) fast tomography-based systems that image the sample from multiple angles.
Early MPM approaches to improve imaging speed aimed to maximize the time that the laser focus is scanning structures of interest. Increasing the imaging speed to kilohertz rates became possible with AOD scanners that allow the laser beam to move within microseconds and hence enable true high-speed random-access imaging (Iyer et al., 2006). For example, a pair of AODs allowed 2D in vivo population calcium imaging with near millisecond precision (Grewe et al., 2010). Moreover, 3D AOD-based systems with fast axial focusing could image populations of neurons at kilohertz rates (Fig. 4A–C) (Katona et al., 2012; Cotton et al., 2013; Nadella et al., 2016). For these reasons, AOD systems are particularly well suited for fast imaging of neuronal voltage signals. Recently, new genetically encoded voltage indicators have been combined with high-speed AOD-based MPM scanning approaches (Chavarha et al., 2018) to achieve fast population voltage imaging and to extract the spiking signals of individual neurons from the optical recordings with millisecond precision.
Figure 4.

Techniques for large-volume and high-speed scanning using AOD scanners and spatiotemporal multiplexing. A, Schematic representation of 3D AOD scanning used in random-access multiphoton (RAMP) microscopy. In AOD MPM systems, the number of neurons within a 3D volume directly determines the temporal sampling rates. B, MPM image overlaid with one of the scan patterns from A. C, Schematic of RAMP microscope. A commercial Ti:Al2O3 laser beam is expanded to fill the AOD aperture. All AODs (X1… Y2) are optically conjugated via 4f telescopes (T1–T4). A dichroic mirror separates the IR excitation and visible light detection paths. GaAsP photomultiplier tubes (PMTs) detect emitted photons (for more technical details, see original publication, Cotton et al., 2013). D, Principle of spatiotemporal multiplexing where a larger FOV is simultaneously scanned with 4 beams to increase data acquisition rate by a factor of 4. E, Simplified layout of the microscope with spatiotemporal multiplexing. The laser emits pulses at 80 MHz corresponding to a 12.5 ns interpulse interval. The initial beam is divided into 4 beams, which are delayed by ∼3 ns. All beams are recombined on a 2D-scanning module with different angular offset. In the imaging plane, this offset translates into a lateral displacement. Beam colors correspond to box colors in D. Emitted fluorescence is detected by PMTs, and photons are assigned to the correct image partition by a custom analog demultiplexing circuit based on their arrival time. For more details on temporal encoding and various techniques of multiplexing, see Cheng et al. (2011), Stirman et al. (2016), and Tsyboulski et al. (2018). Adapted with permission from Cheng et al. (2011) and Cotton et al. (2013).
The second most common approach to increase imaging speed is to divide the FOV into several subfields and to use multiple beams that simultaneously scan these (Fig. 4D,E). Such parallelized raster scanning has been demonstrated using a fast resonant scanning system with four temporally multiplexed beams, resulting in scan rates of up to 250 Hz over ∼400 μm2 FOV (Cheng et al., 2011). Theoretically, many more beams can be used to perform in vivo imaging, even at kHz rates (US Patent 9,820,652). However, such systems would require proportionally more laser power to maintain similar SNR levels for each individual beam, and thus might quickly exceed the heat-absorption-caused damage threshold for neuronal tissues (Podgorski and Ranganathan, 2016). To further increase the imaging bandwidth, more sophisticated multiplexing schemes have been proposed that either rely on a digital micromirror device that encodes a specific digital signature to each pixel by flipping individual micromirrors of the array (Ducros et al., 2013) or using interferometric AOD schemes that encode each beam with a different beat frequency (Tsyboulski et al., 2017). Although promising, these approaches have not yet been used extensively for in vivo recordings.
Another way to achieve kHz sampling rates relies on a tomography-like approach that scans the same probe from multiple angles (Kazemipour et al., 2019). These projections are acquired by four sequential line scans that traverse the FOV at different angles. This tomographic approach demonstrated dendritic imaging at rates >1 kHz across an FOV of 250 × 250 μm2 at a depth of up to 250 μm. However, one caveat of this approach is that the accuracy of signal reconstruction depends on the sparseness of the sample; for example, bright, clustered structures might not be reconstructed well. The tomography approach could be extended to larger FOVs; but as with other tomography methods, the number of reconstructed signal sources is limited to <1000 unless the number of angular projections is increased. Moreover, scanning with a complete line also implies delivering much higher average power to the sample, which might lead to increased thermal load as well as photobleaching and toxicity.
MPM approaches to probe deep cortical and sub-cortical brain areas in awake animals
Measuring complex activity patterns that underlie behavior often requires recording neuronal populations in deep cortical or subcortical brain areas. However, imaging deeper than ∼500 μm requires to adapt standard MPM techniques for an extended imaging depth. It is important to understand that the optical properties of tissue are the fundamental depth-limiting factors because both excitation and emission photons are highly scattered and absorbed by biological tissue. While the scattering problem could be addressed by increasing the laser intensity, this would introduce additional problems, such as sample heating, as well as out-of-focus and near-to-surface fluorescence volume excitation. Indeed, at least for 2P imaging, these issues seem most relevant for restricting the imaging depth (Theer and Denk, 2006), and the technologies that we discuss below attempt to resolve these limitations.
