

ABSTRACT
Health care is increasingly focused on health at the individual level. In the rapidly evolving field of precision nutrition, researchers aim to identify how genetics, epigenetics, and the microbiome interact to shape an individual's response to diet. With this understanding, personalized responses can be predicted and dietary advice can be tailored to the individual. With the integration of these complex sources of data, an important aspect of precision nutrition research is the methodology used for studying interindividual variability in response to diet. This article stands as the first in a 2-part review of current research investigating the contribution of the gut microbiota to interindividual variability in response to diet. Part I reviews the methods used by researchers to design and carry out such studies as well as the statistical and bioinformatic methods used to analyze results. Part II reviews the findings of these studies, discusses gaps in our current knowledge, and summarizes directions for future research. Taken together, these reviews summarize the current state of knowledge and provide a foundation for future research on the role of the gut microbiome in precision nutrition.
Keywords: gut microbiome, personalized nutrition, precision nutrition, methods, interindividual variability, effect modification, prediction, dietary response, metabolism
Introduction
Review outline and scope
Studies investigating the role of the gut microbiota in precision nutrition, rather than focusing on average effects, focus on interindividual variability in response to diet and investigate the potential of the gut microbiota to influence personalized response. The field of gut microbiota research connects many different topics of investigation. This review covers recent studies and methods for assessing the effect of the baseline state of the microbiome on host response to diet. For a more detailed account of search methods and selection criteria for studies included, please refer to Part II of this review.
The first part of this review provides a summary of the methods used to conduct these precision nutrition studies. First, the intersection between precision nutrition and the gut microbiota is introduced. The following sections detail the methods used to conduct studies investigating the gut microbiota's role in precision nutrition, including study design, dietary interventions, and response criteria. Methods used to analyze the data produced by these studies are then summarized, including microbiome and statistical analysis methods. A discussion of future directions for precision nutrition-microbiome research and final remarks then follows. For a summary and discussion of the results of the studies included, please refer to Part II of this review.
Precision nutrition
There will always be variability in how individuals respond to diet, in both direction and magnitude of metabolic response (e.g. weight gain, postprandial glucose, etc.). This interindividual variability has important implications for the efficacy of certain nutrients or dietary patterns in improving or optimizing an individual's health. In other words, what works for one person will not necessarily work as well or in the same way for another. As a result, identifying factors that contribute to an individual's response to diet, as well as devising methods of personalizing dietary recommendations, is critical.
Precision nutrition is a rapidly developing field of research incorporating a multitude of disciplines including nutrition, microbiology, genetics, epigenetics, metabolomics, and others (1) (Figure 1).
Figure 1.
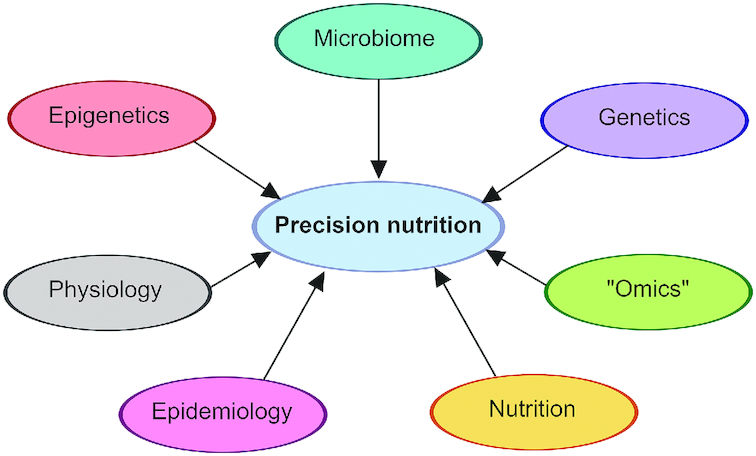
Numerous research fields are integrated and contribute to our overall understanding and study of personalized health and nutrition.
Subfields of precision nutrition include nutrigenetics and nutrigenomics, which study interactions between the human genome and diet (2). These fields examine how genetics, epigenetics, and the microbiome influence dietary response and requirements whilst also being influenced by dietary intake. The result of these interactions is a complex network of metabolic and physiologic processes that define the metabolic phenotype of an individual. Metabolic phenotype can be measured by traditional indicators such as weight, blood pressure, or fasting glucose or by more complex data such as metabolomics, transcriptomics, and proteomics. In combination, these data contribute to our understanding of the processes occurring inside the body as a result of the interconnected influences of diet, the microbiome, genetics, and epigenetics. The potential to predict an individual's response to diet and to optimize an individual's metabolism using diet depends on identifying the biological or physiological features that are relevant to dietary response, determining how these features interact and react to form a response, and understanding how these responses combine to affect human health.
The microbiome: effect compared with effect modification
The body of research on the human microbiome, and particularly the gut microbiome, is growing rapidly. The effects of the gut microbiome extend far beyond the gastrointestinal system, influencing immunity (3, 4), metabolism (5, 6), and brain function (7, 8); in short, everything we require to function as human beings.
However, it is important to distinguish the role of the microbiome as a mediator of the effect of diet on metabolism from the potential of the microbiome to be an effect modifier of response to diet (Figure 2). In the former, diet acts directly on the gut microbiota, altering its composition or function (postintervention microbiome), which then alters host metabolism. In the latter, the effect of diet on metabolism depends on the microbiome, but this effect is not the result of diet-induced changes to the microbiome. For example, preintervention measurements of the microbiome may be used as effect modifiers in an analysis whereas postintervention measurements of the microbiome may serve as mediators. This distinction helps to avoid the circular logic of the effect of the diet on the gut microbiome and the effect of the gut microbiome in response to diet (Figure 3). Although these 2 concepts are inexorably intertwined, they are distinct and require their own independent questions and investigations. This review focuses on the question of effect modification by the gut microbiota and the role this plays in precision nutrition.
FIGURE 2.
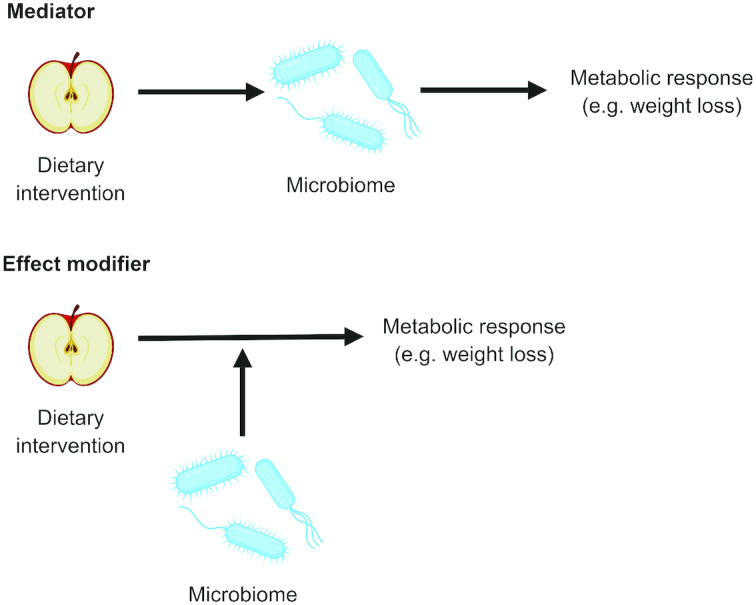
Mediation compared with effect modification. When investigating the effect of diet on the gut microbiome and human health, for example in a study investigating the effect of a dietary intervention on weight loss, the gut microbiome may act as a mediator of effect or as an effect modifier. In the former, the intervention modifies the gut microbiome, which then affects changes in metabolism. In the latter, the intervention may cause changes in metabolism without altering the gut microbiome but these changes may be modified by the preintervention microbiome.
FIGURE 3.
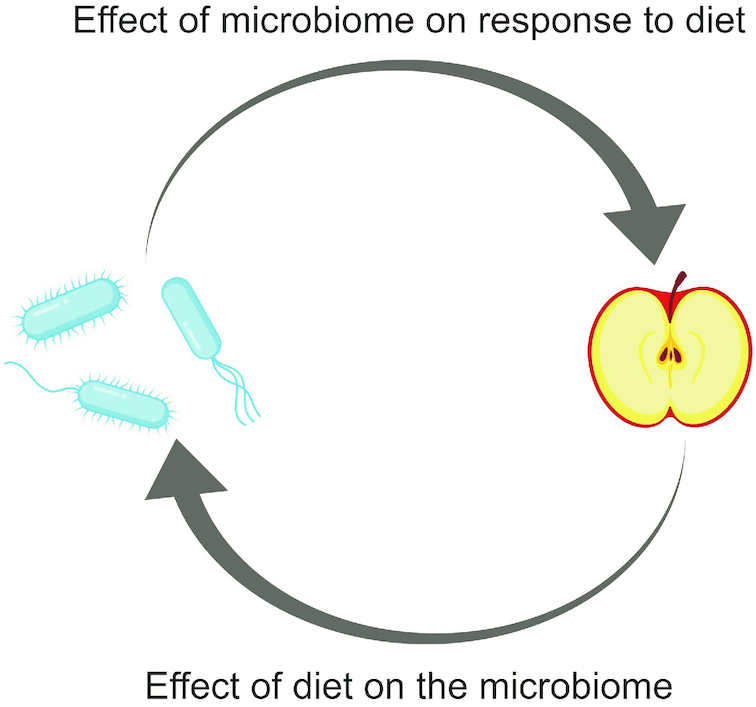
Diet and the microbiota. Dietary factors influence the composition and function of the gut microbiota. Research now shows that the microbiota can also impact the effect of diet on individuals’ health and metabolism. Despite the appearance of circular logic, these are distinct concepts.
Current Status of Knowledge
Methods for conducting precision nutrition studies
Aspects of study design, dietary intervention methods, and response criteria are discussed below and are summarized for recent studies in Table 1.
TABLE 1.
Review of methods used to conduct precision nutrition studies1
| Citation | Dietary intervention | Model | Study design | Biological measures | Responder criteria |
|---|---|---|---|---|---|
| Fiber | |||||
| Korpela et al. (2014) (15) | 3 cohorts: Cohort 1: High-fiber rye bread and whole-grain pasta vs. low-fiber refined wheat bread (12 wk) Cohort 2: Daily dose of 8 g inulin and 8 g oligofructose (12 wk) Cohort 3: RS-enriched diet vs. nonstarch-polysaccharide-enriched diet vs. low-carb/fat, high-pro weight loss diet (10 wk) | Humans (78 adults with metabolic syndrome and/or obesity) | Cohorts 1 & 2: Intervention Randomized Controlled Supplemented diet Cohort 3: Intervention Randomized Cross-over Controlled Standardized diet | TC, insulin sensitivity (HOMA), CRP | Microbiota: Pearson correlation between baseline and postintervention samples. R < 0.87 and NR > 0.92. Host (cholesterol, HOMA, CRP): >10% increase or decrease, <10% change excluded |
| Korem et al. (2017) (16) | Sourdough whole-grain bread vs. white wheat bread (1 wk each) | Humans (20 healthy adults) | Intervention Randomized Cross-over Controlled Supplemented diet | Fasting lipid profile, ALT, AST, GGT, iron, calcium, creatinine, urea, TSH, LDH, CRP, glucose, PPGR, BP, weight, BMR | Lower glycemic response to either white or sourdough bread |
| Smits et al. (2016) (17) | Phase 1: Standardized polysaccharide-rich diet for 4 wk Phase 2: FOS-enriched (10% wt/vol) for 10 d | Mice (gnotobiotic, colonized with human feces from 3 healthy individuals) | Intervention Standardized diet | Metabolomics | Change along principal coordinates (unweighted UniFrac) |
| Hjorth et al. (2017) (18) | NND high in fiber/wholegrain vs. ADD for 6 mo | Humans (62 adults with central obesity and components of metabolic syndrome) | Intervention Randomized Parallel Controlled Standardized diet (plus advice and follow-up after 1 y) | Body fat loss | P/B ratio: High (>0.01) and low (<0.01) Looking at association with fat loss |
| Roager et al. (2014) (19) | NND high in fiber/wholegrain vs. ADD for 6 mo | Humans (62 adults with central obesity and components of metabolic syndrome) | Intervention Randomized Parallel Controlled Standardized diet (plus advice and follow-up after 1 y) | Lipid profile (TC, TAG, HDL-C, LDL-C) | P/B ratio: High (>0.01) and low (<0.01) Looking at association with change in TC |
| Zhao et al. (2018) (20) | WTP diet (high-fiber, whole grains, Chinese medicinal foods, prebiotics) vs. control (standard dietary recommendations of 2013 Chinese Diabetes Society guidelines) for 84 days (3 mo) | Humans (43 adults with T2D) | Intervention Randomized Parallel Controlled Supplemented diet | HbA1c, SCFAs, glucose and insulin, GLP-1, PYY, lipid profiles, body weight, fecal pH | Decrease in HbA1c |
| Kovatcheva- Datchary et al. (2015) (21) | Barley kernel bread (BKB) vs. white wheat flour bread (WWB) for 3 d each | Humans (39 healthy adults, 10 selected as R or NR) and mice (gnotobiotic, colonized with human feces from high or low P/B individuals) | Intervention Randomized Cross-over Controlled Supplemented diet | Humans: Glucose and insulin, SCFAs, breath hydrogen Mice: PPGR, liver glycogen | Change in glycemic response: R (glucose iAUC decreased by ≥25%, total AUC decreased; insulin iAUC decreased by ≥15%). 10 R and 10 NR (showed least or no improvement in glycemic response) selected from initial cohort of 39 |
| Chen et al. (2017) (9) | Arabinoxylans from sorghum bran (SAX) vs. corn arabinoxyan (CAX) vs. fructo-oligosaccharides (FOS) | In vitro fermentation (human stool from 2 individuals) | “Intervention” (nutrient challenge) “Cross-over” | SCFA production | P/B ratio (no cut-off ratio provided) Looking at association with production of SCFAs |
| Salonen et al. (2014) (22) and Walker et al. (2011) (23) | Standard weight maintenance diet (M) vs. controlled diet w/resistant starch (RS, type 3) vs. controlled diet w/nonstarch polysaccharides (NSPs) vs. weight-loss (WL) diet for 10 wk | Humans (14 obese adult males) | Intervention Randomized Cross-over Controlled Standardized diet | Fecal SCFAs, insulin sensitivity (glucose, insulin) | Microbiota stability and clustering in principal coordinates (no discrete cut-off) |
| Tap et al. (2015) (24) | 10 g vs. 40 g dietary fiber for 5 d each | Humans (19 healthy adults) | Intervention Randomized Cross-over Controlled Standardized diet | Cell genotoxicity, SCFAs | Microbiota richness and stability (no discrete cut-off) |
| Martinez et al. (2010) (77) | RS2 (Hi-Maize) vs. RS4 (wheat) vs. control (native starch) each for 3 wk | Humans (10 healthy adults) | Intervention Randomized Cross-over Controlled Supplemented diet | Microbiome only | No identification of aspects that differentiated individuals who responded differently to the intervention |
| Martinez et al. (2013) (25) | Whole-grain barley (WGB, 18.7 g TDF) vs. brown rice (BR, 4.4 g TDF) vs. equal mixture of the 2 (BR + WGB, 11.5 g TDF) for 4 wk each | Humans (28 healthy adults) | Intervention Randomized Cross-over Supplemented diet | Body composition, PPGR, lipid profile, hsCRP, IL-6, fecal SCFAs | Change in plasma IL-6: terciles |
| Venkataramaran et al. (2016) (26) | RS2 (unmodified potato starch) at increasing doses (12 g, 24 g, 48 g) for 10 d | Humans (20 healthy adults) | Intervention Supplemented diet | Fecal SCFAs | Fecal butyrate: Enhanced (9–15 mmol/kg wet feces), high (≥11 mmol/kg), and low (≤8 mmol/kg) |
