

Abstract
Background
Anti-tumorigenic vs pro-tumorigenic roles of estrogen receptor-beta (ESR2) in breast cancer remain unsettled. We investigated the potential of TP53 status to be a determinant of the bi-faceted role of ESR2 and associated therapeutic implications for triple negative breast cancer (TNBC).
Methods
ESR2-TP53 interaction was analyzed with multiple assays including the in situ proximity ligation assay. Transcriptional effects on TP53-target genes and cell proliferation in response to knocking down or overexpressing ESR2 were determined. Patient survival according to ESR2 expression levels and TP53 mutation status was analyzed in the basal-like TNBC subgroup in the Molecular Taxonomy of Breast Cancer International Consortium (n = 308) and Roswell Park Comprehensive Cancer Center (n = 46) patient cohorts by univariate Cox regression and log-rank test. All statistical tests are two-sided.
Results
ESR2 interaction with wild-type and mutant TP53 caused pro-proliferative and anti-proliferative effects, respectively. Depleting ESR2 in cells expressing wild-type TP53 resulted in increased expression of TP53-target genes CDKN1A (control group mean [SD] = 1 [0.13] vs ESR2 depletion group mean [SD] = 2.08 [0.24], P = .003) and BBC3 (control group mean [SD] = 1 [0.06] vs ESR2 depleted group mean [SD] = 1.92 [0.25], P = .003); however, expression of CDKN1A (control group mean [SD] = 1 [0.21] vs ESR2 depleted group mean [SD] = 0.56 [0.12], P = .02) and BBC3 (control group mean [SD] = 1 [0.03] vs ESR2 depleted group mean [SD] = 0.55 [0.09], P = .008) was decreased in cells expressing mutant TP53. Overexpressing ESR2 had opposite effects. Tamoxifen increased ESR2-mutant TP53 interaction, leading to reactivation of TP73 and apoptosis. High levels of ESR2 expression in mutant TP53-expressing basal-like tumors is associated with better prognosis (Molecular Taxonomy of Breast Cancer International Consortium cohort: log-rank P = .001; hazard ratio = 0.26, 95% confidence interval = 0.08 to 0.84, univariate Cox P = .02).
Conclusions
TP53 status is a determinant of the functional duality of ESR2. Our study suggests that ESR2-mutant TP53 combination prognosticates survival in TNBC revealing a novel strategy to stratify TNBC for therapeutic intervention potentially by repurposing tamoxifen.
Triple negative breast cancer (TNBC), most of which is composed of a basal-like breast cancer (BC) molecular subtype, does not express estrogen receptor-alpha (ESR1), progesterone receptor, or human epidermal growth factor 2 receptor (HER2). Therefore, currently available targeted therapies for BC are not effective against these very aggressive tumors. This, coupled with the long-term ineffectiveness of cytotoxic chemotherapy, makes it urgent to discover new therapeutic targets and strategies to treat TNBC. Although ESR1 is not expressed, ESR2 is expressed in about 60%–80% of TNBC (1–3). Furthermore, unlike luminal BCs where TP53 is wild type (WT) in the majority of cases, TP53 is mutated in about 80% of TNBC (4–6). ESR1’s role as a pro-tumorigenic factor in BC is well established, and our studies have shown that ESR1 is capable of binding and functionally inactivating WT TP53 in luminal BC (7–10). ESR1-TP53 crosstalk resulting in cooperation (11) or antagonism (12) has also been reported. However, the role of ESR2 in BC has been elusive (13,14). Although an anti-proliferative role for ESR2 has been proposed primarily based on overexpression of exogenous ESR2 cDNA in cancer cell lines (15–17), including overexpression in a TNBC cell line to show antagonism toward mutant TP53 (18,19), data from other studies do not fit this paradigm. For example, treatment of ovariectomized mice with an ESR2-specific agonist resulted in increased cell proliferation to a similar extent that was observed upon treatment with 17β-estradiol (20). Further, markers of cell proliferation co-localize with ESR2 in mammary epithelial cells (21) and in TNBC (22). Both of these observations are inconsistent with an anti-proliferative role for ESR2. More recently, ESR2 has been shown to have a pro-proliferative role in BC stem cells (23). On the other hand, ESR2 is reported to alleviate the inhibitory effect of ESR1 on TP53-mediated transcriptional regulation (24). Although some retrospective studies on BC tissues showed that ESR2 is an indicator of favorable prognosis (25–31), pro-tumorigenic functions of ESR2 were observed in other studies (32–36). Only some of these divergent effects could be attributed to expression of specific isoforms and their cellular location (37). Such inconsistent observations suggest that ESR2 may have bi-faceted functions depending on the cellular context (1,13,38,39). However, the mechanisms underlying such bi-faceted functions of ESR2 remain unknown.
Somatic mutations in TP53 are very frequent and are clonally dominant compared with other genes in TNBC (5,40). In addition to losing tumor suppressor properties and exerting dominant-negative regulation over any remaining WT TP53, certain mutant TP53s are known to acquire oncogenic gain-of-function properties. Cellular functions mediated by mutant TP53 are context dependent and include increased invasiveness, angiogenesis, abnormal epigenetic regulation, enhancement of “stemness,” and therapeutic resistance (41–49). In this study, we have analyzed the interaction between ESR2 and TP53 signaling and addressed whether TP53 status could be a determinant of pro- vs anti-proliferative functions of ESR2 affecting clinical outcome in basal-like TNBC patients.
Methods
Proximity Ligation Assay (PLA)
PLA is a sensitive and specific in situ protein-protein interaction assay where signals generated by primary and secondary antibodies are amplified by rolling circle replication of circular DNA generated by antibody-liked oligonucleotides, followed by fluorescent labeling to produce punctate fluorescent dots (50). PLA was performed using the Duolink II (Olink Bioscience)/ Millipore-Sigma reagents and protocol.
Immunohistochemical Staining and Scoring of the Human BC Tissue Micro Array (TMA)
TMAs were constructed using three 1-mm tissue cores (triplicates) from tumor specimens and controls from eligible patients who had surgeries performed between 1995 and 2008 at Roswell Park Comprehensive Cancer Center (Roswell), Buffalo, NY. Appropriate Institutional Review Board approval consistent with federal, state, and local requirements was obtained for this project. The TMAs used in this study were generated from pre-existing paraffin blocks, and as per the Institutional Review Board they are “exempt” because they contain no patient identifiers. After staining with TP53 and ESR2 antibodies, TMA slides were digitally scanned using Aperio Scanscope (Aperio Technologies, Inc., Vista, CA), and nuclear ESR2 and TP53 immunohistochemistry (IHC) signals were quantitatively scored. The high or low expression classification was determined by the median of the observed patient-tumor IHC H-scores.
Analysis of Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) Data
METABRIC clinical data including survival data, mRNA expression data, TP53 mutation status, and PAM50 subtype information were previously published (4,40) and downloaded from www.cbioportal.org. Overall survival and BC-specific survival (BCSS), defined as the time of diagnosis to the time of a BC-related death, were used for the survival analyses in the basal-like subgroup (n = 308).
Statistical Analyses
All P values are interpreted as described in the 2016 ASA P Value Statement (51). Statistical significance of comparison of relative gene expression, cell growth, cell cycle, and apoptosis assays was determined by Student t test. Statistical significance of BCSS from METABRIC was tested using univariate Cox analysis (treating ESR2 log2 expression values as a continuous variable) or log-rank test (defining two or three patient groups according to low or high ESR2 expression). All statistical tests were two-sided. P values less than .05 were deemed statistically significant throughout, with no adjustment for multiplicity.
Detailed Methods are in the Supplementary Materials (available online).