The most promising approach to increase the MPM imaging depth has been to shift the laser excitation light to longer wavelengths, for which scattering and absorption are substantially reduced (Lecoq and Schnitzer, 2011). Combined with the newest red-shifted GECIs, such as RCaMP, R-GECO, and jRCaMP (Akerboom et al., 2013; Dana et al., 2016), or related transgenic mice (Bethge et al., 2017), excitation wavelengths >1000 nm can be used to increase the 2P imaging depth down to ∼0.8 mm (Ouzounov et al., 2017). While early approaches (Kobat et al., 2011) used optical parametric oscillators that provided an excitation wavelength of 1280 nm to image vascular structures down to 1.6 mm deep, functional (calcium) imaging beyond a depth of 1 mm (e.g., in mouse CA1) requires even longer wavelengths between 1600 and 1800 nm that still lie within the water absorption minimum. In this regimen, 3-photon (3P) absorption will become the major process of fluorescence excitation. Indeed, out-of-focus and near-surface fluorescence volume excitation, the two biggest depth-limiting factors for 2P imaging, are substantially reduced for 3P imaging (Theer and Denk, 2006). Further increasing the laser excitation wavelength to 1700 nm can be achieved by another laser type, a photonic-crystal pumped by an erbium fiber laser. Using this approach, Horton et al. (2013) demonstrated 3P in vivo calcium imaging of neuronal and vascular structures up a depth of 1.4 mm (Fig. 5). However, 3P imaging also requires an order-of-magnitude higher pulse energies to achieve the same fluorescence signal as 2P excitation. This is because fluorophore absorption cross-sections are substantially smaller for 3P excitation (compare 3P ∼10−83 cm6 s2/photon2 vs 2P ∼10−49 cm4 s/photon for GFP) (Cheng et al., 2014). Such high pulse energies can be achieved through optical parametric amplifiers that provide pulse energies of hundreds of nanojoules within a tunable range of 1000–1600 nm. While the use of optical parametric amplifiers enabled structural in vivo imaging up to a depth of 1.5 mm at 1215 nm wavelength (Miller et al., 2017), more recently functional 3P imaging of mouse hippocampal and deep cortical areas at a depth of 0.7–1.2 mm has been demonstrated (Ouzounov et al., 2017; Yildirim et al., 2019) (Fig. 5).
Figure 5.

Advanced 3P microscopy approaches to record neuronal activity deep in the mouse brain. A, Schematic of the 3P deep-tissue imaging approach. B, Direct comparison of the signal-to-background ratio for 2P and 3P imaging of GCaMP6s-labeled neurons 780 μm deep in mouse cortex. “Background” arrow indicates the out-of-focus fluorescence measured inside a blood vessel. “Background & Neuropil” arrow indicates neuropil and the out-of-focus background. The two images have a comparable number of signal photons and are displayed with the same contrast settings (top 0.4% saturation). Scale bars, 30 μm. C, 3P z stack acquired with 2 μm steps. Green represents GCaMP6s signal. Magenta represents the label-free third harmonic generation signal from the blood vessels in the visual cortex and myelin fibers in the white matter. Scale bar, 100 μm. D, Selection of 2D images from various layers (L2/3, L4, L5, and L6). Scale bar, 50 μm. FOV in all lateral images is 250 μm. Adapted with permission from Yildirim et al. (2019), and Ouzounov et al. (2017).
To summarize, functional 3P microscopy is substantially more demanding than its structural counterpart as it requires fast scanning and short pixel dwell times to adequately sample the neuronal activity in time. In addition, pulse energies must be significantly higher (5–10 nJ compared with 0.3–3 nJ for standard 2P imaging) to gather enough signal during each pixel dwell time. Such high pulse energies also seem to increase photodamage in a nonlinear manner, which is more likely an issue in 3P imaging than heat absorption (Podgorski and Ranganathan, 2016; Yildirim et al., 2019). Another drawback of using optical parametric amplifiers for 3P imaging is that they can lower the pulse repetition rate to ∼400–800 kHz, which limits the number of pixels that can be scanned across the FOV within a certain time interval, thereby effectively reducing the FOV size. However, newer 3P optical parametric amplifiers promise pulse repetition rates of up to 5 MHz. As for other technical developments, a combination of the techniques described above is currently explored to circumvent these limitations. For instance, combining Bessel beam with 3P absorption offers higher volume rate (Chen et al., 2018; Rodríguez et al., 2018).
Comparison of MPM techniques
The core idea of this review is to provide an overview of the most recent MPM developments and applications with the goal to enable researchers to select the best suitable method for their in vivo imaging experiments. However, a direct method comparison is difficult because comparative standards have not yet been established in the field. First, individual MPM approaches cannot be directly compared because experimental conditions vary across studies (e.g., fluorescent indicators, wavelengths, imaging depths, microscope objectives, photomultiplier tubes, and preamplifiers). Second, in addition to the sampling rate and number of neurons, key metrics needed to evaluate the quality of in vivo recordings, such as the ability to discriminate neuronal activity signals from background, are often not reported. Third, typically only 1 or 2 performance data points (sampling rate and number of neurons) are reported per study, and the extrapolation to alternative MPM technologies proves difficult. Although a direct method comparison is hardly possible, it is clear that most recent MPM developments aimed to optimize the following three parameters: (1) the number of neurons that can be recorded; (2) the sampling rate and temporal precision to resolve neuronal activity; and (3) the reliability of detecting calcium events from the fluorescence signals and extracting individual neuronal spikes. While (1) and (2) are straightforward to evaluate, (3) is frequently underappreciated due to the lack of data. In the following section, we will attempt to analytically describe a framework to improve the standardization of reporting (3), the signal detection performance.
While simple metrics, such as the single action potential (1AP) SNR, were initially used to describe the ability to detect neuronal signals from fluorescence traces, recent modeling work suggests the use of more sophisticated metrics, which are commonly known in signal detection theory as the sensitivity index, or d′ (Wilt et al., 2013). d′ quantifies the ability to distinguish individual neuronal calcium events from baseline noise. Under certain assumptions (e.g., normal distribution), d′ can be expressed as , where μ and σ are the mean and the SD for the 1AP and the baseline signal distributions. Since σ is mainly driven by shot noise, one can assume σN = σS, which simplifies the equation to . As a rule of thumb, a d′ of 5 relates to a correct 1AP detection rate of ∼99%.