| Davis et al. (2011) (27) | GOS (increasing dosage: 0, 2.5, 5, 10 g) for 3 wk each | Humans (18 healthy adults) | Intervention Placebo-controlled Cross-over Supplemented diet | Only microbiota response | Compositional shifts in the microbiome (no discrete definition) |
| Bouhnik et al. (2004) (28) | Phase 1: 1 of 7 nondigestible carbohydrates (NDCH) (short-chain fructo-oligosaccharides, soybean oligosaccharides, galacto-oligosaccharides, RS3, lactulose, long-chain inulin, isomalto-oligosaccharides) vs. placebo for 1 wk each Phase 2: One of 4 bifidogenic NDCHs vs. placebo | Humans (200 healthy adults) | Intervention Randomized Placebo-controlled Supplemented diet | Only microbiota response | Change in Bif. abundance (no discrete cut-off) |
| Tuohy et al. (2007) (29) | Partially hydrolyzed guar gum (PHGG) + fructo-oligosacchardes (FOS) vs. placebo for 3 wk each | Humans (31 healthy adults) | Intervention Randomized Placebo-controlled Cross-over Supplemented diet | Only microbiota response | Change in Bif. abundance (no discrete cut-off) |
| Eid et al. (2015) (30) | Palm dates (50 g) vs. placebo (maltodextrin-dextrose, 37.1 g) for 3 wk each | Humans (22 healthy adults) | Intervention Randomized Cross-over Controlled Supplemented diet | SCFA production, ammonia concentrations, genotoxicity, antiproliferation ability | Fiber intake: High (18.5 g/d) and low (6 g/d) Associations found with baseline Bacteroides and microbiota stability |
| Tuohy et al. (2001) (31) | HP-inulin vs. maltodextrin for 2 wk each | Humans (10 healthy adults) | Intervention Placebo-controlled Cross-over Supplemented diet | Only microbiota response | Change in Bif. abundance (no discrete cut-off) |
| Kolida et al. (2007) (32) | (5 g/d & 8 g/d) inulin vs. placebo (8 g/d maltodextrin) for 2 wk each | Humans (30 healthy adults) | Intervention Placebo-controlled Cross-over Supplemented diet | Only microbiota response | Change in Bif. abundance (no discrete cut-off) |
| de Preter et al. (2008) (33) | Lactulose (10 g) vs. oligofructose-enriched inulin (10 g) for 4 wk | Humans (50 healthy adults) | Intervention Randomized (?) Supplemented diet | p-cresol, 15N, and (3H)-PEG | Change in Bif. abundance or metabolite concentrations (no discrete cut-offs) |
| Sonnenburg et al. (2010) (10) | Inulin | In vitro culturing of isolated strains and in vivo 2-member model in gnotobiotic mice | “Intervention” (nutrient challenge) | Inulin metabolism | Increase in relative abundance |
| Holscher et al. (2015) (34) | Agave inulin vs. placebo for 3 wk each | Humans (29 healthy adults) | Intervention Randomized Placebo-controlled Cross-over Supplemented diet | Fecal SCFAs, BCFAs, phenols, indoles, ammonia | Change in Bif. abundance (no discrete cut-off) |
| Fuller et al (2007) (35) | Inulin (10 g/d) vs. no inulin for 3 wk each | Humans (12 healthy adults) | Intervention Randomized Controlled Cross-over Supplemented diet | Allyl mercapturic acid (AMA) | No identification of R/NR |
| Energy restriction and excess | |||||
| Cotillard et al. (2013) (39) | Energy restricted, high protein vs. weight maintenance (6 wk each) | Humans (49 overweight and obese adults) | Intervention Randomized Cross-over Controlled Standardized diet (only supplements provided) | Lipid profile, insulin resistance, inflammatory markers, body composition | Gene count: High (>480,000) and low (<480,000) |
| Shoaie et al. (2015) (40) (using data from Cotillard et al.) | Energy restricted, high protein vs. weight maintenance (6 wk each) | Humans (49 overweight and obese adults) | Intervention Randomized Cross-over Controlled Standardized diet (only supplements provided) | Lipid profile, insulin resistance, inflammatory markers, body composition | Gene count: High (>480,000) and low (<480,000) |
| Kong et al. (2013) (41) | Energy restricted vs. weight maintenance (6 wk each) | Humans (50 overweight and obese adults) | Intervention Randomized Cross-over Controlled Standardized diet (only supplements provided) | Insulin sensitivity, glucose/insulin, adipocyte morphology, body composition, lipid profile, leptin, adiponectin, IL-6, hsCRP | 3 clusters based on weight change trajectories during energy restriction and stabilization periods |
| Griffin et al. (2017) (42) | Chronic calorie restriction w/adequate nutrition (CRON) vs. no dietary restriction (AMER) | Mice (gnotobiotic, colonized with human feces from 1 of 5 CRON or 1 of 5 AMER individuals) Humans (34 CRON, 198 AMER) | Intervention Cross-over Controlled Standardized diet | Body weight | Value and change in community indicator value (CIV) |
| Piening et al. (2018) (43) | Longitudinal intervention including hypercaloric diet (30 d), Eucaloric diet (7 d), calorie-restricted diet (60 d), then follow-up (3 mo postintervention) | Humans (13 overweight insulin-resistant, 10 BMI-matched insulin-sensitive adults) | Intervention Controlled Supplemented diet | Steady state plasma glucose (SSPG), genomics (exome), transcriptomics (RNA-seq), proteomics (LC-MS, targeted assays), metabolomics (LC-MS), anthropometrics, general clinical markers (lipids, creatinine, etc.) | Participants classified as insulin resistant (IR; SSPG > 150) or insulin sensitive (IS; SSPG < 120) at beginning of study |
| Santacruz et al. (2009) (44) | Calorie-restricted diet (10–40%) and increased physical activity (15–23 kcal/kg bw/wk) for 10 wk | Humans (36 overweight adolescents) | Intervention Standardized diet advice provided | BMI, weight loss (WL) | Weight loss: High (>4 kg) and low (<2 kg) |
| Hjorth et al. (2019) (45) | 500 kcal/d deficit diet with high (1500 mg calcium/d) vs. low (≤600 mg calcium/d) in dairy products for 24 wk | Humans (52 overweight adults) | Intervention Randomized Parallel Controlled Dietary advice provided | Weight loss, BMI, body composition | Weight loss (no discrete cut-offs) Groups: High P/B ratio (log (P/B) more than −0.15) Low P/B ratio (log (P/B) less than −0.48) 0-Prevotella |
| Kreznar et al. (2017) (46) | High-fat/high-sucrose vs. control (1st phase: 22 wk, 2nd phase: 16 wk) | Mice (1st phase: 8 different strains, 2nd phase: Transplanted microbiota of 2 metabolically divergent strains) | Intervention Controlled Standardized diet | Fasting insulin and glucose, body weight, hepatic TAG, oGTT | Weight gain, AUC insulin/glucose response to oGTT (no discrete cut-offs) |
| Parks et al. (2013) (47) | High-fat/high-sucrose vs. control chow (8 wk) | Mice (inbred strains) | Intervention Controlled Standardized diet | GWAS, body composition, metabolic rate | Weight gain (no discrete cut-off) |
| Dao et al. (2016) (48) | Calorie-restricted diet vs. weight stabilization (WS) diet for 6 wk each | Humans (49 overweight and obese adults) | Intervention Randomized Cross-over Controlled Standardized diet (only supplements provided) | BMI, waist/hip, body composition, lipid panel, inflammatory markers (hsCRP, IL-6, LPS), ALT, GGT, HOMA, adipocyte morphology, adipose macrophages | A. muciniphila: High (abundance ≥ median) and low (abundance < median) |
| Carmody et al. (2015) (49) | High-fat/high-sugar vs. low-fat, high-plant-polysaccharide (LFPP) (different variations of time) | Mice (inbred and outbred strains) | Intervention Controlled Standardized diet | Body weight | Differentiate responses based on genotype and prior dietary exposure |
| Zou et al (2019) (51) | Calorie restriction (CR, ∼50% normal intake) for 3 weeks | Humans (41 nonobese adults) | InterventionStandardized diet | BMI,change in microbiome composition and function | BMI loss |
| Muñiz Pedrogo et al. (2018) (50) | Volumetric nutritional intervention (larger amounts of fruits/veg and low-energy density foods, lesser intake of high nutrient density foods) with goal to reduce energy intake whilst achieving high food intake volume for 12 mo (outcome at 3 mo) | Humans (26 overweight and obese adults) | Intervention Dietary advice provided | Body weight, fasting glucose, HDL-C, LDL-C, TG | Weight loss ≥5% after 3 months |
| Bioactives, fermented products, and other dietary components | |||||
| Faith et al. (2011) (52) | Phase 1: Varying concentrations of casein (protein) vs. corn oil (fat) vs. cornstarch (polysaccharide) vs. sucrose (simple sugar) (2 wk per diet x 4 diets) Phase 2: New combinations of above (2 wk per diet x 3 diets) Phase 3: Random combinations of above using pureed infant food (1 wk per diet x 6 diets) | Mice (gnotobiotic, 10-strain community) | Intervention Cross-over Controlled Standardized diet | None | Just looking at linear prediction of change |
| Zeevi et al. (2015) (54) | Phase 1: Standardized meals w/50 g carbs (bread, bread + butter, glucose, fructose) for 1 wk Phase 2: “Good” vs. “bad” diet based on PPGR prediction for 1 wk each | Humans (Phase 1: 800 healthy adults, validated on 100 person cohort; Phase 2: 26 healthy adults) | Intervention Cross-over Controlled Phase 1: Supplemented diet Phase 2: Standardized diet | Postprandial glucose response (PPGR) | PPGR (no cut-offs stated) |
| Mendes-Soares et al (2019) (57) | Standardized meals (2 different meals: plain bagel with cream cheese, cereal) | Humans (327 adults without diabetes) | InterventionSupplemented diet | PPGR | PPGR (no cutoffs stated) |
| Le Chatelier et al. (2013) (12) | Not investigating response to intervention | Humans (292 overweight and obese adults) | Cross-sectional | BMI, body fat %, insulin, HOMA, TAG, FFA, leptin, adiponectin, leucocytes, lymphocytes, hsCRP, FIAF, ALT | Gene count: High (>480,000) and low (<480,000). Looking at significant differences in weight gain, metabolic, and inflammatory markers between these groups |
| Bennet et al. (2018) (55) | Low FODMAP diet vs. Traditional IBS diet for 4 wk | Humans (61 adults with IBS) | Intervention Randomized Controlled Standardized diet advice provided | IBS symptom severity score (IBS-SSS) | Change in IBS-SSS: R ≥ 50 (clinically meaningful improvement) and NR < 50. |
| Kolho et al. (2015) (69) | anti-TNF-α medication | Humans (68 children with IBD and 26 controls; 32 received medication) | Intervention Controlled Parallel Supplemented medication only | Calprotectin | Change in calprotectin: R (decrease ∼10-fold or normalization) and NR (no change) |
| Cho et al. (2017) (62) | TMAO (6 oz fish) vs. choline (3 eggs) vs. carnitine (6 oz beef) vs. control (fruit, 2 servings apple sauce) as individual challenge meals | Humans (40 healthy adult males) | Intervention Randomized Controlled Cross-over Supplemented diet | TMAO production, blood chemistry profiles, complete cell counts | TMAO production: High >20% increase in urinary TMAO in response to eggs and beef |
| Suez et al. (2014) (78) | Saccharin (5 mg/kg BW) | Humans (7 healthy adults) | Intervention Supplemented diet | Glycemic response | Glycemic response (significant difference 5–7 d after NAS consumption compared with days 1–4; no discrete cut-off) |
| Kang et al. (2016) (58) | low-CAP (5 mg/d chili powder) vs. high-CAP (10 mg/d chili powder) vs. placebo for 6 wk | Humans (12 healthy adults) | Intervention Placebo-controlled Cross-over Standardized diet | Body weight, SCFA, plasma metabolic and inflammatory markers | P/B enterotype (no discrete cut-off) Looking at association with changes in multiple metabolic/clinical outcomes |
| Possemiers et al. (2007) (59) | Soy (daidzein) and flax (SDG) extracts, isoxanthohumol (IX) from hops | In vitro fermentation (human stool from 100 female subjects) | “Intervention” (nutrient challenge) “Cross-over” | O-DMA, Equol, END, enterolactone (ENL), and 8-prenylnaringenin (8-PN) | Metabolite production: High (H), moderate (M), and low (L) terciles w/significantly different means (cut-off values not stated) END: L (37%), M (51%), H (12%) ENL: L (61%), M (31%), H (8%) O-DMA: L (23%), M (51%), H (23%) Equol: L (33%), M (25%), H (42%) 8-PN: L (30%), M (55%), H (15%) |
| Hullar et al. (2015) (13) | None | Humans (115 adult, premenopausal women) | Cross-sectional | ENL production | ENL excretion: Highest and lowest ENL tertiles (cut-off values not stated) |
| Romo-Vaquero et al. (2019) (60) | Trial 1: 30 g walnuts for 3 d Trial 2: 1 capsule pomegranate extract (450 mg/d) for 3 d Trial 3: 3 capsules of pomegranate extract (1350 mg/d) for 3 d | Humans (249 healthy adults) | Intervention Supplemented diet | Urolithin metabotype (UM) and blood lipid profile | Ellagitannin-metabolizing phenotype (i.e. urolithin metabotype, UM) |
| Li et al. (2011) (61) | Standardized meal containing 200 g of cooked broccoli | Humans (23 healthy adults) and ex vivo fermentation with 50 µM glucoraphanin (from 5 highest and lowest excretors) | Intervention Supplemented diet | Isothiocyanate (ITC) excretion | High- and low-ITC excretors (no cut-off) |
| Zmora et al. (2018) (63) | Probiotic (Bifidobacterium bifidum, Lactobacillus rhamnosus, Lactobacillus lactis, Lactobacillus casei, Bifidobacterium breve, Streptococcus thermophilus, Bifidobacterium longum sbsp. longum, Lactobacillus paracasei, Lactobacillus plantarum, Bifidobacterium longum sbsp. infantis) vs. placebo for 4 wk | Humans (29 healthy adults, 10 naïve, 19 intervention) | Intervention Randomized Controlled Parallel Supplemented diet | Probiotic colonization in gut mucosa | Probiotic colonization |
