

Abstract
Colorectal cancer (CRC) remains a leading cause of cancer-related deaths in the United States. Although immunotherapy has dramatically changed the landscape of treatment for many advanced cancers, the benefit in CRC has thus far been limited to patients with microsatellite instability high (MSI-H):DNA mismatch repair–deficient (dMMR) tumors. Recent studies in the refractory CRC setting have led to US Food and Drug Administration approvals for pembrolizumab as well as nivolumab (with or without ipilimumab) for tumors harboring an MSI-H:dMMR molecular profile. Several randomized controlled trials are underway to move immunotherapy into the frontline for metastatic cancer (with or without chemotherapy) and the adjuvant setting. Awareness of these studies is critical given the relatively low incidence (approximately 3%–5%) of MSI-H:dMMR in advanced or metastatic CRC to support study completion, because the results could be potentially practice changing. The real challenge in this disease is related to demonstrating the benefit of immunotherapy for the vast majority of patients with CRC not harboring MSI-H:dMMR. Given the rapid pace of scientific changes, this article provides a narrative review regarding the current landscape of immunotherapy for CRC. Particular attention is paid to the currently available data that inform today’s clinical practice along with upcoming randomized controlled trials that may soon dramatically change the treatment landscape for CRC.
Colorectal cancer (CRC) is the third leading cause of cancer-related death in the United States, with an estimated 135 430 new cases and 50 260 cancer-related deaths annually (1). Although the incidence and disease-specific mortality has gradually declined over the past two decades, recent studies describe a disturbing trend of an increased incidence in younger (<50 years) individuals (2,3). The majority of patients diagnosed with metastatic colorectal cancer (mCRC) have incurable disease, with the exception of those with oligometastatic disease, for which successful surgical or ablative interventions and systemic therapy has yielded 5-year and 10-year survival rates of approximately 40% and 20%, respectively (4–6). For all other patients with mCRC, the use of combination systemic therapies and optimal supportive care has produced meaningful improvements in mortality, with the median overall survival (OS) now exceeding 30 months (7). However, with an overall 5-year survival of only approximately 20%, there remains much room for improvement with therapeutic strategies.
In recent years, there have been substantial advancements in our understanding of the intersection between host immune surveillance and tumorigenesis. As a result, clinically beneficial pharmacologic interventions have led to the approval of immunotherapeutic agents for all advanced microsatellite instability high (MSI-H):DNA mismatch repair–deficient (dMMR) solid tumors, including mCRC (Table 1). The demonstration of durable clinical responses and improved survival outcomes in these select situations has spurred a renewed interest in using the immune system as an antineoplastic biological weapon. Unfortunately, for the vast majority of patients with mCRC whose tumors are not MSI-H:dMMR (>95%), immunotherapy currently offers little to no clinical benefit.
Table 1.
Recent clinical trials supporting checkpoint inhibitor use in MSI-H mCRC*
| Immuno-oncology agent (target) | Study (design) | Reference | No. | ORR (95% CI) | Secondary endpoints |
|---|---|---|---|---|---|
| Pembrolizumab (PD-1) | KEYNOTE-016 (Phase II) | Le et al. 2015 (8) | 25 | 40.0% (12% to 74%) | Median PFS = NR Median OS = NR DCR = 90% (95% CI = 55% to 100%) |
| Nivolumab (PD-1) | CheckMate 142 (Phase II) | Overman et al. 2017 (9) | 74 | 31.1% (20.8% to 2.9%) | 1-year PFS = 50% (95% CI = 38% to 61%) 1-year OS = 73% (95% CI = 62% to 82%) DCR ≥12 weeks = 69% (95% CI = 57% to 69%) |
| Nivolumab + ipilimumab (PD-1 + CTLA-4) | CheckMate 142 (Phase II) | Overman et al. 2018 (10) | 119 | 54.6% (45.2% to 3.8%) | 1-year PFS = 71% (95% CI = 61.4% to 78.7%) 1-year OS = 85% (95% CI = 77.0% to 90.2%) DCR ≥12 weeks = 80% (95% CI = 71.5% to 86.6%) |
CI = confidence interval; CTLA-4 = cytotoxic T-lymphocyte–associated protein 4; DCR = disease control rate; mCRC = metastatic colorectal cancer; MSI-H = microsatellite instability (high); ORR = overall response rate; OS = overall survival; PD-1 = programmed cell death 1; PFS = progression-free survival; NR = not reached.
The aims of this comprehensive review are to examine what is known about the immunological microenvironment in mCRC and summarize the available evidence regarding the use of immunotherapy in current treatment paradigms. Moreover, through an updated analysis of the existing literature and anticipated results of ongoing clinical trials, we discuss pragmatic strategies for future investigation into novel immunotherapy targets and discuss current obstacles in navigating the immunological landscape of mCRC.
Rationale for Immunotherapy in mCRC
Vogelstein et al. set the foundation for our current understanding of the molecular evolution of CRC (11). Researchers have continued to build on this foundation, which has led to impactful targeted biologic therapies (ie, anti-vascular endothelial growth factor [VEGF] and anti-epidermal growth factor [EGF] receptor) that has improved the OS of patients with mCRC primarily by complementing active classic cytotoxic therapy. However, these systemic therapies control mCRC only for a period of time instead of eradicating the disease and curing patients.
The tumor microenvironment (TME) refers to the setting in which cancer cells interact with their surroundings, including tumor-related immune cells, blood vessels, cytokines, stroma, and other signaling molecules, such as EGF, transforming growth factor-beta (TGF-β), fibroblast growth factor, and tumor necrosis factor-alpha (TNF-alpha) (12). The close interplay between a tumor and its TME is bidirectional, with tumors affecting their TME via the extracellular signals released and the TME driving tumorigenesis (12,13). The TME also supports tumor heterogeneity, adding another level of interpatient and intratumoral complexity (14). Tumors with a greater infiltrate of T cells have increased chemokine concentrations with activation of the innate immune system. This increased T-cell infiltrate correlates with an improved prognosis, namely a longer disease-free interval in patients with CRC (15).
A number of immunotherapeutic agents rely on tumor cell exploitation of major histocompatibility complex (MHC)-T-cell receptor (TCR)–dependent signaling pathways to suppress the immune system and promote anergy through upregulation of immune checkpoint expression, including programmed cell death 1 (PD-1), PD-1 ligand (PD-L1), cytotoxic T-lymphocyte–associated protein 4 (CTLA-4), indoleamine 2, 3-dioxygenase, and lymphocyte-activation gene 3.
PD-1 is a transmembrane protein expressed on the surface of multiple hematopoietic cell linages (eg, T cells, B cells, dendritic cells, and natural killer [NK] cells) and is specifically overexpressed within inflammatory microenvironments and on tumor cells (16). This inhibitory molecule binds with PD-L1 to induce a signaling cascade that directly inhibits tumor cell apoptosis and stimulates conversion of effector T cells to regulatory T cells (Tregs). The PD-1/PD-L1 interaction functions primarily to promote anergy in peripheral effector T cells via inhibition of downstream kinases and decreased cytokine production. PD-1 has two ligands, PD-L1 (B7-H1/CD274) and PD-L2 (B7-DC/CD273), both inhibiting downstream proliferation of T cells and cytokine production (17). PD-L1 is recognized as the primary ligand upregulated by tumor cells binding PD-1 and CD80 on T cells, whereas PD-L2 is selectively expressed on activated monocytes, macrophages, and dendritic cells. Although high PD-L2 expression has been associated with various B-cell lymphomas, its immunomodulatory function in solid tumors has yet to be elucidated. The distinct molecular mechanisms of PD-L1 interactions, including different binding affinities, conformational receptor changes, and the lack of interaction between PD-L2 and CD80 (coinhibitory TCR), illuminate potential strategies for developmental immunotherapy targets (18).
The coinhibitory molecule CTLA-4 is a well-known regulator of signal transduction pathways modulating T-cell function and activation and as such has been therapeutically targeted to augment the antitumor host response. CTLA-4 functions as an immune checkpoint through binding B7-1 (CD80) and B7-2 (CD86) ligands on antigen-presenting cells (APC) to downregulate tumor-reactive T-cell activation, clonal expansion, and subsequent antitumor rejection (19).
