

Abstract
Knee osteoarthritis (OA) is a leading cause of chronic disability worldwide, but no diagnostic or prognostic biomarkers are available. Increasing evidence supports epigenetic dysregulation as a contributor to OA pathogenesis. In this pilot study, we investigated epigenetic patterns in peripheral blood mononuclear cells (PBMCs) as models to predict future radiographic progression in OA patients enrolled in the longitudinal Osteoarthritis Initiative (OAI) study. PBMC DNA was analyzed from baseline OAI visits in 58 future radiographic progressors (joint space narrowing at 24 months, sustained at 48 months) compared to 58 non-progressors. DNA methylation was quantified via Illumina microarrays and beta- and M-values were used to generate linear classification models. Data were randomly split into a 60% development and 40% validation subsets, models developed and tested, and cross-validated in a total of 40 cycles. M-value based models outperformed beta-value based models (ROC-AUC 0.81 ± 0.01 vs. 0.73 ± 0.02, mean ± SEM, comparison p = 0.002), with a mean classification accuracy of 73 ± 1% (mean ± SEM) for M- and 69 ± 1% for beta-based models. Adjusting for covariates did not significantly alter model performance. Our findings suggest that PBMC DNA methylation-based models may be useful as biomarkers of OA progression and warrant additional evaluation in larger patient cohorts.
Subject terms: Prognostic markers, Osteoarthritis
Introduction
Osteoarthritis (OA) is the leading cause of chronic disability in the United States, and is the third most rapidly rising chronic medical condition associated with disability worldwide1,2. Despite its importance and economic impact, there are no disease-modifying anti-osteoarthritic drugs (DMOADs) approved by the US Food and Drug Administration (FDA) or European Medicines Agency, in stark contrast to the multitude of biologic and nonbiologic disease-modifying treatments available in other forms of arthritis3. This lag in the development of DMOADs is due in no small part to a lack of easily accessible radiographic and/or biochemical biomarkers to diagnose OA and discriminate patient phenotypes, including prediction of future progressors.
Accordingly, much attention has recently been focused on the development of diagnostic and prognostic biomarkers for OA. Several groups have described biomarker candidates for OA diagnosis, discriminating OA patients from healthy controls based on peripheral blood analytes. In 2014, Ramos et al. described an mRNA-based peripheral blood signature which could discriminate OA patients from matched controls4, with a c-statistic of 0.97. In 2018, Li et al. published a reanalysis of these data using a different machine learning technique that performed at an equally high sensitivity and specificity on a reduced subset genes5. In 2018, Huang and colleagues used data and biospecimens from the DOXY (doxycycline for treatment of OA) clinical trial to examine four plasma biomarkers6, and found baseline levels of lipopolysaccharide binding protein (LBP) were associated with future radiographic progression (TIC over 18 months OR = 1.418).
A more difficult but perhaps more clinically relevant biomarker task, however, is the classification of OA patients into distinct phenotypes. The largest study yet to take on the task of OA phenotype discrimination was an analysis of serum and urine biochemical biomarkers performed by the Foundation for the National Institutes of Health (FNIH) OA Biomarkers Consortium (OABC-FNIH), using data and biospecimens from the longitudinal, US-based Osteoarthritis Initiative (OAI) study7,8. In this study, the authors produced a model combining three serum and urine biomarkers that could discriminate radiographic and pain progressors from nonprogressors with a receiver operator characteristic area under the curve (AUC-ROC, c-statistic) of 0.6319. Baseline values of these parameters did not offer substantial predictive capability; rather, models utilized a time-integrated concentration (TIC) approach tracking analyte measurements over a period of years.
A drawback of traditional biochemical biomarkers such as those used in the OABC-FNIH is their inherent variability. Epigenetic assays offer theoretical advantages as biomarkers. Most notably, an epigenetics-based assay has the potential to offer prognostic information based on a single time point reflecting early, relatively stable gene regulatory changes that precede gene transcription and subsequent protein translation. Early epigenetic changes in peripheral blood have been found to be useful biomarkers in several rheumatic diseases including rheumatoid arthritis10 and systemic lupus erythematosus11, as well as several chronic low-level inflammatory diseases, including type-2 diabetes mellitus risk12 and cardiovascular disease13. Several recent studies, including our own, have demonstrated alterations in joint tissue epigenetic patterns associated with OA development and progression14–18; however, no analyses of blood epigenetic changes have yet been published.
In this pilot study, we aimed to evaluate the potential of peripheral blood mononuclear cell (PBMC)-based epigenetic models to predict future knee OA radiographic progression in a well-matched cohort of patients from the Osteoarthritis Initiative (OAI), using similar definitions to those previously published in OABC-FNIH studies9.
Results
Baseline patient demographic, disease, and PBMC composition characteristics were well matched. Models developed using only patient characteristics were not predictive of future progression
We first identified a group of 58 OA radiographic progressors with baseline Kellergren-Lawrence (K/L) radiographic grade 2–3, symptomatic knee OA who exhibited ≥0.7 mm of joint space width (JSW) loss over the first 24 months of follow-up and persistent JSW loss at 48 months from the longitudinal, US-based Osteoarthritis Initiative (OAI) study. We then matched 58 nonprogressors with ≤0.5 mm of joint space width loss over 48 months of follow-up by age category, sex, BMI category, ethnicity, and baseline KL radiographic grade (Table 1, Supplementary Table 1). Although not included in our matching criteria, there were no statistical differences in baseline JSW, NSAID use, or smoking history between the two groups. We did note a statistically significant increase in baseline Western Ontario and McMaster (WOMAC) pain subscale among progressors (21.5 ± 2 mean ± SEM vs. 17.0 ± 1.5 points on a 0–100 point normalized scale, P = 0.05).
Table 1.
Patient group characteristics.
| Radiographic progressors (cases) (n = 58), mean ± SEM | Nonprogressors (controls) (n = 58), mean ± SEM | 2-tailed P value | |
|---|---|---|---|
| Baseline characteristics | |||
| Age | 60 ± 1 | 60 ± 1 | 0.90 |
| Sex (% female) | 53.4% | 60.3% | 0.13 |
| BMI | 30.5 ± 0.5 | 30.9 ± 0.6 | 0.61 |
| Ethnicity (% caucasian) | 88% | 88% | 1.00 |
| Smoking (% positive) | 46% | 43% | 0.71 |
| NSAID use (% positive) | 29% | 17% | 0.13 |
| Mean WOMAC pain (0–100 point normalized scale) | 21.5 ± 2 | 17.0 ± 1.5 | 0.05 |
| Mean JSW (mm) | 3.9 ± 0.1 | 4.0 ± 0.1 | 0.34 |
| Baseline K/L grade 2 | 25 | 35 | 0.09 |
| Baseline K/L grade 3 | 33 | 23 | |
| Baseline estimated PBMC composition | |||
| CD8 + T cells | 8.5 ± 0.7% | 8.6 ± 0.6% | 0.9 |
| CD4 + T cells | 21 ± 0.9% | 21 ± 1% | 0.9 |
| NK cells | 7.4 ± 0.5% | 7.6 ± 0.6% | 0.9 |
| B cells | 9.6 ± 0.5% | 9.8 ± 0.5% | 0.8 |
| Monocytes | 7.4 ± 0.3% | 6.7 ± 0.3% | 0.12 |
| Granulocytes | 51 ± 1% | 51 ± 1% | 0.9 |
| Baseline comorbidities | |||
| Type 2 diabetes | 4 (patients/58) | 6 (# patients/58) | 0.74 |
| History of heart attack | 0 | 0 | n/a |
| History of heart failure | 2 | 3 | 1.0 |
| History of stroke | 2 | 4 | 0.68 |
| History of lung disease | 0 | 0 | n/a |
| History of cancer | 1 | 4 | 0.36 |
Recent studies have reported that clinical characteristics alone can be used to model future OA progression19. To ensure that our models were not being affected by patient characteristics, we first developed models using baseline patient characteristics data alone, without including DNA methylation data. These models included age, sex, BMI, baseline JSW, baseline WOMAC pain, smoking history, and NSAID use. Baseline models were not able to discriminate the two groups (receiver operator characteristic area under the curve ROC-AUC, c-statistic = 0.49 ± 0.01 mean ± SEM, accuracy = 50 ± 0.7%) (Fig. 1). Although infrequent, we then added data regarding comorbidities to these baseline models, including history of heart attack, heart failure, diabetes, lung disease, and cancer (Table 1). This did not improve the discriminatory capability of baseline patient characteristic models (p = 0.28 for comparison with models not including comorbidity data).