Results
Direct Interaction of ESR2 With Both WT and Mutant TP53
We have demonstrated ESR2 binds directly to TP53 (52,53), and this finding was subsequently confirmed in an independent study (19). PLA (50) showed that ESR2 is in complex with WT TP53 in situ in MCF-7, ZR-75–1 (luminal BC cells), and CAL-51 (TNBC cells) (Figure 1A–C) as well as with mutant TP53 in MDA-MB-231, MDA-MB-468 (TNBC cells), and SK-BR-3 (HER2-overexpressing cells) (Figure 1D–F), and T-47D (luminal BC cells) (Supplementary Figure 1A, available online). Such interaction was considerably reduced when ESR2 was silenced (Figure 1A–F, right panels; Supplementary Figure 1, B and C, available online). In MCF-7, MDA-MB-231, and SK-BR-3 cells transfected with FLAG-ESR2, PLA signals were specifically localized to the cells expressing FLAG-ESR2 (Supplementary Figure 2A–C, available online). Co-IP assays further confirmed the specific interaction of endogenous mutant and WT TP53 with the endogenous ESR2 and the exogenously expressed FLAG-ESR2 (Figure 1G–K).
Figure 1.

Interaction of estrogen receptor-beta (ESR2) with both wild-type (WT) and mutant TP53. A–F) ESR2-TP53 interaction was analyzed by proximity ligation assay (PLA) in (A) MCF-7, (B) ZR-75-1, (C) CAL-51 (cell lines expressing endogenous WT TP53), (D) MDA-MB-231, (E) MDA-MB-468, and (F) SK-BR-3 (cells expressing endogenous mutant TP53). Cells were transfected with either control (non-specific siRNA [si-NS]) (left panels) or ESR2-specific siRNA (si-ESR2) (right panels) for 48 hours. Interaction assayed by PLA is noted in white font (inset). Scale bar for Figure 1A and D = 20 μm; scale bar for Figure 1B, C, E, and F = 10 μm. G–K) Co-immunoprecipitation (Co-IP) of endogenous ESR2 and WT TP53 was performed in G) MDA-MB-468 and in H) MCF-7, followed by immunoblotting with TP53 and ESR2 antibodies. I) Co-IP of endogenous TP53 and exogenously expressed FLAG-ESR2 was performed in MCF-7 cells, followed by immunoblotting with TP53 and FLAG antibodies. J) Co-IP of endogenous ESR2 and WT TP53 in MDA-MB-231 cells was performed, followed by immunoblotting with TP53 and ESR2 antibodies. K) Co-IP of endogenous TP53 and exogenously expressed FLAG-ESR2 in MDA-MB-231cells was performed with TP53 antibody, followed by immunoblotting with TP53 and FLAG antibodies. L) Schematic diagram of WT and truncated mutant TP53 proteins and their ability to bind ESR2 in a Glutathie S-Transferase. (GST) pull-down assay. (See also Supplementary Figure 3A–C, available online.) TA = transactivation domain; DBD = DNA binding domain; TD = tetramerization domain; REG = regulatory domain; * = point mutation; − = no binding; + = strong binding; Numbers = position of amino acid residues. M) Schematic diagram of WT and truncated mutant ESR2 proteins and their ability to bind TP53 in a GST pull-down assay. (See also Supplementary Figure 3, D and E, available online). A/B = activation function 1 (AF1); C = DNA binding domain (DBD); D = hinge region; E/F = activation function 2 (AF2)/Ligand binding domain (LBD) = glutatione S-transferase.
Reciprocal cell-free Glutathie S-Transferase-pull down assays showed that the region of ESR2 containing the C and D domains (amino acid residues: 149–248) was necessary and sufficient to bind to TP53, whereas the C-terminal regulatory domain of TP53 (amino acid residues: 361–393) was the minimal region required for interaction with ESR2 (Figure 1, L and M, and Supplementary Figure 3A–E, available online). Collectively, these protein-protein interaction data demonstrate that ESR2 and TP53 are found in one complex and are capable of binding directly to one another.
Effect of ESR2 on TP53-Target Gene Expression and Cell Proliferation in the WT TP53 Context
We hypothesized that the direct protein-protein interaction between ESR2 and WT TP53 might inhibit TP53’s function as a transcriptional regulator. First, we confirmed with quantitative real-time polymerase chain reaction and immunoblotting that ESR2 is expressed in measurable levels in multiple BC cell lines expressing either WT TP53 or mutant TP53 and can be reproducibly knocked down with multiple RNAi approaches and be overexpressed (Supplementary Figure 4A–E, available online). Upon silencing ESR2 expression in MCF-7 cells, the mRNA levels of CDKN1A (p21), BBC3 (PUMA), and PMAIP1 (NOXA) (genes activated by TP53) were induced, whereas the levels of CD44 (gene repressed by TP53) mRNA were reduced (Figure 2, A and B). Similar data were obtained in ZR-75–1, another luminal BC cell line expressing WT TP53 (Figure 2, G and H). Opposite effects on transcript levels were observed when ESR2 was overexpressed in these cells (Figure 2F, I, J). These data show that endogenous ESR2 is capable of blocking both the transcriptional activation and repression by TP53.
Figure 2.
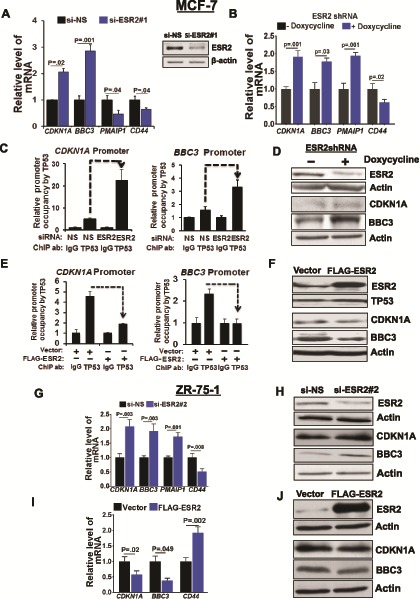
Functional effect of estrogen receptor-beta (ESR2) on wild-type TP53. A) TP53-target gene expression in MCF-7 cells with or without knocking down ESR2. Transcripts in MCF-7 cells treated with non-specific siRNA (si-NS) or ESR2 #1 siRNA for 48 hours were measured by quantitative real-time polymerase chain reaction (qRT-PCR. Inset: Reverse transcriptase (RT)-PCR analysis of ESR2 mRNA was performed to monitor ESR2 knockdown after 48 hours. B) TP53- target gene expression in MCF-7ESR2shRNA stable cells treated with or without 1 μg/mL doxycycline for 48 hours to induce ESR2 shRNA expression was determined by qRT-PCR. C) Quantitative chromatin immunoprecipitation (qChIP) for TP53 on CDKN1A and BBC3 promoters was performed in MCF-7 cells with or without ESR2 knockdown with siRNA #1 for 48 hours. D) Expression of ESR2, CDKN1A, and BBC3 proteins in MCF-7ESR2shRNA cells treated with or without 1 μg/mL doxycycline for 48 hours was analyzed by immunoblotting. E) qChIP was performed for TP53 at CDKN1A and BBC3 promoters in MCF-7 cells 48 hours post-transfection with vector or FLAG-ESR2 cDNA. F) ESR2, TP53, CDKN1A, and BBC3 protein expression in MCF-7 cells with or without FLAG-ESR2 transfection for 48 hours was analyzed by immunoblotting. G) TP53-target gene expression in ZR-75-1 cells with or without knocking down ESR2 with siRNA for 48 hours was determined by qRT-PCR. H) Expression of ESR2, CDKN1A, and BBC3 proteins in ZR-75-1 cells 48 hours post-transfection with ESR2 siRNA was analyzed by immunoblotting. I) TP53-target gene expression in ZR-75-1 cells 48 hours post-transfection with vector or FLAG-ESR2 was determined by qRT-PCR. J) Expression of ESR2, CDKN1A, and BBC3 proteins in ZR-75-1 cells 48 hours post-transfection with vector or FLAG-ESR2 was analyzed by immunoblotting. All P values were determined by two-tailed Student t test. Error bars represent SD.
Quantitative chromatin immunoprecipitation (qChIP) assays showed that knocking down of ESR2 in MCF-7 cells resulted in enhanced TP53 recruitment to CDKN1A and BBC3 promoters (Figure 2C), resulting in their increased expression (Figure 2D). On the contrary, there was three-to four-fold reduction in TP53 recruitment to these promoters following overexpression of FLAG-tagged-ESR2 or HA-tagged ESR2 (Figure 2E;Supplementary Figure 5A, available online, respectively) and concomitant reduction in CDKN1A and BBC3 protein levels (Figure 2F).
