

Bacterial species in the microbiome are often represented by multiple genetically and phenotypically different strains, making insight into subspecies diversity critical to a full understanding of the microbiome, especially with respect to opportunistic pathogens. However, methods allowing efficient high-throughput clonal typing are not currently available. This study combines a conventional E. coli typing method with deep amplicon sequencing to allow analysis of many samples concurrently. While our method was developed for E. coli, it may be adapted for other species, allowing microbiome researchers to assess clonal strain diversity in natural samples. Since assessment of subspecies diversity is particularly important for understanding the spread of antibiotic resistance, we applied our method to the study of a pandemic multidrug-resistant E. coli clone. The results we present suggest that this clone could be highly competitive in healthy carriers and that the mechanisms of colonization by such clones need to be studied.
KEYWORDS: Escherichia coli, ST131, antibiotic resistance, bladder colonization, gut microbiome
ABSTRACT
While microbiome studies have focused on diversity at the species level or higher, bacterial species in microbiomes are represented by different, often multiple, strains. These strains could be clonally and phenotypically very different, making assessment of strain content vital to a full understanding of microbiome function. This is especially important with respect to antibiotic-resistant strains, the clonal spread of which may be dependent on competition between them and susceptible strains from the same species. The pandemic, multidrug-resistant, and highly pathogenic Escherichia coli subclone ST131-H30 (H30) is of special interest, as it has already been found persisting in the gut and bladder in healthy people. In order to rapidly assess E. coli clonal diversity, we developed a novel method based on deep sequencing of two loci used for sequence typing, along with an algorithm for analysis of the resulting data. Using this method, we assessed fecal and urinary samples from healthy women carrying H30 and were able to uncover considerable diversity, including strains with frequencies at <1% of the E. coli population. We also found that, even in the absence of antibiotic use, H30 could completely dominate the gut and, especially, urine of healthy carriers. Our study offers a novel tool for assessing a species’ clonal diversity (clonobiome) within the microbiome, which could be useful in studying the population structure and dynamics of multidrug-resistant and/or highly pathogenic strains in their natural environments.
IMPORTANCE Bacterial species in the microbiome are often represented by multiple genetically and phenotypically different strains, making insight into subspecies diversity critical to a full understanding of the microbiome, especially with respect to opportunistic pathogens. However, methods allowing efficient high-throughput clonal typing are not currently available. This study combines a conventional E. coli typing method with deep amplicon sequencing to allow analysis of many samples concurrently. While our method was developed for E. coli, it may be adapted for other species, allowing microbiome researchers to assess clonal strain diversity in natural samples. Since assessment of subspecies diversity is particularly important for understanding the spread of antibiotic resistance, we applied our method to the study of a pandemic multidrug-resistant E. coli clone. The results we present suggest that this clone could be highly competitive in healthy carriers and that the mechanisms of colonization by such clones need to be studied.
INTRODUCTION
Microbiomes, in terms of both function and diversity, have recently been a topic of considerable interest. The gut microbiome has gotten special attention due to its high complexity and importance to health (1–9). So far, studies have almost exclusively focused on species or higher-level diversity. However, this paints an incomplete picture, since strains within the same species can be of distinct clonal origins and have vastly different metabolic, pathogenic, and antibiotic resistance profiles (10–19). Importantly, multidrug-resistant bacterial strains have been found competing with commensal strains in the gut, even without antibiotic pressure (18–23). Thus, there is a pressing need to identify strains in the human microbiome for species of critical health importance.
Escherichia coli is one of the most common residents of the gut. While primarily a commensal colonizer, extraintestinal pathogenic E. coli clones are implicated in a variety of diseases, including urinary tract infections (UTIs), a leading cause of human antibiotic use (24–28). The spread of multidrug-resistant E. coli is now a major health concern, especially the pandemic fimH30 subclone of sequence type 131 (ST131) (H30). Though recently emerged, H30 is now globally distributed and comprises up to half of all urinary and bloodstream isolates of E. coli that are fluoroquinolone resistant and produce extended-spectrum beta-lactamases (ESBL) (29–31).
Additionally, H30 is strongly associated with “drug-bug” mismatches and adverse outcomes in elderly and immunocompromised individuals (29, 30). Somewhat paradoxically, H30 is also a persistent gut colonizer of healthy people and frequently causes asymptomatic bacteriuria (ABU) in such carriers (31). However, the relative clonal predominance of H30 strains among E. coli strains colonizing the gut or bladder in healthy carriers remains unknown. Answering these questions could have a significant impact on understanding the spread of antibiotic resistance and its reservoirs.
Currently, microbiome diversity is studied by sequencing the 16S rRNA gene, but this cannot capture clonal diversity (32, 33). Conventional methods for assessing clonal diversity, such as metagenomic sequencing and single-colony typing, are costly and labor-intensive. For reliable clonal diversity analysis, metagenomic sequencing requires very high coverage per sample, while single-colony typing requires handpicking large numbers of colonies for multilocus sequence typing (MLST) (34–37). In E. coli, MLST requires assessment of 7 genes per isolate, which is analytically complex, costly, and labor-intensive, and therefore difficult to implement. Previously, we reported an alternative clonotyping method that requires sequencing regions of only 2 genes: fumC, which is part of the MLST scheme, and fimH, which encodes a rapidly evolving fimbrial adhesin (38). The fumC-fimH-based (CH) typing of E. coli is widely accepted due to its simplicity and ability to not only identify specific STs, but subdivide them into smaller subclones (38). Specifically, H30 is identified using the allele combination fumC40-fimH30, while other, less resistant ST131 strains have the same fumC but different fimH alleles.
Here, we report a high-throughput method for clonal typing of E. coli strains by combining CH typing and deep amplicon sequencing. We developed a new algorithm—population-level allele profiler (PLAP)—for detecting alleles and predicting the relative prevalence of each allele in a sample. We were able to assess the prevalences of clonal groups (including H30) in multiple fecal and urine samples concurrently, with a limit of relative abundance detection at <1% of the total population.
RESULTS
Deep amplicon sequencing of defined samples.
To validate our approach and establish a limit of detection for strain presence, we first tested our deep amplicon sequencing procedure on a set of defined samples. To create the defined samples, we first selected a fecal sample from our laboratory collection known to contain H30 and ST101. Next, we isolated a single colony of each and confirmed them to be strains of H30 (fumC40-fimH30) and ST101 (fumC41-fimH86) by using CH typing. From these single colonies, we first created H30-only and ST101-only mixtures of fumC and fimH amplicons. We also created four ST101-H30 mixed samples by combining the fumC and fimH amplicons from ST101 and H30 in ST101/H30 ratios of 1:1, 1:4, 1:100, and 1:1,000.
Analysis of raw sequencing data from H30-only and ST101-only samples showed the average coverage of erroneous bases was 0.08% ± 0.09% for both strains. Erroneous bases were observed in both genes across most nucleotide positions. The highest coverages for an erroneous base were 0.66% of aligned reads in fumC and 0.45% in fimH for H30 and 0.68% of reads in fumC and 0.46% in fimH for ST101. The frequency distribution for erroneous base coverage is presented in Fig. S1 in the supplemental material.