Figure 1.
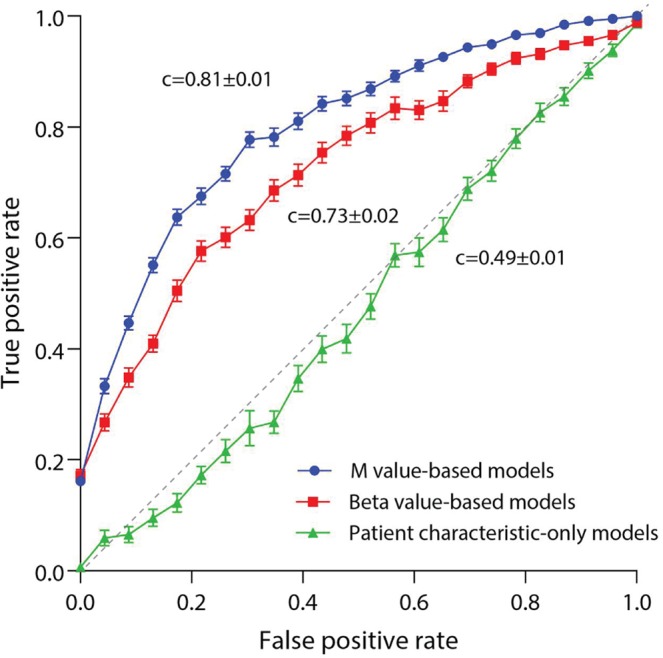
Mean receiver operator characteristic (ROC) curves for patient characteristic-only models, Beta value-based models, and M value-based models when tested on unseen (lockbox) data. Curves represent mean values over 40 cycles of development, error bars represent SEM. ROC-AUC (c-statistic) values are given as mean ± SEM.
DNA methylation data from mixed PBMC samples can be skewed by underlying differences in cellular composition among groups20. As the OAI dataset does not include data on individual blood sample cellular composition, we estimated this using a computational approach21. There were no statistically significant differences in estimated PBMC composition between the two groups; however, we did note a trend towards increased monocyte counts in cases (progressors 7.4 ± 0.3% mean ± SEM vs. nonprogressors 6.7 ± 0.3%, P = 0.12) (Table 1). Accordingly, we corrected our dataset for PBMC composition differences using frozen surrogate variable analysis (FSVA), a technique previously demonstrated to be robust in correcting cellular composition differences in genome-wide DNA methylation data22,23, before developing our epigenetic models.
Models developed based on PBMC DNA methylation data are predictive of radiographic progression
Traditionally, epigenome-wide association studies have reported DNA methylation data as beta values, defined as the fraction methylation (0–1 scale) of each CpG site included in the array. However, beta values are characterized by high heteroscedasticity (most beta values fall within extreme high- and low-percent methylation levels). Therefore, we also analyzed M values (the log2 ratio of methylated:unmethylated probe intensities for a given CpG site), which are approximately homoscedastic, in our models24. In our analysis, models based on PBMC DNA methylation data were consistently capable of discriminating those patients who would go on to experience radiographic progression from nonprogressors (Figs 1, 2, Table 2). Models based on M values outperformed those based on beta values (M value models c = 0.81 ± 0.01, mean ± SEM, vs. beta value models 0.75 ± 0.01, comparison p = 0.002), with corresponding accuracies of 73 ± 1%, mean ± SEM, for M and 69 ± 1% for beta based models. The mean number of CpGs selected for inclusion during model development and optimization was 22 ± 2 (mean ± SEM) for M-based and 19 ± 2 for beta value-based assays (Supplementary Fig. 1), this was not statistically significantly different between the two DNA methylation measures (p = 0.32).
Figure 2.

OA rapid progressor PBMC DNA methylation-based machine learning discriminant model development plan.
Table 2.
Performance of PBMC DNA methylation models to predict future radiographic progression in OA patients when evaluating previously unseen data.
| M-value based models (mean ± SEM) | Beta value-based models (mean ± SEM) | |
|---|---|---|
| ROC-AUC (c-statistic) | 0.81 ± 0.01 | 0.75 ± 0.01 |
| Accuracy | 73 ± 1% | 69 ± 1% |
| Odds ratio | 11 ± 2 | 9 ± 2 |
| Sensitivity | 74 ± 1% | 73 ± 2% |
| Specificity | 70 ± 1% | 70 ± 1% |
We then added patient characteristic covariate data to model development, including age, sex, BMI, baseline JSW, baseline WOMAC pain, smoking history, and NSAID use. Including these data did not alter model performance (M value models c = 0.80 ± 0.01, beta value models c = 0.78 ± 0.01, both nonsignificant compared to models without covariates). Finally, as our analysis included data from two methylation assay types, we then analyzed model performance for 450k and 850k chips separately; both types demonstrated equivalent performance in our models (p = 0.27).
A subset of DNA methylation CpG sites were selected frequently in multiple rounds of model development and were enriched in functional pathways previously associated with OA development
Although 969 CpGs were selected in at least one round of model development in either M or Beta based models, a subset of CpG sites were selected several times (Table 3, complete list of CpGs selected in Supplementary Table 2). Many of these most frequently selected CpGs in both M value-based and Beta value-based model development were shared; for example, 7 of the top 20 most frequently selected CpGs in both M- and Beta models were identical. The majority of the top 20 CpGs selected during modeling were associated with CpG islands (13 of 20 M- and 16 of 20 Beta-based models), including all but one of the CpG sites shared among the two methylation measures, suggesting these sites may play a role in gene regulation25.
Table 3.