Consistent with the repressive effect of ESR2 on TP53 transcriptional targets, induction of ESR2 knockdown resulted in increased apoptosis in MCF-7 compared with control cells (Figure 3A) and levels of pro-apoptotic proteins such as BAX, uncleaved and cleaved BID, BCL2L11/BIM, and cleaved poly (ADP-ribose) polymerase (PARP1) were increased (Figure 3B). Similar results were obtained with ZR-75–1 cells (Figure 3, C and D).
Figure 3.
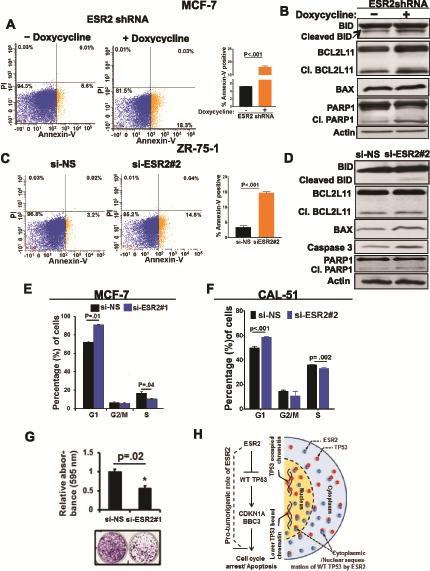
Effect of estrogen receptor-beta (ESR2) on cell proliferation in the wild-type (WT TP53 context. A) Apoptosis in MCF7shESR2 stable cells treated with or without 1 µg/mL doxycycline (to induce ESR2 shRNA) for 48 hours was analyzed by flow cytometry after double-staining with Annexin V-FITC and propidium iodide (PI). Bar graph (right panel) shows fold change of Annexin +/PI – cells. B) Expression of active apoptosis markers: cleaved BID, cleaved BCL2L11, BAX, and cleaved PARP1 proteins in MCF-7ESR2 shRNA stable cells with or without 1 μg/mL doxycycline for 48 hours to induce ESR2 shRNA expression was analyzed by immunoblotting. C) Apoptosis in ZR-75-1 cells following ESR2 #2 siRNA knockdown for 48 hours was analyzed by flow cytometry after double-staining the cells with Annexin V-FITC and PI. Bar graph (far right panel) shows fold change of Annexin +/PI – cells. D) Expression of active apoptosis markers: cleaved BID, cleaved BIM, BAX, and cleaved PARP proteins in ZR-75-1 with or without ESR2 #2 siRNA knockdown for 48 hours was analyzed by immunoblotting. E) Quantification of flow cytometry analysis of MCF-7 cells stained with PI for cell cycle distribution, with or without knockdown with ESR2 #1 siRNA for 48 hours. F) Quantification of flow cytometry analysis of CAL-51 cells stained with PI for cell cycle distribution, with or without knockdown with ESR2 #2 siRNA for 48 hours was performed. G) MCF-7 cells with or without knockdown with ESR2 #1 siRNA were subjected to clonogenic assay. Top panel: Bar graph shows quantification of average absorbance of three independent experiments. Bottom panel: Representative image of colonies stained with crystal violet are shown. H) A model for the pro-tumorigenic role of ESR2 in the WT TP53 setting is shown. Showing two prototypic TP53-targets, CDKN1A and BBC3, does not imply these are the only proteins participating in the ESR2-TP53 signaling crosstalk. All P values were determined by two-tailed Student t test. Error bars represent SD. si-NS = non-specific siRNA; siESR2 = ESR2-specific siRNA; Cl. BCL2L11 = cleaved BCL2L11; Cl. PARP1 = cleaved PARP.
Furthermore, cell cycle arrest in the G1-phase was increased, whereas the S-phase was decreased when ESR2 expression was silenced in MCF-7 and CAL-51 (Figure 3, E and F). Colony formation assays showed that cell proliferation was considerably reduced in ESR2-knocked down cells (Figure 3G). Collectively, these data show that ESR2 has a pro-proliferative effect in BC cells expressing WT TP53 (Figure 3H).
Effect of ESR2 on TP53-Target Gene Expression and Cell Proliferation in the Mutant TP53 Context
Unlike in the case of BC cells expressing WT TP53 where increased expression of TP53-target genes CDKN1A (control group mean [SD] = 1 [0.13] vs ESR2 depletion group mean [SD] = 2.08 [0.24], P = .003) and BBC3 (control group mean [SD] = 1 [0.06] vs ESR2 depleted group mean [SD] = 1.92 [0.25], P=.003) was observed (Figure 2G), depleting ESR2 in MDA-MB-231 TNBC cells expressing mutant TP53 resulted in decreased expression of TP53-target genes (Figure 4, A and B; Supplementary Figure 5C, available online). For example, expression of CDKN1A (control group mean [SD] = 1 [0.21] vs ESR2 depleted group mean [SD] = 0.56 [0.12], P = .02) and BBC3 (control group mean [SD] = 1 [0.03] vs ESR2 depleted group mean [SD] = 0.55 [0.09], P = .008) was decreased (Figure 4A). Similar data were obtained from MDA-MB-468, SK-BR-3, and T-47D cells (Figure 4E, G, H, respectively). Consistent with the effect on gene expression, there was decreased apoptosis (Figure 4, C and F) and decreased cell cycle arrest (Figure 4D) upon depletion of ESR2. Opposite results were obtained when exogenous FLAG-ESR2 was overexpressed in T-47D (Supplementary Figure 5B, available online) and MDA-MB-231 (Supplementary Figure 5, D and E, available online). Thus, contrary to the pro-proliferative effect of ESR2 in BC cells expressing WT TP53, ESR2 is anti-proliferative in BC cells expressing mutant TP53.
Figure 4.
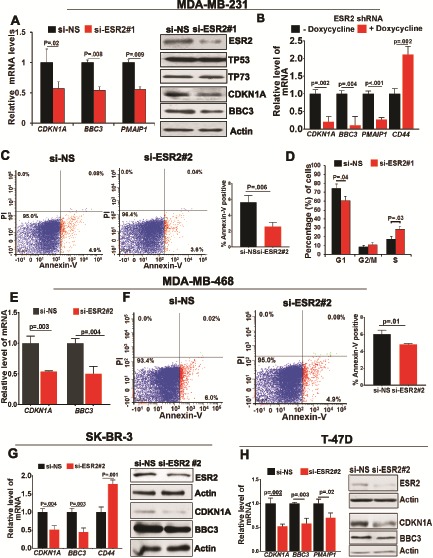
Effect of estrogen receptor-beta (ESR2) on cell proliferation in the mutant TP53 context. A) TP53 target gene expression in MDA-MB-231 cells with or without knocking down ESR2 with ESR2 siRNA#1 for 48 hours was determined by quantitative real-time polymerase chain reaction (qRT-PCR). Right panel: Expression of ESR2, TP53, TP73, CDKN1A, and BBC3 proteins in MDA-MB-231 cells post-ESR2 knockdown with siRNA #1 for 48 hours was analyzed by immunoblotting. B) TP53 target gene expression in MDA-MB-231ESR2shRNA cells treated with or without 1 μg/mL doxycycline for 48 hours to induce ESR2 shRNA expression was determined by qRT-PCR. C) MDA-MB-231 cells with or without knocking down ESR2 with siRNA #2 for 48 hours were double-stained with Annexin V-FITC and propidium iodide (PI) for flow cytometry analysis of apoptosis. Right panel: Quantification of flow cytometry analysis of MDA-MB-231 cells with or without knocking down ESR2 with siRNA for 48 hours. D) MDA-MB-231 cells with or without ESR2 knockdown with ESR2siRNA #1 for 48 hours were stained with PI for analyzing cell cycle with flow cytometry. E) TP53-target gene expression in MDA-MB-468 cells with or without ESR2 knockdown with ESR2 siRNA #2 for 48 hours was determined by qRT-PCR. F) MDA-MB-468 cells with or without knocking down ESR2 with siRNA #2 for 48 hours were double-stained with Annexin V-FITC and PI for assaying apoptosis by flow cytometry. Bar graph (far right panel) shows fold change of Annexin +/PI– cells. G) TP53-target gene expression in SK-BR-3 cells with or without ESR2 knockdown with ESR2 siRNA #2 for 48 hours was determined by qRT-PCR. Right panel: Expression of ESR2, p21, and PUMA proteins in SK-BR-3 cells with or without knocking down ESR2 with siRNA #2 for 48 hours was analyzed by immunoblotting. H) TP53-target gene expression in T-47D cells with or without ESR2 knockdown with ESR2 siRNA #2 for 48 hours was determined by qRT-PCR. Right panel: Expression of ESR2, CDKN1A, and BBC3 proteins in T-47D cells with or without knocking down ESR2 with siRNA #2 for 48 hours was analyzed by immunoblotting. All P values were determined by two-tailed Student t test. Error bars represent SD. siESR2 = ESR2-specific siRNA.