Analysis of raw sequencing data from ST101-H30 mixtures showed that both H30 and ST101 alleles were detectable in the 1:1, 1:4, and 1:100 mixtures. In the 1:1,000 mixture, only alleles of the dominant H30 strain were observed. In the 1:1, 1:4, and 1:100 mixtures, the input and observed allele prevalence were highly correlated for both fumC and fimH (R2 = 0.996 and 0.997, respectively [see Fig. S2 in the supplemental material]). Erroneous bases were observed at 0.09% ± 0.1% and 0.08% ± 0.09% of aligned reads in fumC and fimH, respectively (see Fig. S1). The highest coverages for erroneous bases among all mixtures were 0.79% of aligned reads for fumC and 0.57% of aligned reads for fimH.
Since 0.79% of aligned reads was the highest coverage for an erroneous base, we established 0.8% as a cutoff for correct base calling in both genes. This cutoff was used for all further PLAP analysis.
Deep sequencing of study samples and allele prediction.
Next, we applied PLAP to 67 participant samples (43 fecal and 24 urine) collected from a previous study (31). A total of 128 fumC and 129 fimH alleles were predicted across all the samples, of which 123 (96.1%) and 125 (96.9%) were previously known fumC and fimH alleles, respectively. Five novel fumC and 4 novel fimH alleles were potentially detected. All the novel fumC and fimH alleles were phylogenetically distant from other alleles predicted in the sample, indicating that the alleles are not artifacts of sequencing (see Fig. S3 and S4 in the supplemental material). These novel alleles nonetheless clustered with other E. coli fumC and fimH alleles, indicating that they are novel E. coli alleles rather than alleles belonging to other species.
The average number of alleles predicted per sample was 1.91 ± 0.96 for fumC and 1.93 ± 1.01 for fimH. Forty-three samples had the same numbers of predicted fumC and fimH alleles; 24 samples had different numbers of predicted fumC and fimH alleles (Fig. 1). Overall, the number of predicted fumC alleles correlated with the number of predicted fimH alleles, with an R2 value of 0.88 (Fig. 1).
FIG 1.
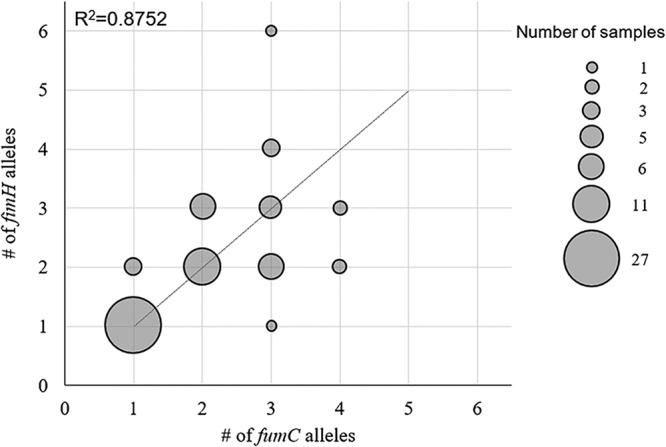
Congruency of fumC and fimH allele counts in fecal and urine samples. The sizes of the circles correspond to the numbers of samples with designated fumC or fimH allele counts (i.e., 1 sample with one fumC allele and three fimH alleles). The linear fit with the Pearson square correlation index is shown.
To assess the performance of PLAP for predicting alleles, we used samples containing criterion clones—strains previously identified by single-colony typing. PLAP detected criterion fimH and fumC alleles in 52 of the samples (90%). In the 6 samples where a criterion allele(s) was not found, the criterion clones were ciprofloxacin resistant, but their isolation from the sample required ≥2 plating attempts. This leads us to believe that these alleles were not detected because they were absent in the MacConkey agar-plated population prior to deep sequencing.
A total of 72 noncriterion (previously unidentified) fumC and 71 noncriterion fimH alleles were predicted by PLAP across all 67 samples. To assess the performance of PLAP on noncriterion alleles, we analyzed 14 samples (10 fecal and 4 urine) predicted to contain 22 noncriterion fumC and 22 noncriterion fimH alleles. Twelve of these samples had at least one noncriterion allele alongside criterion alleles; the remaining 2 had multiple noncriterion alleles only in each gene. For each sample, ≥40 single colonies were isolated, and the CH type was determined using 7-single-nucleotide polymorphism (SNP) quantitative PCR (qPCR), with each CH type verified by sequencing. With these data, we confirmed 19 (86%) predicted noncriterion alleles for each gene. They included one predicted novel fumC allele. Of the unconfirmed alleles, one was not distinguishable by 7-SNP qPCR and had a predicted prevalence of 1%; therefore, we did not attempt to locate it. The remaining unconfirmed alleles had predicted prevalences of <3% and therefore may have been missed due to insufficient sampling. Additionally, all the criterion alleles in these samples, 12 per gene, were predicted by PLAP.
Prediction of allele prevalence in multiallele samples.
We have also designed PLAP to predict the within-sample prevalence of each allele. The average allele prevalence in fecal samples was 47.3% ± 4.3% (standard error of the mean [SEM]) (range, 0.88% to 100%) for fumC and 48.4% ± 4.22% (SEM) (range, 1% to 100%) in fimH. The average allele prevalence in urine samples was 64.8% ± 6.91% (SEM) (range, 1.4% to 100%) for fumC and 58.3% ± 7.18% (SEM) (range, 1% to 100%) in fimH.
In order to verify that the prevalences predicted by PLAP were accurate, we compared the predictions to actual in-sample prevalences using two different methods.
In the first method, we used H30, since ascertaining its prevalence is relatively simple. By plating the sample on MacConkey agar and then patching onto LB-ciprofloxacin, it is possible to compare the number of ciprofloxacin-resistant (H30) colonies to the total number of E. coli colonies. The ratio of these two numbers provides the H30 load in a sample. We compared the predicted prevalences of fumC40 and fimH30 to the H30 load in 17 fecal samples containing ciprofloxacin-resistant H30.
The correlations between the H30 load and the predicted prevalences of fumC40 and fimH30 were 0.86 and 0.84, respectively (Fig. 2), indicating that the prevalences given by PLAP were representative of actual allele prevalences. To determine whether outliers were present, we calculated the 99% confidence interval (CI) range for every sample (see Materials and Methods). Three outlier samples were identified (Fig. 2, open circles). Since it is possible that these outliers contain ciprofloxacin-sensitive non-H30 fimH30-containing clones, fumC-null or fimH-null clones, and/or ciprofloxacin-sensitive H30, we decided to employ screening of a large number of single colonies.
FIG 2.