Top 20 CpGs selected for supervised model development. CpGs shared by both Beta-value-based and M-value-based models are highlighted in bold (n = 7).
| M value-based algorithm feature | # of development rounds selected (out of 40) | Associated gene | CpG location (regulatory region) | Location within CpG island | Beta value-based algorithm feature | # of development rounds selected (out of 40) | Associated gene | CpG location (regulatory region) | Location within CpG island |
|---|---|---|---|---|---|---|---|---|---|
| cg15974085 | 15 | C18orf55; FBXO15 | TSS200 | Island | cg11865413 | 15 | N_Shelf | ||
| cg26384229 | 14 | ALG10B | TSS200 | Island | cg21643086 | 15 | |||
| cg21643086 | 12 | cg17745251 | 11 | C10orf140 | Body | Island | |||
| cg03687650 | 11 | OSBPL5 | Body | S_Shelf | cg06409741 | 11 | RASA3 | Body | Island |
| cg06409741 | 11 | RASA3 | Body | Island | cg03870777 | 10 | KRT18 | TSS200 | N_Shore |
| cg18111500 | 10 | cg17956079 | 10 | PLEKHB1 | TSS200 | ||||
| cg07772660 | 9 | MST1P9 | Body | Island | cg26384229 | 9 | ALG10B | TSS200 | Island |
| cg11865413 | 9 | N_Shelf | cg03687650 | 7 | OSBPL5 | Body | S_Shelf | ||
| cg03212634 | 9 | cg10306485 | 7 | RASA3 | Body | S_Shelf | |||
| cg00142933 | 8 | LIMS2 | Body | N_Shore | cg19559392 | 7 | MORN2; DHX57 | 5′UTR; TSS1500 | S_Shore |
| cg16001460 | 8 | PRIM2 | TSS1500 | cg05587853 | 7 | MSL2 | TSS1500 | Island | |
| cg17745251 | 7 | C10orf140 | Body | Island | cg14616423 | 7 | |||
| cg14330460 | 7 | cg08728848 | 6 | ||||||
| cg03870777 | 6 | KRT18 | TSS200 | N_Shore | cg07379140 | 5 | ALG14 | TSS200 | Island |
| cg02215141 | 6 | N_Shore | cg02962630 | 5 | DLL4 | Body | Island | ||
| cg20200361 | 6 | THADA | Body | cg16790849 | 5 | NEU1 | Body | N_Shore | |
| cg07258847 | 6 | cg00143249 | 5 | MNX1 | Body | Island | |||
| cg23226134 | 5 | CLCN6;MTHFR | 1st Exon | Island | cg10609068 | 4 | ANO6 PLEKHA9 | TSS1500; 5′UTR | N_Shore |
| cg16121685 | 5 | UHMK1 | TSS200 | Island | cg14710040 | 4 | EEF2 | TSS200 | Island |
| cg10966582 | 5 | MST1P9 | Body | Island | cg17310773 | 4 | SLC25A17 | 1stExon | Island |
Although we did not perform a differential methylation analysis in the traditional sense, we reasoned that CpG sites chosen for inclusion in our models might still cluster in functional pathways important in OA. To investigate this, we performed gene ontology analysis using the Ingenuity Pathway Analysis (IPA) system of genes associated with CpG sites chosen in at least one round of model development. The most significantly enriched canonical pathways enriched in this gene set included the antigen presentation pathway (n = 6 genes, p = 7E-4), adenosine monophosphate kinase (AMPK) signaling (n = 15, p = 1E-3, and sonic hedgehog signaling (n = 4 genes, p = 9E-3), among others (Table 4). The top upstream regulators identified by IPA include the transcription factor PITX2 (n = 14 genes, p = 1E-5), histone H3 (n = 29 genes, p = 7E-5) and H4 (n = 14,p = 9.6E-5), miR-141 (n = 9, p = 1.9E-4), miR-9 (n = 5, p = 3.4E-4), miR-137 (n = 2, p = 8.2E-4), and bone morphogenic protein 2 (BMP2, n = 15, p = 9.0E-4), a TGF-β superfamily member, among others (Table 4).
Table 4.
Ontology analysis of genes associated with rapid OA progressor DNA methylation models.
| Canonical pathway | p-value | Genes associated | |
|---|---|---|---|
| Antigen presentation | 7.08E-04 | CIITA,HLA-DPA1,HLA-DPB1,PSMB8,PSMB9,TAP2 | |
| AMPK signaling | 1.05E-03 | AK8,CFTR,CHRM2,CHRNA2,CHRNA9,EEF2,FOXG1,INS,PDPK1,PPM1E,PRKACB,RAB7A,RPTOR,SMARCA4,SMARCD3 | |
| Sonic hedgehog signaling | 9.33E-03 | GLIS1,GLIS2,PRKACB,SUFU | |
| Synaptogenesis Signaling Pathway | 1.58E-02 | AP2A2,CACNB4,CDH22,CDH23,CDH4,EIF4EBP3,EPHA8,EPHB3,FGR,GRIN2B,PRKACB,SHC2,STX1B,SYN2,SYT15,SYT5 | |
| Aryl Hydrocarbon Receptor Signaling | 1.95E-02 | ALDH1A3,ARNT,CDK6,CDKN2A,NCOA2, NR2F1,RXRG,SMARCA4,TP73 | |
| Endocannabinoid Neuronal Synapse Pathway | 2.75E-02 | CACNB4,CACNG3,CACNG8,CNR2,GNG7,GRIN2B,PRKACB,PTGS2 | |
| Autophagy | 2.82E-02 | ATG7,CTSO,LAMP1,SQSTM1,WIPI1 | |
| Upstream regulator | Molecule type | p-value | Number of associated genes |
| PITX2 | transcription regulator | 1.34E-05 | 14 |
| Bvht | long noncoding RNA | 5.48E-05 | 10 |
| histone H3 | group | 6.93E-05 | 29 |
| histone H4 | group | 9.63E-05 | 14 |
| miR-141 | microRNA | 1.91E-04 | 9 |
| ASCL1 | transcription regulator | 3.09E-04 | 10 |
| miR-9 | microRNA | 3.37E-04 | 5 |
| LHX6 | transcription regulator | 4.42E-04 | 5 |
| miR-137 | microRNA | 8.23E-04 | 2 |
| BMP2 | growth factor | 8.96E-04 | 15 |
Discussion
Biomarkers hold great potential for improving clinical outcomes in common chronic diseases including OA. The early identification of OA patients who are likely to progress radiographically, clinically, or both, will allow for the enrichment of clinical trials of disease-modifying antiosteoarthritic drugs (DMOADs) with appropriate patients. Biomarkers of progression may also allow clinicians to stratify early OA patients, offering personalized care by taking into account an individual’s likelihood to respond to a particular therapy. In this pilot study, we sought to test whether baseline peripheral blood mononuclear cell DNA methylation data could be used to develop models to discriminate those patients who would go on to experience radiographic progression from nonprogressors. While based on a relatively small number of patients from a single cohort, our results nonetheless suggest that peripheral blood-based epigenetic models may be useful for OA subtype discrimination and should be evaluated in future larger studies of heterogeneous OA patients from additional cohorts.