Diametrically Opposite Effects of ESR2 in TNBC Cells Expressing Mutant vs WT TP53
It could be possible that the opposite effects elicited by ESR2 in the context of WT vs mutant TP53 are due to expression of other proteins and signaling pathways that are different in these cell lines representing different subtypes of BC, rather than determined by the TP53 context per se. To rule out this possibility, we analyzed the TP53-dependent duality of ESR2 function in the following TNBC cell lines where ESR2 is the only ER expressed: MDA-MB-231 expressing mutant TP53 (R280K); isogenic MDA-MB-231-TP53 knockout with TP53 knockout (generated by CRISPR/Cas-9) (Supplementary Figure 6, A and B, available online); BC3-WT TP53 cells expressing WT TP53; isogenic BC3-shTP53 cells where TP53 was stably knocked down; and CAL-51 cells expressing endogenous WT TP53.
Unlike in the parental MDA-MB-231 cells (Figure 5A), ESR2 depletion did not have a statistically significant effect on transcription of TP53-target genes in TP53KO cells (Figure 5B). When the isogenic TP53KO cells were transfected with WT TP53 cDNA, transcription of both CDKN1A and BBC3 (Figure 5C) was increased, and when ESR2 was depleted, there was further increase in these transcripts, reminiscent of luminal BC cells expressing endogenous WT TP53. Knocking down ESR2 in either null-TP53 or WT TP53 cells did not affect TP53 protein levels (Supplementary Figure 6C, available online), ruling out the possibility that changes in TP53 levels caused by ESR2 depletion could have contributed to the ESR2-mediated effects on transcription of CDKN1A and BBC3.
Figure 5.
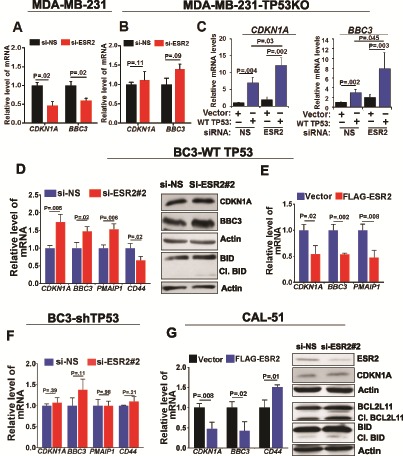
Comparison of estrogen receptor-beta (ESR2) effects in triple negative breast cancer cells expressing mutant vs wild-type (WT) TP53. A) TP53-target gene expression in MDA-MB-231 with or without knocking down ESR2 with siRNA #2 for 48 hours was determined by quantitative real-time PCR (qRT-PCR). B) TP53-target gene expression in MDA-MB-23-TP53KO with or without knocking down ESR2 with siRNA #2 for 48 hours was determined by qRT-PCR. C) TP53 target gene expression in MDA-MB-231-TP53KO cells following ESR2 knockdown along with or without transfection with WT TP53 cDNA for 48 hours was determined by qRT-PCR. D) TP53-target gene expression in BC3-WT TP53 cells with or without knocking down ESR2 with ESR2 siRNA #2 for 48 hours was determined by qRT-PCR. Right panel: Expression of CDKN1A, BBC3, and BID proteins in BC3-WT TP53 cells transfected with or without knocking down ESR2 with siRNA #2 for 48 hours was analyzed by immunoblotting. E) TP53-target gene expression in BC3-WT TP53 cells 48 hours post-transfection with or without FLAG-ESR2 was determined by qRT-PCR. F) TP53-target gene expression in BC3-shTP53 cells with and without knocking down ESR2 with siRNA #2 for 48 hours was analyzed by qRT-PCR. G) TP53-target gene expression in CAL-51 cells 48 hours post-transfection with or without FLAG-ESR2 was determined by qRT-PCR. Right panel: Expression of CDKN1A1 and BBC3 proteins in BC3-WT TP53 cells transfected with or without knocking down ESR2 with ESR2 siRNA #2 for 48 hours was analyzed by immunoblotting. All P values were determined by two-tailed Student t test. Error bars represent SD. si-NS = non-specific siRNA; siESR2 = ESR2-specific siRNA; Cl.BID = cleaved BID; Cl.BCL2L11 = cleaved BCL2L11.
Compared with MDA-MB-231, the BC3 TNBC cell line expressing endogenous WT TP53 (BC3-WT TP53) (54,55) showed complementary response to manipulating ESR2 levels (depletion vs overexpression) (Figure 5, D and E). Furthermore, similar to the isogenic MDA-MB-231-TP53KO cells, isogenic BC3-shTP53 cell line where WT TP53 was stably depleted did not show any effect on TP53-target gene expression when ESR2 was depleted (Figure 5F). Moreover, CAL-51 cells the showed similar effects as observed in BC3-WT TP53 cells (Figure 5G). These data show that ESR2 is pro-proliferative in TNBC cells expressing WT TP53 and anti-proliferative in those expressing mutant TP53. Both of these effects are TP53 dependent.
Effect of Sequestration of Mutant TP53 by ESR2 on Mutant TP53-TP73 Complex and TP73 Activation
One of the major tumorigenic gain-of-functions of mutant TP53 is its ability to bind and inactivate tumor suppressor TP73, a member of the TP53 family (Figure 6A) (49,56–62). CDKN1A, BBC3, and PMAIP1 have been reported to be transcriptional targets of TP73 (63–66). Because TP73 deregulation is a frequent event in BC (67–69) and is expressed in MDA-MB-231 cells (70), we hypothesized that ESR2 may sequester and prevent mutant TP53 from binding and inhibiting tumor suppressor TP73, leading to TP73 activation and its enhanced recruitment to TP53/TP73-target gene promoters. PLA showed interaction between endogenous TP73 and mutant TP53 in MDA-MB-231, MDA-MB-468, and SK-BR-3 cells, and the interaction was increased when ESR2 was depleted (Figure 6B, G, and I) whereas the opposite was observed in ESR2-overexpressing cells (Supplementary Figure 7, A and B, available online). Furthermore, co-immunoprecipitation assay showed that TP73 binding to mutant TP53 was increased in ESR2-depleted cells (Figure 6C), indicating sequestration of mutant TP53 by ESR2 leads to reduced TP73-mutant TP53 interaction. Interaction of endogenous ESR2 or exogenously overexpressed FALG-ESR2 with endogenous TP73 was considerably low (Supplementary Figure 8A-C, available online). The ability of ESR2 to discriminate between mutant TP53 and TP73 for binding could be a result of the lack of conservation in TP73 of the carboxy-terminal domain of TP53 (71,72) (Figure 6A) because this domain is minimally required for binding to ESR2 (Figure 1L;Supplementary Figure 3, D and E, available online). Indeed, qChIP assays showed that TP73 recruitment to CDKN1A and BBC3 promoters in MDA-MB-231 cells is decreased when ESR2 was depleted (Figure 6D). Consistent with these observations, depleting TP73 decreased transcription of TP53 target genes (Figure 6E, F, H). These data demonstrate that by sequestering mutant TP53, ESR2 derepresses TP73, leading to increased expression of its target genes that are anti-tumorigenic (Figure 6J).
Figure 6.