If the sampling rate f is substantially higher than the decay time constant τind of the calcium indicator (f × τind ≫ 1), d′ can be approximated as (Wilt et al., 2013). Here, F0 is the somatic background fluorescence in photons/s and ΔF/F is the peak fluorescence change detected for a single AP. This means that, to achieve a d′ of 5 with GCaMP6f (1AP ΔF/F ∼15%, τind ∼0.2 s), the laser intensity or signal integration times need to be adjusted so that at least F0 >11,000 photons/s/cell are detected under baseline conditions. If the main interest is to reliably detect short bursts of, for example, 5 APs with the same d′, at least F0 > 2200 photons/s/cell are required assuming a linear signal summation. In the following paragraph, we use standardized data acquired at the Allen Brain Observatory (de Vries et al., 2018) to provide an idea of how to calculate typical F0 numbers and associated theoretical d′ values for standard MPM systems.
The standard MPM systems at the Allen Institute for Brain Science use resonant galvanometric scanners, capable of scanning a single excitation laser beam across a 400 × 400 μm2 FOV (512 × 512 pixels) at 30 Hz (de Vries et al., 2018). Taking into account the gain and offset of the data acquisition (photomultiplier tubes and preamplifiers), we calculated the median photon flux per pixel directly from the motion-corrected fluorescence data (for code to apply this method to any MPM data, see https://github.com/AllenInstitute/QC_2P) to be, on average, 0.89 photons per pixel per dwell time (n = 2080 experiments across both excitatory and inhibitory transgenic cre lines expressing GCaMP6f in all layers of the mouse visual cortex). In this case, a cellular soma spans ∼100 pixels2 that provide a F0 ≅ 2670 photons/s/cell or a theoretical d′ value of 2.4, which is well within a range where ≥5 APs are reliably detected (d′ ∼ 5).
A simple way to increase d′ is to decrease the FOV size from 500 × 500 μm2 to 180 × 180 μm2, which would result in a 5× increase of the number of pixels per neuron and thus a 5× higher number of detected photons. This would result in F0 > 13,000 photons/s/cell and thus a theoretical d′ of 5.4. However, shrinking the FOV will also decrease the number of recorded neurons by a factor of 5. Alternatively, slower versions of GCaMP6 (i.e., GCaMP6s) can be used, providing an approximate 1AP ΔF/F change of 25% and a τind ∼ 0.6 s (Chen et al., 2013). Because of the larger ΔF/F and τind, GCaMP6s can achieve a d′ of 5 with 1.7 times less recorded pixels per cell compared with GCaMP6f. However, using slower GECIs comes at the cost of a reduced temporal precision of detecting neuronal signals.
The best temporal precision can be achieved when using genetically encoded voltage indicators that directly report fast changes of the neuronal membrane voltage. For example, using an improved voltage indicator, ASAP3, in vivo population voltage imaging of 3–5 neurons was recently demonstrated at a rate of ∼3 kHz, which is ∼8 times faster than the temporal dynamics of the recorded voltage transients (Chavarha et al., 2018). The approximation introduced above (Wilt et al., 2013) will only hold if f × τind ≫ 1. Thus, if lower sampling frequencies are used, even higher photon counts are required to achieve a d′ of 5. However, even when imaging at 3 kHz, imaging with a d′ value of 5 can still falsely detect ∼10 spikes per second, since each imaging frame can yield false positives. Thus, for fast voltage imaging, higher d′ values are required. For example, sampling at 3 kHz, Chavarha et al. (2018) reported a d′ of 9, which independent of the actual neuronal activity resulted in a false positive spike detection rate of one spike every 200 s and a false negative detection rate of 1.5 × 10−6 spikes per second. Given the reported 1AP ΔF/F of 9% and a decay time of ∼4 ms, the number of baseline photons that should be collected are F0 > 5000 photons/ms/cell. These simple calculations illustrate that voltage imaging of large populations (>50 neurons) will require substantially more advanced MPM solutions or alternative imaging approaches that provide at least 1–2 orders of magnitude better imaging capabilities than current high-speed MPM approaches.
We summarize the properties of recent MPM developments in Table 1 and Figure 6, which shows that the number of recorded neurons is generally inversely related to the imaging rate. In this figure, we only report features and numbers that have been published or, in some cases, that we obtained by contacting the authors to clarify their method. Our comparison of the different microscopy systems is not optimal because the reported data have been acquired under different imaging conditions (e.g., differently labeling techniques, expression levels, and approaches to quantify and report SNR). The difficulty in comparing these methods also underlines the increasing need to develop a set of standardized evaluation metrics to report advances in MPM.
Table 1.
