

Abstract
Due to their structural and mechanical properties, 1D helical protein assemblies represent highly attractive design targets for biomolecular engineering and protein design. Here we present a designed, tetrameric protein building block, Zn8R4, which assembles via Zn coordination interactions into a series of kinetically stable, crystalline, helical nanotubes whose widths can be controlled by solution conditions. X-ray crystallography and transmission electron microscopy (TEM) measurements indicate that all three classes of protein nanotubes are constructed through the same 2D arrangement of Zn8R4 tetramers held together by Zn coordination. The mechanical properties of these nanotubes are correlated with their widths. All Zn8R4 nanotubes are found to be highly flexible despite possessing crystalline order, owing to their small inter-building-block interaction surfaces that are mediated solely by metal coordination.
Graphical Abstract
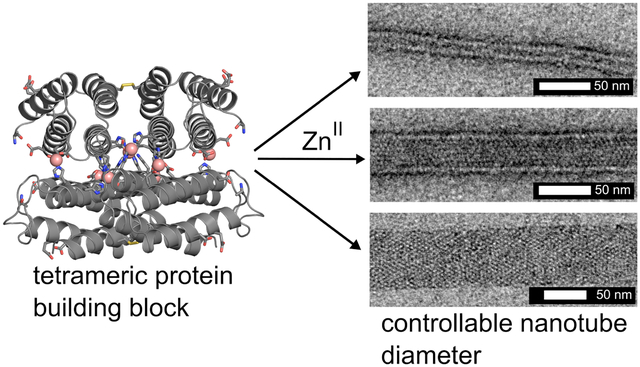
A major goal in nanotechnology is the bottom-up design and construction of self-assembled materials that combine the structural order, dynamicity, and functional properties of natural protein assemblies.1,2 Of particular interest are one-dimensional, helical architectures with hollow interiors, which in nature fulfill a large number of biomechanical roles such as the formation of the cyto-skeleton,3 molecular transport and cell division (microtubules),4,5 cell motility (bacterial flagella),6 infection (type III secretion needles),7,8 endocytosis (dynamin),9,10 and compartmentalization (tubular virus capsids).11 Invariably, all of these biological architectures are assembled from small (<10 nm) protein building blocks that polymerize through non-covalent interactions in a helical symmetry. This mode of assembly–as opposed to, for example, linear stacking of larger, ring-like components–endows natural, 1D protein architectures with the ability to rapidly polymerize or depolymerize and to adapt their structures in response to external stimuli while retaining high mechanical/chemical stability. These properties of biological nanotubes, along with their inherent directionality, chirality, long-range and short-range periodicity, and high surface area-to-volume ratios, render them as highly attractive molecular templates and design targets.12,13
While peptide-based building blocks have shown promise for constructing helical superstructures,3,12,14–19 successes in the design of tubular assemblies from protein synthons have been limited to the use of physical methods (e.g., layer-by-layer assembly on solid templates),20–23 assembly under harsh conditions that alter the structure of the protein subunits,24 or to the use of natively ring-shaped proteins which can be manipulated to stack into tubes.25 We recently established that the simultaneous strength, directionality and reversibility of metal coordination interactions can be exploited to direct the formation of small protein building blocks into discrete oligomers or highly ordered 1-, 2- and 3D architectures.26–31 These assemblies are distinguished from many other designed supramolecular protein architectures by their stimuli-responsiveness. Because metal-protein interactions are inherently tunable (through metal concentration, identity, oxidation state or solution pH), it follows that the structures and assembly states of metal-directed protein architectures can also be modulated by external stimuli. Accordingly, we present here the metal-directed assembly of a designed protein building block into a series of crystalline, helical nanotubes, whose diameters and structure-dependent mechanical properties can be varied through solution conditions that modulate metal-protein interactions.