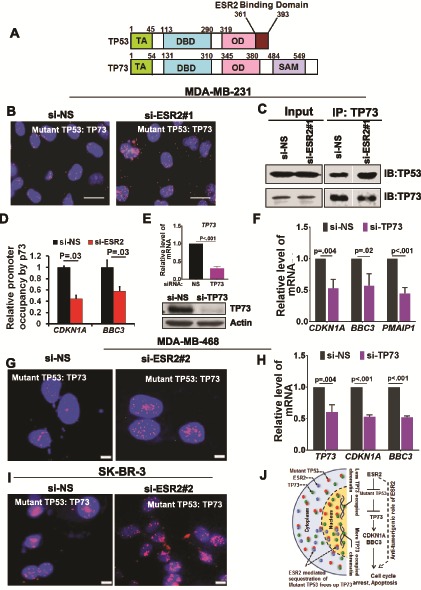
Effect of sequestration of mutant TP53 by estrogen receptor-beta (ESR2) on mutant TP53-TP73 complex and TP73 activation. A) Schematic diagram comparing domains of TP53 and TP73, showing the N-terminal transactivation domain, DNA binding domain, oligomerization domain, and sterile alpha motif domain. Proteins are not aligned proportionately to the length of each domain. Numbers correspond to the amino acid residues that mark different domains. Note that the C-terminus of TP53 domain (aa 361–393) that binds to ESR2 is not conserved in TP73. B) Proximity ligation assay (PLA) for TP53-TP73 interaction was performed in MDA-MB-231 cells with or without knocking down ESR2 with ESR2-specific siRNA si-ESR2#1 for 48 hours. Scale bar = 20 μm. C) Co-IP of TP73 and TP53 was performed with MDA-MB-231 cells following ESR2 knockdown with si-ESR2 #1 for 48 hours. D) Quantitative immunoprecipitation for TP73 on CDKN1A and BBC3 gene promoters was performed in MDA-MB-231 cells following ESR2 knockdown with si-ESR2 #1 for 48 hours. E) Endogenous TP73 mRNA in MDA-MB-231 cells transfected with or without TP73 siRNA for 48 hours was analyzed by quantitative real time polymerase chain reaction (qRT-PCR). Bottom panel: TP73 protein levels in the cell lysates were analyzed by immunoblotting. F) TP53-target gene expression in MDA-MB-231 cells transfected with or without TP73 siRNA for 48 hours was determined by qRT-PCR. G) PLA for interaction between mutant TP53 and TP73 was performed in MDA-MB-468 cells with or without knocking down ESR2 with si-ESR2 #2 for 48 hours. Scale bar = 5 μm. H) TP53-target gene expression in MDA-MB-468 cells transfected with and without TP73 siRNA for 48 hours was determined by qRT-PCR. I) PLA for interaction between mutant TP53 and TP73 was performed in SK-BR-3 cells with or without knocking down ESR2 with si-ESR2 #2 for 48 hours. Scale bar = 5 μm. J) A model for the anti-tumorigenic role of ESR2 in the mutant TP53 context is shown. Showing two prototypic TP53-targets, p21 and PUMA, does not imply these are the only proteins participating in the ESR2-TP53 signaling crosstalk. All P values were determined by two-tailed Student t test. Error bars represent SD. IB = immunoblot.
Effect of Tamoxifen on ESR2-Mutant TP53 Interaction and TP73 Activity
Tamoxifen is a competitive inhibitor of ESR1 and is widely used to treat ESR1-positive BC (73,74), whereas it is not standard-of-care for TNBC. However, there have been isolated reports on beneficial effects of tamoxifen therapy in certain cohorts of ESR1-negative BC. It has been reported that high levels of ESR2 in ESR1-negative BC (28) and TNBC (25) patient tumors were associated with good clinical outcome in response to tamoxifen therapy. In another study, expression of ESR2 along with its co-regulator was found to be predictive for benefit from tamoxifen therapy (75). Furthermore, fulvestrant was reported to synergize with tamoxifen to upregulate ESR2 in BC cells (76), and ESR2 enhanced the sensitivity of ESR1-positive BC cells to the anti-estrogenic effects of endoxifen, a metabolite of tamoxifen (77). However, the mechanistic basis for such beneficial effects of tamoxifen in TNBC remains undefined. Here, we show that, surprisingly, treatment with 4-hydroxy tamoxifen leads to increased ESR2-mutant TP53 interaction in MDA-MB-231, MDA-MB-468, and SK-BR-3 cells (Figure 7A, C, and D, respectively). Concomitantly, there was decreased mutant TP53-TP73 interaction (Figure 7B), which in turn led to enhanced recruitment of TP73 to the PUMA gene promoter (Supplementary Figure 9A, available online). ESR2 protein levels were increased after tamoxifen treatment (Figure 7, E and H). Importantly, tamoxifen failed to enhance transcription of CDKN1A and BBC3 in the absence of ESR2 or TP73 in MDA-MB-231 cells (Figure 7, F and G), MDA-MB-468 (Figure 7, I and J) and SK-BR-3 cells (Supplementary Figure 9, B and C, available online).
Figure 7.

Effect of tamoxifen on estrogen receptor-beta (ESR2)-mutant TP53 interaction and TP73 activity. A) Proximity ligation assay (PLA) for interaction between ESR2 and mutant TP53 was performed in MDA-MB-231 cells following treatment with 5 μM TAM. Scale bar = 20 μM. B) PLA for interaction between mutant TP53 and TP73 was performed in MDA-MB-231 cells following treatment with 5 μM TAM. Scale bar = 20 μM. C) PLA for interaction between ESR2 and mutant TP53 was performed in MDA-MB-468 following treatment with 5 μM TAM. Scale bar = 5 μM. D) PLA for interaction between ESR2 and mutant TP53 in SK-BR-3 cells following treatment with 5 μM TAM. Scale bar = 10 μM. E) Expression of ESR2 and TP53 in MDA-MB-231 cells following treatment with 5 μM TAM was analyzed by immunoblotting. F) TP53-target gene expression in MDA-MB-231 with or without ESR2 knockdown (with ESR2-specific siRNA [si-ESR2#1]) for 48 hours and with or without treatment with 5 μM TAM for 24 hours was determined by qRT-PCR). G) TP53 target gene expression in MDA-MB-231 with or without TP73 knockdown for 48 hours and with or without treatment with 5 μM TAM for 24 hours was determined quantitative real-time polymerase chain reaction (by qRT-PCR. H) Expression of ESR2 and TP53 proteins in MDA-MB-468 cells following treatment with 5 μM TAM was analyzed by immunoblotting. I) TP53-target gene expression in MDA-MB-468 with or without ESR2 knockdown for 48 hours and with or without treatment with 5 μM TAM for 24 hours was determined by qRT-PCR. J) TP53-target gene expression in MDA-MB-231 with or without TP73 knockdown for 48 hours and with or without treatment with 5 μM TAM for 24 hours was determined by qRT-PCR. All P values were determined by two-tailed Student t test. Error bars represent SD. si-NS = non-specific siRNA; si-TP73 = siRNA specific to TP73; VEH = vehicle; TAM = 4-OH tamoxifen.
Impact of TP53 Status on the Prognostic Role of ESR2 in Patients with Basal-like TNBC Tumors
As in the case of TNBC cell lines, TP53 and ESR2 protein expression (IHC) and their interaction (PLA) were observed in representative TNBC tumor tissues (Figure 8A). To confirm the findings from the in vitro experiments with cell lines, we analyzed data from 308 patients with basal-like tumors in the METABRIC cohort of 1904 BC patients (4,6,40). In the mutant analysis TP53 subgroup of the basal-like tumors (n = 259), low levels of ESR2 mRNA were associated with poor prognosis for BCSS (log-rank test P = .001 [Figure 8B]; univariate Cox regression hazard ratio [HR] = 0.32, 95% confidence interval (CI) = 0.08 to 1.26, P = .10 (Supplementary Table 3, available online) and overall survival (log-rank test P < .001; univariate Cox regression analysis HR = 0.26, 95% CI = 0.08 to 0.84, P = .02) [Supplementary Table 3, available online]. However, in the WT TP53 subgroup (n = 49), lower levels of ESR2 were not associated with better prognosis (Supplementary Figure 10, B and C, available online). Importantly, ESR2 expression levels per se did not appear substantially affected by TP53 status in the basal-like subgroup (Figure 8C).
Figure 8.