Several groups have previously investigated biomarkers predictive of future OA progression. The largest biochemical study in this regard was the OAI biomarker studies including the osteoarthritis biomarker consortium of the Foundation for the National Institutes of Health (OABC-FNIH) which investigated a targeted set of biochemical biomarkers as predictors of OA pain and radiographic progression. Their analysis identified a combination of three biochemical biomarkers for discrimination of pain and radiographic progression from nonprogressors with a c-statistic of 0.6319, although they used a time-integrated concentration (TIC) approach, requiring multiple biomarker measurements over time as input into their prognostic model. This same study also evaluated the predictive capability of baseline biomarkers alone, but these not demonstrate robust differentiation of progressors (c = 0.586)9.
Radiographic biomarkers predicting future disease progression have also been evaluated. In 2016, Collins et al. reported models based on changes in MRI semi-quantitative markers over 24 months to predict the likelihood of radiographic progression at 48 months as part of the OABC-FNIH study26; the best model achieved a c-statistic of 0.74. Like the OABC-FNIH biochemical biomarker study, this study leveraged changes in imaging parameters over time to predict future progression. Trabecular bone texture (TBT) is perhaps the best-studied single imaging biomarker for prediction of future OA progression. Kraus et al. evaluated this in FNIH patients, with a best composite model using baseline TBT to predict future radiographic progression with a c-statistic of 0.62427. This is somewhat lower than previous reports using TBT, Janvier and colleagues, for example, reported a c-statistic of AUC 0.77 in 201728.
There have also been recent reports using machine learning to model future progression based on combinations of baseline imaging and demographic data. Joseph and colleagues in 2018 published the results of three such models, with a maximum c-statistic of 0.72 to predict progression to moderate-severe OA over 8 years including demographic data, risk factors, K/L score, cartilage WORMS score, meniscal tear, and cartilage MRI T2 imaging data29 from the OAI cohort. Also in 2018, Halilaj and colleagues published the results of a mixed-effects mixture model within the OAI cohort to predict joint space narrowing in 1243 subjects. They described an impressive c-statistic of 0.86 for prediction of future progression using a combination of baseline and year 1 follow-up variables, using a 10-cycle, 90% development/10% lockbox validation data splitting approach with 10-fold internal cross-validation, not unlike our methodology. Models based solely on baseline data, however, did not predict future radiographic progression (c ≤ 0.6)30.
Several recent studies have also identified mitochondrial haploptypes as biomarkers which can discriminate OA phenotypes. For example, in 2012 Fernandez-Moreno et al. identified significant increases in cartilage-specific biomarkers of OA in patients carrying mitochondrial haplogroup H associated with the onset of OA31. Similarly, this group later identified a decrease in incident OA risk in patients with mitochondrial haplogroup J compared to hpalogroup H (HR 0.7, p < 0.05) in both the OAI and CHECK cohorts32, as well as slowing of the rates of radiographic progression rate among OA patients with haplogroup T compared to other haplogroups within the OAI cohort33. Future studies should investigate potential complementarity between mitochondrial haplogroups and DNA methylation as predictive OA biomarkers.
Much work has been done over the past several years linking epigenetic changes, particularly DNA methylation, with OA. Our group and others have noted differential DNA methylation in inflammatory cell pathways in both cartilage and subchondral bone from OA patients14–17,34,35. A subgroup of end-stage OA patients’ cartilage demonstrates a fingerprint of epigenetic changes within inflammatory genes18,36. Given chondrocyte and subchondral bone DNA methylation aberrancy in both knee and hip OA, along with the known contributions of chronic inflammation and inflammatory gene epigenetic changes to OA, the DNA methylation patterns within PBMCs associated with OA progression we describe in this report are intuitively consistent with previously published data.
Our PBMC DNA methylation-based approach has a few potential advantages over previously published biomarkers. Most notably, our approach offers predictive capability based on a single baseline blood draw. Further, our models demonstrated higher c-statistic values than previously described biochemical biomarkers, although this should be interpreted cautiously given our relatively small group sample size and lack of non-OAI cohort confirmation. We also performed a traditional DNA methylation analysis of progressors and nonprogressors and did not identify significantly differentially methylated positions (DMPs) with a false data rate-corrected p-value ≤ 0.05. This is not particularly surprising given our mixed PBMC population. Our approach utilized the combined effects of multiple CpG sites when developing DNA methylation-based predictive models.
Relatively few of the CpG sites selected most frequently during model development have been previously associated with OA. A CpG near the transcription start site of KRT18 was selected in both M- and Beta-model development. Reductions in KRT18 expression have been associated with lumbar spine OA37. LIMS2, selected in M-development, is a focal adhesion molecule differentially expressed in cartilage and subchondral bone of frizzled-related protein (Frzb) knockout mice38. A CpG site selected in M-based assay development was located near CLCN6, which has been associated with life expectancy and aging39. This same CpG site is also associated with the gene MTHFR, which is a rheumatoid arthritis (RA) risk allele40, and the protein encoded by MTHFR is the target of the RA disease-modifying antirheumatic drug methotrexate. Only one published association has been found linking MTHFR gene mutations and OA of the knee, hand, and hip in a Turkish population41. A CpG within DLL4 was selected in Beta-based models. Dll4, a component of the Notch pathway, has been previously associated with the development of OA of the temporomandibular joint42. EEF2, also selected during Beta-based model development, has been shown to play a role in intervertebral disc degeneration and is the target of the OA-associated microRNA-143-5p43. This paucity of previously published OA association in model-selected CpGs is not surprising, as our study is the first report of peripheral blood epigenetic patterns in OA.
Despite the lack of previous OA associations among the specific CpG sites included in our models, gene ontology analysis revealed several canonical gene pathways and upstream regulators that have been previously described in OA. Unfortunately, there have not been previous epigenetic analyses of peripheral blood cells in OA in humans or mice to compare our data to. However, many pathways and regulators previously described in articular tissues overlap with our findings. For example, AMP kinase (AMPK) deficiency in chondrocytes accelerates both age- and trauma-associated OA in mice44, whereas a pharmacological stimulator of AMPK attenuates post-traumatic OA in rats45. The endocannabinoid system within synovial tissue has been associated with OA pain46. Autophagy has been widely associated with OA. Age-related loss of autophagy has been linked with OA severity in human47 and murine48 joint tissue. Furthermore, cartilage-specific deletion of mammalian target of rapamycin (mTOR) increases autophagy signaling and protects against post-traumatic OA induction in mic49. Among the topstream regulators identified in our ontology analysis was PITX2, a component of the Wnt signaling pathway key to mensenchymal stem cell function which is altered in OA patients50. MicroRNA-141 is dysregulated in human OA and contributes to pathogenesis by augmentation of lipid metabolism51, and is important in maintaining the appropriate expression of key osteoblast differentiation proteins including Runx2, Sclerostin, ALP, and Dlx552. MicroRNA-9 has been shown to directly augment the expression of the key catabolic enzyme matrix metalloproteinase-13 (MMP13) in OA patients53 and animal models54. MicroRNA-137 regulates chondrocyte metabolism via targeting Runx255. Finally, bone morphogenic protein-2 (BMP2) upregulation has been linked to OA development and increased matrix turnover56. It is a key regulator of chondrocyte metabolism57 and has been suggested as a novel OA therapeutic58. Future studies of circulating inflammatory cells and cell subsets in OA patients and/or OA animal models will be critical to confirmation of our findings and will help elucidate whether common pathway deficiencies, i.e. autophagy or Wnt signaling, are defective globally in OA rather than acting in a jont-specific manner.