Tumors characterized by MSI-H:dMMR mechanisms harbor a high level of somatic mutations, resulting in the generation of multiple neopeptides (also referred to as neoantigens), which may be recognized as “foreign.” Antigen presentation by MHC-I molecules has been the major focus of studies; however, recent evidence revealed that MHC-I is thought to be of less affinity and therefore less effective compared with MHC-II (20). Ongoing investigation into MHC-II–restricted neoantigen is a potential future target (21). The accumulation of neoantigens elicits a robust host immune response, associated with increased density of Tumor-Infiltrating Lymphocytes (TILs) and upregulation of immune checkpoint expression (22). Immunotherapy targeting blockade of the PD-1/PD-L1 axis can activate peritumoral lymphoid cells to recognize and attack cancer cells (23).
Dendritic cells serve as a biologic immune intermediate for neoantigen delivery and have an ability to augment the immune response through cytokine release. The clinical significance of regulatory cytokines within the TME has been another emerging area of interest. Interleukin-12 (IL-12) is a pro-inflammatory cytokine produced by macrophages and dendritic cells promoting differentiation and activation of CD8+ T cells and NK cells (24). Murine models have demonstrated synergistic antitumor activity in lung cancer using PD-1 blockade combined with IL-12 therapy (25). TGF-β is an anti-inflammatory cytokine generated by both tumor and host immune cells, favoring an immunosuppressive axis through inhibition of TCR signaling, T-cell differentiation, and upregulating production and function of Tregs (26). Overexpression of TGF-β signaling pathway genes within the TME has been associated with a poorer prognosis in a subset of CRC patients. Preclinical studies using patient-derived CRC tumor organoid and xenograft models have demonstrated TGF-β signaling blockade is an effective therapeutic strategy that resulted in cessation of tumor progression (27). In CRC murine models, combination anti-TGF-β and anti-PD-L1 monoclonal antibodies (mAbs) induced a strong antitumor immune response, leading to a statistically significant increase in the number of CD8+ TILs (28).
Quantitively measuring cytotoxic CD8+ T-cell gene signatures related to antigen presentation, chemokine expression, cytotoxic activity, and adaptive immune resistance within the TME elucidates the overall functional activity of TILs and is a potential predictive marker for checkpoint inhibitors currently under investigation (29).
Early studies demonstrated antitumor activity using selective, nonmodified adoptive transfer of TILs prepared through ex vivo expansion, but its application was ultimately limited because the TILs could recognize tumor epitopes presented only by patient-specific MHC (30). This drawback led to the development of chimeric antigen receptor T cells, which are genetically engineered to express artificial receptors that recognize antigen independent of MHC presentation.
Immunotherapy has been an evolving field of oncological research focusing on harnessing the host immune system to combat tumor progression and metastasis with efforts focusing both on active and passive antitumor immunity (31). Studies in unselected patients with advanced solid tumors reported poor response rates to any immunotherapy intervention in mCRC. However, more recent studies have demonstrated an increased signal of activity in mCRC patients with certain molecular profiles, specifically with MSI-H:dMMR tumors. Currently, there are three US Food and Drug Administration (FDA)-approved mAbs (pembrolizumab and nivolumab ± ipilimumab) for patients with MSI-H:dMMR mCRC (32–34). The trial-level evidence backing these approvals not only highlights the clinical benefit for this subgroup of mCRC patients but, importantly, has ushered in an exciting era of scientific discovery and clinical trials aimed to improve outcomes for all patients with CRC (35).
Consensus Molecular Subtypes (CMS)
The understanding that genomic expression is closely related to cellular phenotype and biological tumor activity has led to extensive international efforts to define a transcriptomics-based classification system of CRC subtypes. Comprehensive analyses of genomic and epigenomic features recently enabled researchers to categorize a majority of CRC into one of four CMS subtypes based on their distinguishing features (Table 2): CMS1 (MSI-like), CMS2 (canonical), CMS3 (metabolic), and CMS4 (mesenchymal) (36).
Table 2.
Consensus molecular subtypes of colorectal cancer
| CMS | Frequency | Prognosis | Molecular features |
|---|---|---|---|
| CMS-1 (MSI-like) | 14% | Good | High level of somatic mutations, CIMP high, immune activation, BRAF mutations, high-level TILs |
| CMS-2 (canonical) | 37% | Intermediate | High somatic copy number alterations, WNT and MYC activation, low TILs |
| CMS-3 (metabolic) | 13% | Intermediate | Metabolic deregulation, mixed MSI status, KRAS mutations, low TILs |
| CMS-4 (mesenchymal) | 23% | Poor | High somatic copy number alterations, stromal infiltration, angiogenesis, and TGF-β activation, high-level TILs |
Adapted from Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015 (36). BRAF = B-Raf proto-oncogene; CIMP = CpG island methylator phenotype; CMS = consensus molecular subtypes; KRAS = KRAS proto-oncogene; MSI = microsatellite instability; MYC = MYC proto-oncogene; TGF-B = transforming growth factor beta; TIL = tumor-infiltrating lymphocytes; WNT = Wingless/Integrated signaling pathway.
The CMS1 group has increased expression of genes specific to cytotoxic lymphocytes and is associated with a good prognosis. Importantly, the influential immunosuppressive signature of CMS4 tumors, characterized by overexpression of cancer-associated fibroblasts and their coregulatory chemokines (VEGF, hepatocyte growth factor, and platelet-derived growth factor), result in a TME favoring tumor-associated inflammation, angiogenesis, and activation of TGF-β, conveying the poorer prognosis (22,37).
Although these molecular subtypes have not been established as a therapeutic stratification tool at this time, comprehensive genomic databases have been constructed to facilitate further understanding of distinct biological CRC entities and their potential to respond to immunotherapy. There are ongoing efforts to characterize local and systemic antitumor immunity more closely, including immunophenotyping of the immune compartment and studying the interplay between immunotherapy and host gut microbiome (37).
Biomarkers of Immune Response
Although promising immunotherapy advancements in CRC continue to evolve and generate enthusiasm, to optimize treatment efficacy, overcome resistance to immune checkpoint blockade, and appropriately select for patients who will likely benefit from immunotherapy, the development of rational therapeutic combinations remains critical. There are several ongoing studies investigating potential targetable pathways involved in the host immune response to CRC. For this field to substantively evolve, correlative studies from clinical trials will be essential to identify biomarkers that can serve as immune response surrogates in mCRC. The following section briefly reviews a limited number of candidate biomarkers and molecular classification systems that have demonstrated the potential to exploit inherent tumor biology and immunophenotype to further our knowledge in this area. A more detailed review on the topic of immunotherapy biomarker selection, validation, and development is beyond the scope of this article and has been recently published elsewhere (38–41).
MSI-H:dMMR
Patients harboring MSI-H:dMMR tumors represent a unique population of mCRC patients that currently appear to benefit the most from immune-based therapies (42). High DNA microsatellite instability, defined as instability at two or more loci (or ≥30% of loci if a larger panel of markers is used), results in a high number of DNA replication errors and represents hallmark consequences both of hereditary and sporadic alterations in MMR genes (43).
The utility of MSI-H:dMMR as a predictive biomarker to anti-PD-1 therapy (pembrolizumab) in mCRC was highlighted in the KEYNOTE-016 trial, which showed a notably higher overall response rate (ORR) in MSI-H:dMMR compared with microsatellite stable (MSS):proficient MMR (pMMR) tumors: 40% (95% confidence interval [CI] = 12% to 74%) vs 0% (95% CI = 0% to 19%), respectively (8). One molecular rationale for the discrepancies between these subgroups is differences in tumor mutational burden. Using whole-exome sequencing, investigators identified an average of 1782 somatic mutations in MSI-H:dMMR tumors vs 73 in MSS:pMMR (P = .01). Interestingly, a high tumor mutational burden was associated with prolonged progression-free survival (PFS) in MSI-H patients (hazard ratio [HR] = 0.63, 95% CI = 0.42 to 0.93, P = .02).
These data, in addition to similar data for nivolumab with or without ipilimumab, led to the recent FDA approval of these agents in MSI-H:dMMR–refractory mCRC patients (Table 1). However, despite MSI-H:dMMR accounting for 15%–20% of all sporadic CRC, this subset of patients represents only a minority (∼5%) of mCRC, and ongoing efforts to expand the application of immunotherapy in MSS:pMMR mCRC are eagerly awaited.