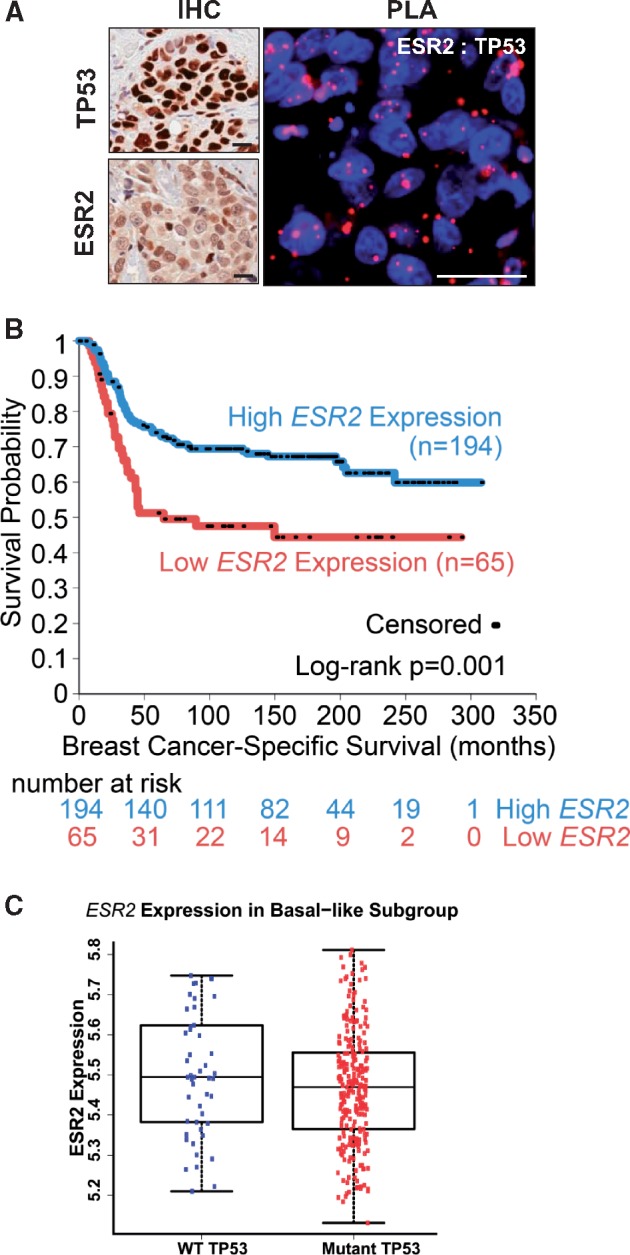
Impact of TP53 status on the prognostic role of estrogen receptor-beta (ESR2) in patients with basal-like triple negative breast cancer (TNBC) tumors. A) Images of immunohistochemistry (IHC) staining for TP53 and ESR2 (left panels) and proximity ligation assay (PLA) for ESR2-TP53 interaction (right panels) in representative TNBC patient tumors are shown. Scale bar = 20μm. B) Kaplan-Meier survival curves for breast cancer-specific survival in patients with mutant TP53, basal-like breast tumors of the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) cohort, stratified into high ESR2 expression (expression above 25th percentile), and low ESR2 expression (below 25th percentile) are shown. Number of patients at risk at each time point is listed below the x-axis. P = .001 by log-rank test, comparing bottom 25% of ESR2 expression vs other cases. C) Distribution of ESR2 expression in basal-like breast tumors of the METABRIC cohort stratified by TP53 mutation status is shown. Kruskal-Wallis test was used for the analysis. There was no considerable difference in ESR2 expression in WT TP53 vs mutant TP53 groups. Error bars represent SD.
The data from the METABRIC cohort was complemented with those from a small TNBC patient cohort (n = 46) from Roswell. The patient tumors were stratified into high and low ESR2 as well as high and low TP53 expression based on median cut-off of continuous H-scoring. Because there was not sufficient tumor tissue available for sequencing TP53, intensity of IHC staining of TP53 was used as a surrogate for determining TP53 mutation status (78–80). Tumors with mutant TP53 and higher levels of ESR2 were smaller in size and lower in stage (Supplementary Figure 10, D and E, available online). Consistent with these data, TNBC patients with tumors with WT TP53 and high ESR2 levels had worse progression-free survival and overall survival (Supplementary Figure 10, F and G, available online).
Discussion
Although bi-faceted functioning has been proposed to reconcile some of the disparate, complex, and often contradicting findings on ESR2 functions, the mechanistic basis for such duality has remained largely unknown. In this study, we discovered that TP53 status is an important determinant of pro- vs anti-proliferative activity of ESR2 in BC cells. TP53 mutations have different clinical relevance depending on the subtypes of BC (6). Clinical significance of our cellular and molecular data on TP53-dependent differential function of ESR2 is validated by our findings from the METABRIC TNBC patient cohort. In the mutant TP53-expressing basal-like tumors, those with high ESR2 levels have better survival. In the case of WT TP53-expressing tumors, those with high ESR2 levels were trending toward worse survival, although the difference was not statistically significant. Like most basal-like TNBC patient cohorts, only 16% of the METABRIC cohort was expressing WT p53, and a such low number of patients likely contributed to the statistically nonsignificant survival in this subcategory. Patient data from the Roswell cohort, although based on TP53 status determined by surrogate IHC staining, are consistent with those obtained from the much larger METABRIC cohort. These findings suggest that the ESR2-TP53 combination can be used to stratify TNBC into therapeutically actionable subgroups.
Our data show that treatment with tamoxifen can lead to sequestration of mutant TP53 away from TP73 and thereby reactivate tumor suppressor activities of TP73 provide, for the first time to our knowledge, a strong rationale for suggesting that tamoxifen therapy could be beneficial to basal-type and TNBC patients expressing mutant TP53. On the other hand, based on our data that ESR2 is pro-proliferative in the WT TP53 context, we predict that in TNBCs expressing WT TP53, tamoxifen or other agents that increase ESR2-WTP53 interaction may not only be ineffective as anti-tumor agents but also may contribute to adverse outcome.
There are limitations to our study. First, the unavailability of a sufficient number of basal-like TNBC patient cases with WT TP53 has hampered demonstration of a statistically significant difference in survival of this group based on ESR2 levels. Second, analysis of the role of ESR2 in ESR1 (ESR1)-expressing or ER+ tumors has proven to be much more complex, likely because of confounding effects of treatment with tamoxifen, which is standard-of-care for ER+ luminal BC. One might speculate that any effect of ESR2 in these tumors is potentially overcome by the strong positive response to tamoxifen therapy targeting ESR1, especially in the case of patients carrying WT TP53. These observations are consistent with our conclusion in this study that ESR2-TP53 interaction is likely to be more relevant in the case of TNBC.
In conclusion, our observations provide at least one explanation for the disparate reports on opposite functions of ESR2 in BC. These findings suggest that depending on the WT vs mutant TP53 status, downregulating or upregulating ESR2 alone as well as in combination with compounds capable of disrupting the WT TP53-ESR2 interaction or enhancing mutant TP53-ESR2 interaction could be promising therapeutic approaches. Most importantly, our findings on the novel effects of tamoxifen have important translational significance, especially because of the attractive prospect of relatively fast repurposing of tamoxifen for precision medicine to treat basal-like TNBC tumors stratified based on TP53 status.
Funding
This work was supported in part by grants to GMD from the National Cancer Institute (NCI) at the National Institutes of Health (NIH) (grant number CA079911), Susan G. Komen for the Cure Grant BCTR0600180, the Jayne & Phil Hubbell Family and Roswell Park Alliance Foundation Grant, and Breast Cancer Coalition of Rochester; the NIH Ruth L. Kirchstein National Research Service Award Institutional Research Training Grant 5323CA009072, and the Graduate Student Association at University at Buffalo (SUNY) Mark Diamond Research Fund SP-14–15 (to WMS); and the University at Buffalo (SUNY) Presidential Fellowship (to CA). The work was also supported by the NCI at the NIH (grant number P30CA016056) involving the use of Roswell Park Comprehensive Cancer Center’s Pathology Network, Biostatistics, Biomedical Data Science, Flow and Image Cytometry, and Onsite Supply Center Shared Resources. BAK was supported by funds from the Dan L Duncan Comprehensive Cancer Center, Baylor College of Medicine. CJC was supported in part by NIH grant CA125123.