Validation of predicted H30 allele prevalence. Shown are the PLAP-predicted prevalences of H30 alleles versus actual H30 loads in H30-containing fecal samples. The predicted prevalences of fumC40 (A) and fimH30 (B) are shown. The predicted prevalences of fumC40 and fimH30 are expressed as percentages of all E. coli bacteria in each sample. The experimentally confirmed H30 load is expressed as the percentage of H30 (ciprofloxacin-resistant) single colonies among all plated E. coli single colonies. At least 130 colonies were tested per sample. Outliers (open circles) were outside the 99% confidence interval of the number of colonies tested.
In the second method, we used single-colony typing for the in-depth characterization of 14 multiallele samples described above, alongside 4 additional single-allele samples (2 fecal and 2 urine) for which only one allele per gene was predicted. This set of 18 samples included 11 of the 17 fecal samples used for the H30-based analysis described above, including one of the outlier samples. For all 18 samples, we used CH typing of ≥40 single colonies per sample to determine the prevalence of each fumC and fimH allele. Correlation between the PLAP-predicted prevalence and the experimental allele prevalence was 0.98 for both fumC and fimH alleles (Fig. 3). As in the H30 analysis described above, we determined whether outliers were present by using the 99% CI range for every sample. Only one outlier was detected, corresponding to the only sample that contained colonies from which fimH could not be amplified (fimH-null colonies). Furthermore, the sample that was an outlier in the H30-based analysis was found to contain a relatively rare ciprofloxacin-sensitive H30.
FIG 3.

Validation of predicted fumC and fimH allele prevalences. (A) PLAP-predicted versus experimental within-sample fumC and fimH allele prevalences in 18 samples. Experimental allele prevalence was determined by CH typing of at least 40 single bacterial colonies per sample. Outliers (open circles) were outside the 99% confidence interval of the number of colonies sampled. (B) Predicted prevalence of fumC versus fimH alleles from the same CH type in 11 samples where no sharing of alleles between strains was present.
Matching fumC and fimH alleles to predict sample strain content.
In CH typing, unique combinations of fumC and fimH alleles are used to determine the identities of the strains in a sample. Since a strain contains one copy of fumC and fimH, the prevalences of alleles of the two genes in the sequencing data should be identical. For example, in a sample containing 30% H30 (fumC40-fimH30) and 70% ST101 (fumC41-fimH86), we expect to see 30% of fumC reads to be fumC40 and 30% of fimH reads to be fimH30. In reality, however, the prevalences will be slightly different due to PCR and sequencing errors. To establish an acceptable difference between the prevalences of same-strain fumC and fimH alleles, we looked at 11 samples containing unique CH types (i.e., without allele sharing). In these 11 samples, the predicted prevalences of fumC and fimH were highly correlated (0.99) (Fig. 3). First, we calculated the absolute difference between the predicted fumC and fimH prevalences for each matched pair of alleles. Next, each absolute difference was divided by the predicted fumC or fimH prevalence to obtain a relative deviation (Fig. 4). Finally, we used the relative deviations to derive an equation for the maximum acceptable difference between matching fumC and fimH alleles (Fig. 4).
FIG 4.
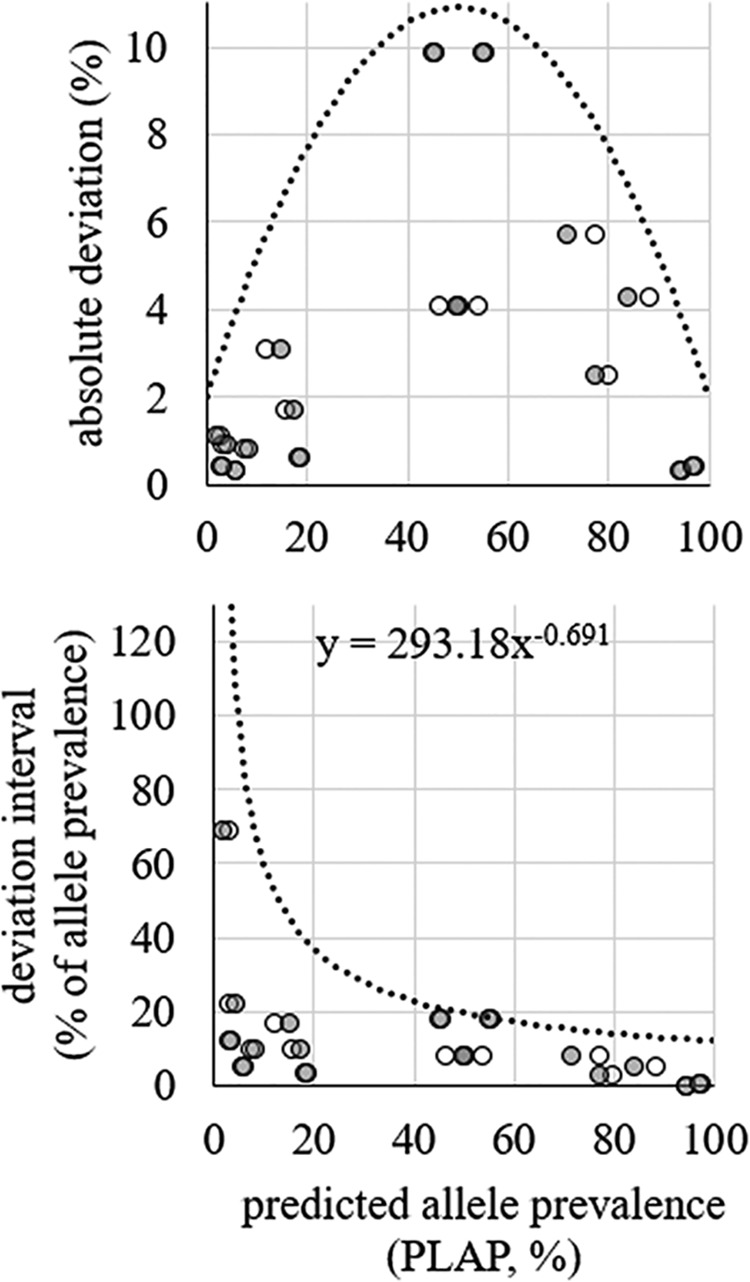
Difference in predicted prevalence between fumC and fimH alleles from the same E. coli strain. Deviation in absolute numbers (top) and deviation as a percentage of the prevalence of the allele (bottom) are shown. The open circles indicate fimH data points. The shaded circles indicate fumC data points. Trend lines and equations were used to determine intervals for matching (i.e., belonging to the same CH type) fumC and fimH alleles.
While some samples, like those discussed above, contain only unique CH types, others contain CH types with shared alleles. For example, in a sample containing 30% H30 and 70% ST131, which share fumC40, the prevalence of fumC40 is not representative of either H30 or ST131 prevalence. For such samples, the minority rule was applied to resolve the strain content. Thus, under the minority rule, the percentage of H30 in the example above would be determined by fimH30 rather than fumC40, since the fimH30 prevalence is lower. We tested this approach on both the H30 and the 18-sample analyses described above to see if it resolved outliers. In both cases, using the minority rule removed outliers and improved the correlation between predicted and experimental prevalence (see Fig. S5 in the supplemental material). Thus, we were able to assign strain content and strain prevalence to all samples, including samples with allele sharing.