From a retrosynthetic perspective, a 1D helical tube can be considered as an anisotropic (i.e., rectangular, not square) 2D sheet wrapped around a cylinder with longitudinal and lateral growth axes (Figure S1). Such an anisotropic 2D sheet can be constructed from self-assembling D2 symmetric building blocks that similarly possess bi-directional symmetry in the 2D plane. If the interactions between these building blocks can be controlled (thermodynamically or kinetically) through external means, it should in principle be possible to modulate the magnitude of anisotropy between the longitudinal and lateral growth directions, thereby controlling the widths or the aspect ratios of the resulting tubes. Previously, we reported on the construction of D2 symmetric assemblies of the monomeric protein cytochrome cb562 through Zn2+ coordination.32,33 Here, we considered that these tetrameric scaffolds themselves can be used as building blocks for assembling anisotropic 2D sheets (and thereby 1D nanotubes), as they feature two sets of weakly metal chelating motifs on their external surfaces to promote bidirectional growth: Motif 1) the bidentate combination of Glu8 and Asp12 carboxylates; Motif 2) the tridentate combination of Ala1 N-terminal amine and carbonyl oxygen and Glu39 carboxylate. (Figures 1 and 2) Both motifs were observed in several crystal structures to be capable of mediating latticepacking interactions through Zn2+ coordination (PDB IDs 4JEB, 3TOM, 3QVY, 3M4B, 3M4C).
Figure 1.

Proposed Zn-mediated assembly of a disulfide-linked protein dimer (R2) into a closed, D2 symmetric tetramer (Zn8R4), which acts as a synthon for larger supramolecular architectures upon further Zn coordination. Heme cofactors are shown as green sticks; they have been omitted in later figures for clarity.
Figure 2.

Crystallographic characterization of Zn8R4. (a) Zn-mediated protein interactions observed in the crystal lattice. Internal Zn2+ sites (salmon spheres) stabilize individual Zn8R4 tetramers. Motif 1 (cyan spheres) and Motif 2 (magenta spheres) coordination sites promote intertetramer assembly and the formation of higher order arrays. (b) Arrangement of Zn8R4 building blocks in the contiguous, Zn-mediated 2D sheets within the 3D lattice. Protein tetramers are alternatively colored to show 2D arrangement.
As a starting point for building a stable D2 symmetric building block, we used a cyt cb562 variant (RIDC3) which was previously designed to form weak Zn-mediated dimers that further assembled into 1-, 2- and 3D arrays.26,27 RIDC3 was engineered with a Cys residue at position 96 (informed by earlier work),33 such that it could be prepared as a covalent C96-C96 linked dimer (Figures 1a and S2). A His residue was then incorporated in position 59 in addition to pre-existing metal-coordinating residues on RIDC3, such that the disulfide-linked dimers would lock into the desired D2 tetramer via coordination by a total of 8 Zn2+ ions (Zn8:H59/C96RIDC34, Figure 1b). The dimeric, metal-free dimer is hereafter referred to as R2 and the metal-bound tetramer as Zn8R4.
To probe whether Zn8R4 properly forms and can self-assemble into planar sheets, we first set out to produce single 3D crystals by Zn-directed self-assembly. Our work with RIDC3 had shown that the growth of large Zn-directed 2D sheets and their subsequent stacking into 3D crystal lattices can be promoted by the inclusion of high concentrations of the weakly metal-coordinating buffer TRIS, which lowers the effective free Zn concentration and slows the nucleation rate.26 Accordingly, we were able to obtain hexagonal, diffraction-quality crystals of Zn8R4 from bulk solutions that contained 50 μM Zn8R4, 2.5 mM Zn2+ and 100 mM TRIS (Figure S3). The 2.3-Å resolution crystal structure (Table S1, P6122, 52.9 × 52.9 × 257.1 Å, PDB ID 5BU7) confirmed the formation of the desired D2 symmetric tetramers, the pair of C96-C96 disulfide bonds and the two sets of four, internal Zn-coordination sites (Figures 2 and S4). The examination of the lattice revealed that the Zn8R4 units could indeed form 2D arrays through Zn coordination by Motif1 and Motif2 (Figure 2). While these 2D arrays are not flat (owing to the 61 screw axis that runs along the 2D bc plane) and not every tetramer has its external Zn-coordination motifs occupied, the sheets are contiguously linked by Zn2+ ions and the two motifs propagate self-assembly in orthogonal directions as intended. Further growth of these 2D arrays into 3D crystals is directed by Zn2+ ions oriented perpendicular to their surfaces (Figure S5).