PD-L1
The utility of PD-L1 expression as a surrogate of tumor immunogenicity and predictor of response to checkpoint inhibitors remains controversial, but despite conflicting evidence of its clinical significance, it is recognized as one of the most extensively studied candidate biomarkers to date. Studies have shown that upregulation of PD-L1 in the TME is associated with increased effector T-cell infiltration, and that PD-L1–“positive” tumors have a higher likelihood of clinical benefit with checkpoint inhibitors. In a large meta-analysis of 21 trials (non-CRC primary tumors), the ORR in PD-L1–positive tumors was 34.1% (95% CI = 27.6% to 41.3%) vs 19.9% (95% CI = 15.4% to 25.3%) in PD-L1– negative tumors (44). However, in mCRC the available data have demonstrated no predictive role of PD-L1 expression with regard to clinical outcomes. This is in contrast to other tumor sites (ie, melanoma and non-small cell lung cancer), for which PD-L1 expression has been shown to predict response to immune checkpoint inhibition.
A small subgroup analysis (n = 23) from a multicohort phase-Ib trial of pembrolizumab in advanced solid tumors (KEYNOTE-028) reported a poor ORR of 4% with only one PD-L1–positive mCRC patient, who was also noted to be MSI-H:dMMR, achieving a partial response (45). In the phase II trial of pembrolizumab (KEYNOTE-016) for patients with refractory mCRC (included both MSI-H and MSS), no statistically significant association was found between PD-L1 expression and PFS or OS (8). A recent update from the CheckMate-142 trial demonstrated a promising ORR for MSI-H:dMMR patients both in the monotherapy (nivolumab alone) and combination immunotherapy arm (nivolumab + ipilimumab); however, ORR appeared irrespective of PD-L1 expression level (46).
The observed discordance between PD-L1 expression and response to immune checkpoint blockade may be related to the dynamic nature of this cell surface biomarker, with variable expression at any one time point depending on interactions within the local TME. It is also important to note that different pharmDx kits were used in these studies (IHC 22C3 for pembrolizumab, IHC 28–8 for nivolumab). In addition, the lack of standardized metrics and consensus on “positivity” thresholds may have also obfuscated the true clinical potential of this predictive biomarker.
Immunoscore
The density and distribution of the immune response to tumor cells, represented by TILs, is another potential predictive biomarker in CRC. Some studies have shown peritumoral immune infiltration to be a more useful and predictive surrogate marker of disease progression than the standardized American Joint Committee on Cancer TNM staging system (15,23). Researchers developed the Immunoscore as a risk stratification tool performed by quantification of two lymphocyte population densities in the core of the tumor and at the invasive margin. Although the role of this scoring system to predict response to immunotherapy agents has not yet been widely accepted (owing in part to the central pathologic review and score calculation), multivariable analysis revealed that Immunoscore was a superior prognostic biomarker compared with MSI-H status in predicting disease-specific recurrence and survival in mCRC (47). Additional evidence supporting the consensus Immunoscore as a reliable biomarker to estimate the risk of recurrence in stage I–III CRC was provided by Pagès et al., who recently published the results from a large (n = 2648 tumor samples) multicenter study conducted by an international consortium of expert pathologists and immunologists (48). The prognostic value of the Immunoscore was validated to predict statistically significant (P < .0001) differences in time to recurrence (HR = 0.33, 95% CI = 0.23 to 0.47), DFS (HR = 0.50, 95% CI = 0.39 to 0.64), and OS (HR = 0.56, 95% CI = 0.43 to 0.73). Importantly, the Immunoscore had a larger relative prognostic value than pathologic TNM staging, lymphovascular invasion, tumor differentiation, and MSI status (and based on predictive models, appears independent of those factors as well). The relevance of the Immunoscore in mCRC has been less studied, though limited studies have suggested its continued prognostic significance in patients with metastases.
Treatment With Immune Checkpoint Inhibitors
A growing body of translational and clinical research has identified multiple molecular regulators of lymphocyte activation and suppression that can be therapeutically targeted (Figure 1). The most notable checkpoints in mCRC under active clinical investigation include those that inhibit T-cell activation (ie, CTLA-4, PD-1, and PD-L1), those that promote T-cell activation (ie, LAG-3, OX40, and glucocorticoid-induced TNF receptor family-related protein), and those involved in T-cell metabolism (ie, indoleamine 2, 3-dioxygenase) (Figure 1) (49). Blockage of the suppressive checkpoints (ie, PD-1, PD-L1, CTLA-4) have thus far demonstrated clinical benefit only in patients with MSI-H:dMMR mCRC.
Figure 1.
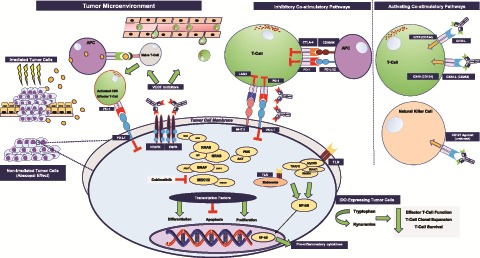
Select targets of immunotherapy therapeutics currently under investigation in colorectal cancer clinical trials. AKT = protein kinase B kinase (AKT8 virus oncogene cellular homolog); APC = antigen-presenting cell; CTLA-4 = cytotoxic T-lymphocyte-associated protein 4; EGFR = epidermal growth factor receptor; ERK = extracellular signal-regulated kinase; GITR = glucocorticoid-induced TNF receptor family-related protein; IDO = indoleamine 2, 3-dioxygenase; LAG3 = lymphocyte-activation gene 3; MEK = mitogen-activated protein extracellular signal-regulated kinase; MHC = major histocompatibility complex; NF-kB = nuclear factor kappa-light-chain-enhancer of activated B cells; PD-1 = programmed cell death 1; PD-L1 = PD-1 ligand; PI3K = phosphoinositide 3-kinase; RAF = rapidly accelerated fibrosarcoma; RAS = rat sarcoma; TLR = Toll-like receptor; VEGF = vascular endothelial growth factor.
PD-1
Despite the FDA approval for a variety of cancers based on improvements in disease control rate (DCR) and OS, including the landmark tumor-agnostic approval of pembrolizumab for all MSI-H:dMMR solid tumors, most studies of checkpoint inhibitors in unselected mCRC populations have shown limited efficacy of this therapeutic option (50). In the seminal phase I study of nivolumab in 39 patients with advanced-stage solid tumors, including 14 mCRC patients, one mCRC patient (7%) achieved a durable complete response at 6 months, which remained ongoing at 3 years (51). Importantly, the one responder in the trial was found to have MSI-H:dMMR mCRC as well as high expression of PD-L1 on TILs. In a similar phase I study of nivolumab across multiple cancers (n = 296), no objective responses (0 of 19, 0%) were seen in an unselected mCRC population (50).
The utility of checkpoint inhibitors for MSI-H:dMMR mCRC was further highlighted in the aforementioned KEYNOTE-016 trial of pembrolizumab (10 mg/kg every 14 days) in patients with refractory mCRC (≥2 prior systemic therapies) (8). An updated data analysis for an expanded cohort of 54 mCRC patients (28 of 54 MSI-H:dMMR, 52.0%) was presented at the 2016 Annual American Society of Clinical Oncology meeting (52). At a median follow-up of 8.7 months, ORR and DCR were 50% (95% CI = 31% to 69%) and 89% (25 of 28) for MSI-H:dMMR mCRC compared with 0% (95% CI = 0% to 14%) and 16% (4 of 25) for MSS:pMMR mCRC, respectively.
Further clinical benefit of targeting PD-1 was illustrated in CheckMate 142, a nonrandomized phase II trial of nivolumab with or without ipilimumab in heavily pretreated patients with MSI-H:dMMR mCRC (NCT02060188). In the previously reported interim analysis of 74 patients in the nivolumab monotherapy arm, investigator-assessed ORR was 31.1% (95% CI = 20.8% to 42.9%), with a complete response in 3%, DCR 12 or more weeks in 69% (95% CI = 57% to 79%), and 1-year OS of 73% (95% CI = 62% to 82%) (46). In an updated analysis of all treated patients (median follow-up 21 months), Overman et al. reported a similar ORR (34%, 95% CI = 23.2% to 45.7%), with 24% (18 of 74) having a PR as their best response. Of note, the rate of complete response increased from 3% (2 of 74) at 13 months to 9% (7 of 74). The DCR remained durable: 47% at 13 months and 46% at 21 months (9). The benefits of PD-1 monotherapy were seen across all subgroups regardless of tumor or TIL PD-L1 expression, mutational status (BRAF, KRAS), or the presence of germline dMMR.