Notes
Affiliations of authors: Department of Pharmacology and Therapeutics, Roswell Park Comprehensive Cancer Center, Buffalo, NY (UKM, CCO, CA, NW, SB, RM, WMS, AM, AC, GMD); Department of Bioinformatics and Biostatistics, Roswell Park Comprehensive Cancer Center, Buffalo, NY (AM); Department of Pathology, Roswell Park Comprehensive Cancer Center, Buffalo, NY (AO, WB); Department of Genetics, Institute for Cancer Research, Oslo University Hospital Radiumhospitalet, Norway (LSP, ALBD); Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX (JHP); Aurora Research Institute, Aurora Health Care, Milwaukee, WI (SDK); Department of Medicine, Baylor College of Medicine, Houston, TX (CJC)
The study sponsors had no role in the study design; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication.
The authors have no disclosures.
We thank Drs Arnie Levine, Masami Muramatsu, Carolyn Smith, and John Hawse for generously providing plasmids. We thank Drs John Ebos and Robert Kerbel for MDA-MB-231-LM-4LUC+ cells; Dr Helen Piwnica-Worms for the generous gift of BC3-WT TP53, BC3-shTP53; Dr Nitai Hait for providing MCF-10A cells; Drs Boyko Atanassov and Muthusami Thangaraju for providing SK-BR-3 and CAL-51 cells, respectively; John Hawse for ESR2 MC10 antibody; and Drs Shondra Miller and Patrick Connelly for help with CRISPR for generating isogenic cell lines. We thank Drs Dean Tang and David Goodrich for critically reading the manuscript and Nishant Gandhi for input and active discussion.
Supplementary Material
References
- 1. Leung YK, Lee MT, Lam HM, et al. Estrogen receptor-beta and breast cancer: translating biology into clinical practice. Steroids. 2012;777:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marotti JD, Collins LC, Hu R, et al. Estrogen receptor-beta expression in invasive breast cancer in relation to molecular phenotype: results from the Nurses' Health Study. Mod Pathol. 2010;232:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skliris GP, Leygue E, Watson PH, et al. Estrogen receptor alpha negative breast cancer patients: estrogen receptor beta as a therapeutic target. J Steroid Biochem Mol Biol. 2008;109(1–2):1–10. [DOI] [PubMed] [Google Scholar]
- 4. Pereira B, Chin SF, Rueda OM, et al. The somatic mutation profiles of 2, 433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;4867403:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Silwal-Pandit L, Vollan HK, Chin SF, et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin Cancer Res. 2014;2013:3569–3580. [DOI] [PubMed] [Google Scholar]
- 7. Konduri SD, Medisetty R, Liu W, et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci USA. 2010;10734:15081–15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu W, Ip MM, Podgorsak MB, et al. Disruption of estrogen receptor alpha-p53 interaction in breast tumors: a novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res Treat. 2009;1151:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu W, Konduri SD, Bansal S, et al. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J Biol Chem. 2006;28115:9837–9840. [DOI] [PubMed] [Google Scholar]
- 10. Sayeed A, Konduri SD, Liu W, et al. Estrogen receptor alpha inhibits p53-mediated transcriptional repression: implications for the regulation of apoptosis. Cancer Res. 2007;6716:7746–7755. [DOI] [PubMed] [Google Scholar]
- 11. Menendez D, Inga A, Resnick MA.. Estrogen receptor acting in cis enhances WT and mutant p53 transactivation at canonical and noncanonical p53 target sequences. Proc Natl Acad Sci USA. 2010;1074:1500–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bailey ST, Shin H, Westerling T, et al. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc Natl Acad Sci USA. 2012;10944:18060–18065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haldosen LA, Zhao C, Dahlman-Wright K.. Estrogen receptor beta in breast cancer. Mol Cell Endocrinol. 2014;3821:665–672. [DOI] [PubMed] [Google Scholar]
- 14. Hartman J, Strom A, Gustafsson JA.. Estrogen receptor beta in breast cancer--diagnostic and therapeutic implications. Steroids. 2009;748:635–641. [DOI] [PubMed] [Google Scholar]
- 15. Lazennec G, Bresson D, Lucas A, et al. ER beta inhibits proliferation and invasion of breast cancer cells. Endocrinology. 2001;1429:4120–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paruthiyil S, Parmar H, Kerekatte V, et al. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004;641:423–428. [DOI] [PubMed] [Google Scholar]
- 17. Strom A, Hartman J, Foster JS, et al. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci USA. 2004;1016:1566–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas CG, Strom A, Lindberg K, et al. Estrogen receptor beta decreases survival of p53-defective cancer cells after DNA damage by impairing G(2)/M checkpoint signaling. Breast Cancer Res Treat. 2011;1272:417–427. [DOI] [PubMed] [Google Scholar]
- 19. Bado I, Nikolos F, Rajapaksa G, et al. ERbeta decreases the invasiveness of triple-negative breast cancer cells by regulating mutant p53 oncogenic function. Oncotarget. 2016;712:13599–13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng G, Weihua Z, Warner M, et al. Estrogen receptors ER alpha and ER beta in proliferation in the rodent mammary gland. Proc Natl Acad Sci USA. 2004;10111:3739–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saji S, Jensen EV, Nilsson S, et al. Estrogen receptors alpha and beta in the rodent mammary gland. Proc Natl Acad Sci USA. 2000;971:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reese JM, Suman VJ, Subramaniam M, et al. ERbeta1: characterization, prognosis, and evaluation of treatment strategies in ERalpha-positive and -negative breast cancer. BMC Cancer. 2014;14:749. doi.org/10.1186/1471-2407-14-749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma R, Karthik GM, Lovrot J, et al. Estrogen receptor beta as a therapeutic target in breast cancer stem cells. J Natl Cancer Inst. 2017;1093:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu W, Katzenellenbogen BS.. Estrogen receptor-beta modulation of the ERalpha-p53 loop regulating gene expression, proliferation, and apoptosis in breast cancer. Horm Canc. 2017;84:230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Honma N, Horii R, Iwase T, et al. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J Clin Oncol. 2008;2622:3727–3734. [DOI] [PubMed] [Google Scholar]
- 26. Nakopoulou L, Lazaris AC, Panayotopoulou EG, et al. The favourable prognostic value of oestrogen receptor beta immunohistochemical expression in breast cancer. J Clin Pathol. 2004;575:523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saji S, Hirose M, Toi M.. Clinical significance of estrogen receptor beta in breast cancer. Cancer Chemother Pharmacol. 2005;56(suppl 1):21–26. [DOI] [PubMed] [Google Scholar]
- 28. Gruvberger-Saal SK, Bendahl PO, Saal LH, et al. Estrogen receptor beta expression is associated with tamoxifen response in ERalpha-negative breast carcinoma. Clin Cancer Res. 2007;137:1987–1994. [DOI] [PubMed] [Google Scholar]
- 29. Borgquist S, Holm C, Stendahl M, et al. Oestrogen receptors alpha and beta show different associations to clinicopathological parameters and their co-expression might predict a better response to endocrine treatment in breast cancer. J Clin Pathol. 2007;612:197–203. [DOI] [PubMed] [Google Scholar]
- 30. Esslimani-Sahla M, Simony-Lafontaine J, Kramar A, et al. Estrogen receptor beta (ER beta) level but not its ER beta cx variant helps to predict tamoxifen resistance in breast cancer. Clin Cancer Res. 2004;1017:5769–5776. [DOI] [PubMed] [Google Scholar]
- 31. Hopp TA, Weiss HL, Parra IS, et al. Low levels of estrogen receptor beta protein predict resistance to tamoxifen therapy in breast cancer. Clin Cancer Res. 2004;1022:7490–7499. [DOI] [PubMed] [Google Scholar]