| Zhang et al. (2016) (64) | Fermented milk product (FMP) | Rats | Intervention Standardized diet | Only microbiota response | Persistence of L. lactis: Resistant/noncarrier (elimination of L. lactis similar to transit marker) and Permissive/carrier (longer persistence of L. lactis)—cut-offs not stated |
| Senan et al. (2015) (65) | Probiotic (lassi, Streptococcus thermophilus and Lactobacillus helveticus) vs. placebo for 4 wk each | Humans (16 geriatric subjects) | Intervention Randomized Placebo-controlled Cross-over Supplemented diet (?) | Total cholesterol (TC, primary outcome), lipid profile, IgG, IgM, TNF-α, INF-γ, IL-2 | Total cholesterol: R (no change or <1.72 mg/dL change in TC) and NR (increase in TC of ≥2.509 mg/dL) |
| Veiga et al. (2010) (66) | Fermented milk product (BFMP, containing Bifidobacterium lactis) vs. nonfermented milk product (MP) for 4 wk at age 4 wk and 12 wk | Mice (T-bet-/-Rag2-/- versus control Rag2-/-) | Intervention Controlled Standardized diet | Cecal pH, SCFA, colitis scores | Colitis score: R (0–3) and NR (≥4) Also increased SCFA and decreased pH (no cut-offs stated) |
| Volokh et al (2019) (68) | Fermented dairy product (FDP, containing Bif. animalis subsp. lactis BB-12) for 30 days | Humans (150 adults) | InterventionSupplemented diet | Only microbiota response | Coefficient of change in lactose-fermenting microbial taxa (LFTs) |
| Mobini et al. (2017) (67) | Placebo vs. low (108 CFU/d) vs. high (1010 CFU/d) Lactobacillus reuteri DSM 17938 for 12 wk | Humans (46 adults with T2D) | Intervention Randomized Parallel Supplemented diet | HbA1c, insulin sensitivity, bile acids, liver fat content, body composition, body fat distribution | Increase in insulin sensitivity index (ISI) |
| Chumpitazi et al. (2015) (56) | Low FODMAP diet vs. typical American childhood diet (TACD) for 48 h each | Humans (33 children with IBS) | Intervention Randomized Controlled Cross-over Standardized diet | Abdominal pain frequency | Abdominal pain frequency: R (≥50% decrease on low FODMAP diet only) and NR (no improvement during either intervention) |
| Spencer et al. (2011) (53) | Baseline (550 mg/70 kg bw, 10 d) vs. depletion (<50 mg/70 kg bw, 42d) vs. repletion (850 mg/70 kg bw, 10 d) choline | Humans (15 female adults) | Intervention Parallel Standardized diet | Liver fat to spleen fat ratio (LF/SF), PEMT promoter SNP rs12325817 | Change in liver fat content |
1ADD, average Danish diet; ALT, alanine aminotransferase; AMA, allyl mercapturic acid; AMER, no dietary restriction; AST, aspartate aminotransferase; AUC, area under the receiver operating characteristic curve; BCFA, branched-chain fatty acid; Bif, Bifidobacteria; BMR, basal metabolic rate; BP, blood pressure; BR, brown rice; bw, body weight; CIV, community indicator value; CRON, chronic calorie restriction with adequate nutrition; CRP, C-reactive protein; END, enterodiol; ENL, enterolactone; FIAF, fasting induced adipose factor; FMP, fermented milk product; FODMAP, fermentable oligosaccharides, disaccharides, monosaccharides, and polyols; FOS, fructo-oligosaccharides; GGT, gamma-glutamyl transpeptidase; GLP-1, glucagon-like peptide-1; GOS, galacto-oligosaccharides; GWAS, genome wide association study; HbA1c, hemoglobin A1c; HDL-C, HDL-cholesterol; [3H]-PEG, [3H]-polyethylene glycol; HOMA, homeostatic model assessment; HP-inulin, high performance inulin; hsCRP, high-sensitivity C-reactive protein; iAUC, incremental AUC; IBD, inflammatory bowel disease; IBS, irritable bowel syndrome; IR, insulin resistant; IS, insulin sensitive; ITC, isothiocyanate; IX, isoxanthohumol; LDH, lactate dehydrogenase; LDL-C, LDL-cholesterol; LFPP, low-fat, high-plat-polysaccharide; M, maintenance; NDCH, nitrogen-15 (15N), nondigestible carbohydrate; NND, new Nordic diet; NR, nonresponders; O-DMA, O-desmethylangolensin; oGTT, oral glucose tolerance test; P/B ratio, Prevotella to Bacteroides ratio; PEMT, phosphatidylethanolamine N-methyltransferase; PHGG, partially hydrolyzed guar gum; PPGR, postprandial glucose response; PYY, peptide YY; R, responders; RS, resistant starch; RS2/3/4, resistant starch type-2, -3, -4; SDG, secoisolariciresinol diglucoside; SNP, single nucleotide polymorphism; SSPG, steady state plasma glucose; SSS, IBS symptom severity score; T2D, type 2 diabetes; TACD, TAG, or TG, triacylglycerides or triglycerides typical American childhood diet; TC, total cholesterol; TDF, total dietary fiber; TMAO, trimethylamine N-oxide; TSH, thyroid stimulating hormone; UM, urolithin metabotype; WGB, whole-grain barley; WL, weight-loss; WS, weight stabilization; WTP, diet of whole grains, traditional Chinese medicinal foods, and prebiotics; 8-PN, 8-prenylnaringenin.
Study design
Table 2 summarizes key features of the design of dietary intervention studies. The most common approach for investigating personalized response to diet is a controlled intervention cohort study. For example, 51 out of the 55 studies conducted between 2000 and 2019, were intervention cohort studies in living organisms (humans or rodents). Three studies examined the effect of supplementing nutrients to in vitro cultures from human stool (9–11). Just 2 were observational, cross-sectional studies (12, 13).
TABLE 2.
Features of dietary intervention design
| Study design | Observational | Intervention |
|---|---|---|
| Intervention | Parallel | Cross-over |
| Control | Placebo (e.g. placebo capsule vs. polyphenol capsule) | “Typical” diet option (e.g. white bread vs. whole-grain bread) |
| Diet intervention type | Diet standardization (e.g. all food provided) | Diet supplementation (e.g. dietary component of interest is provided) |
| Diet intervention content | Targeted dietary component (e.g. source of fiber, polyphenols, etc.) | Broad dietary pattern (e.g. Mediterranean diet, Western diet, etc.) |
Intervention studies allow researchers to standardize the dietary stimulus, providing every participant the same amount (or relative amount) and type or quality of ingredients. Interventions involving humans or animals supplement participants’ habitual diets or completely standardize diets by providing all meals during the intervention period. Although standardizing participants’ diets reduces variability, it is both expensive and intensive, and requires a much greater commitment from subjects compared with supplementing the diet. Studies that choose to supplement participant diets collect dietary recall information in order to account for this source of variability. In addition, a control group is typically included in the design to control for, or identify, variability unrelated to the intervention. If the supplement takes the form of nutrient capsules or pills, then a placebo control group that is indistinguishable from the true intervention should be used. On the other hand, if the supplement is a type of food or a dietary pattern, then a decision on the control group is not straightforward. Ideally, the control food or diet should physically resemble the intervention food and have a similar taste profile. When this is not possible, contrasting foods may be used, but it must be acknowledged that this is not a true control, but rather a separate intervention.
Both parallel and cross-over study designs are used in dietary intervention studies. In a parallel design, different participants are assigned to each intervention group; in a cross-over design the same participants receive each intervention. There is typically greater natural variation between the control and intervention groups in a parallel design, requiring a larger sample size to identify significant treatment effects. In contrast, a cross-over study reduces the uncontrolled variation between the intervention and control arms, but introduces the potential for a carry-over effect. In both cases, a randomization scheme is implemented, randomizing either the order of treatments (cross-over) or the assignment of individuals to separate treatments (parallel).
Randomized, controlled clinical trials with a standardized diet are generally considered the “gold standard” (14) in dietary intervention trials because the potential for confounding is minimized and the estimated intervention effect can be interpreted causally. Identifying the appropriate control is challenging with diet interventions that are designed with whole foods, as a true placebo diet is not possible. For example, the effect of a standardized diet may vary depending on how different it is from the habitual diet of the study participants. Thus, in the attempt to control one source of variation, another is introduced. Therefore, when considering individual nutrients or dietary components, it may be more advantageous to allow participants to continue eating their habitual diets so that any change during the study period within the individual can be attributed to the specific change introduced.
Ultimately, the study design must take into account the dietary intervention being introduced, the resources available, and the responses being measured.
Dietary intervention
Three main categories of dietary interventions have been used in precision nutrition-microbiome studies: fiber; energy restriction and excess; and bioactives, fermented products, and other dietary components. Many studies investigating the effect of the microbiota on metabolic response to diet have employed interventions involving dietary fiber or other nondigestible dietary components (9, 10, 15–35), as these compounds cannot be hydrolyzed by endogenous human enzymes (36) and are thus able to pass through the upper gastrointestinal tract largely intact and enter the large intestine where they can be fermented by the gut microbiota. These compounds serve as a primary source of fuel for this community (36). As the microbiota has also been shown to influence energy harvest and predisposition to obesity (37, 38), a number of studies have also looked at the effect of the gut microbiota on response to an energy-restricted or high-energy diet (39–51). The effect of the gut microbiome on metabolic response to macronutrient distribution (52), micronutrient content (53), dietary patterns (54–57), polyphenols and other plant-based compounds (13, 58–61), animal products (62), probiotics (63–68), as well as antibiotics and certain drugs (69–72) has also been investigated. Dietary interventions may be targeted, such as supplementing an isolated nutrient in the participants’ diets (e.g. fiber), or broad, such as altering the participants’ entire dietary pattern (e.g. low fermentable oligosaccharides, disaccharides, monosaccharides and polyols [FODMAP] diet).
Response criteria
Similar to the choice of dietary intervention, how studies define “response” may also be targeted or broad (e.g. lipid profile or HDL-cholesterol), continuous or categorical (e.g. bodyweight compared with overweight/obese). Different studies have used different biological indicators of “response,” even studies implementing the same dietary intervention (Table 1, Responder Criteria). However, there are some general trends. For instance, all 12 studies using an energy-restricted or high-energy diet analyzed differences in weight gain/loss or changes in body composition (39–50). Changes in circulating lipids (39–41, 43, 48, 50), insulin sensitivity/resistance (39–41, 43), or inflammatory markers (39–41, 48) were also common measured responses. Fiber-intervention studies (9, 10, 15–35) measure many of these same variables but also often include SCFAs (9, 20–24, 25, 26, 30, 34), which are one of the major metabolites produced by the gut microbiota during fermentation of dietary carbohydrates and have many biological effects (73). Changes within the gut microbial community as well as metabolites and clinical markers produced and/or influenced by the gut microbiota have also been used as indicators of responsiveness (9, 13, 15, 17, 23, 24, 27–29, 31–34, 42, 48, 59, 62, 68). Thresholds used to define response or differentiate individuals vary based on the variables being measured and are generally not standardized. Ideally, such thresholds should be based on clinically relevant effects or standardized health recommendations. If there are no standardized recommendations or limits for the variables being measured, it is important that studies clarify the clinical relevance of their findings.
Methods for analyzing precision nutrition data
Laboratory analytical methods for gut microbiome data
The microbiome can be measured using a variety of methods (Table 3, Microbiome Measures), which are subject to their own inherent limitations and biases (74), making comparisons between studies using different methods difficult. Additionally, differences in sample collection, sample preparation, PCR amplification, and bioinformatics pipeline (75) contribute additional variability, further complicating comparisons. Thus, it can be difficult to make broad conclusions regarding the effect of the microbiota when looking at findings in the literature. A brief overview of the different methods and their protocols, advantages, and disadvantages is provided here but, for further discussion, readers are directed to additional reviews of this topic (74, 76).
TABLE 3.