Our study does have several weaknesses, the two largest being our small sample size and lack of external validation in a non-OAI cohort. We chose samples carefully in order to minimize group differences and reduce the possibility of batch effects, which we further reduced via fsva correction. Our findings are preliminary and should certainly be confirmed in a larger and more heterogeneous patient cohort. Unfortunately, our precise JSW measurement requirement59 and use of baseline peripheral blood DNA limits the possibility of direct comparison with other cohorts which are not as well characterized. These factors have led several previous studies to include only samples from the OAI7,9,19,28–30,60–62. Future studies will no doubt relax our inclusion criteria and expand the potential sources for additional samples to include larger sample sizes and additional cohorts.
A major concern with machine learning is generally the problem of overfitting. We reduced this possibility through extensive cross-validation and report the accuracy of models when tested on data unseen during model development. We included in our analysis data from both Illumina 450 k and 850 k chips, as the 450 k assays became unavailable from the manufacturer during this project. All data from 850k chips were subset to only those sites included on the 450k array and were batch corrected. We saw no evidence for differences in predictive capability with the two chip types (see above). We did see differences comparing M value-based to beta value-based models; however, this is not unsurprising given previous reports of variations in statistical measures of these values24 owing to differences in scedasticity. Finally, we are unable to draw substantial conclusions about underlying pathophysiological associations of altered PBMC DNA methylation patterns with OA given our mixed PBMC samples; this would be best investigated by future studies focusing on individual immune cell subsets. There is an emerging literature in the OA field which has demonstrated the importance of activated macrophages in disease development and progression63–65. We are unable to comment specifically on whether the nearly-statistically-significant increase in monocytes detected by computational estimation of PBMC composition which we found among progressors reflects this activated phenotype subset; however, future studies should certainly examine this possibility. It is important to highlight that the gene ontology analyses we performed herein were based on CpG sites selected following PBMC cell composition correction. Therefore, there is a possibility that additional, particularly macrophage- or monocyte-related, functional pathways and/or upstream regulators may have been missed by our analysis. These concerns do not, however, prevent us from performing predictive and prognostic biomarker analysis, as a clinically viable test would most easily include DNA from mixed PBMCs or whole blood.
Our data raise several questions which highlight potential avenues for future investigation. For example, it is unclear when epigenetic patterns change in the natural history of OA development and how stable they are over time; a longitudinal DNA methylation analysis in an incident OA cohort would be helpful in this regard. Our approach could also be used to evaluate PBMC epigenetic models for prediction of pain and combined pain and radiographic progression in larger OA cohorts. In this study, we did not seek to produce a consensus model by training on our entire dataset, as this would no doubt have produced significant model overfitting given our small sample size; rather, we sought to determine the feasibility and capability of PBMC methylation-based models for OA subtype discrimination more generally. Future studies on larger cohorts will no doubt have the ability to produce such a consensus model that would be of direct clinical relevance.
In summary, our pilot study provides the first analysis of peripheral blood epigenetic patterns as predictive models for knee OA radiographic progression. Our use of linear modeling applied to a large genome-wide DNA methylation dataset from well-matched case and control cohorts offer the first glimpse into the potential of future OA epigenetic classification. If confirmed, our approach could offer advantages over traditional biochemical biomarkers, including requiring a single baseline blood sample. Our results will certainly require additional validation in larger datasets but offer hope for the future development of easily accessible predictive biomarkers to personalize and improve the care of adults with knee OA.
Methods
Study design
This nested case-control study (116 total patients, 58 progressors, 58 nonprogressors) used data and biospecimens from the Osteoarthritis Initiative (OAI) database, which is available for public access at https://data-archive.nimh.nih.gov/oai/. All Osteoarthritis Initiative (OAI) participants provided written informed consent, and the study was carried out in accordance with the OAI data user agreement, approved by the Committee on Human Research of the Institutional Review Board (IRB) for the University of California, San Francisco (UCSF). The IRBs of the University of Oklahoma Health Sciences Center and Oklahoma Medical Research Foundation also reviewed and approved the project. Detailed case and control criteria are presented in Supplementary Document 1. Participants had baseline and yearly follow-up knee radiographs and PBMC DNA available. Kellgren-Lawrence Grade (KLG) and quantitative joint space width (JSW)59 were assessed by the central reading site using non-fluoroscopic fixed-flexion knee radiographs with a Synaflexer positioning device (Synaflexor, Synarc, Newark, CA). All participants had baseline KLG of 2–3 in at least one knee without a history of total knee joint replacement through 48-months. Participants with a tibial plateau rim distance of 6.5 mm at baseline, or with a change in the rim distance of >2.0 mm between baseline and follow-up were excluded due to unreliable radiographic positioning. Our case and control selection was limited by the availability of matched cases and controls, as below.
Definition of radiographic progression, case and control group characteristics
Our definition of radiographic progression was similar to the case definitions of the OABC-FNIH9. Cases had radiographic progression in the medial tibiofemoral compartment by a longitudinal loss in the minimum JSW of at least 0.7 mm from baseline to 24-month follow-up in one index knee, with persistent narrowing in the same index knee at 48 months based on radiographs obtained from a non-fluoroscopic fixed flexion protocol (Synaflexor, Synarc, Newark, CA). In each case, the contralateral (non-index) knee had less progression than the index knee, or no progression over the follow-up period. Participants with a tibial plateau rim distance of 6.5 mm at baseline, or with a change in the rim distance of >2.0 mm between baseline and follow-up were excluded due to inappropriate and/or unreliable radiographic positioning. Non-progressors were defined as those with ≤0.5 mm of JSW loss from baseline to 48 months in either knee. These thresholds were set to coincide with previous OAI biomarker studies9,60,62,66, as defined in the Foundation for the National Institutes of Health biomarker consortium project (OABC-FNIH) and were set based on the distribution of 1-month change in minimum JSW in normal knees of OAI control participants and estimated to have ≤10% probability of change due to measurement error8,67.
Controls were frequency matched with cases by age category, sex, race, and BMI category as shown in Table 1 and Supplementary Table 1 and were also frequency matched with the larger OAI cohort to broaden the applicability of our findings. Group differences were calculated using a Student t-test and statistically analyzed using a 2-tailed P value. There were no differences among the two groups in any demographic category with the exception of mean baseline Western Ontario and McMaster (WOMAC) pain subscale (Table 1), although it should be noted that there was a trend towards fewer females included among cases (53% vs. 60%, p = 0.13), more NSAID use at baseline among cases (29% vs. 17%, p = 0.13), and an increased mean baseline K/L grade among cases (2.2 vs. 2.0, p = 0.09), although no difference in mean baseline JSW was noted in cases (3.9 vs. 4.0, p = 0.34). We also evaluated the presence of various comorbid conditions among data collected by the OAI, including history of heart attack, failure, stroke, pulmonary disease, diabetes, and cancer (Table 1). We adjusted our models for these variables as part of our analysis (see below).