Predicted strain diversity of fecal and urine samples.
Using the equation described above, we were able to classify all the samples in our study into 4 categories (Fig. 5): samples with only one CH type (uniclonal); samples with multiple unique CH types (unambiguous); samples with one dominant unique CH type and multiple minor, nonunique CH types (ambiguous-simple); and samples where the dominant CH type was not unique (ambiguous-complex). Fecal samples were 33% uniclonal, 23% unambiguous, 21% ambiguous-simple, and 23% ambiguous-complex. Urine samples were 54% uniclonal, 8% unambiguous, 25% ambiguous-simple, and 12.5% ambiguous-complex.
FIG 5.

Representative examples of each sample category defined by within-sample breakdown of prevalence for fumC and fimH alleles. The numbers of fecal and urine samples belonging to each category are listed below.
Overall, 107 fecal and 48 urine strains were predicted, corresponding to 68 clones in fecal samples and 33 clones in urine samples. Of these clones, 50 (73.5%) and 24 (73%), respectively, were found in Enterobase, an online repository of E. coli genomes and MLST types (https://enterobase.warwick.ac.uk).
Out of the 155 total strains predicted, 6 were fumC null (3.9%) and 2 were fimH null (1.3%). This is congruent with the occurrence of null alleles in our 18-sample subset, where 1 (3%) out of 35 total strains predicted was a null-allele strain.
The average number of strains per sample was 2.47 ± 1.32 for fecal samples and 1.96 ± 1.40 for urine samples. Based on Enterobase’s ST-phylogroup data, we determined that B2 was the most common (14 out of 47; 30%) among noncriterion fecal strains. Other phylogroups included A (26%), B1 (19%), C (8.5%), D (11%), E (2%), and F (4%). Noncriterion strains in urine samples included strains from phylogroups B2 (8 out of 16; 50%), B1 (19%), D (19%), and A and F (6% each).
Novel clones.
Seventeen fecal samples (40%) and 8 urine samples (33%) in our study were found to contain at least one novel CH type. They included 19 fecal and 9 urine CH types not found in Enterobase. Of these, 5 fecal and 3 urine CH types included at least one novel allele, and 14 fecal and 6 urine CH types were combinations of fumC and fimH that were not previously observed (novel CH combinations). Both CH types involving novel alleles and novel CH combinations were observed to be primarily low-frequency clones. The average predicted prevalence for novel CH combinations was 8.7% ± 3.5% (SEM) (range, 1% to 64.2%), and 13 out of 20 novel CH combinations had predicted prevalences of <5%. One such combination was confirmed in our set of 14 characterized samples, consisting of fumC24 and fimH9, with a predicted prevalence of 1.6% and experimental prevalence of 1.2%.
Similarly, 7 out of 8 novel allele-containing CH types had predicted prevalences of <2%. The remaining CH type had a predicted prevalence of 70.7% and was detected using single-colony typing. The novel fumC allele was paired with fimH47 and was verified to be 8 SNPs away from the closest known allele. The remaining MLST gene alleles for the strain were adk46, icd260, mdh160, gyrB266, purA1, and recA221.
Clones below the error threshold.
To ascertain if we could identify alleles at prevalences below our defined error threshold of 0.8%, we ran PLAP on the set of 14 multiallele samples, using an error threshold of 0.5%. In 8 and 6 samples, respectively, the prevalence of fumC and fimH alleles was <0.8%. None of the alleles corresponded to known fumC or fimH alleles. These apparently novel alleles clustered alongside known alleles identified in the sample (see Fig. S6 and S7 in the supplemental material), leading us to conclude that they arose due to sequencing or amplification errors rather than belonging to clonally different strains.
Predicted strain diversity in urine and fecal samples.
Strain diversity in the first fecal samples was comparable to the diversity in the second fecal samples (paired t test; P > 0.1). Distinguishing between H30-containing and non-H30 samples showed that there was no statistical difference in strain diversity between H30-containing and non-H30 fecal samples of either kind (unpaired t test; P > 0.1) and that there was no difference in diversity between first and second fecal samples in either non-H30 or H30-containing samples (Fig. 6) (paired t test; P > 0.1). Both H30 and non-H30 urine samples were less diverse than corresponding fecal samples (paired t test; P < 0.01 and 0.02, respectively). However, H30 urine samples were less diverse than non-H30 urine samples (t test; P = 0.04).
FIG 6.

Diversity of E. coli in individual fecal/urine samples. H30 content was determined by PLAP and/or culturing.
It is also noteworthy that in 6 out of 23 H30-containing fecal samples, H30 was the only strain predicted, indicating that it may be fully dominant in the gut niche in these participants.
Strain turnover in fecal samples.
There was no correlation between the numbers of strains in the first and second fecal samples, as well as no correlation between the numbers of strains in the urine sample and either fecal sample (Fig. 7). When comparing the strain contents of the first and second fecal samples, we found that 92% of noncriterion strains appeared to be transient, i.e., were detected in one of the fecal samples only. Transient noncriterion strains were also skewed toward lower-frequency strains (t test; P < 0.001) (Fig. 8B). It is possible that these strains were present in both fecal samples but were below our limit of detection in one. However, we found that in one participant (P2 [see the supplemental material]) the first fecal sample contained 3 ciprofloxacin-sensitive noncriterion strains while the second fecal sample contained only ciprofloxacin-resistant H30, as verified by single-colony testing. This leads us to believe that there may have been significant strain turnover in our fecal samples overall.
FIG 7.

Counts of E. coli strains in fecal and urine samples. Shown are the numbers of strains detected by PLAP in the first fecal sample versus urine (A), second fecal sample versus urine (B), and first fecal sample versus second fecal sample (C). Each circle represents participants with the corresponding number of E. coli strains in the designated sample. The circle size indicates the number of participants with the determined number of strains. The linear fit with the Pearson square correlation index shown.
FIG 8.

Persistence of E. coli strains in fecal samples. (A) Prevalences of criterion fecal strains in first versus second fecal samples. The open data points represent H30 strains, while the shaded data points represent non-H30 strains. The circled cluster represents 4 strains present at 100% prevalence in both samples. The dotted lines indicate the mean prevalence for strains in the first and second fecal samples. The distribution of prevalences in both first and second fecal samples is not significantly different from random (t test; P > 0.05). (B) Prevalences of noncriterion fecal strains in first versus second fecal samples. The dotted lines indicate the mean prevalence for transient strains in the first and second fecal samples. Transient strains are defined as strains that are present in only one of the two fecal samples from the same participant. The distribution of prevalences in both first and second fecal samples is significantly skewed toward lower prevalences (t test; P < 0.01).