Review of methods used to analyze precision nutrition studies1
| Citation | Microbiome measures | Association or prediction | Statistical methods |
|---|---|---|---|
| Fiber | |||
| Korpela et al. (2014) (15) | Microbiota composition (HITChip microarray) | Prediction | Linear models Logistic regression Independent test cohorts AIC Correlation criterion |
| Korem et al. (2017) (16) | Microbiota composition and function (16S and metagenomics) | Prediction | Linear mixed model PCA, PCoA (Bray–Curtis) Stochastic gradient boosting regression Internal leave-one-out cross-validation AUC criterion |
| Smits et al. (2016) (17) | Microbiota composition (16S) | Prediction | PCA, PCoA (Unweighted UniFrac) Mann–Whitney–Wilcoxon test Wilcoxon rank test Mantel's Pearson test Procrustes transformation Random forests LDA |
| Hjorth et al. (2017) (18) | Microbiota enterotype (Prevotella to Bacteroides (P/B) ratio, qPCR) | Association | Parametric (t-test) or nonparametric (Wilcoxon rank-sum) 2-sample test Chi-square test Linear mixed models |
| Roager et al. (2014) (19) | Microbiota enterotype (Prevotella to Bacteroides (P/B) ratio, qPCR) | Association | Mann–Whitney U test or unpaired t-test Wilcoxon signed-rank test or paired t-test Chi-square test PCA |
| Zhao et al. (2018) (20) | Microbiota composition and function (metagenomics) | Association | 2-way and 1-way ANOVA Mann–Whitney test PCoA (Bray–Curtis) Procrustes analysis Wilcoxon matched-pair signed-rank test Network plots |
| Kovatcheva-Datchary et al. (2015) (21) | Microbiota composition (16S and 16S qPCR) and function (shotgun metagenomics, MG-RAST) | Association | Spearman's correlation 2 group comparison: Student's t-test, Wilcoxon matched-pairs signed-rank test, and Mann–Whitney U test 3 + comparison: ANOVA PCoA (Unweighted UniFrac) |
| Chen et al. (2017) (9) | Microbiota enterotype (Prevotella or Bacteroidetes, 16S) | Association | RDA PCoA (weighted Unifrac) Chi-square test Procrustes analysis |
| Salonen et al. (2014) (22) and Walker et al. (2011) (23) | Microbiota composition (HITChip microarray, qPCR) | Association | Linear mixed models (random effects regression, hierarchical generalized linear models) Pearson's correlation ANOVA |
| Tap et al. (2015) (24) | Microbiome composition (qPCR, 16S) and function (metatranscriptomics) | Association | Co-inertia using RV coefficient Wilcoxon signed-rank test Spearman correlation |
| Martinez et al. (2010) (77) | Microbiome composition (16S, DGGE, Bif.-specific qPCR) | Association | 1-way ANOVA |
| Martinez et al. (2013) (25) | Microbiota composition (16S) | Association | Pearson's correlation Linear model |
| Venkataramaran et al. (2016) (26) | Microbiome composition (16S) | Association | k-means clustering Random forests LEfSe |
| Davis et al. (2011) (27) | Microbiome composition (plate counts, PCR-DGGE, qPCR) | Association | ANOVA |
| Bouhnik et al. (2004) (28) | Microbiota composition (plate counts) | Association | ANOVA Wilcoxon signed-rank test Bonferroni test |
| Tuohy et al. (2007) (29) | Microbiome composition (FISH) | Association | Paired t-test |
| Eid et al. (2015) (30) | Microbiota composition (FISH) | Association | Linear model ANOVA Paired t-test |
| Tuohy et al. (2001) (31) | Microbiome composition (FISH) | Association | Paired t-test |
| Kolida et al. (2007) (32) | Microbiota composition (FISH) | Association | Paired t-test |
| de Preter et al. (2008) (33) | Microbiota composition (DGGE, qPCR) | Association | Paired and unpaired t-tests Pearson correlation |
| Sonnenburg et al. (2010) (10) | Microbiota composition (qPCR) and function (protein expression) | Association | Paired t-test |
| Holscher et al. (2015) (34) | Microbiota composition (16S) | Association | Linear mixed models Mann–Whitney test Pearson's correlation PCoA (Unweighted UniFrac) |
| Fuller et al. (2007) (35) | Microbiota composition (Bifidobacteria abundance by qPCR) | Association | ANOVA, ANCOVA |
| Energy restriction and excess | |||
| Cotillard et al. (2013) (39) | Microbiota richness and composition (metagenomic sequencing) | Prediction | PCA DBA score Internal cross-validation + independent test cohort AUC criterion |
| Shoaie et al. (2015) (40) (using data from Cotillard et al.) | Microbiota richness and composition (metagenomic sequencing) | Prediction | CASINO |
| Kong et al. (2013) (41) | Microbiota composition (qPCR of 7 bacteria) | Prediction | BN analysis Internal leave-one-out cross-validation AUC criterion |
| Griffin et al. (2017) (42) | Microbiota richness, composition (16S) | Prediction and association | Random forests Internal cross-validation |
| Piening et al. (2018) (43) | Microbiota composition (16S and metagenomics) | Prediction | t-tests Random forests AdaBoost Classification ANOVA General linear models LASSO |
| Santacruz et al. (2009) (44) | Microbiota composition (qPCR) | Association | Mann–Whitney U test Wilcoxon signed-rank test |
| Hjorth et al. (2019) (45) | Microbiota composition (16S) | Association | 1-way ANOVA Pearson's chi-squared test Pearson's correlation Linear mixed models Post hoc t-tests |
| Kreznar et al. (2017) (46) | Microbiota composition and function (16S, metagenomics) | Association | ANOVA 2-tailed unpaired Student's t-test (Mann–Whitney U test for nonnormally distributed samples) PCA, PCoA (Unweighted UniFrac) PERMANOVA Pearson correlation |
| Parks et al. (2013) (47) | Microbiota composition (16S) | Association | Linear mixed models Biweight midcorrelation PCoA (Unweighted UniFrac) |
| Dao et al (2016) (48) | Microbiota composition and richness (quantitative metagenomics, (Akkermansia muciniphila abundance also by qPCR) | Association | BN BIC Paired t-tests or ANCOVA Wilcoxon rank-sum or Kruskal–Wallis Spearman correlation |
| Carmody et al. (2015) (49) | Microbiota composition (16S) and function (PICRUSt) | Association | Wilcoxon rank-sum test LEfSe PCoA (Bray–Curtis, Unweighted UniFrac) PERMANOVA |
| Zou et al (2019) (51) | Microbiota composition and function (metagenomics) | Association | Wilcoxon rank-sum testPearson's chi-square testPERMANOVA |
| Muñiz Pedrogo et al. (2018) (50) | Microbiota composition (16S) and function (PICRUSt) | Association | Wilcoxon rank-sum test LEfSe PERMANOVA Wilcoxon signed-rank |
| Bioactives, fermented products, and other dietary components | |||
| Faith et al. (2011) (52) | Microbiota composition (shotgun sequencing) and gene expression (RNA-seq) | Prediction | Linear model Internal cross-validation + multiple independent test cohorts |
| Zeevi et al. (2015) (54) | Microbiota composition and function (16S, metagenomics) | Prediction | PCoA (Bray–Curtis) Pearson correlation Linear regression Gradient boosting regression Internal cross-validation + independent test cohort PDPs iAUC |
| Mendes-Soares et al. (2019) (57) | Microbiota composition and function (metagenomics) | Prediction | Pearson correlationAUC criterionStochastic gradient boosting regression |
| Le Chatelier et al. (2013) (12) | Microbiota richness, composition, and function (metagenomics, microarray) | Prediction | DBA score Internal cross-validation + independent test cohort AUC criterion |
| Bennet et al. (2018) (55) | Microbiota compostion (GA-map Dysbiosis Test) | Prediction | OPLS-DA Internal leave-one-out cross-validation |
| Kolho et al. (2015) (69) | Microbiome composition (microarray and qPCR) | Prediction | Linear mixed effect models PCoA (Bray–Curtis) Internal cross-validation AIC AUC criterion |
| Cho et al. (2017) (62) | Microbiota composition (16S) | Association | ANOVA ANOSIM Unpaired t-test PCoA (Unweighted UniFrac) |
| Suez et al. (2014) (78) | Microbiota composition (16S) | Association | ANOVA Unpaired t-test G-test PCoA (Weighted UniFrac) Pearson and Spearman correlation Mann–Whitney U test |
| Kang et al. (2016) (58) | Microbiota composition (16S) and function (PICRUSt) | Association | ANOVA ANCOVA PCoA (JSD; Euclidean; Bray–Curtis; UniFrac, weighted and unweighted) Clustering (Calinski-Harabasz pseduo F-statistic; Rousseeuw's Silhouette internal cluster quality index) |
| Possemiers et al. (2007) (59) | Microbiota composition (qPCR) | Association | Spearman correlation Kruskal–Wallis |
| Hullar et al. (2015) (13) | Microbiome composition (16S) | Association | Linear regression Multivariate regression ANOVA PCoA (UniFrac, weighted and unweighted) PAM clustering |
| Romo-Vaquero et al. (2019) (60) | Microbiome composition (16S) | Association | Shapiro–Wilk test Wilcoxon signed-rank test Bonferroni t-test Kruskal–Wallis Dunn's test PCA HCA Multinomial logit model Spearman's rank correlation |
| Li et al. (2011) (61) | Microbiome composition (T-RFLP 16S, qPCR) | Association | Student's t-test Fisher's exact test NMS MRPP MRBP Cluster analysis Linear regression |
| Zmora et al. (2018) (63) | Microbiota composition and function (16S, qPCR, metagenomics, RNA-Seq) of stool as well as lumen and mucosa samples | Association | Kruskal–Wallis Dunn's test PCA PCoA (weighted and unweighted UniFrac) Bray–Curtis dissimilarity Spearman's rank correlation 2-way and 1-way ANOVA Dunnett's test Sidak test Mann–Whitney PERMANOVA |
| Zhang et al. (2016) (64) | Microbiota composition (qRT-PCR, 16S) | Association | LEfSe, Kruskal–Wallis LMM PLSDA PCA, PCoA (Weighted UniFrac) CAP PERMANOVA ROC analysis |
| Senan et al. (2015) (65) | Microbiota composition (16S, metagenomics) and Lactobacilli abundance (plating, qPCR) | Association | Paired t-tests |
| Veiga et al. (2010) (66) | Microbiota composition (16S, qPCR, RT-qPCR, metagenomics) | Association | Mann–Whitney U test 1-Way ANOVA Wilcoxon signed-rank test Hierarchical cluster analysis |
| Volokh et al. (2019) (68) | Microbiota composition (16S) | Association | Mann-Whitney testk-means clustering (average silhouette width to determine number of clusters)MaAsLinBenjamini-Hochberg |
| Mobini et al. (2017) (67) | Microbiota composition (16S) | Association | Repeated-measures ANCOVA Wilcoxon signed-rank test Mann–Whitney test and Manny Whitney U-test Kruskal–Wallis Spearman's correlation |
| Chumpitazi et al. (2015) (56) | Microbiota composition (16S) and function (PICRUSt) | Association | LEfSe PCoA (UniFrac, weighted and unweighted) |
| Spencer et al. (2011) (53) | Microbiota composition and function (metagenomics) | Association | paired t-test Welch's t test PCA Linear model |
1AdaBoost, adaptive boosting; ADD, average Danish diet; AIC, Akaike information criterion; ANOSIM, analysis of similarities; BIC, Bayesian information criterion; Bif, Bifidobacteria; BN, Bayesian network; CAP, canonical analysis of principal coordinates; CASINO, community and systems-level interactive optimization; DBA, decisive bacterial abundance; DGGE, denaturing gradient gel electrophoresis; FISH, fluorescence in situ hybridization; HCA, hierarchical clustering analysis; HITChip, human intestinal tract chip; iAUC, incremental AUC; JSD, Jensen-Shannon divergence; LASSO, least absolute shrinkage and selection operator; LDA, linear discriminant analysis; LEfSe, linear discriminant analysis effect size; LMM, linear mixed models; MG-RAST, metagenomic rapid annotations using subsystems technology; MRBP, blocked multi-response permutation procedure; MRPP, multi-response permutation procedure; NMS, nonmetric multidimensional scaling ordination; NND, new Nordic diet; OPLS-DA, orthogonal projections to latent structures discriminant analysis; P/B ratio, Prevotella to Bacteroides ratio; PAM, partitioning around medoids; PCA, principal components analysis; PCoA, principal coordinates analysis; PDP, partial dependence plot; PERMANOVA, permutational analysis of variance; PICRUSt, phylogenetic investigation of communities by reconstruction of unobserved states; PLSDA, partial least squares discriminant analysis; RDA, redundancy analysis; ROC, receiver operating characteristic; T-RFLP, terminal restriction fragment length polymorphism.
In early studies of the gut microbiome, culturing was the primary method used to investigate the growth and activity of microbes. However, the majority of gut microbes have not been cultured successfully (74) and this technique is labor intensive. Thus, advances in sequencing technology have led to rapid adoption of culture-independent techniques.
Most of these techniques are based on analysis of the 16S ribosomal RNA (rRNA) gene, which provides a phylogenetic marker for different bacterial taxa. The 16S rRNA gene is ubiquitous among bacterial species with extreme sequence conservation, which can be targeted for amplification, as well as variable domains that can be used to classify taxa (79). Some methods also utilize the recombinant protein A (recA) gene, which has been suggested as a potential marker to identify higher taxonomic ranks because of its ubiquity and house-keeping function in bacteria (80). Protocols such as multilocus sequence typing (MLST) (81) may also rely on multiple house-keeping genes. This technique, however, requires either a completely sequenced bacterial genome or one that contains all the loci necessary for MLST (typically 7 loci) (81) and, due to the variability of house-keeping genes, does not provide sufficient discrimination except between closely related bacteria (82).
Before any of these techniques are implemented, samples are first subjected to PCR, amplifying the genetic material in the sample. Sequence identification of PCR amplicons of the 16S rRNA gene can only be used to provide relative abundance of the gut microbial taxa. In contrast, qPCR, or real-time PCR, measures amplification of DNA in real-time using fluorescence of hybridized probes specific for individual taxa, allowing for more accurate quantification (74).
Once amplified, DNA can be used for a variety of methods to compare and identify samples. Denaturing gradient gel electrophoresis and terminal restriction fragment length polymorphism (T-RFLP) are fingerprinting techniques that separate mixtures of 16S rRNA gene amplicons into bands of various sizes based on enzymatic cut sites. However, although these methods can be useful for checking the stability of dominant members and clustering communities according to these dominant members, they do not provide direct phylogenetic identification and it is difficult to relate banding patterns to changes in particular species (83, 84). When coupled with 16S rRNA sequencing, these methods can provide more specific composition information. DNA microarrays, which utilize oligonucleotide probes immobilized onto a glass slide to hybridize to complementary nucleotide sequences, provide phylogenetic identification. However, this method is subject to potential cross-hybridization (hybridization of multiple probes to single targets), can have difficulty detecting low abundance taxa, and is limited to identifying species that will hybridize to the probes provided on the slide (74).
Sequencing has become the most widely adopted method for taxonomic identification. Direct sequencing of the 16S rRNA gene provides only taxonomic (i.e. composition) information. Alternatively, metagenomic sequencing provides information regarding all genes present in the sample (i.e. composition and function). Although advances have led to a substantial reduction in the cost of direct sequencing (85), metagenomic technologies and other “omics” techniques, such as metatranscriptomics, that require much greater sequencing depth (86, 87) remain costly. A challenge for both methods is that they are computationally intense and require expertise to analyze the data that are generated. Additionally, the bioinformatics techniques used for analysis can have a significant effect on the results and interpretation of raw data (75).