Representative publications for multiple recent advanced MPM techniquesa
| Technique | No. of neurons, range | Recording frequency range, Hz | Depth, μm | Spatial resolution: lateral × axial, μm | Reported application | Features and highlights | Reference |
|---|---|---|---|---|---|---|---|
| eMS2PM: encoded multisite 2PM | 3–15 | 4100–1040 | 300 | 0.7 × 1.4 | Voltage imaging in cultured cells, fast OGB calcium imaging in vivo | Simultaneous recording; less phototoxic; noise increase with increased number of recording sites; limited to 2D scanning; sensitive to motion | Ducros et al., 2013 |
| RAPS: random-access pattern scanning | 16–100 | 490–180 | 300 | 1.0 × 4.1 | High SNR in vivo recordings of calcium transients | 2D-AOD scanning; 1AP detection; sensitive to motion | Grewe et al., 2010 |
| SLAP: Tomographic TPM | 100 | 1016 | 300 | 0.44 × 1.62 | Single-cell voltage imaging, dendritic and axonal calcium recording | Constant frame rate independent of number of sources; tomographic image reconstruction; varying accuracy depending on label distribution; 2D FOV | Kazemipour et al., 2019 |
| ULOVE: 2D-AOD pattern scanning | 1–150 | 15000–100 | 500 | 0.5 × 1.5* | Single-cell imaging of membrane activity in deep neurons during awake behavior | Resilience to motion; high SNR tracking of spikes; 2D FOV | Chavarha et al., 2018 |
| Bessel 2PM: extended focus scanning | 50–400 | 40–10 | 400 | 0.5, 15–400 | High-speed volumetric calcium imaging in vivo in sparsely labeled tissue | Volumetric imaging with extended focus; volume size limited by power; axial dimension reconstructed post hoc | Lu et al., 2017 |
| RAMP: 3D-AOD | 27–411 | 1800–120 | 500 | 0.5 × 1.5* | High-speed calcium recordings from hundreds of neurons | Highest-speed sequential 3D recordings; sensitive to sample motion | Cotton et al., 2013; Nadella et al., 2016 |
| Galvo-Galvo: raster scan with galvanometers | 100–300 | 10–3 | 500 | 0.5 × 1.5* | Calcium imaging in vivo | Well-established technique; available from commercial companies | Denk et al., 1990 |
| TPM-Mux: multibeam multiplexing | 200–800 | 250–60 | 300 | 0.5 × 1.5* | Calcium imaging in vivo in multiple regions | Increased recording throughput by using multiple beams; crosstalk between beams | Stirman et al., 2016; Cheng et al., 2011 |
| Dual-beam TPM: dual-area scan, two beams | 200–600 | 10–3 | 700 | 1 × 9 | Calcium imaging recordings in vivo in multiple regions | Simultaneous imaging of two areas decoupled in X/Y/Z; minimum separation is ∼1 mm center-to-center; requires optical alignment for each experiment | Lecoq et al., 2014; Yang et al., 2016 |
| s-Te-Flo: encoded multibeam scanning | 200–4000 | 160–3 | 500 | 5 × 10 | High-speed calcium recordings in populations of neurons | PSF sculpted to size of neuronal cell body-low spatial resolution; synchronizing pixels to laser pulses | Prevedel et al., 2016 |
| Trepan2p: large FoV and beam multiplexing | 400–5000 | 40–0.1 | 700 | 1.2 × 12 | Dual beam calcium recordings over >9 mm2 FOV | Independent positioning of two beams within 9 mm2; highly reconfigurable in terms of scan size and frame rate; crosstalk between beams | Stirman et al., 2016 |
| 2P-RAM: 25 mm2 FOV | 200–3000 | 44–1.9 | 500 | 1.2 × 5 | Mesoscale imaging of calcium activity in neuronal populations | High collection efficiency; fast z scanning; highly flexible configuration of imaged regions and frame rates; requires prep adaptation to a bulky objective | Sofroniew et al., 2016 |
| Resonant scanner, fast axial scanner | 300–13000 | 30–2.5 | 500 | 0.5 × 1.5* | Calcium recordings in vivo in neuronal populations | Improved frame rate but lower SNR compared with Galvo-Galvo; available from commercial companies | Fan et al., 1999; Pachitariu et al., 2017 |
aRecording capabilities and microscope parameters are listed along with the associated references.
*Spatial resolution values correspond to theoretical values for the objective lenses used in system designs, as PSF sizes were not reported.
Figure 6.
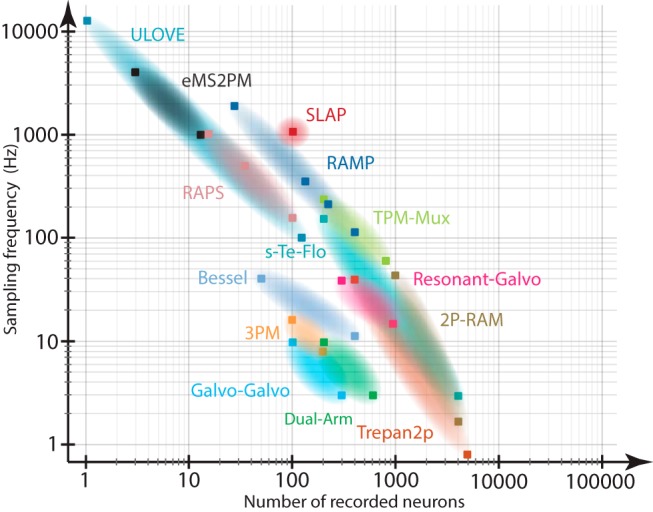
Comparison of MPM techniques in terms of number of neurons and recording speed. Mapping of recently developed MPM systems onto a 2D space (number of imaged neurons vs sampling rate). For some cases, we converted the imaging FOV into number of neurons assuming a densely labeled GCaMP mouse line, such as Slc17a7:Ai148 where, on average, 200 labeled neurons are detected in a 500 × 500 μm2 FOV. Conventional galvo-galvo and resonant-galvo systems are included to emphasize the progress in technology development over the last decade. With the increasing number of scanning beams, the plot shifts toward higher speed or number of neurons, depending on the microscope configuration. Square markers represent published data. Clouds represent hypothetical interpolation between the reported data points.
Discussion
The future of large-scale neuronal recording most certainly rests upon a combination of the above-mentioned techniques. Newly designed objectives lenses offer an extended view into neuronal tissues that can only be leveraged with high-bandwidth recording techniques. SLMs are promising tools providing flexibility to create multiple beams and even allow us to look deeper into tissue using concepts of adaptive optics (Ji et al., 2010; Liu et al., 2018). Similarly, multiplexing techniques combined with sophisticated scanning technologies, large FOV objective lens designs, and excitation beam shaping offer the opportunity to further increase the number of simultaneously recorded neurons to many thousands of neurons (Tsyboulski et al., 2018; Weisenburger et al., 2019). It is reasonable to expect that, in mice, entire cortical areas could be recorded in the very near future. As the sophistication of our instruments increases, the need for comparative quantitative standards becomes central to the development of future technologies. With integrated optical and mechanical computer-aided design tools, such as Zemax, Solidworks (Dassault Systèmes), and Creo (Parametric Technology), it becomes incredibly important to predict all performance aspects of new instruments before substantial financial investments are made.