DNA methylation assays, PBMC composition assessment and adjustment
Five hundred nanograms of DNA was treated with sodium bisulfite (EZ DNA methylation kit, Zymo) and loaded onto Illumina Infinium HumanMethylation450k (n = 62 samples: 30 cases, 32 controls) or 850k (n = 54 samples: 28 cases, 26 controls) arrays. As no direct measures of PBMC cell type composition were available from the OAI, these data were estimated using the estimateCellCounts function of minfi. No statistically significant differences in estimated PBMC cell composition were found between groups (Table 1, Supplementary Table 2), although there was a trend towards increased monocytes among cases (7.4 ± 0.3% vs. 6.7 ± 0.3%, cases vs. controls mean ± SEM, p = 0.12). DNA methylation data were corrected for cell count variation using frozen surrogate variable analysis22 via the sva package (v. 3.28.0). This method has been previously shown to robustly correct for cell count variation and other batch effects in large-scale epigenomic studies22,23,68.
Data preprocessing
Statistical analysis was performed using R (v. 3.5.0). Raw.IDAT files were processed using the minfi package (v. 1.26.2). Illumina 850k (EPIC) array data were subset to only those sites also included in the 450k array. Preprocessing and normalization were performed using the preprocessFunnorm function. All samples passed internal controls included on each chip and had detection p ≤ 0.01 for ≥97% of cytosine-guanosine dinucleotide (CpG) positions included on each array. Epigenome-wide association studies traditionally have used DNA methylation beta values, defined as the fraction methylation (0–1 scale) for a particular CpG site. Beta values, however, are characterized by high heteroscedasticity (most beta values fall within extreme high- and low-percent methylation levels), and questions have been raised regarding statistical validity of beta value analysis24. Therefore, we also analyzed M values (the log2 ratio of methylated:unmethylated probe intensities for a given CpG site), which are approximately homoscedastic. We excluded from analysis CpG probes located on sex chromosomes, probes with known single nucleotide polymorphisms (SNPs) with minor allele frequency of ≥5%, and probes not detected in all samples, leaving a final n = 435,118 CpG sites for analysis in each sample.
Modeling
Elastic-net regularized generalized linear models were developed using the glmnet package (v. 2.0–16). As model overfitting is a frequently-occurring problem when developing classifiers on high-dimensional data, we implemented three strategies to reduce potential overfitting. First, we performed 7-fold internal cross-validation during development via the cv.glmnet function, utilizing the ‘one standard error rule’ when selecting lambda values69. Second, we tested models on lockbox data not used for training (Fig. 2). Finally, we repeated our model development and testing with 40 cycles of random splits of data into development and lockbox (validation) subsets. During each cycle of development, data were first randomly split into 60% development and 40% lockbox sets. Development data were then used to generate regularized cross-validated models and these models were tested on lockbox data and performance characteristics recorded. Model performance was assessed by the mean c-statistic (area under the receiver operator characteristic curve AUC-ROC), diagnostic odds ratio, accuracy, sensitivity, and specificity. The identity of DNA methylation sites (features) selected by gmnet for inclusion in each model, and the total number of sites required by each model, were recorded and compared. The performance of models by chip type (450k vs. 850k) was assessed by comparing accuracies using Fisher’s exact test of a 2 × 2 contingency table.
Ontology analysis
Genes associated with CpG sites selected at least once during model development (n = 969) were analyzed using Ingenuity Pathway Analysis (IPA, Qiagen) v. 48207413 build 2019-06-16 using default settings.
Supplementary information
Acknowledgements
This work was supported by NIH grant K08-AR070891, and by a grant from the Oklahoma Rheumatic Diseases Research Cores Center based at the University of Oklahoma and Oklahoma Medical Research Foundation, NIH grant P30-AR053483. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding source had no involvement in the writing of this article. This work was supported by a Physician Scientist Development Award (PDSA) from the Presbyterian Health Foundation, and an Oklahoma Center for the Advancement of Science and Technology (OCAST) Oklahoma Health Research Grant. The OAI is a public-private partnership comprised of five contracts (N01-AR-2-2258; N01-AR-2-2259; N01-AR-2-2260; N01-AR-2-2261; N01-AR-2-2262) funded by the National Institutes of Health, a branch of the Department of Health and Human Services, and conducted by the OAI Study Investigators. Private funding partners include Merck Research Laboratories; Novartis Pharmaceuticals Corporation, GlaxoSmithKline; and Pfizer, Inc. Private sector funding for the OAI is managed by the Foundation for the National Institutes of Health. This manuscript was prepared using an OAI public use data set and does not necessarily reflect the opinions or views of the OAI investigators, the NIH, or the private funding partners. We gratefully acknowledge the assistance and advice of Dr. Richard Pelikan PhD, Oklahoma Medical Research Foundation, regarding algorithm development design and implementation.
Author contributions
M.A.J. and C.M.D. performed DNA methylation and statistical analysis, wrote the main manuscript text, prepared figures and tables. M.J., M.N. and J.L. designed patient selection criteria and selected cases and controls. All authors reviewed and revised the manuscript.
Data availability
The datasets generated during during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-53298-9.
References
- 1.Vos T, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention (CDC) Prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation–United States, 2010–2012. MMWR Morb Mortal Wkly Rep. 2013;62:869–873. [PMC free article] [PubMed] [Google Scholar]
- 3.Singh JA, et al. American College of Rheumatology Guideline for the Treatment of Rheumatoid. Arthritis. Arthritis Rheumatol. 2015;2016(68):1–26. doi: 10.1002/art.39480. [DOI] [PubMed] [Google Scholar]
- 4.Ramos YFM, et al. Genes expressed in blood link osteoarthritis with apoptotic pathways. Ann Rheum Dis. 2014;73:1844–1853. doi: 10.1136/annrheumdis-2013-203405. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Lan C-N, Kong Y, Feng S-S, Huang T. Identification and Analysis of Blood Gene Expression Signature for Osteoarthritis With Advanced Feature Selection Methods. Front Genet. 2018;9:246. doi: 10.3389/fgene.2018.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang Z.Y., Perry E., Huebner J.L., Katz B., Li Y.-J., Kraus V.B. Biomarkers of inflammation – LBP and TLR— predict progression of knee osteoarthritis in the DOXY clinical trial. Osteoarthritis and Cartilage. 2018;26(12):1658–1665. doi: 10.1016/j.joca.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunter D, et al. Longitudinal validation of periarticular bone area and 3D shape as biomarkers for knee OA progression? Data from the FNIH OA Biomarkers Consortium. Ann Rheum Dis. 2016;75:1607–1614. doi: 10.1136/annrheumdis-2015-207602. [DOI] [PubMed] [Google Scholar]
- 8.Hunter DJ, Nevitt M, Losina E, Kraus V. Biomarkers for osteoarthritis: current position and steps towards further validation. Best Pract Res Clin Rheumatol. 2014;28:61–71. doi: 10.1016/j.berh.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kraus Virginia Byers, Collins Jamie E, Hargrove David, Losina Elena, Nevitt Michael, Katz Jeffrey N, Wang Susanne X, Sandell Linda J, Hoffmann Steven C, Hunter David J. Predictive validity of biochemical biomarkers in knee osteoarthritis: data from the FNIH OA Biomarkers Consortium. Annals of the Rheumatic Diseases. 2016;76(1):186–195. doi: 10.1136/annrheumdis-2016-209252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Andres MC, et al. Assessment of global DNA methylation in peripheral blood cell subpopulations of early rheumatoid arthritis before and after methotrexate. Arthritis Res Ther. 2015;17:233. doi: 10.1186/s13075-015-0748-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao M, et al. IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann Rheum Dis. 2016;75:1998–2006. doi: 10.1136/annrheumdis-2015-208410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toperoff G, et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum Mol Genet. 2012;21:371–383. doi: 10.1093/hmg/ddr472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakatochi M, et al. Epigenome-wide association of myocardial infarction with DNA methylation sites at loci related to cardiovascular disease. Clin Epigenetics. 2017;9:54. doi: 10.1186/s13148-017-0353-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rushton MD, et al. Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol. 2014;66:2450–2460. doi: 10.1002/art.38713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.den Hollander W, et al. Knee and hip articular cartilage have distinct epigenomic landscapes: implications for future cartilage regeneration approaches. Ann Rheum Dis. 2014;73:2208–2212. doi: 10.1136/annrheumdis-2014-205980. [DOI] [PubMed] [Google Scholar]