Based on the data reported in these studies, the FDA extended the approval both for pembrolizumab and nivolumab, with or without ipilimumab, for MSI-H:dMMR mCRC refractory to fluoropyrimidine (5-FU), oxaliplatin, and irinotecan-based treatment (34). The National Comprehensive Cancer Network lists PD-1 antibodies (ie, nivolumab and pembrolizumab, with or without ipilimumab) as suggested second-line therapy for MSI-H:dMMR mCRC (53).
A crucial study to assess the role of anti-PD-1 in the front-line setting is the phase III international KEYNOTE-177 RCT (NCT02563002), in which investigators will evaluate pembrolizumab vs investigator’s choice of chemotherapy for mCRC in the first-line setting. This trial has completed accrual and the results are highly anticipated.
PD-L1
Although there have been no clinical studies directly comparing PD-1 and PD-L1 inhibitors to date, indirect comparative analyses suggest similar outcomes in terms of tumor response and toxicity profile. In a phase I dose-escalation study of the anti-PD-L1 mAb atezolizumab (MPDL3280A), one of four unselected mCRC patients achieved a durable partial response (54). In contrast, in a phase I study of anti–PD-L1 mAb (BMS-936559) in 207 patients with advanced solid tumors, no clinical response was observed among 18 unselected mCRC (55).
The most logical next step in improving outcomes with immunotherapy in MSI-H:dMMR mCRC is in the treatment-naive patient population. The highly anticipated NRG-GI004/S1610 COMMIT trial (NCT02997228) is an ongoing phase III study whereby patients with newly diagnosed mCRC are randomly assigned between mFOLFOX with bevacizumab (control arm) with or without atezolizumab or atezolizumab monotherapy. The incorporation of immunotherapy with standard first-line cytotoxic agents or in comparison with it alone will represent a potential landmark change in treatment paradigms for patients with MSI-H:dMMR mCRC.
However, moving immunotherapy earlier in the stage of disease may offer the opportunity to improve both DFS and OS. The ALLIANCE 021502 (ATOMIC) trial (NCT02912559) is an ongoing phase III randomized controlled trial (RCT) allocating patients with stage III MSI-H:dMMR CRC to either standard adjuvant chemotherapy alone (6 months mFOLFOX6) or combined with atezolizumab, with continuation of anti–PD-L1 therapy for an additional 6 months (total 12 months of immunotherapy). It is hoped the results of this trial will elucidate whether immunotherapy can effectively eradicate minimal residual disease in a high-risk patient population.
CTLA-4
Ipilimumab, an anti-CTLA-4 mAb, was the first checkpoint inhibitor to attain FDA approval based on its ability to achieve durable responses and prolong survival in unresectable and/or metastatic melanoma (56,57). The role of dual checkpoint inhibition (PD-1/CTLA-4) in mCRC was investigated in the CheckMate 142 trial (NCT02060188), which is the largest study of combination immunotherapy in this population to date. In the first interim analysis presented at American Society of Clinical Oncology (ASCO 2016, Overman et al. reported modest activity in MSI-H:dMMR mCRC both with single-agent (nivolumab) and combination immunotherapy (four doses of nivolumab 3 mg/kg plus ipilimumab 1 mg/kg every 3 weeks, followed by nivolumab alone 3 mg/kg every 2 weeks), with an ORR of 27% and 15%, respectively (58). The updated findings for the combination arm presented at the 2018 Gastrointestinal Cancer Symposium suggested MSI-H:dMMR patients may achieve greater clinical benefit with dual checkpoint blockade, although the two study arms were not prospectively designed for direct comparison (10). After a median follow-up of 13.4 months (range, 9–25), the ORR was 54.6% (95% CI = 45.2% to 63.8%), DCR 12 or more weeks of 80% (95% CI = 71.5% to 86.6%), 12-month PFS and OS of 71% (95% CI = 61.4% to 78.7%) and 85% (95% CI = 77.0% to 90.2%), respectively. Interestingly, the reported ORR in a subset of 16 patients (13%) who discontinued treatment because of immune-mediated toxicity was 63%, comparable to the overall population. The indirect comparison of outcomes in this trial suggests combination immunotherapy provides high response rates, durable disease control, encouraging survival, and clinically meaningful improvements in key patient-reported quality-of-life measures, albeit with more toxicity than seen in the monotherapy nivolumab arm. Based on these encouraging results, the FDA in July 2018 granted accelerated approval to the immunotherapy combination for the treatment of MSI-H mCRC following progression on standard chemotherapy (34).
Although single-agent nivolumab appears to have activity in MSI-H:dMMR mCRC, it is important to note that a single-arm phase II study of anti–CTLA-4 monotherapy (tremelimumab) in 47 heavily pretreated mCRC patients failed to demonstrate clinical benefit when used alone (59). Thus, for now, CTLA-4 therapy in mCRC is reserved for use in combination with anti–PD-1 therapy for patients with MSI-H:dMMR. The results of these ongoing pivotal studies will define the role and benefit of chemoimmunotherapy in advanced MSI-H:dMMR CRC. However, many questions will remain including the appropriate duration of immunotherapy treatment, sequencing of immunotherapy agents, interchangeable class effects of the checkpoint inhibitors, and the utility of alternative immunotherapy drugs in the salvage setting in patients who have previously been exposed to checkpoint inhibitors.
Role of Immunotherapy in MSS:pMMR mCRC
Although the available evidence clearly supports the use of immunotherapy for the small subset (3%–5%) of patients with dMMR:MSI-H stage IV CRC, unfortunately, analyses of MSS:pMMR mCRC cohorts in the checkpoint inhibitor trials have failed to demonstrate any clinically meaningful response or survival benefit with either PD-1 monotherapy or dual checkpoint blockade (35). To enhance the activity of these agents in MSS:pMMR tumors, investigators have evaluated the potential synergistic approach of checkpoint inhibitor and tumor signal transduction pathways. Preclinical models demonstrated targeting the mitogen-activated protein kinase pathway through the inhibition of MEK (mitogen-activated protein kinase kinase) increases tumor cell expression of MHC-I, thereby stimulating clonal expansion of peritumoral T cells and enhancing anti–PD-L1 activity (60). This biologic synergy was further investigated combining immunotherapy (anti–PD-L1) and cobimetinib, an oral, highly selective, and reversible small molecule inhibitor of MEK1/2, and central components of the RAS/RAF/MEK/ERK signaling pathway.
Unfortunately, the highly anticipated results of the randomized phase III IMblaze370 study reported that the combination (cobimetinib and atezolizumab) failed to meet its primary OS endpoint. In an analysis of 363 patients with refractory mCRC (92% MSS:pMMR), the immunotherapy combination and atezolizumab monotherapy did not demonstrate a statistically significant OS benefit vs regorafenib in the intention-to-treat population (61). The median OS in the atezolizumab + cobimetinib and regorafenib arms was 8.9 vs 8.5 months (HR = 1.00, 95% CI = 0.73 to 1.38, P = .99), and 7.1 months with atezolizumab monotherapy (HR vs regorafenib = 1.19, 95% CI = 0.83 to 1.71). In addition, PFS and ORR were similar across treatment groups. While we await the final peer-reviewed manuscript as well as correlative and subset analyses, it is important to note the incredibly rapid speed at which this study enrolled, a reflection of the desire for and still unmet need in the mCRC patient population.
Biological Combinations
Another pragmatic approach is to exploit the inherent immunomodulatory properties of conventional CRC therapies in combination with immune-stimulatory agents. These combinations include chemotherapy, small molecule tyrosine kinase inhibitors, targeted mAbs, and radiotherapy (RT).
Chemoimmunotherapy
Numerous cytotoxic agents (ie, anthracyclines and oxaliplatin) have been shown to induce cell death through immunogenic mechanisms resulting in cellular fragmentation that is taken up by APCs and presented to T cells (62). Intuitively, myeloablative chemotherapy would be assumed to work in an antagonistic fashion by suppressing the immune response; however, chemotherapy-induced bone marrow suppression decreases immune suppressive cells (ie, Tregs) to a greater extent as well as induces proliferation of homeostatic T-cell populations, providing a complementary potential partner for immune stimulation (63).