DISCUSSION
We combined conventional fumC-fimH typing with deep amplicon sequencing to assess E. coli clonal diversity in a high-throughput manner. Our method has several advantages over existing protocols. First, our method has high sequencing resolution for target species. Since we sequence only E. coli fumC and fimH, we can generate ≥0.5 million reads per sample, yielding ≥5,000 reads per base. In contrast, metagenomic sequencing, which is nonspecific for target species, yields only 20 reads per base per genome (assuming a 5-Mb genome). Secondly, our method assessed up to 46 samples per sequencing run. In contrast, MLST requires typing ≥100 single colonies per sample to capture the low-prevalence strains that PLAP detects. Finally, while we developed PLAP for E. coli CH typing, PLAP is not limited to E. coli clonotyping and may be generalized to other MLST schemes. For those attempting to use or adapt our approach, we have provided guidelines for both the experimental and algorithm portions on PLAP’s website (http://www.github.com/marade/PLAP).
Despite studies showing that the healthy gut E. coli population typically includes multiple clones, we show that the pandemic multidrug-resistant subclone H30 can dominate the gut in healthy women, sometimes as the only detectable clone (42, 44–47). This builds upon previous research that has found multidrug-resistant bacteria in healthy people and healthy people who appear to harbor only one gut clone (42, 46). Total dominance is especially concerning, since antibiotic pressure was absent, indicating that H30 is potentially outcompeting other clones by alternative means. Whether these mechanisms are metabolic or whether certain virulence factors give H30 an advantage is unclear, though previous studies have speculated that some virulence factors may be beneficial for E. coli gut survival (40, 43, 44). Additionally, our study involved a small number of participants in whom H30 was present in the gut and bladder. Therefore, it is possible that host differences play a significant role. Another novel observation was that H30 was the sole detected urinary strain more frequently than other clones, regardless of H30 gut dominance or nondominance. This may indicate that H30 is an especially well-adapted uropathogen, potentially explaining its association with UTI. Since it is unknown how ABU converts to UTI, further study of H30 dominance in both ABU and UTI is needed.
We also uncovered substantial diversity in our samples. This included significant E. coli diversity in non-H30 urine samples from healthy women. Reports of multistrain bacteriuria are rare, likely due to the convention of selecting one isolate per urine sample (41, 42). Therefore, it is unknown how common multistrain bacteriuria may truly be. Remarkably, we also detected low-prevalence strains in the gut, some of which were novel clones, with up to 6 clones in a single sample. Gut E. coli diversity of this magnitude is supported by studies typing >200 single colonies per sample (42). Studies using smaller counts usually report fewer clones, indicating that there may be undescribed E. coli diversity when manageable numbers of colonies are used (40, 41, 47). Therefore, we believe that microbiome-like approaches to E. coli diversity are necessary to fully understand intraspecies dynamics in both the gut and bladder.
Our approach does have limitations. First, our lowest detectable strain prevalence is 0.8% of the E. coli population. This limit may be addressed in several ways, including use of a high-fidelity polymerase and preferential selection of E. coli colonies. However, we also recognize that detection of rare strains may still prove difficult and that methods like ours may not fully replace current techniques. Secondly, our method relies on subculturing E. coli. We are aware that, theoretically, some strains could be suppressed during growth on selective media, forming no or smaller colonies and skewing prevalence results. However, we did not encounter this during our study. While amplification of fumC and fimH may be applied to urine samples without culturing, attempts at doing this directly from fecal samples were unsuccessful, possibly due to E. coli comprising <1% of the gut microbiome, making E. coli DNA too rare to effectively amplify. Therefore, we used culturing for all samples and believe that evaluating target species abundance using 16S rRNA sequencing is warranted in such cases. Lastly, we used antibiotic resistance for validation, which is not possible with clones/species where antibiotic resistance is absent or not strongly clonal. In these cases, validation using single-colony typing should be considered. These issues lower the reliability of our approach, but we believe that it remains an important step toward development of comprehensive clonal diversity (clonobiome) assessment tools for any species of interest.
MATERIALS AND METHODS
Study design and sample processing.
We selected a subset of participants from a previous study carried out by Kaiser Permanente Washington and the University of Washington (Seattle, WA) (31). That study identified healthy gut carriers of ciprofloxacin-resistant E. coli, including E. coli H30. These E. coli strains were found in initial fecal samples by plating on LB-ciprofloxacin and CH typing of 1 to 8 single colonies. After the initial fecal sample was analyzed, the H30 carriers, as well as carriers of some other strains, were asked to provide urine samples. These were received on average 152 ± 55.9 days after the initial sample (85% responded). The respondents were then asked to provide follow-up fecal samples, which were received on average 82 ± 41.1 days after the urine sample (84% responded). All the fecal and urine samples were tested for ciprofloxacin-resistant E. coli as with the initial samples. For this study, we chose 28 individuals who supplied all three samples. For 11 participants, H30 was identified in all three samples; for 4 additional participants, H30 was isolated in two samples. For 8 participants, ciprofloxacin-resistant ST1193 was found in at least two samples. For 5 participants, the same ciprofloxacin-susceptible clone was found in at least two samples. The sample types, strain clonal identities, and sampling times for all participants are shown in Fig. S8 in the supplemental material. The average age of participants was 66.7 ± 15.7 years.
Preparation of predefined control samples.
For control experiments, two predefined strains were chosen: H30 (E. coli FESS614.ds6) and clonal group ST101 (E. coli FESS614.ds4). DNA from these strains was extracted, and fumC and fimH were amplified by PCR under the following conditions: 3 min denaturation (95°C), 35 cycles of annealing (95°C for 45 s, 57°C for 45 s, and 72°C for 45 s), 5 min extension (72°C), and 4°C hold. The primers (10 μM) used were as follows: 5′-TCACAGGTCGCCAGCGCTTC-3′ (fumC forward), 5′-GTACGCAGCGAAAAAGATTC-3′ (fumC reverse), 5′-TCAGGGAACCATTCAGGCA-3′ (fimH forward), and 5′-ACAAAGGGCTAACGTGCAG-3′ (fimH reverse). The amount of PCR product was measured with Qbit. To create H30-only and ST101-only samples, the corresponding fumC and fimH PCR products were pooled at a 1:1 ratio. To create mixtures, H30 and ST101 amplicons of fumC were mixed in ST101/H30 ratios of 1:1, 1:4, 1:10, 1:100, and 1:1,000. The same was performed with fimH amplicons. The fumC and fimH mixtures were then pooled by ratio type to create mixtures that had equal concentrations of total fumC and fimH. The DNA mixtures were prepared for sequencing with a Nextera XT DNA library preparation kit using the standard protocol. The resulting library was sequenced on the Illumina MiSeq (v3 kit). All the mixtures, except 1:10, reached coverage of ≥9,000× and were analyzed.
Deep sequencing and allele analysis of fecal and urine samples.