- 16.Jeffries, M. A, Donica, M. & Baker, L. W. Genome‐Wide DNA Methylation Study Identifies Significant Epigenomic Changes in Osteoarthritic Subchondral Bone and Similarity to Overlying Cartilage. Arthritis. Available, https://onlinelibrary.wiley.com/doi/abs/10.1002/art.39555 (2016). [DOI] [PMC free article] [PubMed]
- 17.Jeffries, M. A., Donica, M. & Baker, L. W. Genome‐wide DNA methylation study identifies significant epigenomic changes in osteoarthritic cartilage. Arthritis. Available, https://onlinelibrary.wiley.com/doi/abs/10.1002/art.38762 (2014). [DOI] [PubMed]
- 18.Fernández-Tajes J, et al. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann Rheum Dis. 2014;73:668–677. doi: 10.1136/annrheumdis-2012-202783. [DOI] [PubMed] [Google Scholar]
- 19.LaValley MP, et al. Development of a clinical prediction algorithm for knee osteoarthritis structural progression in a cohort study: value of adding measurement of subchondral bone density. Arthritis Res Ther. 2017;19:95. doi: 10.1186/s13075-017-1291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Titus AJ, Gallimore RM, Salas LA, Christensen BC. Cell-type deconvolution from DNA methylation: a review of recent applications. Hum Mol Genet. 2017;26:R216–R224. doi: 10.1093/hmg/ddx275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houseman EA, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker HS, Corrada Bravo H, Leek JT. Removing batch effects for prediction problems with frozen surrogate variable analysis. PeerJ. 2014;2:e561. doi: 10.7717/peerj.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaushal A, et al. Comparison of different cell type correction methods for genome-scale epigenetics studies. BMC Bioinformatics. 2017;18:216. doi: 10.1186/s12859-017-1611-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du P, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamashita Riu, Suzuki Yutaka, Sugano Sumio, Nakai Kenta. Genome-wide analysis reveals strong correlation between CpG islands with nearby transcription start sites of genes and their tissue specificity. Gene. 2005;350(2):129–136. doi: 10.1016/j.gene.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Collins JE, et al. Semiquantitative Imaging Biomarkers of Knee Osteoarthritis Progression: Data From the Foundation for the National Institutes of Health Osteoarthritis Biomarkers Consortium. Arthritis Rheumatol. 2016;68:2422–2431. doi: 10.1002/art.39731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kraus VB, et al. Predictive Validity of Radiographic Trabecular Bone Texture in Knee Osteoarthritis: The Osteoarthritis Research Society International/Foundation for the National Institutes of Health Osteoarthritis Biomarkers Consortium. Arthritis Rheumatol. 2018;70:80–87. doi: 10.1002/art.40348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janvier T, et al. Subchondral tibial bone texture analysis predicts knee osteoarthritis progression: data from the Osteoarthritis Initiative: Tibial bone texture & knee OA progression. Osteoarthritis Cartilage. 2017;25:259–266. doi: 10.1016/j.joca.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Joseph GB, et al. Tool for osteoarthritis risk prediction (TOARP) over 8 years using baseline clinical data, X-ray, and MRI: Data from the osteoarthritis initiative. J Magn Reson Imaging. 2018;47:1517–1526. doi: 10.1002/jmri.25892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halilaj E, Le Y, Hicks JL, Hastie TJ, Delp SL. Modeling and predicting osteoarthritis progression: data from the osteoarthritis initiative. Osteoarthritis Cartilage. 2018;26:1643–1650. doi: 10.1016/j.joca.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernández-Moreno M, et al. Mitochondrial haplogroups define two phenotypes of osteoarthritis. Front Physiol. 2012;3:129. doi: 10.3389/fphys.2012.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernández-Moreno M, et al. Mitochondrial DNA haplogroups influence the risk of incident knee osteoarthritis in OAI and CHECK cohorts. A meta-analysis and functional study. Ann Rheum Dis. 2017;76:1114–1122. doi: 10.1136/annrheumdis-2016-210131. [DOI] [PubMed] [Google Scholar]
- 33.Soto-Hermida A, et al. Mitochondrial DNA (mtDNA) haplogroups influence the progression of knee osteoarthritis. Data from the Osteoarthritis Initiative (OAI) PLoS One. 2014;9:e112735. doi: 10.1371/journal.pone.0112735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aref-Eshghi E, et al. Genome-wide DNA methylation study of hip and knee cartilage reveals embryonic organ and skeletal system morphogenesis as major pathways involved in osteoarthritis. BMC Musculoskelet Disord. 2015;16:287. doi: 10.1186/s12891-015-0745-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, et al. Identification of DNA methylation changes associated with disease progression in subchondral bone with site-matched cartilage in knee osteoarthritis. Sci Rep. 2016;6:34460. doi: 10.1038/srep34460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reynard Louise N. Analysis of genetics and DNA methylation in osteoarthritis: What have we learnt about the disease? Seminars in Cell & Developmental Biology. 2017;62:57–66. doi: 10.1016/j.semcdb.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 37.Minogue BM, Richardson SM, Zeef LA, Freemont AJ, Hoyland JA. Transcriptional profiling of bovine intervertebral disc cells: implications for identification of normal and degenerate human intervertebral disc cell phenotypes. Arthritis Res Ther. 2010;12:R22. doi: 10.1186/ar2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lodewyckx L, Cailotto F, Thysen S, Luyten FP, Lories RJ. Tight regulation of wingless-type signaling in the articular cartilage - subchondral bone biomechanical unit: transcriptomics in Frzb-knockout mice. Arthritis Res Ther. 2012;14:R16. doi: 10.1186/ar3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Q, et al. DNA methylation levels at individual age-associated CpG sites can be indicative for life expectancy. Aging. 2016;8:394–401. doi: 10.18632/aging.100908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inanir A, Yigit S, Tekcan A, Tural S, Kismali G. IL-4 and MTHFR gene polymorphism in rheumatoid arthritis and their effects. Immunol Lett. 2013;152:104–108. doi: 10.1016/j.imlet.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 41.Inanir A, Yigit S, Tural S, Cecen O, Yildirim E. MTHFR gene C677T mutation and ACE gene I/D polymorphism in Turkish patients with osteoarthritis. Dis Markers. 2013;34:17–22. doi: 10.1155/2013/658654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dong Y, et al. VEGF promotes cartilage angiogenesis by phospho-ERK1/2 activation of Dll4 signaling in temporomandibular joint osteoarthritis caused by chronic sleep disturbance in Wistar rats. Oncotarget. 2017;8:17849. doi: 10.18632/oncotarget.14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Q, Guo X-P, Cheng Y-L, Wang Y. MicroRNA-143-5p targeting eEF2 gene mediates intervertebral disc degeneration through the AMPK signaling pathway. Arthritis Res Ther. 2019;21:97. doi: 10.1186/s13075-019-1863-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou S, et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci Rep. 2017;7:43245. doi: 10.1038/srep43245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Luyao, Shan Haojie, Wang Bin, Wang Nan, Zhou Zubin, Pan Chenhao, Wang Feng. Puerarin Attenuates Osteoarthritis via Upregulating AMP-Activated Protein Kinase/Proliferator-Activated Receptor-γ Coactivator-1 Signaling Pathway in Osteoarthritis Rats. Pharmacology. 2018;102(3-4):117–125. doi: 10.1159/000490418. [DOI] [PubMed] [Google Scholar]
- 46.Richardson D, et al. Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Res Ther. 2008;10:R43. doi: 10.1186/ar2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meulenbelt, I. & Bos, S. D. Faculty of 1000 evaluation for Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. F1000 - Post-publication peer review of the biomedical literature, 10.3410/f.2687957.2351056 (2010).