The appropriate methods to use depend on the resources available as well as the features of the microbiome being investigated. The aspects of the microbiota (e.g. composition, function, relative compared with absolute abundance, etc.) that are most informative depend on the dietary intervention and response variables being investigated.
Normalization and transformation methods for gut microbiome data
Microbiota sequence count data often require normalization and/or rarefaction to avoid biases due to uneven sequencing depth (88) and to allow the comparison of data from different samples (89). The most common normalization technique, total sum scaling (TSS), divides the number of reads assigned to a certain taxa by the total number of reads in the sample (90). This method, although straightforward, has been shown to bias estimates in some data sets due to differential amplification efficiency of certain taxa (88). Cumulative-sum scaling (CSS) divides raw counts by the cumulative sum of counts up to a percentile determined by the nature of the data set (88). Data can also be rarefied, rather than scaled. Rarefying draws randomly without replacement from each sample such that all samples then have the same total count and excludes samples with total counts below that defined threshold (89). Rarefaction curves can provide guidance to determine proper rarefaction depth for a specific data set but, depending on how much data is removed, rarefying can reduce statistical power (89) and has been denounced by some (91). The choice of normalization method depends on data characteristics such as library size (89).
Another issue is the distribution of the data. If studies plan to utilize parametric statistical tests, such as a t-test or ANOVA, data must sometimes be transformed to make the distribution more Gaussian. Data can be transformed using an arcsine square root transformation (92), which is the transformation used by MaAsLin (multivariate association with linear models) (93), a bioinformatics tool used by researchers to discover associations between clinical variables and the microbiome. For a more comprehensive review of such tools, please refer to Mallick et al. (94). Log transformation is another commonly used method, but as it cannot be applied to zeros, this method also requires that zeros be replaced with small values (i.e. pseudocounts). The choice of pseudocount can influence results and there is currently no consensus on how to decide this value (89).
Levels of microbiome classification
There are many different levels at which the microbiome may be analyzed, from broad classifications such as enterotypes to targeted species identification. This complexity leads to many different ways in which responsive groups may be differentiated.
Arumugam et al. (95) first introduced the concept of “enterotypes”, clustering individuals into 3 groups according to their dominant genera: Bacteroides, Prevotella, and Ruminococcus. Although enterotypes have since been shown to exist more on a continuous scale (96), the Bacteroides and Prevotella enterotypes have been consistently replicated across cohorts (97). The identification of the Ruminococcus enterotype is more dependent on clustering and modeling approaches (97). Studies, such as Hjorth et al. (18, 45), Roager et al. (19), Kovatcheva-Datchary et al. (21), Gu et al. (72), and Chen et al. (9), highlight measures such as enterotype or the Prevotella to Bacteroides ratio. Diversity and richness are also common measures used to highlight differences between groups (12, 13, 21–24, 39, 40, 42, 44, 48, 62, 64, 65).
In addition to these broad groupings, individuals can also be classified according to the presence or absence of specific taxa, taxonomic abundance, richness and diversity, functional genes, or other features. For example, the abundance of individual taxa, such as Bifidobacterium, has been shown to be a significant indicator of response to fiber-based interventions (27–29, 32–35) and combinations of taxa, Moryella, Acetanaerobacterium, Fastidiosipila, and Streptobacillus, were found to be associated with a higher production of the phytoestrogen metabolite enterolactone (13). Studies such as Zeevi et al. (54), Kovatcheva-Datchary et al. (21), Sonnenburg et al. (10), and Chumpitazi et al. (56) also highlight differences in the abundance of functional genes, attempting to get closer to the activity of bacteria. These levels of microbiome classification may be combined in order to give a comprehensive picture of the interactions that contribute to response to diet.
Association compared with prediction studies
When a study moves towards the analysis phase of bringing together microbiome, diet, and metabolic response, there are 2 steps in the process towards development of precision nutrition recommendations: 1) identifying associations between microbiome features and dietary responsiveness and 2) predicting and validating individuals’ response to dietary interventions and/or advice (Figure 4). Some studies carry out only the first step (“Association” studies, Table 3), identifying baseline differences in the microbiota that differentiate groups of individuals who respond differently to dietary interventions. Other studies go further to complete the second step (“Prediction” studies, Table 3), using an individual' pretreatment microbiome (sometimes in addition to other baseline characteristics) to devise models to predict how that individual will respond to a dietary intervention, or to design dietary advice suitable for the individual.
FIGURE 4.

Steps of precision nutrition research. The 2 steps of personalized nutrition research involve 1) identifying associations between baseline features of the individual and their response to diet and 2) testing these associations in a population to determine if predicted responses are correct and/or if personalized recommendations based on these predictions result in better health outcomes.
Association and prediction studies share the above methods of conducting the study and preparing the gut microbiome data. Where they differ, is in the statistical analysis of these data and the use of models to predict response and validate associations found in the first step of precision nutrition-microbiome research. It is important to note that these goals (association and prediction) are not mutually exclusive and several methods may be utilized within the same study to address different scientific goals and analyze different parts of the data set. The complexity of the task of analyzing the microbiome and its various effects means that this is often the case. Each of these methods has its own advantages and limitations, which should be recognized when presenting or comparing the results of studies.
Association analysis methods
Association methods define and compare groups, find correlations, and determine significant differences (Table 4). These methods differ in the number of groups being compared, the nature of the data being compared (i.e. categorical or continuous), and assumptions regarding the distribution of the data (i.e. normal, nonnormal). The methods shown below provide a rough introduction to statistical approaches that are currently in use for these types of analyses. Detailed information on commonly used methods can be found in Van Belle et al. (98). Additional references are given as needed.
TABLE 4.
Statistical methods1
| Dependent variable | ||
|---|---|---|
| Independent variable | Continuous | Categorical |
| Categorical = 2 | t-test (paired/unpaired)2 Mann–Whitney U (Wilcoxon rank-sum) Wilcoxon signed-rank | G-test Chi-square test Fisher's exact test |
| Categorical >2 | ANOVA2 PERMANOVA ANOSIM Kruskal–Wallis PCA/PCoA k-means clustering Calinski-Harabasz pseudo-F Rousseeuw Silhouette index Partitioning around medoids Hierarchical cluster analysis | |
| Continuous | Spearman correlation Pearson correlation Biweight midcorrelation Procrustes | |
| Continuous AND categorical | ANCOVA2 CAP2 Linear regression2 Linear mixed models (random effects regression, hierarchical models)2 Random forests2 RDA2 | Logistic regression2Generalized linear mixed models2Random forests2 |
1ANOSIM, analysis of similarities; CAP, canonical analysis of principal coordinates; PCA, principal components analysis; PCoA, principal coordinates analysis; PERMANOVA, permutational analysis of variance; RDA, redundancy analysis.
2Methods may also be used for prediction.
Comparing 2 groups in association studies
The t-test compares the mean responses of 2 groups to determine whether they are significantly different. This would be appropriate when comparing the abundance of a single taxa between 2 groups at 1 time point. The t-test may be conducted on paired data (e.g. comparing means within the same individuals at different time points as in a cross-over design) or unpaired data (e.g. comparing means between unrelated individuals as in a parallel design). This approach assumes that the data are normally distributed (parametric) or the sample size is “large.” The t-test does not account for potential confounding factors. The Mann–Whitney U (also called the Wilcoxon rank-sum) test is a nonparametric (does not assume the data are normally distributed) alternative to the t-test. When the spread and shape of the 2 distributions are the same, the Mann–Whitney U test compares the median of 2 unpaired samples (e.g. between groups). The Wilcoxon signed-rank test is used to determine if the median of the differences between paired responses is different from zero (e.g. within individuals). For example, comparing the abundance of a single taxa within 1 individual before and after a dietary intervention. When the dependent variable is categorical (e.g. abundance above or below the median), a G-test or chi-square (or Fisher's exact test) test may be used when the data are unpaired or a McNemar's test may be used if the data are paired. Both methods compare the relative frequency of taxa between 2 groups to determine whether they are significantly different.
Comparing >2 groups in association studies
When the number of comparison groups exceeds 2, methods such as ANOVA, permutational multivariate analysis of variance (PERMANOVA), analysis of similarities (ANOSIM), or Kruskal–Wallis may be used. ANOVA analyzes the differences between group means and can examine the effects of multiple (categorical) independent variables on a single (univariate) or multiple (multivariate) dependent variables (i.e. 2-way, 3-way). This is useful when comparing multiple time points or when the number of groups exceeds 2, as in Bouhnik et al. (28), which tested 7 different types of nondigestible carbohydrates to determine their effect on the gut microbiota. Kruskal–Wallis is the nonparametric equivalent of the 1-way ANOVA and has been used to develop tools such as linear discriminant analysis effect size (LEfSe) (99). PERMANOVA is a nonparametric alternative to multivariate ANOVA (MANOVA) (100, 101). ANOSIM is also used to compare multiple independent variables with a multivariate dependent variable (101). It differs from PERMANOVA in that, instead of using the raw data, ANOSIM ranks values based on their similarity/dissimilarity. This minimizes the effects of outliers in the data, making it useful when data are highly skewed. Whereas classical ANOVA and multivariate ANOVA methods use Euclidean distance, both PERMANOVA and ANOSIM may be implemented with any distance/dissimilarity metric.
Dimension reduction and clustering methods in association studies
Methods such as principal components analysis (PCA) or principal coordinates analysis (PCoA) are used as dimension reduction techniques that form new variables (the PCs) using combinations of the original variables. The hope is that a limited number of these new PCs can represent the data almost as well as the many original variables. Additionally, investigators often look for clustering of samples to identify groups. PCA and PCoA both utilize the raw data and are identical when the distance metric is Euclidean, though PCoA can be used with other distance metrics, such as UniFrac (weighted or unweighted) (102) or Bray–Curtis (103) that are used for microbiome data. Typically, observations are plotted on a scatterplot of the first 2 principal coordinates and data points are labeled to identify biologically significant groups (e.g. disease, no disease).
Clustering methods such as k-means clustering (104), partitioning around medoids (PAM) (105), Calinski–Harabasz pseudo-F-statistic (106), hierarchical cluster analysis (HCA) (107), and Rousseeuw Silhouette index (108) are used to discover groups within the data. k-means clustering attempts to identify groups within the data by minimizing the distances between points within each group (defined by the mean of points within that group), and maximizing the distances between groups. Venkataraman et al. (26) used k-means clustering to identify responsive and nonresponsive groups based on butyrate production before and during the fiber intervention. PAM is similar but, instead of taking the mean of groups, chooses one datapoint to serve as the “center” or medoid around which groups are formed. This method was used in Wu et al. (109) to identify enterotype clusters. Both k-means clustering and PAM require the input of a desired or suspected number of clusters and use Euclidian distances. In contrast, both the Calinski–Harabasz pseudo-F and Rousseeuw Silhouette indices are used to determine the optimal number of clusters in a data set. HCA starts with the correlation matrix and sequentially groups variables and clusters, progressing from smaller, less inclusive groups, to larger, more inclusive groups until one large cluster is formed. This produces a dendrogram, showing the relation among the clusters, allowing the researcher to observe the clustering structure. However, in all of these clustering methods, groups are statistically defined and may not indicate any biological significance.
Correlation of continuous variables in association studies
Variables may also be continuous values along a scale (e.g. weight, blood pressure), rather than categorical groups within a population. In these cases, various correlation methods, such as Spearman, Pearson, Biweight midcorrelation (110), Procrustes analysis (111), or Co-inertia using the RV coefficient (112) can be used to assess the association of the independent variable with a continuous outcome. An important point to remember is that correlation, as opposed to regression, makes no distinction between the 2 variables being used, meaning that it does not identify one as the independent/predictor variable and the other as the dependent/outcome variable.
Pearson (based on the raw data) and Spearman (based on ranks and, therefore, robust) correlation coefficients both evaluate the relation between 2 continuous variables. However, whereas the Pearson correlation assumes a linear relation and normally distributed variables, the Spearman correlation only assumes a monotonic relation (i.e. continually increasing or decreasing) and makes no assumption about distributions. Thus, the Spearman correlation may be preferred for highly skewed outcomes. This may be useful when considering microbiota abundance data, which is often highly skewed with an excess of zeros (113). Biweight midcorrelation also measures the correlation of pairs of univariate measurements but differs from both Pearson and Spearman correlations as it is based on the median of the data, rather than the mean, making it less sensitive to outliers (114).
Procrustes analysis (111) and Co-inertia using the RV coefficient (115) extend this idea to pairs of multivariate measurements. Procrustes analysis, originally developed for comparing shapes, computes the distances between pairs of high-dimensional samples after centering and scaling. Co-intertia analysis is a global measure of covariation between pairs of multivariate measurements. Co-inertia is high when the 2 sets of observations vary simultaneously (or inversely), and low when they vary independently. These methods are useful when comparing the similarity, or dissimilarity, of whole microbial communities.
Combining continuous and categorical variables in association studies
Some methods may be used when the independent/explanatory variables are a mix of both continuous and categorical variables. These include ANCOVA, regression, linear mixed models (an extension of regression to dependent observations), canonical analysis of principal coordinates (CAP) (116), redundancy analysis (RDA) (117), and random forests (118). ANCOVA compares the means of a continuous dependent variable across levels of a categorical independent variable (i.e. experimental groups) whilst also controlling for the effects of other continuous variables (i.e. covariates such as BMI, cholesterol, etc.). This could be used to compare weight loss between groups on different diets, whilst controlling for baseline measures such as BMI or habitual fiber intake.
Regression refers to the process of modeling the relation between a dependent variable and one or more independent variables. Indeed, ANOVA and ANCOVA may be viewed as special cases of regression. Regression models can take several forms depending on the nature of the data. For example, linear regression models a relation between a continuous dependent variable that changes at a constant rate with change in the independent variable. Generalized linear models extend this idea to other types of dependent variables such as binary (logistic regression) or count (log regression) variables. Logistic regression models the relation between a binary dependent variable and one or more independent variables by estimating the probability of obtaining the outcome of interest. This can be used to determine the association between responder status and the abundance of one or more bacterial taxa.