Finally, given today's remarkable technological and experimental possibilities, the next generation of MPM instruments should be driven by neurobiological questions arising in the field. Indeed, multiphoton microscopes have matured from their initial design to showcase different, complementary capabilities. Instruments to image vast populations of neurons are designed very differently than those that aim to image dendritic processes.
Given the increased complexity of MPM instruments, their limited availability should not prevent us from concentrating on important neuroscience questions. In this regard, an academic observatory model currently used in high-energy physics and astronomy offers a unique opportunity to gain access to complex instrumentation and experimental techniques while encouraging data standardization. The idea of a brain observatory model is to open access to advanced experimentation based on unbiased scientific reviews of submitted proposals. Supportive staff can then perform the experiments while meeting high-quality standards and standardized operating procedures. Such an observatory approach is currently explored for systems neuroscience at the Allen Institute for Brain Science through the OpenScope project.
While monitoring the activity of thousands of neurons chronically was a distant vision just a few decades ago, it is now routinely performed in many laboratories through MPM of genetically encoded calcium reporters of neuronal activity. Significant technological developments have recently broadened our access to neuronal tissues and greatly increased the number of different techniques available. These allow for sampling larger populations of neurons, accessing neuronal networks located deep in the neuronal tissue, or achieving faster recording rates. Future developments will likely combine these techniques to further broaden our access to neuronal activity. Tight integration of these developments with neuroscience research is bound to transform our understanding of brain function.
Footnotes
We thank Kevin Takasaki and Peter Saggau (Allen Institute for Brain Science) for providing helpful comments on the manuscript; and Bénédicte Rossi for providing scientific illustrations. The authors thank the Allen Institute for Brain Science founders, Paul G. Allen and Jody Allen, for their vision, encouragement, and support.
The authors declare no competing financial interests.
References
- Adrian ED. (1936) The spread of activity in the cerebral cortex. J Physiol 88:127–161. 10.1113/jphysiol.1936.sp003427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J, Carreras Calderón N, Tian L, Wabnig S, Prigge M, Tolö J, Gordus A, Orger MB, Severi KE, Macklin JJ, Patel R, Pulver SR, Wardill TJ, Fischer E, Schüler C, Chen TW, Sarkisyan KS, Marvin JS, Bargmann CI, Kim DS, et al. (2013) Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci 6:2. 10.3389/fnmol.2013.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann ML, Kerlin AM, Reid RC (2010) Chronic cellular imaging of mouse visual cortex during operant behavior and passive viewing. Front Cell Neurosci 4:3. 10.3389/fncel.2010.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attardo A, Fitzgerald JE, Schnitzer MJ (2015) Impermanence of dendritic spines in live adult CA1 hippocampus. Nature 523:592–596. 10.1038/nature14467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu DR, Davison IG, Bifano TG, Mertz J (2018) Simultaneous multiplane imaging with reverberation multiphoton microscopy. arxiv. Available at https://arxiv.org/abs/1812.05162. [DOI] [PMC free article] [PubMed]
- Bethge P, Carta S, Lorenzo DA, Egolf L, Goniotaki D, Madisen L, Voigt FF, Chen JL, Schneider B, Ohkura M, Nakai J, Zeng H, Aguzzi A, Helmchen F (2017) An R-CaMP1.07 reporter mouse for cell-type-specific expression of a sensitive red fluorescent calcium indicator. PLoS One 12:e0179460. 10.1371/journal.pone.0179460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botcherby EJ, Smith CW, Kohl MM, Débarre D, Booth MJ, Juskaitis R, Paulsen O, Wilson T (2012) Aberration-free three-dimensional multiphoton imaging of neuronal activity at kHz rates. Proc Natl Acad Sci U S A 109:2919–2924. 10.1073/pnas.1111662109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavarha M, Villette V, Dimov I, Pradhan L, Evans S, Shi D, Yang R, Chamberland S, Bradley J, Mathieu B, St-Pierre F, Schnitzer MJ, Bi G, Toth K, Ding J, Dieudonne S, Lin M (2018) Fast two-photon volumetric imaging of an improved voltage indicator reveals electrical activity in deeply located neurons in the awake brain. bioRxiv. Advance online publication. Retrieved October 17, 2018. doi: 10.1101/445064 10.1101/445064 [DOI] [Google Scholar]
- Chen B, Huang X, Gou D, Zeng J, Chen G, Pang M, Hu Y, Zhao Z, Zhang Y, Zhou Z, Wu H, Cheng H, Zhang Z, Xu C, Li Y, Chen L, Wang A (2018) Rapid volumetric imaging with Bessel-beam three-photon microscopy. Biomed Opt Express 9:1992–2000. 10.1364/BOE.9.001992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300. 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Tiago Gonçalves JT, Golshani P, Arisaka K, Portera-Cailliau C (2011) Simultaneous two-photon calcium imaging at different depths with spatiotemporal multiplexing. Nat Methods 8:139–142. 10.1038/nmeth.1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LC, Horton NG, Wang K, Chen SJ, Xu C (2014) Measurements of multiphoton action cross sections for multiphoton microscopy. Biomed Opt Express 5:3427–3433. 10.1364/BOE.5.003427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton RJ, Froudarakis E, Storer P, Saggau P, Tolias AS (2013) Three-dimensional mapping of microcircuit correlation structure. Front Neural Circuits 7:151. 