In the proof-of-concept GOLFIG-1 trial of 46 mCRC patients (74% had ≥1 prior systemic treatment) (64), the combination of granulocyte macrophage colony stimulating factor and IL-2 following chemotherapy (gemcitabine, oxaliplatin, levofolinic acid, and 5-FU) was associated with manageable toxicity and promising antitumor activity with an ORR of 56.5% (95% CI = 42.1% to 69.8%) and DCR of 91.3% (95% CI = 79.6% to 96.4%). Of note, there was a pronounced survival benefit observed in the six patients who developed therapy-related self-limiting autoimmunity, associated with a mean time to progression of 24 months and OS of 32 months (65). Validation of this approach was explored in a subsequent phase III RCT comparing GOLFIG to FOLFOX-4 in first-line mCRC (66); however, the trial was unfortunately closed prematurely because of poor accrual despite allowing MSS patients.
The MODUL trial (NCT02291289) is a randomized multicenter, parallel-group trial investigating immunotherapy maintenance after first-line chemotherapy (FOLFOX + bevacizumab) in mCRC. Cohort two of this trial enrolled 445 patients with wild-type BRAF mCRC and treated them with fluoropyrimidine and bevacizumab with or without atezolizumab, with no difference in PFS (primary endpoint) seen with the addition of atezolizumab (P = .48) (67).
The use of chemoimmunotherapy in mCRC has shown early promise; however, with numerous limiting factors including small sample sizes and heterogeneity of previous systemic therapies, it is difficult at this time to make practice-changing conclusions. Additional studies with large sample sizes are required to elucidate and further characterize this effect.
Immunotherapy With Targeted mAbs
The combination of immunotherapy and targeted mAbs blocking growth factor receptors is one such strategy to enhance the host immune response (68). Because these mAbs have the potential to induce antibody-dependent cell-mediated cytotoxicity (ADCC), there is preclinical justification to combine these with immunotherapies to enhance or potentiate that response, particularly with agents already proven to be active in mCRC (ie, anti-EGF receptor mAbs cetuximab and panitumumab). In a recently presented phase Ib/II trial of cetuximab plus pembrolizumab in nine RAS wild-type mCRC patients, toxicity was manageable and six patients (67%) maintained stable disease for at least 16 weeks (69). An ongoing phase II study of 33 additional patients will use dual primary endpoints (ORR and 6-month PFS) to further assess the efficacy of this approach (NCT02713373).
Stimulation of NK cells represents an alternative ideal target for such a molecular approach because they are mediators of ADCC when tumor cells are bound by antitumor mAbs. This hypothesis-generating immunomodulatory approach in mCRC is currently under early investigation in a phase Ib study of cetuximab in combination with urelumab (CD137 agonist), which is designed to bind and activate both NK cells and cytotoxic T cells (NCT02110082). If validated, these and other approaches to leverage ADCC may have an impact with agents already widely in use.
Immunotherapy With VEGF Inhibition
The role of anti-angiogenic agents to enhance immunomodulation of the TME has been supported by early-phase trials in metastatic melanoma (70) and renal cell carcinoma (71,72). Preclinical data suggest this could also be a therapeutic strategy in mCRC (73). VEGF inhibition has been shown to suppress activation of tumor-associated macrophages, enhance interactions between APCs and dendritic cells, as well as augment vasculature endothelium to enhance lymphocyte chemotaxis and T-cell tumor infiltration (Figure 1) (74).
The first phase Ib study evaluating this approach for MSI-H:dMMR mCRC was presented at ASCO 2017, reporting outcomes of 10 pretreated patients (70% had ≥2 prior chemotherapies) who received a combination of atezolizumab and bevacizumab (75). At 11 months, the median OS had not been reached, ORR was 30% (95% CI = 6.7% to 65.3%), median duration of response was 7.8 months (95% CI = 5.5 to 7.8 months), and DCR was 90% (30% PR).
However, the previously mentioned MODUL trial demonstrated no benefit with the addition of atezolizumab combined with 5-FU and bevacizumab after initial oxaliplatin-based therapy in BRAF wild-type mCRC. The randomized phase-2 BACCI trial is further exploring the efficacy of this biologic combination, adding atezolizumab to capecitabine and bevacizumab in refractory mCRC (NCT02873195).
Also, as mentioned previously, the NRG-GI004/S1610 (“COMMIT”) trial (NCT02997228) will investigate the role of bevacizumab added to chemoimmunotherapy (mFOLFOX6 and atezolizumab) in the first-line management of MSI-H:dMMR mCRC. In addition to the primary survival endpoint (PFS), therapeutic arms will undergo comparative analysis using multiple simultaneous stratification variables, including BRAF mutation status, history of prior adjuvant therapy for CRC, and site of metastatic disease. The results will help to further define the optimal way to incorporate immunotherapy in MSI-H:dMMR mCRC, particularly in the initial treatment planning for this patient population.
Because not every patient with MSI-H:dMMR mCRC derives durable benefit from immunotherapy, several studies are in development or are ongoing that test reexposure of the patient to anti–PD-(L)1 therapy in conjunction with novel immune-oncology targets (eg, T-cell activators). As an example, the Glucocorticoid-induced TNF receptor family-related protein-agonist (INCAGN01876) is being tested in several combinations and sequences in immunotherapy-naive patients with relapsed MSI-H:dMMR mCRC (NCT03126110). This is but one of several studies looking to overcome the immunoediting associated with checkpoint inhibitor resistance in this patient population. Clinical signals in these early studies will leverage correlative biomarker assessments to identify a population of patients in whom to validate the results in expansion cohorts.
Immunotherapy With RT
In keeping with a multidisciplinary approach to CRC management, there is a growing body of evidence suggesting the combination of immunotherapy and RT enhances our ability to harness the abscopal effect. The abscopal effect is characterized by delivering RT to a solitary site of cancer with resultant immune activation against tumor cells at more distant (nonirradiated) sites, extending the antitumor treatment effects of RT both to local and metastatic disease (Figure 1). There is anecdotal evidence in melanoma of combining ipilimumab with localized RT, resulting in dramatic tumor regression of unirradiated metastatic sites (76). Our current understanding of this phenomenon is based on preclinical work highlighting the concept of immunogenic cell death with subsequent release of damage-associated molecular patterns (77,78), which include cancer-associated neoantigens, inflammatory cytokines, upregulation of immunogenic cell surface markers on tumor cells and stroma, and increased antigen presentation by APCs (Figure 1) (79–81).
The clinical utility of this proposed synergistic treatment modality was recently presented by Segal and colleagues in a nonrandomized phase II study (NCT02437071) of pembrolizumab plus RT for patients with MSS:pMMR mCRC (n = 22) (82). Palliative RT was delivered to a metastasis followed by the first dose of pembrolizumab (200 mg every 3 weeks), with objective response in a nonradiated lesion as the primary endpoint. Despite the combination being very well tolerated, only one patient achieved a partial response (ORR = 4.5%), suggesting the single-agent immunotherapy with RT used in this trial is not enough to induce a systemic anticancer effect. As a logical next step, NSABP FC-9 is currently enrolling patients with MSS:pMMR refractory mCRC (NCT02701400). This phase II single-arm study tests dual checkpoint inhibitor with durvalumab (PD-L1) plus tremelimumab (CTLA-4) following palliative hypofractionated RT. Similarly, a phase II trial of durvalumab plus tremelimumab with either standard RT or local ablation in mCRC is underway (NCT03122509). Results from both studies are eagerly awaited.
Enhancing Tumor-Specific Immunogenicity
Adoptive T-Cell Therapies
Another novel treatment option with the potential to augment the host antitumor immune response and enhance the therapeutic efficacy of vaccines is adoptive T-cell therapy. This approach to stimulate tumor immunity involves transfusion of genetically engineered autologous T cells directed at tumor-specific antigens and has demonstrated promising clinical results in a number of hematologic and solid organ malignancies, including a case of metastatic cholangiocarcinoma effectively treated with bioengineered CD4+ T cells (83). The limitation of this selective, nonmodified adoptive transfer is the restriction to tumor neoepitopes presented by patient-specific MHCs. Recognition of this therapeutic drawback led to the development of T cells genetically designed to express artificial receptors that recognize cancer-specific epitopes independent of MHC presentation, termed CARTs. The use of adoptive T-cell therapy in CRC was first evaluated in a phase I trial in three patients with treatment-refractory mCRC (84). Patients were transfused with autologous TCRs genetically engineered to express anti-human carcinoembryonic antigen (CEA) epitopes. A response was noted with a substantial decrease in serum CEA levels (74%–99%) and objective tumor regression of liver and lung metastasis in one patient. Of note, all three patients developed severe transient inflammatory colitis.