Generalized linear mixed models (119) are an extension of generalized linear models and may include both fixed and random effects. Random effects represent factors with levels that are considered to be randomly sampled from some larger population. For example, individuals in a study are randomly sampled from a larger population. Random effects cannot be controlled experimentally. This is beneficial when analyzing longitudinal or repeated measures (i.e. cross-over studies) or multivariate outcomes (i.e. studies with multiple endpoints). In the first case, each individual contributes multiple measurements on the same outcome. In the second case, each individual contributes measurements on more than one outcome. In either case, the results are not independent of one another.
CAP utilizes a distance matrix and PCoA to determine significant differences in principal components between groups (i.e. categorical) as well as along a scale (i.e. continuous). This can be used to quantify the distance between the microbiota composition of different groups or individuals over time, as in Zhang et al. (64). RDA extends multiple linear regression by summarizing the linear relations between multiple dependent variables and multiple independent variables in a matrix, which is then incorporated into PCA. RDA could be used to examine the effect of different components of a dietary intervention on multiple taxa to identify microbes that are influenced by the diet, as done in Chen et al. (9). Random forests is a machine-learning technique that uses multiple decision trees, each built on random samples of the data, to estimate the mean value of the dependent variable. Each node in the decision tree represents a condition or question about the sample or datapoint (e.g. did butyrate increase or decrease in this sample?). The branches extending from these nodes represent the relevant data pertaining to the sample (e.g. yes/no, degree of change, etc.), eventually leading to a conclusion regarding an outcome of interest (e.g. responder/nonresponder). This method may be used for classification (i.e. categorical outcomes) or regression (i.e. continuous outcomes). For instance, Venkataraman et al. (26) used random forest regression to identify associations between operational taxonomic unit abundances and butyrate concentration, both before and during fiber supplementation. Classification could be used to identify taxa associated with either an increase or decrease in butyrate concentration (categorical).
Correction for multiple hypothesis testing
When analyzing the gut microbiota, a highly complex biological system, it is often necessary to correct for multiple comparisons. This is done to avoid false positives when conducting many comparisons simultaneously. Some common methods used to do this are the Bonferroni correction (120) and the Benjamini–Hochberg procedure (121). The Bonferroni correction takes the typical error rate (usually 0.05) and divides it by the number of tests to find the critical value (α). The Benjamini–Hochberg procedure instead controls the false discovery rate by ranking the raw P values (i.e. 1, 2, …n), dividing the rank by the number of tests, and multiplying this value by the false discovery rate (Q), which is set by the researcher. This produces a set of adjusted P values, which are then compared with the raw P values. The largest raw P value that is less than the adjusted P value is set as the threshold of significance so all P values less than or equal to this are significant.
Prediction analysis methods
In principle, models used for prediction require accuracy, not biological plausibility. In practice, many models used for prediction are developed by taking population-level associations found using the approaches discussed above and using these to classify and predict response. There are 2 general steps in this process: 1) model fitting and 2) validation (Table 5).
TABLE 5.
Steps of predicting response1
| Model-fitting phase | Explanation |
|---|---|
| Linear and linear mixed effect models | Estimates continuous response variable as a function of one or more predictor variables by fitting a best line Linear models are “fixed-effects-only” whereas mixed effect models add one or more random effects |
| Logistic regression | Estimates categorical values as a function of a set of independent variables by fitting to a logit function and predicting probability |
| Random forests | Builds deep decision trees in parallel, randomly sampling from the data set for each tree (bagging) dividing population into groups |
| Stochastic gradient boosting regression | Sequentially builds shallow decision trees to reduce residual, randomly sampling from the data set for each tree (bagging) dividing population into groups |
| Bayesian network analysis | Builds model representing the probabilistic relations/dependences between variables |
| k-means clustering | Observations are clustered according to the nearest mean value, minimizing sum of squares distances within clusters (i.e. variance), and maximizing distance between clusters (i.e. separation) |
| OPLS-DA | Determines best predictor variables for groups defined by the user (contrast to PCA which determines best discriminating variables for unknown groups) |
| Decisive bacterial abundance score | Sums the abundances of taxa more frequent in one group (e.g. HGC) and subtracts the sum of the abundances of taxa more frequent in the other group (e.g. LGC) |
| Validation phase | |
| Internal cross-validation | Model validated on the same cohort used to construct the model. Leave-one-out method uses all data minus one subject repeatedly to construct the best-fit model |
| Independent test cohort | Model validated on an independent cohort from the one used to construct the model |
| Performance criteria | |
| Akaike information criterion (AIC) | Estimates quality of model relative to other models in terms of goodness of fit as well as complexity of the model |
| Bayesian information criterion (BIC) | Related to AIC, larger penalty for increased number of parameters in model (i.e. increased complexity of model) |
| Area under ROC curve (AUC) criterion | Plots true positive rate against the false positive rate to illustrate the accuracy of a model |
| Correlation criterion | Plots observed response against predicted response to illustrate the accuracy of a model |
1DBA, decisive bacterial abundance score; HGC, high gene count; LGC, low gene count; OPLS-DA, orthogonal projections to latent structures discriminant analysis; PCA, principal components analysis; ROC, receiver operating characteristic.
Step 1: model fitting
In the model-fitting step, methods such as regression, decision trees, network analysis, and clustering can be used to incorporate information learned in the association phase to predict response in an individual. Many of the methods used in the association phase may also be used in this step (see footnote in Table 4). Here, we describe additional methods that tend to be used exclusively when the modeling goal is prediction.
Stochastic gradient boosting regression (122) uses decision trees on random samples of the data, similar to random forests. However, instead of building multiple deep trees in parallel (i.e. at the same time), it builds many shallow trees, called weak learners, sequentially (i.e. one after the other) (Figure 5). Each new tree improves upon the classification of previous trees in an additive manner to reduce “loss” or error. This method performs best when dimensionality is low (i.e. fewer input variables) whereas random forests may perform better when dimensionality is high (i.e. many input variables) (123).
FIGURE 5.

Decision trees. Various methods of prediction such as random forests and stochastic gradient boosting use decision trees. In random forests, decision trees are built simultaneously and independently of one another, using more data to build deeper trees. In contrast, stochastic gradient boosting builds smaller trees sequentially, one after the other, with each subsequent tree learning from previous ones.
Bayesian network (BN) analysis (124) is a graphical model that depicts the relations between variables and their probabilities or dependencies. For instance, a BN could depict the probability of a connection between a certain baseline symptom or state (e.g. abundance of a certain bacterial taxa), to one or many responses (e.g. SCFA production, glycemic response) (that also may be connected to one another within the network).
Linear discriminant analysis (LDA) (125, 126) is similar to PCA/PCoA. However, in LDA, the groups of interest are known a priori and the axes represent the linear combinations of the measurements that best describe the separation between the groups. This can be used to separate dietary groups based on metabolic or microbiome data. In contrast, PCA/PCoA finds the linear combinations of the measurements that maximize variance (but may or may not separate the groups a posteriori).
Partial least squares discriminant analysis (PLS-DA) and orthogonal projections to latent structures discriminant analysis (OPLS-DA) (127) are extensions of LDA. Both PLS-DA and OPLS-DA may utilize weaker sources of variation to separate groups but OPLS-DA also eliminates variation that is unrelated to the separation of groups, creating a less complex model (128). Although these are powerful methods, they may force group separation at the expense of model validity, relying on weaker sources of variability in the data set (128).
Some studies have even devised their own methods for prediction. Le Chatelier et al. (12) devised the decisive bacterial abundance (DBA) score to predict individual responses based on the abundance of taxa known to be associated with previously identified groups (i.e. high-gene-count and low-gene-count individuals, referring to bacterial richness) that have been shown to have different metabolic phenotypes.
The use of these methods, as with the methods used in the association phase, depends on the nature and complexity of the data as well as the desired format of prediction (i.e. group, continuous value, etc.).
Step 2: validation
In the validation step, the model is used to predict response in the individuals used to fit the model (internal cross-validation) or in new individuals (external validation) to evaluate model performance (129). When utilizing internal cross-validation, a common practice is “leave-one-out” in which the model is fitted in an iterative process that utilizes all data minus one to predict the response of the left-out data point. The advantage of internal cross-validation is that the data are already available and new data does not have to be collected or obtained from another study. However, this test is not as rigorous as testing in an independent cohort and the model may not perform as well when applied to new data sets. Testing a model in an independent cohort allows for a better assessment of the applicability of the model in the broad population.
Model performance criteria
To assess the accuracy of the model, several performance criteria may be used (Table 5). The Akaike information criterion (AIC) (130) and Bayesian information criterion (BIC) (131) refine the selection of multiple models and assess their performance. Both methods provide an assessment of relative performance, rather than absolute accuracy. BIC includes a larger penalty for increased complexity (i.e. numbers of parameters) in the model, typically resulting in a smaller model than AIC.
In contrast, area under the receiver operating characteristic (ROC) (132, 133) curve (for models that predict binary outcomes) and proportion of variance explained by the model (R2) (for models predicting continuous outcomes) both assess the absolute accuracy of the model but do not have any penalty for increased complexity of the model. This may lead to increasingly complex models that, although they are highly accurate on the current data set, are over-fitted and will not perform well on an independent data set.
Future directions
Use of animal and in vitro models
Mice offer a model in which differences in the gut microbiota may be studied in a controlled experimental context, allowing for the determination of causality and development of mechanistic hypotheses (134). For example, transplantation of isolated strains or communities of bacteria into germ-free mice is one of the best models in use to demonstrate causal relations between the gut microbiota and host metabolism (135). This approach has been used in studies such as Kovatcheva-Datchary et al. (21), which used stool samples from individuals who did (responders) or did not (nonresponders) exhibit improved glucose tolerance in response to barley kernel supplementation to colonize germ-free mice. Colonized mice recapitulated the response phenotype of their respective donors, suggesting that the gut microbiome played a causal role in the improvement in glucose tolerance. This study also investigated the effect of monocolonization by taxa, overrepresented in responders (Prevotella copri) and nonresponders (Bacteroides thetaiotaomicron), and found differential gene expression in the colonized mice that altered hepatic glycogen storage, allowing for a mechanistic understanding of observed differences in glucose metabolism phenotypes.
In vitro studies take place in an isolated system, allowing increased control over manipulation of conditions. This strategy has been utilized to investigate gut microbiome communities from humans (9, 59) and isolated strains (10) to determine the effect that differences in these taxa or communities have on the production of metabolites and fermentation of compounds.
Both animal and in vitro models have limitations when it comes to drawing conclusions about human health and nutrition as they lack the specific interactions present in the human supraorganism. For further information, readers are directed to additional reviews of animal (136, 137) and in vitro (138–140) models for human nutrition research. However, these techniques used in combination with human clinical trials will be crucial in the development of precision nutrition research and our understanding of the interaction between the host and the gut microbiota.
Differences in methodology
As discussed above, studies investigating the contribution of the gut microbiota to personalized health and metabolism differ in what features of the microbiome are highlighted. Two meta-analyses have attempted to integrate results from multiple studies that have investigated the effects of the gut microbiota on human health using different laboratory methods and analysis strategies (141, 142). Such work is important to improve our understanding of how the results of these studies complement each other and how the features or methods used affect the results.
In order to facilitate the comparison of results, some standardization of protocols would be helpful. Obviously, there are limits to standardization as different studies have different scientific aims, which dictate the outcomes measured and methods of microbiota analysis. The technology used for microbiota analysis is still developing but some work has been done to attempt to standardize protocols (76). However, more work is needed to develop procedures that minimize nonbiological variability due to laboratory procedures, analysis pipelines, and analytical methods. It may also be of interest to the field to determine how to combine data collected under different protocols rather than developing overarching standardized procedures.
Differences in microbiota features and responses examined
Additionally, there has been no assessment of how microbiome features that have been found useful in prediction of response to a certain dietary intervention translate to prediction of response to other nutrients or for broad dietary patterns. This is important as the taxa or features of the microbiome that may be responsible for variability in response to one nutrient may not be involved in or indicative of metabolism of other nutrients. In addition to applicability in a variety of dietary contexts, research should aim to determine which features of the microbiome are relevant in the context of different responses. This will allow researchers to devise a network of effects to better understand the ways in which the microbiota impacts our health as well as the ways in which we may manipulate the microbiota to improve our health.
Conclusions
As the focus of nutrition research shifts to a more individualized view of health and diet, appropriate methods must be developed to adequately test, detect, and validate the features that determine individual metabolic response to dietary components. As seen in this review, considerable progress has been made in recent years, but there is much that remains to be done. Current data give us only an incomplete understanding of the complexity of the human and microbial interaction. To get a full picture of the individual that captures this complexity requires researchers to learn how to design, conduct, and analyze studies that focus on the detailed characterization of the individual and their metabolic phenotype. Without the fine-scale resolution that optimization of methods and addition of mechanistic studies provide, we will be unable to interpret interindividual differences in response to diet. Additionally, without some way of comparing and combining the results of these studies, we will be unable to integrate these data and apply them. Further integration and consolidation of knowledge from the fields of microbiology, genetics, epigenetics, and nutrition will allow a more coherent picture to emerge.
Acknowledgments
The author's contributions were as follows—RLH: performed the literature review, and conceived and composed the manuscript. MLM, JPH, NLK, and MEK: provided content and formatting advice, and edited the final manuscript, and all authors: read and approved the final manuscript.
Notes
Supported by the University of California Innovation Institute for Food and Health, Agricultural Research Service CRIS Projects 2032-51530-022-00D and 2032-53000-001-00D, Arcadia Biosciences and Ardent Mills. The funding agencies did not participate in data collection, or interpretation of the data. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA. The USDA is an equal opportunity provider and employer. Figures were created using BioRender (https://biorender.com).
Author disclosures: RLH, MLM, JPH, NLK, and MEK, no conflicts of interest.
Abbrevations used: AIC, Akaike information criterion; ANOSIM, analysis of similarities; BIC, Bayesian information criterion; BN, Bayesian network; CAP, canonical analysis of principal coordinates; CSS, cumulative-sum scaling; HCA, hierarchical cluster analysis; LDA, linear discriminant analysis; MLST, multilocus sequence typing; OPLS-DA, orthogonal projections to latent structures discriminant analysis; PAM, partitioning around medoids; PCA, principal components analysis; PCoA, principal coordinates analysis; PERMANOVA, permutational multivariate analysis of variance; PLS-DA, partial least squares discriminant analysis; RDA, redundancy analysis; ROC, receiver operating characteristic; rRNA, ribosomal RNA.