Each fecal and urine sample was plated on MacConkey agar to reach ∼1,000 E. coli single colonies per plate. All the colonies were swabbed from the agar, and DNA was extracted using a Qiagen blood and tissue kit. From this pooled DNA, fumC and fimH genes were amplified by PCR using the same primers and conditions described above for control samples. The amplicons were then purified and pooled by sample using a Qiagen gel extraction kit and then prepared for sequencing with a Nextera XT DNA library preparation kit using the standard protocol except for the use of 52.5 μl of resuspension buffer (RSB) in the final magnetic-bead cleanup step. The resulting library was sequenced on the Illumina MiSeq (v3 kit). Sequencing data were analyzed using a Python program of our construction, PLAP, and have been made available for public use on GitHub (http://www.github.com/marade/PLAP). The process is described below (see Fig. S9 in the supplemental material).
For each sample, adapter sequences were removed using Trim-Galore, and the resulting trimmed reads were aligned to a list of all known fumC and fimH alleles using KMA with strict 99.99% identity matching (https://github.com/FelixKrueger/TrimGalore) (48, 49, 50). For each KMA-detected allele per sample, trimmed reads were again aligned to the sequence using Minimap2 and SAMtools (49, 50). Any candidate allele that had at least 1 base supported by <0.8% of reads was removed from consideration. False positives were filtered using a moving 10-bp window for each allele as follows. Reads of ≥100 bp with 100% identity within the window were counted. Alleles with low initial coverage, unstable coverage (high average deviation from the mean), and high similarity in the coverage pattern to an allele with more stable coverage were removed from consideration. If >3 alleles were left for consideration for a gene, 10-bp moving-window analysis was repeated with ≥200-bp reads. If for any interval in this second analysis >60% of coverage was lost compared to the first moving-window coverage, the allele was discarded. Heterogeneity at any positions that remained undescribed by surviving alleles was recorded. The relative abundances of all alleles were determined using the minimum coverage found during the first moving-window analysis. In samples found by PLAP to be ≥50% made up of <100-bp reads (overtagmented samples), allele prevalence was calculated manually by ascertaining the base(s) unique to each allele and using the coverage of the base(s) to calculate prevalence.
Out of the 28 total sets of fecal and urine samples chosen for this study, at least one sample failed PCR amplification or sequencing library preparation in 4 sets, and therefore, all the samples from these sets were dropped. From the remaining 24 sets, we were able to sequence fumC and fimH in all three samples. Of those, 67 (89%) samples—22 first fecal, 24 urine, and 21 second fecal—reached ≥9,000× coverage per gene and were included in the analysis.
Determining within-sample clonal group breakdown.
The identities of strains present in a sample were determined by combining fumC and fimH allele numbers and determining the ST using Enterobase. In uniclonal and unambiguous samples, every allele had one match supported by the equation for maximum acceptable difference between same-strain fumC and fimH. Therefore, these alleles formed a CH type based on which ST type was determined.
For ambiguous-simple samples, the most prevalent fumC and fimH alleles formed an equation-supported CH type. Any alleles that also had a single-equation-supported match were assigned to form a CH type. For all other alleles, Enterobase was consulted to determine which allele combinations had been observed. If the CH type(s) produced was between alleles that had different prevalences according to the equation, the “remaining” prevalence was calculated for the allele with the greater prevalence. This allele was then paired with an allele(s) for which an Enterobase-logged CH type was not available and/or any novel alleles until the remaining prevalence was consumed. If any allele(s) remained after this step, it was paired with the major allele of the opposite gene.
For ambiguous-complex samples, the most prevalent fumC and most prevalent fimH allele were assigned to the same CH type. The remaining prevalence was calculated for the allele with the greater prevalence and treated as an unmatched allele. From this step, we proceeded as with ambiguous-simple samples.
Determining prevalence of clonal groups by culturing.
The prevalence of ciprofloxacin-resistant clones in each sample was determined by diluting ∼1 μl of sample with ≥300 μl of H2O, plating 40 μl of this dilution on MacConkey agar, picking >130 single E. coli colonies, patching on HardyCHROM UTI chromogenic agar (Hardy Diagnostics) to verify E. coli identity, and then patching colonies on LB-ciprofloxacin. The prevalences of other clonal groups were validated by plating on MacConkey agar and subsequent patching of single colonies onto HardyCHROM UTI agar to distinguish E. coli. The fumC and fimH alleles of these colonies were then determined by 7-SNP clonotyping and Sanger sequencing (51).
Statistical and phylogenetic analysis.
To determine the 99% CI for the prevalence of ciprofloxacin-resistant strains, the number of resistant colonies was treated as the number of successes, and the total number of picked colonies was treated as the total. To determine the 99% CI for the prevalence of ciprofloxacin-sensitive strains, the number of colonies of that strain was treated as the number of successes, and the total number of picked colonies was treated as the total. Confidence intervals were calculated using Stata (52). All t tests were run using GraphPad.
Phylogenetic trees were constructed using MEGA7 (53). An erroneous base coverage graph was generated using seaborn (54). E. coli fumC alleles were downloaded from Enterobase MLST allele data. The E. coli fimH alleles used are publicly available (55). Escherichia fergusonii and Escherichia albertii fumC alleles were downloaded from NCBI. Klebsiella pneumoniae and Enterobacter aerogenes alleles of fimH were downloaded from the PATRIC database (http://www.patricbrc.org).
Supplementary Material
ACKNOWLEDGMENTS
We thank the personnel of KPWARI for assistance in collection of samples and Sifang Chen for proofreading the manuscript.
This work was supported by the National Institutes of Health (grant numbers R01AI106007 and R42 AI116114-02 to E.V.S.).
E.V.S. conceived the project and designed the experiments. D.K. performed control sample sequencing and analysis. All other sequencing, validation, and analysis were performed by S.G.S. V.T. provided study data and samples. M.R. programmed the algorithm; M.R. and S.G.S. tested and calibrated it. S.G.S. and E.V.S. wrote the manuscript with input from all of us.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01866-19.