Expansion of this novel scientific technology was recently demonstrated with the development of “armored” CARTs, allowing modification of T cells to express proteins of potential therapeutic targeting, such as inhibitory ligands that bind PD-1 receptors (85).
Another noteworthy evolutionary product of the bioengineering era includes bispecific T-cell engagers, which are artificial antibodies composed of a fusion protein containing two individual single-chain variable regions. These two distinct antibody fragments work by simultaneously binding CD3 and tumor-specific surface molecules. Although the therapeutic efficacy of this technology has primarily been evaluated in hematologic malignancies (86), bispecific T-cell engagers specific to a CRC-associated epitope (CEA) were recently developed, and clinical trial validation is expected (87).
Despite the success in the treatment of lineage-restricted hematologic malignancies, failure to identity targetable cellular epitopes has limited the utility thus far of chimeric antigen receptor T-cell therapy for solid tumors, although it remains an area of active investigation (88).
Vaccine-Based Therapy
Deficiencies in DNA MMR proteins can cause insertion or deletion mutations, resulting in genomic instability at microsatellite coding sequences and subsequent translation of frameshift peptide antigens. These shared antigens, resulting directly from driver mutations in gene-encoded DNA segments, are considered to be highly immunogenic stimulators of T cells, making them an optimal target for therapeutic vaccines (89). However, despite this innate biologic opportunity, attempts to establish the role of therapeutic vaccines in CRC through any number of vaccine delivery methods (eg, dendritic cells, autologous tumor cells, recombinant viral vectors, and peptides) have shown mixed results, with limited efficacy in improving clinical outcomes (90). In an early study of historical interest, 254 patients with surgically resected CRC received adjuvant therapy with active specific immunotherapy (ASI), a vaccine consisting of irradiated autologous tumor cells and Bacillus Calmette-Guérin bacteria (91). The conclusion from the original investigation was that patients with stage II CRC treated with adjuvant ASI had a recurrence-free survival benefit, which was not observed in patients with stage III disease, initially attributed to differences in tumor burden between the two groups. However, in a recent retrospective analysis, investigators revisited 196 preserved CRC tumor specimens from this study (34/196 dMMR:MSI-H, 17.3%) to assess outcomes relative to MSI status (92). When compared with surgery alone, patients administered adjuvant vaccine therapy had an improved 15-year recurrence-free survival, irrespective of MSI status and tumor stage (HR = 0.57, 95% CI = 0.34 to 0.94, P = .03). Patients with dMMR:MSI-H CRC were found to have statistically significantly improved rates of 15-year recurrence-free survival compared with pMMR:MSS patients irrespective of treatment arm: 85% vs 64% (HR = 0.45, 95% CI = 0.24% to 0.86%, P = .02). However, the authors failed to find a statistically significant difference in recurrence rates between treatment arms (surgery alone vs ASI) for dMMR:MSI-H patients, suggesting this tumor type has an inherently favorable prognosis.
To evaluate the immunogenic potential of the dMMR genotype, a small phase I–II trial was conducted (NCT01461148) using a peptide vaccine consisting of three frameshift neoantigens commonly associated with dMMR CRC combined with an adjuvant emulsion to promote immunogenicity. The preliminary results of this study reported novel measurable induction of cell-mediated and humoral immunity against at least one frameshift peptide in all 16 vaccinated patients with a favorable toxicity profile (93). Although no overall clinical outcomes data have been published, preliminary results presented at ASCO 2015 reported stable CEA levels and disease control for more than 7 months after initiating the vaccination protocol in a patient with mCRC.
Further exploration of the interaction between tumor cells and our innate, or “nonspecific,” immune system is being investigated through targeting of Toll-like receptors (TLRs), which are cell surface recognition molecules activated by motifs in bacterial DNA, referred to as pathogen-associated molecular patterns. The role of TLR agonists in mCRC is undergoing further testing in multiple early-phase trials, including a TLR3 agonist (poly-ICLC) with pembrolizumab in pMMR:MSS mCRC (NCT02834052), a TLR8 agonist (VTX-2337) in combination with cyclophosphamide (NCT02650635), and a TLR9 agonist (MGN1703) combined with ipilimumab (NCT02668770) and as maintenance following chemotherapy (NCT02077868).
As previously mentioned, identification of tumors with a high CD45RO+ cell (memory T-cell isoform) density is a discriminatory prognostic index associated with improved clinical outcomes and survival (15,94). Investigators are further evaluating the clinical significance of this marker in a pilot study of an allogeneic CRC vaccine (GVAX) combined with cyclophosphamide and SGI-110, an immunomodulatory agent shown to recruit peritumoral CD45RO+ T-cells.
Although the current evidence for vaccine therapeutics in mCRC represents a mechanistically feasible approach, finding the right antigenic stimulant (or combination thereof) coupled to the right delivery system remains currently elusive.
Immunotherapy for mCRC is rapidly evolving with the potential to revolutionize the treatment of this common disease. As our knowledge of the immune system and its intricacies continues to grow, so will our ability to harness its potential. We have already begun to see the potential of immunotherapy with the breakthrough of anti–PD-1 therapy for the MSI-H:dMMR patient population, such as pembrolizumab and nivolumab. The challenge remains making the therapies effective for all patients, regardless of MSI:MMR status. Efforts are underway to exploit the immune system using traditional and novel therapies, recognizing that one approach may not work consistently for each patient. As highlighted in Table 3, there is an array of promising trials under investigation with the hope to bring these therapies to the clinic.
Table 3.
Ongoing or future randomized phase II–III clinical trials of immunotherapy in mCRC*
| Study | Design | MSI status | Patient population | Arms | Primary endpoint |
|---|---|---|---|---|---|
| NCT03186326 | Phase II | MSI-H | Second-line after progression on 5-FU–based chemotherapy | FOLFOX or FOLFIRI+ targeted therapy vs avelumab (anti-PD-L1) | PFS |
| NCT02291289 MODUL trial | Phase II | MSI testing not performed | First-line mCRC | Multiple targeted and immunotherapy arms based on molecular profiling | PFS; ORR |
| NCT02888743 | Phase II | MSS | Refractory mCRC | Tremelimumab + durvalumab with high or low-dose RT | ORR |
| NCT02870920 | Phase II | MSI testing not performed | Refractory mCRC | Tremelimumab + durvalumab + BSC vs BSC alone | OS |
| NCT02873195 | Phase II | MSI testing NP | Refractory mCRC | Capecitabine + bevacizumab + atezolizumab vs placebo | PFS |
| NCT02563002 Keynote 177 | Phase III | MSI-H | First-line mCRC | Pembrolizumab monotherapy vs chemotherapy | PFS |
| NCT02997228 NRG-GI004/SWOG-S1610 | Phase III | MSI-H | First-line mCRC | Atezolizumab monotherapy vs atezolizumab + mFOLFOX6/bevacizumab vs mFOLFOX + bevacizumab | PFS |
| NCT02077868IMPALA | Phase III | MSI testing not performed | Maintenance after 1st line induction chemotherapy | Chemotherapy vs TLR9 agonist (MGN1703) | OS |
BSC = best supportive care; 5-FU = 5-flourouracil; FOLFOX = 5-flourouracil, leucovorin, oxaliplatin; FOLFIRI = 5-flourouracil, leucovorin, irinotecan; mCRC = metastatic colorectal cancer; MSI-(H) = microsatellite instability-(high); MSS = microsatellite stable; ORR = overall response rate; OS = overall survival; PD-L1 = programmed cell death ligand-1; PFS = progression-free survival; RT = radiotherapy; TLR9 = Toll-like receptor.
Alternative attempts to augment host antitumor immune response and enhance the therapeutic efficacy including adoptive T-cell therapies, vaccine-based therapy, TLR agonists, and cell surface recognition molecules activated by motifs in bacterial DNA (ie, pathogen-associated molecular patterns) are each promising areas of active research. Although still in early phases of clinical research, these novel approaches in conjunction with the ongoing pragmatic trials outlined in this article offer many reasons to be optimistic about future immunotherapies for patients with CRC.
Funding
This work was supported by the Gatorade Trust through funds distributed by the University of Florida Department of Medicine.
Notes
Affiliations of authors: Division of Hematology & Oncology, Department of Medicine, University of Florida, Gainesville, FL (AJF, WPS, JSS, HP, CA, TJG); Division of Hematology/Oncology, Moffitt Cancer Center/University of South Florida, Tampa, FL (AJF); Division of Hematology/Oncology, Mayo Clinic, Jacksonville, FL (JSS); Department of Medicine, University of Pittsburgh Cancer Institute, Pittsburgh, PA (JJL); Department of GI Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX (MJO).