10.3389/fncir.2013.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Chen TW, Hu A, Shields BC, Guo C, Looger LL, Kim DS, Svoboda K (2014) Thy1-GCaMP6 transgenic mice for neuronal population imaging in vivo. PLoS One 9:e108697. 10.1371/journal.pone.0108697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, Patel R, Macklin JJ, Bargmann CI, Ahrens MB, Schreiter ER, Jayaraman V, Looger LL, Svoboda K, Kim DS (2016) Sensitive red protein calcium indicators for imaging neural activity. Elife 5:312727. 10.7554/eLife.12727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Sun Y, Mohar B, Hulse BK, Kerlin AM, Hasseman JP, Tsegaye G, Tsang A, Wong A, Patel R, Macklin JJ, Chen Y, Konnerth A, Jayaraman V, Looger LL, Schreiter ER, Svoboda K, Kim DS (2019) High-performance GFP-based calcium indicators for imaging activity in neuronal populations and microcompartments. Nat Methods 16:649–657. 10.1038/s41592-019-0435-6 [DOI] [PubMed] [Google Scholar]
- de Vries SEJ, Lecoq J, Buice MA, Groblewski PA, Ocker GK, Oliver M, Feng D, Cain N, Ledochowitsch P, Millman D, Roll K, Garrett M, Keenan T, Kuan L, Mihalas S, Olsen S, Thompson C, Wakeman W, Waters J, Williams D, et al. (2018) A large-scale, standardized physiological survey reveals higher order coding throughout the mouse visual cortex. bioRxiv. Available at https://europepmc.org/abstract/PPR/PPR7988. [DOI] [PMC free article] [PubMed]
- Denk W, Strickler JH, Webb WW (1990) Two-photon laser scanning fluorescence microscopy. Science 248:73–76. 10.1126/science.2321027 [DOI] [PubMed] [Google Scholar]
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, Tank DW (2007) Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron 56:43–57. 10.1016/j.neuron.2007.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducros M, Goulam Houssen Y, Bradley J, de Sars V, Charpak S (2013) Encoded multisite two-photon microscopy. Proc Natl Acad Sci U S A 110:13138–13143. 10.1073/pnas.1307818110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duemani Reddy G, Kelleher K, Fink R, Saggau P (2008) Three-dimensional random access multiphoton microscopy for functional imaging of neuronal activity. Nat Neurosci 11:713–720. 10.1038/nn.2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan GY, Fujisaki H, Miyawaki A, Tsay RK, Tsien RY, Ellisman MH (1999) Video-rate scanning two-photon excitation fluorescence microscopy and ratio imaging with chameleons. Biophys J 76:2412–2420. 10.1016/S0006-3495(99)77396-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göbel W, Kampa BM, Helmchen F (2007) Imaging cellular network dynamics in three dimensions using fast 3D laser scanning. Nat Methods 4:73–79. 10.1038/nmeth989 [DOI] [PubMed] [Google Scholar]
- Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, Andermann ML (2014) Removable cranial windows for long-term imaging in awake mice. Nat Protoc 9:2515–2538. 10.1038/nprot.2014.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Huang C, Li JZ, Grewe BF, Zhang Y, Eismann S, Schnitzer MJ (2015) High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor. Science 350:1361–1366. 10.1126/science.aab0810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewe BF, Langer D, Kasper H, Kampa BM, Helmchen F (2010) High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision. Nat Methods 7:399–405. 10.1038/nmeth.1453 [DOI] [PubMed] [Google Scholar]
- Grewe BF, Voigt FF, van 't Hoff M, Helmchen F (2011) Fast two-layer two-photon imaging of neuronal cell populations using an electrically tunable lens. Biomed Opt Express 2:2035–2046. 10.1364/BOE.2.002035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Fee MS, Tank DW, Denk W (2001) A miniature head-mounted two-photon microscope: high-resolution brain imaging in freely moving animals. Neuron 31:903–912. 10.1016/S0896-6273(01)00421-4 [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L (2009) Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4:1128–1144. 10.1038/nprot.2009.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton NG, Kobat D, Wang K, Xu C (2012) In vivo, deep tissue three-photon imaging at the 1700-nm spectral window. In: Biomedical optics and 3-D imaging. Washington, DC: OSA. [Google Scholar]
- Horton NG, Wang K, Kobat D, Clark CG, Wise FW, Schaffer CB, Xu C (2013) In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nat Photonics 7:205–209. 10.1038/nphoton.2012.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V, Hoogland TM, Saggau P (2006) Fast functional imaging of single neurons using random-access multiphoton (RAMP) microscopy. J Neurophysiol 95:535–545. 10.1152/jn.00865.2005 [DOI] [PubMed] [Google Scholar]
- Ji N, Milkie DE, Betzig E (2010) Adaptive optics via pupil segmentation for high-resolution imaging in biological tissues. Nat Methods 7:141–147. 10.1038/nmeth.1411 [DOI] [PubMed] [Google Scholar]
- Ji N, Freeman J, Smith SL (2016) Technologies for imaging neural activity in large volumes. Nat Neurosci 19:1154–1164. 10.1038/nn.4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser W, Garrett CGB (1961) Two-photon excitation in Ca F 2: Eu 2+. Physical review letters 76: 229 [Google Scholar]
- Kannan M, Vasan G, Huang C, Haziza S, Li JZ, Inan H, Schnitzer MJ, Pieribone VA (2018) Fast, in vivo voltage imaging using a red fluorescent indicator. Nat Methods 15:1108–1116. 10.1038/s41592-018-0188-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona G, Szalay G, Maák P, Kaszás A, Veress M, Hillier D, Chiovini B, Vizi ES, Roska B, Rózsa B (2012) Fast two-photon in vivo imaging with three-dimensional random-access scanning in large tissue volumes. Nat Methods 9:201–208. 10.1038/nmeth.1851 [DOI] [PubMed] [Google Scholar]