References
- 1. Wang DD, Hu FB. Precision nutrition for prevention and management of type 2 diabetes. Lancet Diabetes Endocrinol. 2018;6(5):416–26. [DOI] [PubMed] [Google Scholar]
- 2. Kohlmeier M, De Caterina R, Ferguson LR, Gorman U, Allayee H, Prasad C, Kang JX, Nicoletti CF, Martinez JA. Guide and position of the International Society of Nutrigenetics/Nutrigenomics on personalized nutrition: Part 2—Ethics, challenges and endeavors of precision nutrition. J Nutrigenet Nutrigenomics. 2016;9(1):28–46. [DOI] [PubMed] [Google Scholar]
- 3. Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474(7351):327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tremaroli V, Backhed F, Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–9. [DOI] [PubMed] [Google Scholar]
- 6. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host–gut microbiota metabolic interactions. Science. 2012;336(6086):1262–7. [DOI] [PubMed] [Google Scholar]
- 7. Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13(10):701. [DOI] [PubMed] [Google Scholar]
- 8. Dinan TG, Cryan JF. The microbiome-gut-brain axis in health and disease. Gastroenterol Clin. 2017;46(1):77–89. [DOI] [PubMed] [Google Scholar]
- 9. Chen T, Long W, Zhang C, Liu S, Zhao L, Hamaker BR. Fiber-utilizing capacity varies in Prevotella- versus Bacteroides-dominated gut microbiota. Sci Rep. 2017;7(1):2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, Sonnenburg JL. Specificity of polysaccharide use in intestinal Bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141(7):1241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Possemiers S, Rabot S, Espín JC, Bruneau A, Philippe C, González-Sarrías A, Heyerick A, Tomás-Barberán FA, De Keukeleire D, Verstraete W. Eubacterium limosum activates isoxanthohumol from hops (Humulus lupulus L.) into the potent phytoestrogen 8-prenylnaringenin in vitro and in rat intestine. J Nutr. 2008;138(7):1310–6. [DOI] [PubMed] [Google Scholar]
- 12. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S et al.. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–6. [DOI] [PubMed] [Google Scholar]
- 13. Hullar MA, Lancaster SM, Li F, Tseng E, Beer K, Atkinson C, Wahala K, Copeland WK, Randolph TW, Newton KM et al.. Enterolignan-producing phenotypes are associated with increased gut microbial diversity and altered composition in premenopausal women in the United States. Cancer Epidemiol Biomarkers Prev. 2015;24(3):546–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grossman J, Mackenzie FJ. The randomized controlled trial: gold standard, or merely standard?. Perspect Biol Med. 2005;48(4):516–34. [DOI] [PubMed] [Google Scholar]
- 15. Korpela K, Flint HJ, Johnstone AM, Lappi J, Poutanen K, Dewulf E, Delzenne N, de Vos WM, Salonen A. Gut microbiota signatures predict host and microbiota responses to dietary interventions in obese individuals. PLoS One. 2014;9(6):e90702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Korem T, Zeevi D, Zmora N, Weissbrod O, Bar N, Lotan-Pompan M, Avnit-Sagi T, Kosower N, Malka G, Rein M et al.. Bread affects clinical parameters and induces gut microbiome-associated personal glycemic responses. Cell Metab. 2017;25(6):1243–53. e5. [DOI] [PubMed] [Google Scholar]
- 17. Smits SA, Marcobal A, Higginbottom S, Sonnenburg JL, Kashyap PC. Individualized responses of gut microbiota to dietary intervention modeled in humanized mice. mSystems. 2016;1(5): e00098–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hjorth MF, Roager HM, Larsen TM, Poulsen SK, Licht TR, Bahl MI, Zohar Y, Astrup A. Pre-treatment microbial Prevotella-to-Bacteroides ratio, determines body fat loss success during a 6-month randomized controlled diet intervention. Int J Obes (Lond). 2017; 42(3):580–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roager HM, Licht TR, Poulsen SK, Larsen TM, Bahl MI. Microbial enterotypes, inferred by the Prevotella-to-Bacteroides ratio, remained stable during a 6-month randomized controlled diet intervention with the new Nordic diet. Appl Environ Microbiol. 2014;80(3):1142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, Fu H, Xue X, Lu C, Ma J. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359(6380):1151–6. [DOI] [PubMed] [Google Scholar]
- 21. Kovatcheva-Datchary P, Nilsson A, Akrami R, Lee YS, De Vadder F, Arora T, Hallen A, Martens E, Bjorck I, Backhed F. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of Prevotella. Cell Metab. 2015;22(6):971–82. [DOI] [PubMed] [Google Scholar]
- 22. Salonen A, Lahti L, Salojarvi J, Holtrop G, Korpela K, Duncan SH, Date P, Farquharson F, Johnstone AM, Lobley GE et al.. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014;8(11):2218–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A et al.. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5(2):220–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tap J, Furet JP, Bensaada M, Philippe C, Roth H, Rabot S, Lakhdari O, Lombard V, Henrissat B, Corthier G et al.. Gut microbiota richness promotes its stability upon increased dietary fibre intake in healthy adults. Environ Microbiol. 2015;17(12):4954–64. [DOI] [PubMed] [Google Scholar]
- 25. Martinez I, Lattimer JM, Hubach KL, Case JA, Yang J, Weber CG, Louk JA, Rose DJ, Kyureghian G, Peterson DA et al.. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. 2013;7(2):269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Venkataraman A, Sieber JR, Schmidt AW, Waldron C, Theis KR, Schmidt TM. Variable responses of human microbiomes to dietary supplementation with resistant starch. Microbiome. 2016;4(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davis LM, Martinez I, Walter J, Goin C, Hutkins RW. Barcoded pyrosequencing reveals that consumption of galactooligosaccharides results in a highly specific bifidogenic response in humans. PLoS One. 2011;6(9):e25200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bouhnik Y, Raskine L, Simoneau G, Vicaut E, Neut C, Flourié B, Brouns F, Bornet FR. The capacity of nondigestible carbohydrates to stimulate fecal bifidobacteria in healthy humans: a double-blind, randomized, placebo-controlled, parallel-group, dose-response relation study. Am J Clin Nutr. 2004;80(6):1658–64. [DOI] [PubMed] [Google Scholar]
- 29. Tuohy KM, Kolida S, Lustenberger AM, Gibson GR. The prebiotic effects of biscuits containing partially hydrolysed guar gum and fructo-oligosaccharides—a human volunteer study. Br J Nutr. 2007;86(03):341. [DOI] [PubMed] [Google Scholar]
- 30. Eid N, Osmanova H, Natchez C, Walton G, Costabile A, Gibson G, Rowland I, Spencer JP. Impact of palm date consumption on microbiota growth and large intestinal health: a randomised, controlled, cross-over, human intervention study. Br J Nutr. 2015;114(8):1226–36. [DOI] [PubMed] [Google Scholar]
- 31. Tuohy KM, Finlay RK, Wynne AG, Gibson GR. A human volunteer study on the prebiotic effects of HP-inulin—faecal bacteria enumerated using fluorescent in situ hybridisation (FISH). Anaerobe. 2001;7(3):113–8. [Google Scholar]
- 32. Kolida S, Meyer D, Gibson GR. A double-blind placebo-controlled study to establish the bifidogenic dose of inulin in healthy humans. Eur J Clin Nutr. 2007;61(10):1189–95. [DOI] [PubMed] [Google Scholar]
- 33. de Preter V, Vanhoutte T, Huys G, Swings J, Rutgeerts P, Verbeke K. Baseline microbiota activity and initial bifidobacteria counts influence responses to prebiotic dosing in healthy subjects. Aliment Pharmacol Ther. 2008;27(6):504–13. [DOI] [PubMed] [Google Scholar]
- 34. Holscher HD, Bauer LL, Gourineni V, Pelkman CL, Fahey GC Jr., Swanson KS. Agave inulin supplementation affects the fecal microbiota of healthy adults participating in a randomized, double-blind, placebo-controlled, crossover trial. J Nutr. 2015;145(9):2025–32. [DOI] [PubMed] [Google Scholar]
- 35. Fuller Z, Louis P, Mihajlovski A, Rungapamestry V, Ratcliffe B, Duncan AJ. Influence of cabbage processing methods and prebiotic manipulation of colonic microflora on glucosinolate breakdown in man. Br J Nutr. 2007;98(2):364–72. [DOI] [PubMed] [Google Scholar]
- 36. Makki K, Deehan EC, Walter J, Bäckhed F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe. 2018;23(6):705–15. [DOI] [PubMed] [Google Scholar]
- 37. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31. [DOI] [PubMed] [Google Scholar]
- 38. Musso G, Gambino R, Cassader M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu Rev Med. 2011;62:361–80. [DOI] [PubMed] [Google Scholar]
- 39. Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, Almeida M, Quinquis B, Levenez F, Galleron N et al.. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500(7464):585–8. [DOI] [PubMed] [Google Scholar]
- 40. Shoaie S, Ghaffari P, Kovatcheva-Datchary P, Mardinoglu A, Sen P, Pujos-Guillot E, de Wouters T, Juste C, Rizkalla S, Chilloux J et al.. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015;22(2):320–31. [DOI] [PubMed] [Google Scholar]
- 41. Kong LC, Wuillemin PH, Bastard JP, Sokolovska N, Gougis S, Fellahi S, Darakhshan F, Bonnefont-Rousselot D, Bittar R, Dore J et al.. Insulin resistance and inflammation predict kinetic body weight changes in response to dietary weight loss and maintenance in overweight and obese subjects by using a Bayesian network approach. Am J Clin Nutr. 2013;98(6):1385–94. [DOI] [PubMed] [Google Scholar]
- 42. Griffin NW, Ahern PP, Cheng J, Heath AC, Ilkayeva O, Newgard CB, Fontana L, Gordon JI. Prior dietary practices and connections to a human gut microbial metacommunity alter responses to diet interventions. Cell Host Microbe. 2017;21(1):84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Piening BD, Zhou W, Contrepois K, Rost H, Gu Urban GJ, Mishra T, Hanson BM, Bautista EJ, Leopold S, Yeh CY et al.. Integrative personal omics profiles during periods of weight gain and loss. Cell Syst. 2018;6(2):157–70. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Santacruz A, Marcos A, Warnberg J, Marti A, Martin-Matillas M, Campoy C, Moreno LA, Veiga O, Redondo-Figuero C, Garagorri JM et al.. Interplay between weight loss and gut microbiota composition in overweight adolescents. Obesity (Silver Spring). 2009;17(10):1906–15. [DOI] [PubMed] [Google Scholar]
- 45. Hjorth M, Blædel T, Bendtsen L, Lorenzen JK, Holm JB, Kiilerich P, Roager HM, Kristiansen K, Larsen LH, Astrup A. Prevotella-to-Bacteroides ratio predicts body weight and fat loss success on 24-week diets varying in macronutrient composition and dietary fiber: results from a post-hoc analysis. Int J Obes. 2019;43(1):149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kreznar JH, Keller MP, Traeger LL, Rabaglia ME, Schueler KL, Stapleton DS, Zhao W, Vivas EI, Yandell BS, Broman AT et al.. Host genotype and gut microbiome modulate insulin secretion and diet-induced metabolic phenotypes. Cell Rep. 2017;18(7):1739–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ et al.. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 2013;17(1):141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dao MC, Everard A, Aron-Wisnewsky J, Sokolovska N, Prifti E, Verger EO, Kayser BD, Levenez F, Chilloux J, Hoyles L et al.. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut. 2016;65(3):426–36. [DOI] [PubMed] [Google Scholar]
- 49. Carmody RN, Gerber GK, Luevano JM Jr., Gatti DM, Somes L, Svenson KL, Turnbaugh PJ. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe. 2015;17(1):72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Muñiz Pedrogo DA, Jensen MD, Van Dyke CT, Murray JA, Woods JA, Chen J, Kashyap PC, Nehra V. Gut microbial carbohydrate metabolism hinders weight loss in overweight adults undergoing lifestyle intervention with a volumetric diet. Mayo Clin Proc. 2018;93(8):1104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zou H., Wang D., Ren H., Fang C., Shi Z., Zhang P., Chen P., Wang J., Yang H., Cai K. et al.. Nonobese subjects of Bacteroides and Prevotella enterotypes responded differentially to calorie restriction intervention, bioRxiv. 2019; 514596 [Google Scholar]
- 52. Faith JJ, McNulty NP, Rey FE, Gordon JI. Predicting a human gut microbiota's response to diet in gnotobiotic mice. Science. 2011;333(6038):101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology. 2011;140(3):976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, Ben-Yacov O, Lador D, Avnit-Sagi T, Lotan-Pompan M et al.. Personalized nutrition by prediction of glycemic responses. Cell. 2015;163(5):1079–94. [DOI] [PubMed] [Google Scholar]
- 55. Bennet SMP, Bohn L, Storsrud S, Liljebo T, Collin L, Lindfors P, Tornblom H, Ohman L, Simren M. Multivariate modelling of faecal bacterial profiles of patients with IBS predicts responsiveness to a diet low in FODMAPs. Gut. 2018;67(5):872–81. [DOI] [PubMed] [Google Scholar]
- 56. Chumpitazi BP, Cope JL, Hollister EB, Tsai CM, McMeans AR, Luna RA, Versalovic J, Shulman RJ. Randomised clinical trial: gut microbiome biomarkers are associated with clinical response to a low FODMAP diet in children with the irritable bowel syndrome. Aliment Pharmacol Ther. 2015;42(4):418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mendes-Soares H., Raveh-Sadka T., Azulay S., Edens K., Ben-Shlomo Y., Cohen Y., Ofek T., Bachrach D., Stevens J., Colibaseanu D. et al.. Assessment of a Personalized Approach to Predicting Postprandial Glycemic Responses to Food Among Individuals Without Diabetes, JAMA network open. 2019;2(2):e188102–e188102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kang C, Zhang Y, Zhu X, Liu K, Wang X, Chen M, Wang J, Chen H, Hui S, Huang L et al.. Healthy subjects differentially respond to dietary capsaicin correlating with specific gut enterotypes. J Clin Endocrinol Metab. 2016;101(12):4681–9. [DOI] [PubMed] [Google Scholar]
- 59. Possemiers S, Bolca S, Eeckhaut E, Depypere H, Verstraete W. Metabolism of isoflavones, lignans and prenylflavonoids by intestinal bacteria: producer phenotyping and relation with intestinal community. FEMS Microbiol Ecol. 2007;61(2):372–83. [DOI] [PubMed] [Google Scholar]