Whereas the metal-mediated assembly of large 3D crystalline arrays is promoted under slow nucleation/growth conditions (low pH, low effective metal concentration), the formation of 1D nanotubes are expected to be favored when the nucleation is rapid(high pH, high effective metal concentration).26 In initial experiments for forming 1D nanotubes, we first incubated R2 dimers with a 5-fold molar excess of Zn2+ at pH 7.5 in a non-metal chelating buffer (MOPS) to pre-form the Zn8R4 tetramers, which was followed by the addition of another 5-fold excess of Zn2+. This treatment resulted in the rapid formation of uniform, helical protein nanotubes that were 48±3 nm wide (Class I) based on negative-stain TEM (Figures 3, S6 and S7). In contrast, the addition of 10-fold excess Zn2+ in the second step produced significantly thinner, monodisperse nanotubes (Class II) with a diameter of 20±2 nm. Stepwise Zn2+ addition was critical for forming mono-disperse populations of nanotubes. When 10-fold excess of Zn2+ was directly added to the R2 dimer solution without the preincubation step, we observed the formation of amorphous aggregates in addition to Type I nanotubes. When a larger excess of Zn2+ was added without the preincubation step, only amorphous aggregates were observed. The formation of these disordered species is likely due to presence of multiple possible Zn-mediated assembly modes of the R2 dimers and the formation of kinetically trapped amorphous aggregates. The above experiments were repeated at pH 6.5 (in non-coordinating, MES buffer), where the metal-protein coordination interactions (particularly that by the N-terminal amine of Motif2) would be expected to be weaker. As at pH 7.5, the stepwise addition of 5+5-fold excess of Zn2+ to the R2 solution yielded the Class I nanotubes and the direct addition of 20-fold excess ZnII resulted in heterogeneous aggregates. In contrast, the direct addition of 10-fold excess Zn2+ led to the formation of yet another class (Class III) of highly ordered, helical nanotubes that were 68±4 nm wide. The observation that the structural outcome of self-assembly is dependent on the sequence of Zn addition indicates that the formation of different classes of Zn8R4 nanotubes is kinetically governed. We postulate that the decisive, structural-determining steps occur during initial nucleation/growth stages.
Figure 3.

Negative stain TEM images of each of the three classes of Zn8R4 tubes.
For structural analysis of Zn8R4 nanotubes, we first took advantage of the fact that some Class I nanotubes presented frayed ends that possessed a flat, single-layered 2D morphology (Fig. 4). The reconstructed TEM images from both the tubular and the flat regions of the Class I nanotubes revealed compact structures with dimensions similar to the Zn8R4 tetramers (Figures S8 and S9), which were clearly distinct from those observed in reconstructions of RIDC3 nanotubes (Figure S10). These tetramers are arranged into unit cells, each of which consists of six subunits, with dimensions (52 Å× 270 Å, black boxes in Figures 4b and c) that are very similar to those seen in the 3D crystals. Indeed, the 2D packing arrangement of Zn8R4 tetramers observed in the X-ray crystal structure fits reasonably well in the TEM-derived molecular pattern. An analysis of the Class II and Class III nanotubes indicates that they also contain the same arrangement (Figures S11 and S12). These results strongly suggest that the Zn coordination interactions that mediate the formation of the three classes of Zn8R4 nanotubes are the same as those present in the 3D crystal lattice. To provide further evidence that the nanotubes have a similar arrangement of tetramers as in 3D crystals, we constructed a structural model of the thinnest (Class II) tubes. This model indicates that it is possible to build a contiguous, well-packed, helical tube with the expected 15-nm diameter using the crystallographically observed inter-tetramer interaction modalities (Figure 5). According to this model, the Zn-Motif1 interactions point along the lateral tube axis, whereas the Zn-Motif2 interactions are oriented longitudinally, suggesting that the width/aspect ratios of the Zn8R4 nanotubes must be influenced by the differential Zn coordination thermodynamics/kinetics of Motif1- and Motif2- mediated interactions. Specifically, deprotonation of the N-terminal amine of Motif2 at higher pH values apparently results in a larger difference between the interaction strengths of the two coordination motifs and correspondingly thinner nanotubes.