- Kazemipour A, Novak O, Flickinger D, Marvin JS, Abdelfattah AS, King J, Borden PM, Kim JJ, Al-Abdullatif SH, Deal PE, Miller EW, Schreiter ER, Druckmann S, Svoboda K, Looger LL, Podgorski K (2019) Kilohertz frame-rate two-photon tomography. Nat Methods 16:778–786. 10.1038/s41592-019-0493-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JN, Greenberg D, Helmchen F (2005) Imaging input and output of neocortical networks in vivo. Proc Natl Acad Sci U S A 102:14063–14068. 10.1073/pnas.0506029102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Zhang Y, Lecoq J, Jung JC, Li J, Zeng H, Niell CM, Schnitzer MJ (2016) Long-term optical access to an estimated one million neurons in the live mouse cortex. Cell Rep 17:3385–3394. 10.1016/j.celrep.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobat D, Horton NG, Xu C (2011) In vivo two-photon microscopy to 1.6-mm depth in mouse cortex. J Biomed Opt 16:106014. 10.1117/1.3646209 [DOI] [PubMed] [Google Scholar]
- Kong L, Tang J, Little JP, Yu Y, Lämmermann T, Lin CP, Germain RN, Cui M (2015) Continuous volumetric imaging via an optical phase-locked ultrasound lens. Nat Methods 12:759–762. 10.1038/nmeth.3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq J, Schnitzer MJ (2011) An infrared fluorescent protein for deeper imaging. Nat Biotechnol 29:715–716. 10.1038/nbt.1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq J, Savall J, Vucinic D, Grewe BF, Kim H, Li JZ, Kitch LJ, Schnitzer MJ (2014) Visualizing mammalian brain area interactions by dual-axis two-photon calcium imaging. Nat Neurosci 17:1825–1829. 10.1038/nn.3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MZ, Schnitzer MJ (2016) Genetically encoded indicators of neuronal activity. Nat Neurosci 19:1142–1153. 10.1038/nn.4359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Ball N, Brockill J, Kuan L, Millman D, White C, Leon A, Williams D, Nishiwaki S, de Vries S, Larkin J, Sullivan D, Slaughterbeck C, Farrell C, Saggau P (2018) Multi-plane imaging of neural activity from the mammalian brain using a fast-switching liquid crystal spatial light modulator. bioRxiv. Available at https://europepmc.org/abstract/PPR/PPR66128. [DOI] [PMC free article] [PubMed]
- Lu R, Sun W, Liang Y, Kerlin A, Bierfeld J, Seelig JD, Wilson DE, Scholl B, Mohar B, Tanimoto M, Koyama M, Fitzpatrick D, Orger MB, Ji N (2017) Video-rate volumetric functional imaging of the brain at synaptic resolution. Nat Neurosci 20:620–628. 10.1038/nn.4516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, Gu H, Mills M, Cheng A, Tasic B, Nguyen TN, Sunkin SM, Benucci A, Nagy A, Miyawaki A, Helmchen F, et al. (2015) Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85:942–958. 10.1016/j.neuron.2015.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DR, Hassan AM, Jarrett JW, Medina FA, Perillo EP, Hagan K, Shams Kazmi SM, Clark TA, Sullender CT, Jones TA, Zemelman BV, Dunn AK (2017) In vivo multiphoton imaging of a diverse array of fluorophores to investigate deep neurovascular structure. Biomed Opt Express 8:3470–3481. 10.1364/BOE.8.003470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel EA, Nimmerjahn A, Schnitzer MJ (2009) Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63:747–760. 10.1016/j.neuron.2009.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadella KM, Ros H, Baragli C, Griffiths VA, Konstantinou G, Koimtzis T, Evans GJ, Kirkby PA, Silver RA (2016) Random-access scanning microscopy for 3D imaging in awake behaving animals. Nat Methods 13:1001–1004. 10.1038/nmeth.4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Mukamel EA, Schnitzer MJ (2009) Motor behavior activates Bergmann glial networks. Neuron 62:400–412. 10.1016/j.neuron.2009.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Harris JA, Ng L, Winslow B, Cain N, Mihalas S, Wang Q, Lau C, Kuan L, Henry AM, Mortrud MT, Ouellette B, Nguyen TN, Sorensen SA, Slaughterbeck CR, Wakeman W, Li Y, Feng D, Ho A, Nicholas E, et al. (2014) A mesoscale connectome of the mouse brain. Nature 508:207–214. 10.1038/nature13186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oheim M, Beaurepaire E, Chaigneau E, Mertz J, Charpak S (2001) Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J Neurosci Methods 111:29–37. 10.1016/S0165-0270(01)00438-1 [DOI] [PubMed] [Google Scholar]
- Ohki K, Chung S, Ch'ng YH, Kara P, Reid RC (2005) Functional imaging with cellular resolution reveals precise micro-architecture in visual cortex. Nature 433:597–603. 10.1038/nature03274 [DOI] [PubMed] [Google Scholar]
- Ouzounov DG, Wang T, Wang M, Feng DD, Horton NG, Cruz-Hernández JC, Cheng YT, Reimer J, Tolias AS, Nishimura N, Xu C (2017) In vivo three-photon imaging of activity of GCaMP6-labeled neurons deep in intact mouse brain. Nat Methods 14:388–390. 10.1038/nmeth.4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachitariu M, Stringer C, Dipoppa M, Schröder S, Rossi LF, Dalgleish H, Carandini M, Harris KD (2017) Suite2p: beyond 10,000 neurons with standard two-photon microscopy. bioRxiv. Advance online publication. Retrieved July 20, 2017. doi: 10.1101/061507. [DOI] [Google Scholar]