REFERENCES
- 1.Heintz-Buschart A, Wilmes P. 2018. Human gut microbiome: function matters. Trends Microbiol 26:563–574. doi: 10.1016/j.tim.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Caputi V, Giron MC. 2018. Microbiome-gut-brain axis and Toll-like receptors in Parkinson’s disease. Int J Mol Sci 19:1689. doi: 10.3390/ijms19061689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perez-Pardo P, Hartog M, Garssen J, Kraneveld AD. 2017. Microbes tickling your tummy: the importance of the gut-brain axis in Parkinson’s disease. Curr Behav Neurosci Rep 4:361–368. doi: 10.1007/s40473-017-0129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanmiguel C, Gupta A, Mayer EA. 2015. Gut microbiome and obesity: a plausible explanation for obesity. Curr Obes Rep 4:250–261. doi: 10.1007/s13679-015-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De la Cuesta-Zuluaga J, Corrales-Agudelo V, Velásquez-Mejía EP, Carmona JA, Abad JM, Escobar JS. 2018. Gut microbiota is associated with obesity and cardiometabolic disease in a population in the midst of westernization. Sci Rep 8:11356. doi: 10.1038/s41598-018-29687-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roszyk E, Puszczewicz M. 2017. Role of human microbiome and selected bacterial infections in the pathogenesis of rheumatoid arthritis. Reumatologia 55:242–250. doi: 10.5114/reum.2017.71641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bu J, Wang Z. 2018. Cross-talk between gut microbiota and heart via the routes of metabolite and immunity. Gastroenterol Res Pract 2018:6458094. doi: 10.1155/2018/6458094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dzidic M, Boix-Amorós A, Selma-Royo M, Mira A, Collado MC. 2018. Gut microbiota and mucosal immunity in the neonate. Med Sci Basel 6:E56. doi: 10.3390/medsci6030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nunez G. 2017. Linking pathogen virulence, host immunity and the microbiota at the intestinal barrier. Keio J Med 66:14. doi: 10.2302/kjm.66-001-ABST. [DOI] [PubMed] [Google Scholar]
- 10.Tenaillon O, Skurnik D, Picard B, Denamur E. 2010. The population genetics of commensal Escherichia coli. Nat Rev Microbiol 8:207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 11.Gordon DM, O'Brien CL, Pavli P. 2015. Escherichia coli diversity in the lower intestinal tract of humans. Environ Microbiol Rep 7:642–648. doi: 10.1111/1758-2229.12300. [DOI] [PubMed] [Google Scholar]
- 12.Costea PI, Coelho LP, Sunagawa S, Munch R, Huerta-Cepas J, Forslund K, Hildebrand F, Kushugulova A, Zeller G, Bork P. 2017. Subspecies in the global human gut microbiome. Mol Syst Biol 13:960. doi: 10.15252/msb.20177589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Metwaly A, Haller D. 2019. Strain-level diversity in the gut: the P copri case. Cell Host Microbe 25:349–350. doi: 10.1016/j.chom.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Zhang C, Zhao L. 2016. Strain-level dissection of the contribution of the gut microbiome to human metabolic disease. Genome Med 8:41. doi: 10.1186/s13073-016-0304-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leatham MP, Banerjee S, Autieri SM, Mercado-Lubo R, Conway T, Cohen PS. 2009. Precolonized human commensal Escherichia coli clones serve as a barrier to E. coli O157:H7 growth in the streptomycin-treated mouse intestine. Infect Immun 77:2876–2886. doi: 10.1128/IAI.00059-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hecht AL, Casterline BW, Earley ZM, Goo YA, Goodlett DR, Bubeck Wardenburg J. 2016. Clone competition restricts colonization of an enteric pathogen and prevents colitis. EMBO Rep 17:1281–1291. doi: 10.15252/embr.201642282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lam LH, Monack DM. 2014. Intraspecies competition for niches in the distal gut dictate transmission during persistent Salmonella infection. PLoS Pathog 10:e1004527. doi: 10.1371/journal.ppat.1004527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sassone-Corsi M, Nuccio SP, Liu H, Hernandez D, Vu CT, Takahashi AA, Edwards RA, Raffatellu M. 2016. Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature 540:280–283. doi: 10.1038/nature20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno E, Johnson JR, Perez T, Prats G, Kuskowski MA, Andreu A. 2009. Structure and urovirulence characteristics of the fecal Escherichia coli population among healthy women. Microbes Infect 11:274–280. doi: 10.1016/j.micinf.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Bailey JK, Pinyon JL, Anantham S, Hall RM. 2010. Commensal Escherichia coli of healthy humans: a reservoir for antibiotic-resistance determinants. J Med Microb 59:1331–1339. doi: 10.1099/jmm.0.022475-0. [DOI] [PubMed] [Google Scholar]
- 21.Gorrie CL, Mirceta M, Wick RR, Judd LM, Wyres KL, Thomson NR, Strugnell RA, Pratt NF, Garlick JS, Watson KM, Hunter PC, McGloughlin SA, Spelman DW, Jenney AWJ, Holt KE. 2018. Antimicrobial-resistant Klebsiella pneumoniae carriage and infection in specialized geriatric care wards linked to acquisition in the referring hospital. Clin Infect Dis 67:161–170. doi: 10.1093/cid/ciy027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Zhu J. 2017. Targeted metabolic profiling rapidly differentiates Escherichia coli and Staphylococcus aureus at species and strain level. Rapid Commun Mass Spectrom 31:1669–1676. doi: 10.1002/rcm.7949. [DOI] [PubMed] [Google Scholar]
- 23.Galardini M, Koumoutsi A, Herrera-Dominguez L, Cordero Varela JA, Telzerow A, Wagih O, Wartel M, Clermont O, Denamur E, Typas A, Beltrao P. 2017. Phenotype inference in an Escherichia coli strain panel. Elife 6:e31035. doi: 10.7554/eLife.31035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bevan ER, McNally A, Thomas CM, Piddock LJV, Hawkey PM. 2018. Acquisition and loss of CTX-M-producing and non-producing Escherichia coli in the fecal microbiome of travelers to South Asia. mBio 9:e02408-18. doi: 10.1128/mBio.02408-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robin F, Beyrouthy R, Bonacorsi S, Aissa N, Bret L, Brieu N, Cattoir V, Chapuis A, Chardon H, Degand N, Doucet-Populaire F, Dubois V, Fortineau N, Grillon A, Lanotte P, Leyssene D, Patry I, Podglajen I, Recule C, Ros A, Colomb-Cotinat M, Ponties V, Ploy MC, Bonnet R. 2017. Inventory of extended-spectrum-β-lactamase-producing Enterobacteriaceae in France as assessed by a multicenter study. Antimicrob Agents Chemother 61:e01911-16. doi: 10.1128/AAC.01911-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta M, Didwal G, Bansal S, Kaushal K, Batra N, Gautam V, Ray P. 2019. Antibiotic-resistant Enterobacteriaceae in healthy gut flora: a report from north Indian semi-urban community. Indian J Med Res 149:276–280. doi: 10.4103/ijmr.IJMR_207_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson JR, Johnston B, Clabots C, Kuskowski MA, Castanheira M. 2010. Escherichia coli sequence type ST131 as the major cause of serious multidrug-resistant E. coli infections in the United States. Clin Infect Dis 51:286–294. doi: 10.1086/653932. [DOI] [PubMed] [Google Scholar]