The funder had no role in the writing of this review or decision to submit it for publication.
AJF, WPS, JSS, HDP, JJL, MJO, CA, and TJG have no conflicts of interest to disclose.
References
- 1. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;673:177–193. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst. 2017;1098:djw322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bailey CE, Hu C-Y, You YN, et al. Increasing disparities in the age-related incidences of colon and rectal cancers in the United States, 1975-2010. JAMA Surg. 2015;1501:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hornbech K, Ravn J, Steinbrüchel DA.. Outcome after pulmonary metastasectomy: analysis of 5 years consecutive surgical resections 2002-2006. J Thorac Oncol. 2011;610:1733–1740. [DOI] [PubMed] [Google Scholar]
- 5. Simmonds PC, Primrose JN, Colquitt JL, et al. Surgical resection of hepatic metastases from colorectal cancer: a systematic review of published studies. Br J Cancer. 2006;947:982–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tomlinson JS, Jarnagin WR, DeMatteo RP, et al. Actual 10-year survival after resection of colorectal liver metastases defines cure. J Clin Oncol. 2007;2529:4575–4580. [DOI] [PubMed] [Google Scholar]
- 7. Loupakis F, Cremolini C, Masi G, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;37117:1609–1618. [DOI] [PubMed] [Google Scholar]
- 8. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;37226:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;189:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;368:773–779. [DOI] [PubMed] [Google Scholar]
- 11. Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;3199:525–532. [DOI] [PubMed] [Google Scholar]
- 12. Hanahan D, Coussens LM.. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;213:309–322. [DOI] [PubMed] [Google Scholar]
- 13. Korneev KV, Atretkhany K-S, Drutskaya MS, et al. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine. 2017;89:127–135. [DOI] [PubMed] [Google Scholar]
- 14. Junttila MR, de Sauvage FJ.. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;5017467:346–354. [DOI] [PubMed] [Google Scholar]
- 15. Mlecnik B, Tosolini M, Kirilovsky A, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011;296:610–618. [DOI] [PubMed] [Google Scholar]
- 16. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;124:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saunders PA, Hendrycks VR, Lidinsky WA, et al. PD‐L2: PD‐1 involvement in T cell proliferation, cytokine production, and integrin‐mediated adhesion. Eur J Immunol. 2005;3512:3561–3569. [DOI] [PubMed] [Google Scholar]
- 18. Ghiotto M, Gauthier L, Serriari N, et al. PD-L1 and PD-L2 differ in their molecular mechanisms of interaction with PD-1. Int Immunol. 2010;228:651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharma P, Allison JP.. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;1612:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duan F, Duitama J, Al Seesi S, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med. 2014;21111:2231–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun Z, Chen F, Meng F, et al. MHC class II restricted neoantigen: a promising target in tumor immunotherapy. Cancer Lett. 2017;392:17–25. [DOI] [PubMed] [Google Scholar]
- 22. Bever KM, Le DT.. An expanding role for immunotherapy in colorectal cancer. J Natl Compr Canc Netw. 2017;153:401–410. [DOI] [PubMed] [Google Scholar]
- 23. Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;3135795:1960–1964. [DOI] [PubMed] [Google Scholar]
- 24. Vignali DA, Kuchroo VK.. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;138:722.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohs I, Ducimetière L, Marinho J, et al. Restoration of natural killer cell antimetastatic activity by IL12 and checkpoint blockade. Cancer Res. 2017;7724:7059–7071. [DOI] [PubMed] [Google Scholar]
- 26. Sanjabi S, Oh SA, Li MO.. Regulation of the immune response by TGF-β: from conception to autoimmunity and infection. Cold Spring Harb Perspect Biol. 2017;96:a022236.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Calon A, Lonardo E, Berenguer-Llergo A, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;474:320–329. [DOI] [PubMed] [Google Scholar]
- 28. Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;5547693:544.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;1278:2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schumacher TN, Schreiber RD.. Neoantigens in cancer immunotherapy. Science. 2015;3486230:69–74. [DOI] [PubMed] [Google Scholar]
- 31. Schuster M, Nechansky A, Kircheis R.. Cancer immunotherapy. Biotechnol J. 2006;12:138–147. [DOI] [PubMed] [Google Scholar]
- 32.U.S. Food and Drug Administration. FDA approves first cancer treatment for any solid tumor with a specific genetic feature. https://www.fda.gov/news-events/press-announcements/fda-approves-first-cancer-treatment-any-solid-tumor-specific-genetic-feature. Accessed May 23, 2017.
- 33.U.S. Food and Drug Administration. FDA grants nivolumab accelerated approval for MSI-H or dMMR colorectal cancer. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-nivolumab-accelerated-approval-msi-h-or-dmmr-colorectal-cancer.%C2%A0. Accessed August 1, 2017.
- 34.U.S. Food and Drug Administration. U.S. Food and Drug Administration: FDA grants accelerated approval to ipilimumab for MSI-H or dMMR metastatic colorectal cancer. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-ipilimumab-msi-h-or-dmmr-metastatic-colorectal-cancer. Accessed August 21, 2018.
- 35. Segal NH, Saltz LB.. Translational considerations on the outlook of immunotherapy for colorectal cancer. Curr Colorectal Cancer Rep. 2015;112:92–97. [Google Scholar]
- 36. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;2111:1350.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Becht E, de Reyniès A, Giraldo NA, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;2216:4057–4066. [DOI] [PubMed] [Google Scholar]
- 38. Masucci GV, Cesano A, Hawtin R, et al. Validation of biomarkers to predict response to immunotherapy in cancer: volume I—pre-analytical and analytical validation. J Immunother Cancer. 2016;41:76.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dobbin KK, Cesano A, Alvarez J, et al. Validation of biomarkers to predict response to immunotherapy in cancer: volume II—clinical validation and regulatory considerations. J Immunother Cancer. 2016;41:77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gibney GT, Weiner LM, Atkins MB.. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;1712:e542–e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Spencer KR, Wang J, Silk AW, et al. Biomarkers for immunotherapy: current developments and challenges. Am Soc Clin Oncol Educ Book. 2016;36:e493–e503. [DOI] [PubMed] [Google Scholar]
- 42. Price TJ, Thavaneswaran S, Burge M, et al. Update on optimal treatment for metastatic colorectal cancer from the ACTG/AGITG expert meeting: ECCO 2015. Expert Rev Anticancer Ther. 2016;165:557–571. [DOI] [PubMed] [Google Scholar]
- 43. de'Angelis GL, Bottarelli L, Azzoni C, et al. Microsatellite instability in colorectal cancer. Acta Bio Medica Atenei Parmensis. 2018;89(9-S):97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carbognin L, Pilotto S, Milella M, et al. Differential activity of nivolumab, pembrolizumab and MPDL3280A according to the tumor expression of programmed death-ligand-1 (PD-L1): sensitivity analysis of trials in melanoma, lung and genitourinary cancers. PLoS One. 2015;106:e0130142.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Neil B, Wallmark J, Lorente D, et al. 502 Pembrolizumab (MK-3475) for patients (pts) with advanced colorectal carcinoma (CRC): preliminary results from KEYNOTE-028. Eur J Cancer. 2015;51:S103. [Google Scholar]
- 46. Overman MJ, Lonardi S, Leone F, et al. Nivolumab in patients with DNA mismatch repair deficient/microsatellite instability high metastatic colorectal cancer: update from CheckMate 142. In: American Society of Clinical Oncology Annual Meeting. Chicago: American Society of Clinical Oncology; 2017.
- 47. Mlecnik B, Bindea G, Angell HK, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;443:698–711. [DOI] [PubMed] [Google Scholar]
- 48. Pagès F, Mlecnik B, Marliot F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;39110135:2128–2139. [DOI] [PubMed] [Google Scholar]
- 49. Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;51:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 2012;36626:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti–programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;2819:3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Le DT, Uram JN, Wang H, et al. Programmed death-1 blockade in mismatch repair deficient colorectal cancer. In: American Society of Clinical Oncology Annual Meeting. Chicago: American Society of Clinical Oncology; 2016. [Google Scholar]
- 53. Benson A, Venook A, Al-Hawary M.. NCCN Guidelines for Colon Cancer Version 22018. NCCN.org. Accessed December 15, 2018.