- Piyawattanametha W, Barretto RP, Ko TH, Flusberg BA, Cocker ED, Ra H, Lee D, Solgaard O, Schnitzer MJ (2006) Fast-scanning two-photon fluorescence imaging based on a microelectromechanical systems two- dimensional scanning mirror. Opt Lett 31:2018–2020. 10.1364/OL.31.002018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pnevmatikakis EA, Soudry D, Gao Y, Machado TA, Merel J, Pfau D, Reardon T, Mu Y, Lacefield C, Yang W, Ahrens M, Bruno R, Jessell TM, Peterka DS, Yuste R, Paninski L (2016) Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron 89:285–299. 10.1016/j.neuron.2015.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podgorski K, Ranganathan G (2016) Brain heating induced by near-infrared lasers during multiphoton microscopy. J Neurophysiol 116:1012–1023. 10.1152/jn.00275.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevedel R, Verhoef AJ, Pernía-Andrade AJ, Weisenburger S, Huang BS, Nöbauer T, Fernández A, Delcour JE, Golshani P, Baltuska A, Vaziri A (2016) Fast volumetric calcium imaging across multiple cortical layers using sculpted light. Nat Methods 13:1021–1028. 10.1038/nmeth.4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw B, Forbes A, Morison BR (1940) Activity of isocortex and hippocampus: electrical studies with micro-electrodes. J Neurophysiol 3:74–105. 10.1152/jn.1940.3.1.74 [DOI] [Google Scholar]
- Rodríguez C, Liang Y, Lu R, Ji N (2018) Three-photon fluorescence microscopy with an axially elongated Bessel focus. Opt Lett 43:1914–1917. 10.1364/OL.43.001914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew NJ, Flickinger D, King J, Svoboda K (2016) A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. Elife 5:e14472. 10.7554/eLife.14472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Pierre F. (2017) Two-photon imaging of neuronal voltage dynamics using genetically encoded voltage indicators. In: Optics in the Life Sciences Congress. Washington, DC: OSA. [Google Scholar]
- Stirman JN, Smith IT, Kudenov MW, Smith SL (2016) Wide field-of-view, multi-region, two-photon imaging of neuronal activity in the mammalian brain. Nat Biotechnol 34:857–862. 10.1038/nbt.3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A (2003) In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A 100:7319–7324. 10.1073/pnas.1232232100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striker G, Subramaniam V, Seidel CA, Volkmer A (1999) Photochromicity and fluorescence lifetimes of green fluorescent protein. J Phys Chem B 103:8612–8617. 10.1021/jp991425e [DOI] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, Tank DW (1997) In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature 385:161–165. 10.1038/385161a0 [DOI] [PubMed] [Google Scholar]
- Szalay G, Judák L, Katona G, Ócsai K, Juhász G, Veress M, Szadai Z, Fehér A, Tompa T, Chiovini B, Maák P, Rózsa B (2016) Fast 3D imaging of spine, dendritic, and neuronal assemblies in behaving animals. Neuron 92:723–738. 10.1016/j.neuron.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada SI, Kobayashi K, Ohkura M, Nakai J, Matsuzaki M (2018) Super-wide-field two-photon imaging with a micro-optical device moving in post-objective space. Nat Commun 9:3550. 10.1038/s41467-018-06058-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theer P, Denk W (2006) On the fundamental imaging-depth limit in two-photon microscopy. J Opt Soc Am A Opt Image Sci Vis 23:3139–3149. 10.1364/JOSAA.23.003139 [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420:788–794. 10.1038/nature01273 [DOI] [PubMed] [Google Scholar]
- Tsai PS, Mateo C, Field JJ, Schaffer CB, Anderson ME, Kleinfeld D (2015) Ultra-large field-of-view two-photon microscopy. Opt Express 23:13833–13847. 10.1364/OE.23.013833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsyboulski D, Orlova N, Saggau P (2017) Amplitude modulation of femtosecond laser pulses in the megahertz range for frequency-multiplexed two-photon imaging. Opt Express 25:9435–9442. 10.1364/OE.25.009435 [DOI] [PubMed] [Google Scholar]
- Tsyboulski D, Orlova N, Griffin F, Seid S, Lecoq J, Saggau P (2018) Remote focusing system for simultaneous dual-plane mesoscopic multiphoton imaging. bioRxiv. Advance online publication. Retrieved December 20, 2018. doi: 10.1101/503052. [DOI] [Google Scholar]
- Voigt FF, Chen JL, Krueppel R, Helmchen F (2015) A modular two-photon microscope for simultaneous imaging of distant cortical areas in vivo. In: Multiphoton microscopy in the biomedical sciences, Vol XV SPIE BiOS, San Francisco, California. [Google Scholar]
- Weisenburger S, Tejera F, Demas J, Chen B, Manley J, Sparks FT, Martínez Traub F, Daigle T, Zeng H, Losonczy A, Vaziri A (2019) Volumetric Ca2+ imaging in the mouse brain using hybrid multiplexed sculpted light microscopy. Cell 177:1050–1066.e14. 10.1016/j.cell.2019.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wekselblatt JB, Flister ED, Piscopo DM, Niell CM (2016) Large-scale imaging of cortical dynamics during sensory perception and behavior. J Neurophysiol 115:2852–2866. 10.1152/jn.01056.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilt BA, Fitzgerald JE, Schnitzer MJ (2013) Photon shot noise limits on optical detection of neuronal spikes and estimation of spike timing. Biophys J 104:51–62. 10.1016/j.bpj.2012.07.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Miller JE, Carrillo-Reid L, Pnevmatikakis E, Paninski L, Yuste R, Peterka DS (2016) Simultaneous multi-plane imaging of neural circuits. Neuron 89:269–284. 10.1016/j.neuron.2015.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim M, Sugihara H, So PT, Sur M (2019) Functional imaging of visual cortical layers and subplate in awake mice with optimized three-photon microscopy. Nat Commun 10:177. 10.1038/s41467-018-08179-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Xu M, Kamigaki T, Hoang Do JP, Chang WC, Jenvay S, Miyamichi K, Luo L, Dan Y (2014) Selective attention. long-range and local circuits for top-down modulation of visual cortex processing. Science 345:660–665. 10.1126/science.1254126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong W, Wu R, Li M, Hu Y, Li Y, Li J, Rong H, Wu H, Xu Y, Lu Y, Jia H, Fan M, Zhou Z, Zhang Y, Wang A, Chen L, Cheng H (2017) Fast high-resolution miniature two-photon microscopy for brain imaging in freely behaving mice. Nat Methods 14:713–719. 10.1038/nmeth.4305 [DOI] [PubMed] [Google Scholar]