Figure 4.

Negative stain TEM characterization of Zn8R4 arrays. (a) Single Class I nanotube with tubular (bottom) and frayed (top) segments. (b and c) 2D reconstructions of tubular (b) and frayed (c) regions of a single nanotube. The crystallographically characterized 2D arrangement of Zn8R4 molecules are superimposed onto the TEM reconstructions. The slight mismatch between the crystallographic model and TEM reconstruction in (b) is likely due to the curved nature of the 2D arrays which is, to some extent, accounted for by the curvature of the tubes.
Figure 5.

Structural model for Class II nanotubes.
Various structural and derived mechanical properties of the Zn8R4 nanotubes are summarized in Table 1. A comparison of the cryoEM and negative-stain TEM (Figures S13 and S14) analyses indicates that the wide Class III tubes undergo significant flattening by uranyl-acetate staining/drying. In contrast, the thin Class II nanotubes were not greatly affected by this treatment, which can be ascribed to their higher density of protein packing that affords resistance to lateral compression. The persistence lengths of the nanotubes were calculated using the recently published program FiberApp (Figure S15),34 and found to be consistently higher for uranyl-stained samples compared to cryoEM samples. As judged by cryoEM data, the thinnest Class II tubes are also the most flexible with a persistence length (9 μm) that is approximately half that of the widest Class III tubes (18 μm). These values are similar to the persistence lengths of actin filaments (17.7 μm)35 and significantly higher than that of double-stranded DNA (50 nm),36 but considerably lower than that of microtubules (5.2 mm).35 It is notable that the Zn8R4 nanotubes are similar to microtubules (24-nm outer and 12-nm inner diameter) in terms of their dimensions. We posit that the higher stiffness of microtubules arises from their considerably more extensive, highly evolved inter-monomer interfaces (~3000 Å2 buried surface area)37 compared to those in the Zn8R4 nanotubes that are mediated solely by metal coordination with no complementary non-covalent interactions. The Young’s moduli of the Zn8R4 nanotubes can be estimated from the persistence lengths using the Equation 1.38,39
| [1] |
where kB is the Boltzmann constant, T is temperature, P is persistence length, and a is the radius of the tubes. The estimated Young’s moduli serve to roughly understand how these artificial nanotubes compare to those found in nature, with the caveat that additional experiments would need to be performed to more accurately determine these values. The range of values obtained (from 0.3 MPa for Class III to 25 MPa for Class II) are comparable to values determined for soft protein fibers such as fibrin (1–10 MPa) or elastin (1MPa) and much less stiff than microtubules (1000–1500 MPa).38 These data again indicate that the Zn8R4 nanotubes are highly flexible, yet simultaneously possess crystalline order. Under certain conditions, we observed the formation of unique, multi-walled nanotubes alongside Class I nanotubes (Figure S16). Additionally, after incubations of >1 month in solution, we observed the bundling of the Class II nanotubes, reminiscent of actin filament aggregates (Figure S17).40 The formation of both of these superstructures is likely promoted by the presence of unsaturated Zn sites on the surfaces of the nanotubes, and may provide a means to increase their mechanical stiffness. Regardless of their flexibility, Zn8R4 nanotubes are highly stable and persist in solution at room temperature for at least one year (Figure S18).
Table 1.