- 54. Herbst RS, Gordon MS, Fine GD, et al. A study of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic tumors. In: American Society of Clinical Oncology Annual Meeting. Chicago: American Society of Clinical Oncology; 2013. [Google Scholar]
- 55. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;36626:2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;3638:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;3317:1889–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Overman M, Kopetz S, McDermott R, et al. Nivolumab±ipilimumab in treatment (tx) of patients (pts) with metastatic colorectal cancer (mCRC) with and without high microsatellite instability (MSI-H): CheckMate-142 interim results. J Clin Oncol. 2016;3410:3501. [Google Scholar]
- 59. Chung KY, Gore I, Fong L, et al. Phase II study of the anti-cytotoxic T-lymphocyte–associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. JCO. 2010;2821:3485–3490. [DOI] [PubMed] [Google Scholar]
- 60. Ebert PJ, Cheung J, Yang Y, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. 2016;443:609–621. [DOI] [PubMed] [Google Scholar]
- 61. Bendell J, Ciardiello F, Tabernero J, et al. LBA-004 Efficacy and safety results from IMblaze370, a randomised phase III study comparing atezolizumab+ cobimetinib and atezolizumab monotherapy vs regorafenib in chemotherapy-refractory metastatic colorectal cancer. Ann Oncol. 2018;29(suppl 5):mdy208.003 [Google Scholar]
- 62. Kroemer G, Galluzzi L, Kepp O, et al. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. [DOI] [PubMed] [Google Scholar]
- 63. Sun X, Suo J, Yan J.. Immunotherapy in human colorectal cancer: challenges and prospective. World J Gastroenterol. 2016;2228:6362.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Correale P, Tagliaferri P, Fioravanti A, et al. Immunity feedback and clinical outcome in colon cancer patients undergoing chemoimmunotherapy with gemcitabine+ FOLFOX followed by subcutaneous granulocyte macrophage colony-stimulating factor and aldesleukin (GOLFIG-1 trial). Clin Cancer Res. 2008;1413:4192–4199. [DOI] [PubMed] [Google Scholar]
- 65. Correale P, Rotundo MS, Del Vecchio MT, et al. Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J Immunother. 2010;334:435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Correale P, Botta C, Rotundo MS, et al. Gemcitabine, oxaliplatin, levofolinate, 5-fluorouracil, granulocyte-macrophage colony-stimulating factor, and interleukin-2 (GOLFIG) vs FOLFOX chemotherapy in metastatic colorectal cancer patients: the GOLFIG-2 multicentric open-label randomized phase III trial. J Immunother. 2014;371:26–35. [DOI] [PubMed] [Google Scholar]
- 67. Grothey A, Tabernero J, Arnold D, et al. LBA19 Fluoropyrimidine (FP)+ bevacizumab (BEV)+ atezolizumab vs FP/BEV in BRAFwt metastatic colorectal cancer (mCRC): findings from Cohort 2 of MODUL–a multicentre, randomized trial of biomarker-driven maintenance treatment following first-line induction therapy. Ann Oncol. 2018;29(suppl 8):mdy424.020. [Google Scholar]
- 68. Holubec L, Polivka J, Safanda M, et al. The role of cetuximab in the induction of anticancer immune response in colorectal cancer treatment. Anticancer Res. 2016;369:4421–4426. [DOI] [PubMed] [Google Scholar]
- 69. Boland PM, Hutson A, Maguire O, et al. A phase Ib/II study of cetuximab and pembrolizumab in RAS-wt mCRC. In: American Society of Clinical Oncology Annual Meeting. Chicago: American Society of Clinical Oncology; 2018.
- 70. Sznol M, McDermott DF, Jones SF, et al. Phase Ib evaluation of MPDL3280A (anti-PDL1) in combination with bevacizumab (bev) in patients (pts) with metastatic renal cell carcinoma (mRCC). In: American Society of Clinical Oncology Annual Meeting. Chicago: American Society of Clinical Oncology; 2015.
- 71. Hodi FS, Lawrence D, Lezcano C, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;27:632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nadal R, Amin A, Geynisman DM, et al. Efficacy and safety of endothelial growth factor receptor (VEGFR)-tyrosine kinase inhibitors (TKI) after programmed cell death 1 (PD-1) inhibitor treatment in patients with metastatic clear cell renal cell carcinoma (mccRCC). In: American Society of Clinical Oncology Annual Meeting. Chicago: American Society of Clinical Oncology; 2015. [DOI] [PMC free article] [PubMed]
- 73. Limagne E, Euvrard R, Thibaudin M, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX–bevacizumab drug treatment regimen. Cancer Res. 2016;7618:5241–5252. [DOI] [PubMed] [Google Scholar]
- 74. Liu X-D, Hoang A, Zhou L, et al. Resistance to antiangiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic renal cell carcinoma. Cancer Immunol Res. 2015;39:1017–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hochster HS, Bendell JC, Cleary JM, et al. Efficacy and safety of atezolizumab (atezo) and bevacizumab (bev) in a phase Ib study of microsatellite instability (MSI)-high metastatic colorectal cancer (mCRC). In: American Society of Clinical Oncology Annual Meeting. Chicago: American Society of Clinical Oncology; 2017.
- 76. Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;36610:925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Weichselbaum RR, Ishwaran H, Yoon T, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci. 2008;10547:18490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Minn AJ. Interferons and the immunogenic effects of cancer therapy. Trends Immunol. 2015;3611:725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Formenti SC, Demaria S.. Systemic effects of local radiotherapy. Lancet Oncol. 2009;107:718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Formenti SC, Demaria S.. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;1054:256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gaipl US, Multhoff G, Scheithauer H, et al. Kill and spread the word: stimulation of antitumor immune responses in the context of radiotherapy. Immunotherapy. 2014;65:597–610. [DOI] [PubMed] [Google Scholar]
- 82. Segal NH, Kemeny NE, Cercek A, et al. Non-randomized phase II study to assess the efficacy of pembrolizumab (Pem) plus radiotherapy (RT) or ablation in mismatch repair proficient (pMMR) metastatic colorectal cancer (mCRC) patients. In: American Society of Clinical Oncology Annual Meeting. Chicago: American Society of Clinical Oncology; 2016.
- 83. Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;3446184:641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;193:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pegram HJ, Park JH, Brentjens RJ.. CD28z CARs and armored CARs. Cancer J. 2014;202:127.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Topp MS, Kufer P, Gökbuget N, et al. Targeted therapy with the T-cell–engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;2918:2493–2498. [DOI] [PubMed] [Google Scholar]
- 87. Osada T, Hsu D, Hammond S, et al. Metastatic colorectal cancer cells from patients previously treated with chemotherapy are sensitive to T-cell killing mediated by CEA/CD3-bispecific T-cell-engaging BiTE antibody. Br J Cancer. 2010;1021:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Abken H. Adoptive therapy with CAR redirected T cells: the challenges in targeting solid tumors. Immunotherapy. 2015;75:535–544. [DOI] [PubMed] [Google Scholar]
- 89. Reuschenbach M, Kloor M, Morak M, et al. Serum antibodies against frameshift peptides in microsatellite unstable colorectal cancer patients with Lynch syndrome. Familial Cancer. 2010;92:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Quiroga D, Lyerly HK, Morse MA.. Deficient mismatch repair and the role of immunotherapy in metastatic colorectal cancer. Curr Treatment Options Oncol. 2016;178:1–16. [DOI] [PubMed] [Google Scholar]
- 91. Vermorken JB, Claessen AM, van Tinteren H, et al. Active specific immunotherapy for stage II and stage III human colon cancer: a randomised trial. Lancet. 1999;3539150:345–350. [DOI] [PubMed] [Google Scholar]
- 92. de Weger VA, Turksma AW, Voorham QJ, et al. Clinical effects of adjuvant active specific immunotherapy differ between patients with microsatellite stable and microsatellite instable colon cancer. Clin Cancer Res. 2012;183:882–889. [DOI] [PubMed] [Google Scholar]
- 93. Kloor M, Reuschenbach M, Karbach J, et al. Vaccination of MSI-H colorectal cancer patients with frameshift peptide antigens: a phase I/IIa clinical trial. In: American Society of Clinical Oncology Annual Meeting. Chicago: American Society of Clinical Oncology; 2015.
- 94. Pagès F, Kirilovsky A, Mlecnik B, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol. 2009;2735:5944–5951. [DOI] [PubMed] [Google Scholar]