- 60. Romo‐Vaquero M, Cortés‐Martín A, Loria‐Kohen V, Ramírez‐de‐Molina A, García‐Mantrana I, Collado MC, Espín JC, Selma MV. Deciphering the human gut microbiome of urolithin metabotypes: association with enterotypes and potential cardiometabolic health implications. Mol Nutr Food Res. 2019;63(4):e1800958. [DOI] [PubMed] [Google Scholar]
- 61. Li F, Hullar MA, Beresford SA, Lampe JW. Variation of glucoraphanin metabolism in vivo and ex vivo by human gut bacteria. Br J Nutr. 2011;106(3):408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cho CE, Taesuwan S, Malysheva OV, Bender E, Tulchinsky NF, Yan J, Sutter JL, Caudill MA. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: a randomized controlled trial. Mol Nutr Food Res. 2017;61(1). [DOI] [PubMed] [Google Scholar]
- 63. Zmora N, Zilberman-Schapira G, Suez J, Mor U, Dori-Bachash M, Bashiardes S, Kotler E, Zur M, Regev-Lehavi D, Brik RB-Z. Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell. 2018;174(6):1388–405. e21. [DOI] [PubMed] [Google Scholar]
- 64. Zhang C, Derrien M, Levenez F, Brazeilles R, Ballal SA, Kim J, Degivry MC, Quere G, Garault P, van Hylckama Vlieg JE et al.. Ecological robustness of the gut microbiota in response to ingestion of transient food-borne microbes. ISME J. 2016;10(9):2235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Senan S, Prajapati JB, Joshi CG, Sreeja V, Gohel MK, Trivedi S, Patel RM, Pandya H, Singh US, Phatak A et al.. Geriatric respondents and non-respondents to probiotic intervention can be differentiated by inherent gut microbiome composition. Front Microbiol. 2015;6:944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Veiga P, Gallini CA, Beal C, Michaud M, Delaney ML, DuBois A, Khlebnikov A, van Hylckama Vlieg JE, Punit S, Glickman JN. Bifidobacterium animalis subsp. lactis fermented milk product reduces inflammation by altering a niche for colitogenic microbes. Proc Natl Acad Sci. 2010;107(42):18132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mobini R, Tremaroli V, Ståhlman M, Karlsson F, Levin M, Ljungberg M, Sohlin M, Bertéus Forslund H, Perkins R, Bäckhed F. Metabolic effects of Lactobacillus reuteri DSM 17938 in people with type 2 diabetes: a randomized controlled trial. Diabetes Obes Metab. 2017;19(4):579–89. [DOI] [PubMed] [Google Scholar]
- 68. Volokh O., Klimenko N., Berezhnaya Y., Tyakht A., Nesterova P., Popenko A., Alexeev D., Human gut microbiome response induced by fermented dairy product intake in healthy volunteers, Nutrients. 2019;11(3):547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kolho KL, Korpela K, Jaakkola T, Pichai MV, Zoetendal EG, Salonen A, de Vos WM. Fecal microbiota in pediatric inflammatory bowel disease and its relation to inflammation. Am J Gastroenterol. 2015;110(6):921–30. [DOI] [PubMed] [Google Scholar]
- 70. Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci. 2011;108(Suppl 1):4554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Routy B, Le Chatelier E, Derosa L, Duong CP, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–7. [DOI] [PubMed] [Google Scholar]
- 72. Gu Y, Wang X, Li J, Zhang Y, Zhong H, Liu R, Zhang D, Feng Q, Xie X, Hong J. Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment. Nat Commun. 2017;8(1):1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wong JM, De Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol. 2006;40(3):235–43. [DOI] [PubMed] [Google Scholar]
- 74. Fraher MH, O'Toole PW, Quigley EM. Techniques used to characterize the gut microbiota: a guide for the clinician. Nat Rev Gastroenterol Hepatol. 2012;9(6):312–22. [DOI] [PubMed] [Google Scholar]
- 75. Hughes R, Alkan Z, Keim N, Kable M. Impact of sequence variant detection and bacterial DNA extraction methods on the measurement of microbial community composition in human stool. bioRxiv. 2017:212134. [Google Scholar]
- 76. Costea PI, Zeller G, Sunagawa S, Pelletier E, Alberti A, Levenez F, Tramontano M, Driessen M, Hercog R, Jung F-E. Towards standards for human fecal sample processing in metagenomic studies. Nat Biotechnol. 2017;35(11):1069. [DOI] [PubMed] [Google Scholar]
- 77. Martinez I, Kim J, Duffy PR, Schlegel VL, Walter J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS One. 2010;5(11):e15046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O, Israeli D, Zmora N, Gilad S, Weinberger A et al.. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature. 2014;514(7521):181–6. [DOI] [PubMed] [Google Scholar]
- 79. Tringe SG, Hugenholtz P. A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol. 2008;11(5):442–6. [DOI] [PubMed] [Google Scholar]
- 80. Thompson C, Thompson F, Vandemeulebroecke K, Hoste B, Dawyndt P, Swings J. Use of recA as an alternative phylogenetic marker in the family Vibrionaceae. Int J Syst Evol Microbiol. 2004;54(3):919–24. [DOI] [PubMed] [Google Scholar]
- 81. Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Pontén TS, Ussery DW, Aarestrup FM. Multilocus sequence typing of total genome sequenced bacteria. J Clin Microbiol. 2012;50(4):1355–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Maiden MC, Van Rensburg MJJ, Bray JE, Earle SG, Ford SA, Jolley KA, McCarthy ND. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. 2013;11(10):728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hamady M, Knight R. Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 2009;19(7):1141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schütte UM, Abdo Z, Bent SJ, Shyu C, Williams CJ, Pierson JD, Forney LJ. Advances in the use of terminal restriction fragment length polymorphism (T-RFLP) analysis of 16S rRNA genes to characterize microbial communities. Appl Microbiol Biotechnol. 2008;80(3):365–80. [DOI] [PubMed] [Google Scholar]
- 85. Wetterstrand KA. DNA sequencing costs: Data from the NHGRI Genome Sequencing Program (GSP) 2013. [Google Scholar]
- 86. Jovel J, Patterson J, Wang W, Hotte N, O'Keefe S, Mitchel T, Perry T, Kao D, Mason AL, Madsen KL. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front Microbiol. 2016;7:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zaheer R, Noyes N, Polo RO, Cook SR, Marinier E, Van Domselaar G, Belk KE, Morley PS, McAllister TA. Impact of sequencing depth on the characterization of the microbiome and resistome. Sci Rep. 2018;8(1):5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10(12):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, Lozupone C, Zaneveld JR, Vázquez-Baeza Y, Birmingham A. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu Z, Hsiao W, Cantarel BL, Drábek EF, Fraser-Liggett C. Sparse distance-based learning for simultaneous multiclass classification and feature selection of metagenomic data. Bioinformatics. 2011;27(23):3242–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10(4):e1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sokal R, Rohlf F. Biometry. New York: WH Freeman and Company. p. 887, van Heemst HDJ (1985) The influence of weed competition on crop yield. Agric Syst. 1995;18:(2):81–93. [Google Scholar]
- 93. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mallick H, Ma S, Franzosa EA, Vatanen T, Morgan XC, Huttenhower C. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 2017;18(1):228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Knights D, Ward TL, McKinlay CE, Miller H, Gonzalez A, McDonald D, Knight R. Rethinking “enterotypes”. Cell Host Microbe. 2014;16(4):433–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Christensen L, Roager HM, Astrup A, Hjorth MF. Microbial enterotypes in personalized nutrition and obesity management. Am J Clin Nutr. 2018;108(4):645–51. [DOI] [PubMed] [Google Scholar]
- 98. Van Belle G, Fisher LD, Heagerty PJ, Lumley T. Biostatistics: a methodology for the health sciences. Vol 519, John Wiley & Sons, 2004. [Google Scholar]
- 99. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Morrison DF. Multivariate analysis of variance. In: Encyclopedia of Biostatistics. [Google Scholar]; Vol 5. New York: John Wiley & Sons; 2005. p. 1–6. [Google Scholar]
- 101. Anderson MJ, Walsh DC. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing?. Ecological Monographs. 2013;83(4):557–74. [Google Scholar]
- 102. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Beals EW. Bray–Curtis ordination: an effective strategy for analysis of multivariate ecological data, in advances in ecological research, London:Elsevier;1984;14, p. 1–55. [Google Scholar]
- 104. MacQueen J. Some methods for classification and analysis of multivariate observations. In Proceedings of the fifth Berkeley symposium on mathematical statistics and probability. Oakland, CA, USA, 1967. [Google Scholar]
- 105. Kaufman L, Rousseeuw PJ. Partitioning around medoids (program PAM). Finding groups in data: an introduction to cluster analysis, New York: John Wiley; 1990. p. 68–125. [Google Scholar]
- 106. Caliński T, Harabasz J. A dendrite method for cluster analysis. Commun Stat Theory Methods. 1974;3(1):1–27. [Google Scholar]
- 107. Bridges CC., Jr Hierarchical cluster analysis. Psychol Rep. 1966;18(3):851–4. [Google Scholar]
- 108. Rousseeuw PJ. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J Comput Appl Math. 1987;20:53–65. [Google Scholar]
- 109. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R et al.. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wilcox RR. Introduction to robust estimation and hypothesis testing, San Diego (CA)Academic Press; 2011. [Google Scholar]
- 111. Gower JC. Generalized Procrustes analysis. Psychometrika. 1975;40(1):33–51. [Google Scholar]
- 112. Robert P, Escoufier Y. A unifying tool for linear multivariate statistical methods: the RV-coefficient. Appl Stat. 1976;25(3):257–65. [Google Scholar]
- 113. B Sohn M, Li H. A GLM‐based latent variable ordination method for microbiome samples. Biometrics. 2018;74(2):448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Langfelder P, Horvath S. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw. 2012;46(11):i11. [PMC free article] [PubMed] [Google Scholar]
- 115. Dolédec S, Chessel D. Co‐inertia analysis: an alternative method for studying species–environment relationships. Freshwater Biol. 1994;31(3):277–94. [Google Scholar]
- 116. Anderson MJ, Willis TJ. Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology. 2003;84(2):511–25. [Google Scholar]
- 117. Ter Braak CJ, Prentice IC. A theory of gradient analysis, in advances in ecological research, London: Elsevier; 1988. p. 271–317. [Google Scholar]
- 118. Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2(3):18–22. [Google Scholar]
- 119. McCulloch CE, Neuhaus JM. Generalized linear mixed models. New York: Wiley;2014. [Google Scholar]
- 120. Weisstein EW. "Bonferroni correction". From MathWorld—A Wolfram Web Resource. http://mathworld.wolfram.com/BonferroniCorrection.html 2004. [Google Scholar]
- 121. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995; 57(1):289–300. [Google Scholar]
- 122. Friedman JH. Greedy function approximation: a gradient boosting machine. Ann Stat. 2001;29(5):1189–232. [Google Scholar]
- 123. Caruana R, Karampatziakis N, Yessenalina A. An empirical evaluation of supervised learning in high dimensions. In Proceedings of the 25th international conference on machine learning. Helsinki (Finland): ACM; 2008. [Google Scholar]
- 124. Pearl J. Probabilistic reasoning in intelligent systems: networks of plausible inference. San Francisco (CA): Elsevier; 2014. [Google Scholar]
- 125. Fisher RA. The use of multiple measurements in taxonomic problems. Annals of Eugenics. 1936;7(2):179–88. [Google Scholar]
- 126. Izenman AJ. Linear discriminant analysis, in modern multivariate statistical techniques. New York: Springer; 2013. p. 237–80. [Google Scholar]
- 127. Bylesjö M, Rantalainen M, Cloarec O, Nicholson JK, Holmes E, Trygg J. OPLS discriminant analysis: combining the strengths of PLS‐DA and SIMCA classification. J Chemom. 2006;20(8–10):341–51. [Google Scholar]
- 128. Worley B, Powers R. PCA as a practical indicator of OPLS-DA model reliability. Curr Metabolomics. 2016;4(2):97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Miller ME, Hui SL, Tierney WM. Validation techniques for logistic regression models. Stat Med. 1991;10(8):1213–26. [DOI] [PubMed] [Google Scholar]
- 130. Akaike H. Information theory and an extension of the maximum likelihood principle, in Selected Papers of Hirotugu Akaike. Springer; 1998. p. 199–213. [Google Scholar]
- 131. Schwarz G. Estimating the dimension of a model. Ann Stat. 1978;6(2):461–4. [Google Scholar]
- 132. Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143(1):29–36. [DOI] [PubMed] [Google Scholar]
- 133. Bradley AP. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 1997;30(7):1145–59. [Google Scholar]
- 134. Nguyen TLA, Vieira-Silva S, Liston A, Raes J. How informative is the mouse for human gut microbiota research?. Dis Model Mech. 2015;8(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lam YY, Zhang C, Zhao L. Causality in dietary interventions—building a case for gut microbiota. Genome Medicine. 2018;10(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Baker DH. Animal models in nutrition research. J Nutr. 2008;138(2):391–6. [DOI] [PubMed] [Google Scholar]
- 137. Roura E, Koopmans S-J, Lallès J-P, Le Huerou-Luron I, De Jager N, Schuurman T, Val-Laillet D. Critical review evaluating the pig as a model for human nutritional physiology. Nutr Res Rev. 2016;29(1):60–90. [DOI] [PubMed] [Google Scholar]
- 138. Williams C, Walton G, Jiang L, Plummer S, Garaiova I, Gibson GR. Comparative analysis of intestinal tract models. Ann Rev Food Sci Technol. 2015;6:329–50. [DOI] [PubMed] [Google Scholar]
- 139. Venema K, Van den Abbeele P. Experimental models of the gut microbiome. Best Pract Res Clin Gastroenterol. 2013;27(1):115–26. [DOI] [PubMed] [Google Scholar]
- 140. Mortensen A, Sorensen IK, Wilde C, Dragoni S, Mullerová D, Toussaint O, Zloch Z, Sgaragli G, Ovesná J. Biological models for phytochemical research: from cell to human organism. Br J Nutr. 2008;99(E-S1):ES118–26. [DOI] [PubMed] [Google Scholar]
- 141. Sze MA, Schloss PD. Looking for a signal in the noise: revisiting obesity and the microbiome. MBio. 2016;7(4):e01018–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun. 2017;8(1):1784. [DOI] [PMC free article] [PubMed] [Google Scholar]