Structural and derived mechanical properties of the Zn8R4 tubes. (n.s. = negative stain measurements)
| Class I | Class II | Class III | |
|---|---|---|---|
| Width (n.s.) (nm) | 48 ± 3 | 20 ± 2 | 68 ± 4 |
| Width (cryo) (nm) | 25 ± 2 | 15± 1 | 46 ± 3 |
| Persistence length (n.s.) (μm) | 28.2 ± 0.3 | 15.1 ± 0.3 | 64.5 ± 1.1 |
| Persistence length (cryo) (μm) | 16.4 ± 0.2 | 8.8 ± 0.4 | 17.8 ± 0.4 |
| Estimated Young’s modulus (n.s.) (MPa) | 1.4 | 15 | 1.1 |
| Estimated Young’s modulus (cryo) (MPa) | 0.8 | 25 | 0.3 |
In conclusion, we have demonstrated the implementation of metal coordination chemistry to generate multiple well-defined, nanoscale architectures with different structural/mechanical properties from a single, designed protein building block. Typically, protein design approaches have aimed to construct singular structural targets that represent the thermodynamically most favored molecular arrangement formed under equilibrium conditions. This scenario contrasts with many biological self-assembly processes that proceed under non-equilibrium conditions and may yield different structural outcomes based on the environmental conditions or energy input.2 In analogy to such natural, non-equilibrium processes, our study shows that it is possible to kinetically dictate protein self-assembly through the use of externally tunable inter-molecular interactions such as metal coordination.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the US Department of Energy (DOE) (Division of Materials Sciences, Office of Basic Energy Sciences, Award DE-FG02–10ER46677 to F.A.T.). The electron microscopy facilities used in this work are supported by funding to Prof. Timothy S. Baker from the NIH (R37 GM-033050), the Agouron Foundation and UCSD. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC0276F00515.
Footnotes
Supporting Information
Experimental details for protein characterization, table for crystallographic data collection/refinement, and TEM analysis. This material is available free of charge at http://pubs.acs.org.
REFERENCES
- (1).Whitesides GM; Mathias JP; Seto CT Science 1991, 254, 1312. [DOI] [PubMed] [Google Scholar]
- (2).Mann S Angew. Chem. Int. Ed 2008, 47, 5306. [DOI] [PubMed] [Google Scholar]
- (3).Dominguez R; Holmes KC Annu. Rev. Biophys 2011, 40, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Nogales E Annu. Rev. Biophys. Biomol. Struct 2001, 30, 397. [DOI] [PubMed] [Google Scholar]
- (5).Wade RH; Chretien D; Job DJ Mol. Biol 1990, 212, 775. [DOI] [PubMed] [Google Scholar]
- (6).Terashima H; Kojima S; Homma M Int. Rev. Cell Mol. Biol 2008, 270, 39. [DOI] [PubMed] [Google Scholar]
- (7).Loquet A; Sgourakis NG; Gupta R; Giller K; Riedel D; Goosmann C; Griesinger C; Kolbe M; Baker D; Becker S; Lange A Nature 2012, 486, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Basler M; Pilhofer M; Henderson GP; Jensen GJ; Mekalanos JJ Nature 2012, 483, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Stowell MH; Marks B; Wigge P; McMahon HT Nat. Cell Biol. 1999, 1, 27. [DOI] [PubMed] [Google Scholar]
- (10).Mears JA; Ray P; Hinshaw JE Structure 2007, 15, 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Stubbs G; Kendall A Adv. Exp. Med. Biol 2012, 726, 631. [DOI] [PubMed] [Google Scholar]
- (12).Reches M; Gazit E Science 2003, 300, 625. [DOI] [PubMed] [Google Scholar]
- (13).Zhang S Nat. Biotech 2003, 21, 1171. [DOI] [PubMed] [Google Scholar]