- 48.Caramés Beatriz, Taniguchi Noboru, Otsuki Shuhei, Blanco Francisco J., Lotz Martin. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis & Rheumatism. 2010;62(3):791–801. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis. 2015;74:1432–1440. doi: 10.1136/annrheumdis-2013-204599. [DOI] [PubMed] [Google Scholar]
- 50.Tornero-Esteban P, et al. Altered Expression of Wnt Signaling Pathway Components in Osteogenesis of Mesenchymal Stem Cells in Osteoarthritis Patients. PLoS One. 2015;10:e0137170. doi: 10.1371/journal.pone.0137170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park Sujeong, Oh Jinjoo, Kim Yong-Il, Choe Seong-Kyu, Chun Churl-Hong, Jin Eun-Jung. Suppression of ABCD2 dysregulates lipid metabolism via dysregulation of miR-141:ACSL4 in human osteoarthritis. Cell Biochemistry and Function. 2018;36(7):366–376. doi: 10.1002/cbf.3356. [DOI] [PubMed] [Google Scholar]
- 52.Chen Q, Liu W, Sinha KM, Yasuda H, de Crombrugghe B. Identification and characterization of microRNAs controlled by the osteoblast-specific transcription factor Osterix. PLoS One. 2013;8:e58104. doi: 10.1371/journal.pone.0058104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones SW, et al. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-alpha and MMP13. Osteoarthritis Cartilage. 2009;17:464–472. doi: 10.1016/j.joca.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 54.Zhang H, Song B, Pan Z. Downregulation of microRNA-9 increases matrix metalloproteinase-13 expression levels and facilitates osteoarthritis onset. Mol Med Rep. 2018;17:3708–3714. doi: 10.3892/mmr.2017.8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, et al. A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc Natl Acad Sci USA. 2011;108:9863–9868. doi: 10.1073/pnas.1018493108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davidson Esmeralda, Vitters Elly L, van Lent Peter LEM, van de Loo Fons AJ, van den Berg Wim B, van der Kraan Peter M. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Research & Therapy. 2007;9(5):R102. doi: 10.1186/ar2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang C, Silverman RM, Shen J, O’Keefe RJ. Distinct metabolic programs induced by TGF-β1 and BMP2 in human articular chondrocytes with osteoarthritis. J Orthop Translat. 2018;12:66–73. doi: 10.1016/j.jot.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hicks Justin James, Rocha Jorge Luis, Li Hongshuai, Huard Johnny, Wang Yadong, Hogan MaCalus Vinson. Sustained Release of Bone Morphogenetic Protein 2 via Coacervate improves Muscle Derived Stem Cell Mediated Cartilage Regeneration in MIA-induced Osteoarthritis. Orthopaedic Journal of Sports Medicine. 2016;4(7_suppl4):2325967116S0015. doi: 10.1177/2325967116S00152. [DOI] [Google Scholar]
- 59.Wirth W, et al. Direct comparison of fixed flexion, radiography and MRI in knee osteoarthritis: responsiveness data from the Osteoarthritis Initiative. Osteoarthritis Cartilage. 2013;21:117–125. doi: 10.1016/j.joca.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eckstein F, et al. Brief Report: Cartilage Thickness Change as an Imaging Biomarker of Knee Osteoarthritis Progression: Data From the Foundation for the National Institutes of Health Osteoarthritis Biomarkers Consortium. Arthritis Rheumatol. 2015;67:3184–3189. doi: 10.1002/art.39324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruhdorfer A, et al. Between-group differences in infra-patellar fat pad size and signal in symptomatic and radiographic progression of knee osteoarthritis vs non-progressive controls and healthy knees - data from the FNIH Biomarkers Consortium Study and the Osteoarthritis Initiative. Osteoarthritis Cartilage. 2017;25:1114–1121. doi: 10.1016/j.joca.2017.02.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wirth W, et al. Predictive and concurrent validity of cartilage thickness change as a marker of knee osteoarthritis progression: data from the Osteoarthritis Initiative. Osteoarthritis Cartilage. 2017;25:2063–2071. doi: 10.1016/j.joca.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kraus V.B., McDaniel G., Huebner J.L., Stabler T.V., Pieper C.F., Shipes S.W., Petry N.A., Low P.S., Shen J., McNearney T.A., Mitchell P. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthritis and Cartilage. 2016;24(9):1613–1621. doi: 10.1016/j.joca.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Daghestani HN, Pieper CF, Kraus VB. Soluble macrophage biomarkers indicate inflammatory phenotypes in patients with knee osteoarthritis. Arthritis Rheumatol. 2015;67:956–965. doi: 10.1002/art.39006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang, L. et al. Inhibition of Synovial Macrophage Pyroptosis Alleviates Synovitis and Fibrosis in Knee Osteoarthritis. Mediators Inflamm. 2019, 2165918 (2019). [DOI] [PMC free article] [PubMed]
- 66.Kraus VB, et al. Application of biomarkers in the development of drugs intended for the treatment of osteoarthritis. Osteoarthritis Cartilage. 2011;19:515–542. doi: 10.1016/j.joca.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ornetti P, et al. OARSI–OMERACT definition of relevant radiological progression in hip/knee osteoarthritis. Osteoarthritis Cartilage. 2009;17:856–863. doi: 10.1016/j.joca.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McGregor, K. et al. An evaluation of methods correcting for cell type heterogeneity in DNA methylation studies, 10.1101/032185 [DOI] [PMC free article] [PubMed]
- 69.Krstajic D, Buturovic LJ, Leahy DE, Thomas S. Cross-validation pitfalls when selecting and assessing regression and classification models. J Cheminform. 2014;6:10. doi: 10.1186/1758-2946-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during during the current study are available from the corresponding author on reasonable request.