- 28.Johnson JR, Tchesnokova V, Johnston B, Clabots C, Roberts PL, Billig M, Riddell K, Rogers P, Qin X, Butler-Wu S, Price LB, Aziz M, Nicolas-Chanoine MH, Debroy C, Robicsek A, Hansen G, Urban C, Platell J, Trott DJ, Zhanel G, Weissman SJ, Cookson BT, Fang FC, Limaye AP, Scholes D, Chattopadhyay S, Hooper DC, Sokurenko EV. 2013. Abrupt emergence of a single dominant multidrug-resistant clone of Escherichia coli. J Infect Dis 207:919–928. doi: 10.1093/infdis/jis933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burgess MJ, Johnson JR, Porter SB, Johnston B, Clabots C, Lahr BD, Uhl JR, Banerjee R. 2015. Long-term care facilities are reservoirs for antimicrobial-resistant sequence type 131 Escherichia coli. Open Forum Infect Dis 2:ofv011. doi: 10.1093/ofid/ofv011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson JR, Porter S, Thuras P, Castanheira M. 2017. The pandemic H30 subclone of sequence type 131 (ST131) as the leading cause of multidrug-resistant Escherichia coli infections in the United States (2011–2012). Open Forum Infect Dis 4:ofx089. doi: 10.1093/ofid/ofx089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tchesnokova V, Rechkina E, Chan D, Haile HG, Larson L, Schroeder DW, Solyanik T, Shibuya S, Hansen KE, Ralston JD, Riddell K, Scholes D, Sokurenko EV. 2019. Pandemic uropathogenic fluoroquinolone-resistant Escherichia coli have enhanced ability to persist in the gut and cause bacteriuria in healthy women. Clin Infect Dis 4:ciz547. doi: 10.1093/cid/ciz547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ong SH, Kukkillaya VU, Wilm A, Lay C, Ho EX, Low L, Hibberd ML, Nagarajan N. 2013. Species identification and profiling of complex microbial communities using shotgun Illumina sequencing of 16S rRNA amplicon sequences. PLoS One 8:e60811. doi: 10.1371/journal.pone.0060811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen W, Zhang CK, Cheng Y, Zhang S, Zhao H. 2013. A comparison of methods for clustering 16S rRNA sequences into OTUs. PLoS One 8:e70837. doi: 10.1371/journal.pone.0070837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zolfo M, Tett A, Jousson O, Donati C, Segata N. 2017. MetaMLST: multi-locus clone-level bacterial typing from metagenomic samples. Nucleic Acids Res 45:e7. doi: 10.1093/nar/gkw837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, Truong DT, Tett A, Morrow AL, Segata N. 2016. Clone-level microbial epidemiology and population genomics from shotgun metagenomics. Nat Methods 13:435–438. doi: 10.1038/nmeth.3802. [DOI] [PubMed] [Google Scholar]
- 36.Nayfach S, Rodriguez-Mueller B, Garud N, Pollard KS. 2016. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res 26:1612–1625. doi: 10.1101/gr.201863.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer M, Strauch B, Renard BY. 2017. Abundance estimation and differential testing on strain level in metagenomics data. Bioinformatics 33:i124–i132. doi: 10.1093/bioinformatics/btx237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weissman SJ, Johnson JR, Tchesnokova V, Billig M, Dykhuizen D, Riddell K, Rogers P, Qin X, Butler-Wu S, Cookson BT, Fang FC, Scholes D, Chattopadhyay S, Sokurenko EV. 2012. High-resolution two-locus clonal typing of extraintestinal pathogenic Escherichia coli. Appl Environ Microbiol 78:1353–1360. doi: 10.1128/AEM.06663-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reference deleted.
- 40.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson MA, Whitlock JE, Harwood VJ. 2006. Diversity and distribution of Escherichia coli genotypes and antibiotic resistance phenotypes in feces of humans, cattle, and horses. Appl Environ Microbiol 72:6914–6922. doi: 10.1128/AEM.01029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richter TKS, Hazen TH, Lam D, Coles CL, Seidman JC, You Y, Silbergeld EK, Fraser CM, Rasko DA. 2018. Temporal variability of Escherichia coli diversity in the gastrointestinal tracts of Tanzanian children with and without exposure to antibiotics. mSphere 3:e00558-18. doi: 10.1128/mSphere.00558-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diard M, Garry L, Selva M, Mosser T, Denamur E, Matic I. 2010. Pathogenicity associated islands in extraintestinal pathogenic Escherichia coli are fitness elements involved in intestinal colonization. J Bacteriol 192:4885–4893. doi: 10.1128/JB.00804-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le Gall T, Clermont O, Gouriou S, Picard B, Nassif X, Denamur E, Tenaillon O. 2007. Extraintestinal virulence is a coincidental by-product of commensalism in B2 phylogenetic group Escherichia coli strains. Mol Biol Evol 24:2373–2384. doi: 10.1093/molbev/msm172. [DOI] [PubMed] [Google Scholar]
- 45.Nielsen KL, Stegger M, Godfrey PA, Feldgarden M, Andersen PS, Frimodt-Møller N. 2016. Adaptation of Escherichia coli traversing from the faecal environment to the urinary tract. Int J Med Microbiol 306:595–603. doi: 10.1016/j.ijmm.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreno E, Andreu A, Pérez T, Sabaté M, Johnson JR, Prats G. 2006. Relationship between Escherichia coli strains causing urinary tract infection in women and the dominant faecal flora of the same hosts. Epidemiol Infect 134:1015–1023. doi: 10.1017/S0950268806005917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smati M, Clermont O, Le Gal F, Schichmanoff O, Jauréguy F, Eddi A, Denamur E, Picard B. 2013. Real-time PCR for quantitative analysis of human commensal Escherichia coli populations reveals a high frequency of subdominant phylogroups. Appl Environ Microbiol 79:5005–5012. doi: 10.1128/AEM.01423-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Philip TLC, Clausen F, Aarestrup M, Lund O. 2018. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 19:307. doi: 10.1186/s12859-018-2336-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tchesnokova V, Avagyan H, Billig M, Chattopadhyay S, Aprikian P, Chan D, Pseunova J, Rechkina E, Riddell K, Scholes D, Fang FC, Johnson JR, Sokurenko EV. 2016. A novel 7-single nucleotide polymorphism-based clonotyping test allows rapid prediction of antimicrobial susceptibility of extraintestinal Escherichia coli directly from urine specimens. Open Forum Infect Dis 3:ofw002. doi: 10.1093/ofid/ofw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.StataCorp. 2019. Stata statistical software, release 16. StataCorp LLC, College Station, TX. [Google Scholar]
- 53.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waskom M, Botvinnik O, O’Kane D, Hobson P, Lukauskas S, Gemperline DC, Augspurger T, Halchenko Y, Cole JB, Warmenhoven J, de Ruiter J, Pye C, Hoyer S, Vanderplas J, Villalba S, Kunter G, Quintero E, Bachant P, Martin M, Meyer K, Miles A, Ram Y, Yarkoni T, Williams ML, Evans C, Fitzgerald C, Fonnesback C, Lee A, Qalieh A. 2017. Seaborn: statistical data visualization. http://seaborn.pydata.org. Retrieved 5 February 2019.
- 55.Roer L, Tchesnokova V, Allesoe R, Muradova M, Chattopadhyay S, Ahrenfeldt J, Thomsen MCF, Lund O, Hansen F, Hammerum AM, Sokurenko E, Hasman H. 2017. Development of a Web tool for Escherichia coli subtyping based on fimH alleles. J Clin Microbiol 55:2538–2543. doi: 10.1128/JCM.00737-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.