- (14).Egelman EH; Xu C; DiMaio F; Magnotti E; Modlin C; Yu X; Wright E; Baker D; Conticello VP Structure 2015, 23, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ghadiri MR; Choi CJ Am. Chem. Soc 1990, 112, 1630. [Google Scholar]
- (16).Hamley IW Angew. Chem., Int. Ed 2007, 46, 8128. [DOI] [PubMed] [Google Scholar]
- (17).O’Leary LER; Fallas JA; Bakota EL; Kang MK; Hartgerink JD Nat. Chem 2011, 3, 821. [DOI] [PubMed] [Google Scholar]
- (18).Sharp TH; Bruning M; Mantell J; Sessions RB; Thomson AR; Zaccai NR; Brady RL; Verkade P; Woolfson DN Proc. Natl. Acad. Sci. USA 2012, 109, 13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tarabout C; Roux S; Gobeaux F; Fay N; Pouget E; Meriadec C; Ligeti M; Thomas D; Ijsselstijn M; Besselievre F; Buisson DA; Verbavatz JM; Petitjean M; Valery C; Perrin L; Rousseau B; Artzner F; Paternostre M; Cintrat JC Proc. Natl. Acad. Sci. USA 2011, 108, 7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hou S; Wang J; Martin CR Nano Lett. 2005, 5, 231. [DOI] [PubMed] [Google Scholar]
- (21).Lu G; Ai S; Li J Langmuir 2005, 21, 1679. [DOI] [PubMed] [Google Scholar]
- (22).Lu G; Komatsu T; Tsuchida E Chem. Commun 2007, 2980. [DOI] [PubMed] [Google Scholar]
- (23).Qu X; Komatsu T ACS Nano 2010, 4, 563. [DOI] [PubMed] [Google Scholar]
- (24).Lara C; Handschin S; Mezzenga R Nanoscale 2013, 5, 7197. [DOI] [PubMed] [Google Scholar]
- (25).Ballister ER; Lai AH; Zuckermann RN; Cheng Y; Mougous JD Proc. Natl. Acad. Sci. USA 2008, 105, 3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Brodin JD; Ambroggio XI; Tang C; Parent KN; Baker TS; Tezcan FA Nat. Chem 2012, 4, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Brodin JD; Carr JR; Sontz PA; Tezcan FA Proc. Natl. Acad. Sci. USA 2014, 111, 2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Salgado EN; Ambroggio XI; Brodin JD; Lewis RA; Kuhlman B; Tezcan FA Proc. Natl. Acad. Sci. USA 2010, 107, 1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Radford RJ; Brodin JD; Salgado EN; Tezcan FA Coord. Chem. Rev 2011, 255, 790. [Google Scholar]
- (30).Radford RJ; Tezcan FA J. Am. Chem. Soc 2009, 131, 9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Song WJ; Tezcan FA Science 2014, 346, 1525. [DOI] [PubMed] [Google Scholar]
- (32).Salgado EN; Faraone-Mennella J; Tezcan FA J. Am. Chem. Soc 2007, 129, 13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Brodin JD; Medina-Morales A; Ni T; Salgado EN; Ambroggio XI; Tezcan FA J. Am. Chem. Soc 2010, 132, 8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Usov I; Mezzenga R Macromolecules 2015, 48, 1269. [Google Scholar]
- (35).Gittes F; Mickey B; Nettleton J; Howard JJ Cell. Biol 1993, 120, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bloom KS Chromosoma 2008, 117, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Löwe J; Li H; Downing KH; Nogales EJ Mol. Biol 2001, 313, 1045. [DOI] [PubMed] [Google Scholar]
- (38).Guthold M; Liu W; Sparks EA; Jawerth LM; Peng L; Falvo M; Superfine R; Hantgan RR; Lord ST Cell. Biochem. Biophys 2007, 49, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Trachtenberg SHI, In Microscopy: Science, Technology, Applicationsand Education; Mendez-Villas, A. D. JEd.; Formatex Research Center: 2010; Vol. 3. [Google Scholar]
- (40).Bremer A; Aebi U Curr. Opin. Cell Biol 1992, 4, 20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.