- 32. Novelli F, Milella M, Melucci E, et al. A divergent role for estrogen receptor-beta in node-positive and node-negative breast cancer classified according to molecular subtypes: an observational prospective study. Breast Cancer Res. 2008;105:R74.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Skliris GP, Leygue E, Curtis-Snell L, et al. Expression of oestrogen receptor-beta in oestrogen receptor-alpha negative human breast tumours. Br J Cancer. 2006;955:616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jensen EV, Cheng G, Palmieri C, et al. Estrogen receptors and proliferation markers in primary and recurrent breast cancer. Proc Natl Acad Sci USA. 2001;9826:15197–15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O'Neill PA, Davies MP, Shaaban AM, et al. Wild-type oestrogen receptor beta (ERbeta1) mRNA and protein expression in tamoxifen-treated post-menopausal breast cancers. Br J Cancer. 2004;919:1694–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo L, Zhang YU, Yilamu D, et al. ERbeta overexpression results in endocrine therapy resistance and poor prognosis in postmenopausal ERalpha-positive breast cancer patients. Oncol Lett. 2016;112:1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Speirs V. The evolving role of oestrogen receptor beta in clinical breast cancer. Breast Cancer Res. 2008;105:111.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jonsson P, Katchy A, Williams C.. Support of a bi-faceted role of estrogen receptor beta (ERbeta) in ERalpha-positive breast cancer cells. Endocr Relat Cancer. 2014;212:143–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leygue E, Murphy LC.. A bi-faceted role of estrogen receptor beta in breast cancer. Endocr Relat Cancer. 2013;203:R127–R139. [DOI] [PubMed] [Google Scholar]
- 40. Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;4867403:346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Freed-Pastor WA, Prives C.. Mutant p53: one name, many proteins. Genes Dev. 2012;2612:1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Muller PA, Vousden KH.. p53 mutations in cancer. Nat Cell Biol. 2013;151:2–8. [DOI] [PubMed] [Google Scholar]
- 43. Zhang Y, Yan W, Chen X.. Mutant p53 disrupts MCF-10A cell polarity in three-dimensional culture via epithelial-to-mesenchymal transitions. J Biol Chem. 2011;28618:16218–16228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walerych D, Napoli M, Collavin L, et al. The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis. 2012;3311:2007–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Haupt S, Raghu D, Haupt Y.. Mutant p53 drives cancer by subverting multiple tumor suppression pathways. Front Oncol. 2016;6:12. doi.org/10.3389/fonc.2016.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kastenhuber ER, Lowe SW.. Putting p53 in context. Cell. 2017;1706:1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lehmann BD, Pietenpol JA.. Targeting mutant p53 in human tumors. J Clin Oncol. 2012;3029:3648–3650. [DOI] [PubMed] [Google Scholar]
- 48. Levine AJ, Berger SL.. The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017;3112:1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oren M, Rotter V.. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;22:a001107.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soderberg O, Gullberg M, Jarvius M, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;312:995–1000. [DOI] [PubMed] [Google Scholar]
- 51. Wasserstein RL, Lazar Na T.. ASA's statement on p-values: context, process, and purpose. Am Stat. 2016;702:129–133. [Google Scholar]
- 52. Das GM, Mukhopadhya UK, Bansal S, et al. Tumor supressor p53 status as a determinant of estrogen receptor beta signaling in breast cancer. FASEB J Abstracts. 2013;271. [Google Scholar]
- 53. Das GM, Mukhopadhya UK, Bansal S, et al. p53 status as a determinant of estrogen receptor beta function in breast cancer. Cancer Res. 2015;75(Suppl 15). [Google Scholar]
- 54. Powell E, Shao J, Yuan Y, et al. p53 deficiency linked to B cell translocation gene 2 (BTG2) loss enhances metastatic potential by promoting tumor growth in primary and metastatic sites in patient-derived xenograft (PDX) models of triple-negative breast cancer. Breast Cancer Res. 2016;181:13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ma CX, Cai S, Li S, et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest. 2012;1224:1541–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rufini A, Agostini M, Grespi F, et al. p73 in cancer. Genes Cancer. 2011;24:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Strano S, Munarriz E, Rossi M, et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem. 2000;27538:29503–29512. [DOI] [PubMed] [Google Scholar]
- 58. Bergamaschi D, Gasco M, Hiller L, et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell. 2003;34:387–402. [DOI] [PubMed] [Google Scholar]
- 59. Di Como CJ, Gaiddon C, Prives C.. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol. 1999;192:1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marin MC, Jost CA, Brooks LA, et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet. 2000;251:47–54. [DOI] [PubMed] [Google Scholar]
- 61. Stindt MH, Muller PA, Ludwig RL, et al. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene. 2015;3433:4300–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Murphy KL, Dennis AP, Rosen JM.. A gain of function p53 mutant promotes both genomic instability and cell survival in a novel p53-null mammary epithelial cell model. FASEB J. 2000;1414:2291–2302. [DOI] [PubMed] [Google Scholar]
- 63. Vayssade M, Haddada H, Faridoni-Laurens L, et al. P73 functionally replaces p53 in Adriamycin-treated, p53-deficient breast cancer cells. Int J Cancer. 2005;1166:860–869. [DOI] [PubMed] [Google Scholar]
- 64. Flinterman M, Guelen L, Ezzati-Nik S, et al. E1A activates transcription of p73 and Noxa to induce apoptosis. J Biol Chem. 2005;2807:5945–5959. [DOI] [PubMed] [Google Scholar]
- 65. Melino G, Bernassola F, Ranalli M, et al. p73 induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004;2799:8076–8083. [DOI] [PubMed] [Google Scholar]
- 66. Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;2885468:1053–1058. [DOI] [PubMed] [Google Scholar]
- 67. Ahomadegbe JC, Tourpin S, Kaghad M, et al. Loss of heterozygosity, allele silencing and decreased expression of p73 gene in breast cancers: prevalence of alterations in inflammatory breast cancers. Oncogene. 2000;1947:5413–5418. [DOI] [PubMed] [Google Scholar]
- 68. Yamamoto T, Oda K, Kubota T, et al. Expression of p73 gene, cell proliferation and apoptosis in breast cancer: immunohistochemical and clinicopathological study. Oncol Rep. 2002;94:729–735. [PubMed] [Google Scholar]
- 69. Dominguez G, Silva JM, Silva J, et al. Wild type p73 overexpression and high-grade malignancy in breast cancer. Breast Cancer Res Treat. 2001;663:183–190. [DOI] [PubMed] [Google Scholar]
- 70. Zaika AI, Kovalev S, Marchenko ND, et al. Overexpression of the wild type p73 gene in breast cancer tissues and cell lines. Cancer Res. 1999;5913:3257–3263. [PubMed] [Google Scholar]
- 71. Levrero M, De Laurenzi V, Costanzo A, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113(pt 10):1661–1670. [DOI] [PubMed] [Google Scholar]
- 72. Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;904:809–819. [DOI] [PubMed] [Google Scholar]
- 73. Ariazi EA, Ariazi JL, Cordera F, et al. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem. 2006;63:181–202. [PubMed] [Google Scholar]
- 74. Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;23:205–213. [DOI] [PubMed] [Google Scholar]
- 75. Yan Y, Li X, Blanchard A, et al. Expression of both estrogen receptor-beta 1 (ER-beta1) and its co-regulator steroid receptor RNA activator protein (SRAP) are predictive for benefit from tamoxifen therapy in patients with estrogen receptor-alpha (ER-alpha)-negative early breast cancer (EBC). Ann Oncol. 2013;248:1986–1993. [DOI] [PubMed] [Google Scholar]
- 76. Mishra AK, Abrahamsson A, Dabrosin C.. Fulvestrant inhibits growth of triple negative breast cancer and synergizes with tamoxifen in ERa positive breast cancer by up-regulation of ERbeta. Oncotarget. 2016;7(35):56876–56888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wu X, Subramaniam M, Grygo SB, et al. Estrogen receptor-beta sensitizes breast cancer cells to the anti-estrogenic actions of endoxifen. Breast Cancer Res. 2011;132:R27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yemelyanova A, Vang R, Kshirsagar M, et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol. 2011;249:1248–1253. [DOI] [PubMed] [Google Scholar]
- 79. Bartley AN, Ross DW.. Validation of p53 immunohistochemistry as a prognostic factor in breast cancer in clinical practice. Arch Pathol Lab Med. 2002;1264:456–458. [DOI] [PubMed] [Google Scholar]
- 80. Murnyak B, Hortobagyi T.. Immunohistochemical correlates of TP53 somatic mutations in cancer. Oncotarget. 2016;740:64910–64920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.