

Abstract
Rieske oxygenases (ROs) catalyze a large range of oxidative chemistry. We have shown that cis-dihydrodiol-forming Rieske dioxygenases first react with their aromatic substrates via an active site nonheme Fe(III)-superoxide; electron transfer from the Rieske cluster then completes the product-forming reaction. Alternatively, two-electron reduced Fe(III)-peroxo or hydroxo-Fe(V)-oxo activated oxygen intermediates are possible and may be utilized by other ROs to expand the catalytic range. Here, the reaction of a Rieske monooxygenase, salicylate 5-hydroxylase (S5H), which does not form a cis-dihydrodiol is examined. Single-turnover kinetic studies show fast binding of salicylate and O2. Transfer of the Rieske electron required to form the gentisate product occurs through bonds over ~12 Å and must also be very fast. However, the observed rate constant for this reaction is much slower than expected and sensitive to substrate type. This suggests that initial reaction with salicylate occurs using the same Fe(III)-superoxo-level intermediate as Rieske dioxygenases and that this reaction limits the observed rate of electron transfer. A transient intermediate (λmax=700 nm) with an EPR resonance at g=4.3 is observed after product is formed in the active site. Use of 17O2 (I=5/2) results in hyperfine broadening of the g=4.3 signal, showing that gentisate binds to the mononuclear iron via its C5-OH in the intermediate. The chromophore and EPR signal allow study of product release in the catalytic cycle. Comparison of the kinetics of single and multiple turnover reactions shows that re-reduction of the metal centers accelerates product release ~300-fold, providing insight into the regulatory mechanism of ROs.
Graphical Abstract
INTRODUCTION
The nonheme iron Rieske oxygenases (RO) are a diverse family of enzymes that are expressed broadly across Nature.1–11 The first identified RO reactions were the dearomatizing cis-dihydroxylation of benzene, benzoate, naphthalene, and other unactivated aromatics in which both atoms of molecular oxygen were transferred to the substrate (Scheme 1).12
Scheme 1:

Reaction of Benzoate 1,2-Dioxygenase (BZDO), a cis-Dihydrodiol-Forming Rieske Oxygenase
Although this reaction is the only type uniquely catalyzed by ROs, a remarkable array of other reactions are also facilitated.13, 14 These reactions include: arene and non-arene hydroxylation15–18, methyl hydroxylation10, O-/N-demethylation19–22, N-oxygenation23, desaturation24, O- / N-dealkylation25, 26, oxidative carbocyclization1, 27, sterol desaturation28, and C-S bond cleavage29. In some cases, a single RO is capable of carrying out many of these types of reactions depending on the substrates supplied.30 The use of active site RO variants has also been shown to expand the repertoire of wild-type enzyme chemistries.31–33 A key question from a mechanistic perspective is whether the same type of reactive oxygen intermediate is formed by all ROs and for all substrate reactions, or whether several different reactive oxygen intermediates are generated in order to accommodate the diverse reactions catalyzed by the RO family.
The ROs are either two- or three-protein component systems. In vivo, the two electrons required for catalysis are shuttled from NADH to the α3 or α3β3 oxygenase component via an FAD and plant-type (Cys4 [2Fe-2S]) ferredoxin-containing reductase and Rieske (Cys2His2 [2Fe-2S]) ferredoxin in three-component systems,34 or solely by reductase in two-component systems.35, 36 The first X-ray crystal structure of the α3β3 oxygenase component of a RO was of the naphthalene 1,2-dioxygenase (NDO), and it continues to serve as the prototype for the rest of the family.37 In this structure, each α-subunit contains a mononuclear ferrous ion in the active site and a Rieske cluster approximately 44 Å away. This distance is too far for efficient electron transfer between the sites. However, the α3β3 quaternary structure of NDO is assembled such that the Rieske cluster is approximately 12 Å away from the mononuclear iron site in an adjacent α-subunit. This pair of metal centers is connected by a short through-bond pathway for electron transfer,38 and it is considered to be the active unit of the enzyme.37 The mononuclear iron and Rieske cluster cross-subunit interface catalytic unit is also conserved in α3 ROs.16, 39
Studies of NDO and BZDO revealed a complex regulatory scheme designed to ensure efficient coupling of electron transfer for O2 activation and product formation.35, 40 Nearly all as-isolated ROs have an Fe(II) in the active site. This state could potentially bind O2, but no binding is observed in the absence of a substrate and reduction of the Rieske cluster. Structural and spectroscopic studies have shown that substrate binding near (but not to) the mononuclear iron site converts the Fe(II) from 6 to 5 coordinate41, and reduction of the Rieske cluster causes the Fe(II) to move slightly away from the substrate.16, 17, 42, 43 Together these changes open a site for O2 to bind and initiate catalysis. At the end of a single turnover of chemically reduced enzyme, the product remains bound in the active site, suggesting that re-reduction of the mononuclear iron is required for product release.35, 40
The most extensive mechanistic studies of ROs have also been conducted using cis-dihydrodiol forming NDO and BZDO.35, 40 Beginning with both metal centers in the one-electron reduced forms, a single-turnover reaction with nearly stoichiometric product yield occurred after addition of an aromatic substrate and O2. Both metal centers were oxidized in the reaction, showing that two electrons are required for product formation. This finding led to the proposal that a two-electron reduced form of oxygen such as an Fe(III)-peroxo or hydroxo-Fe(V)-oxo species is generated (Scheme 2, path A). Electronically equivalent species are formed in the reaction cycles of cyt. P45044, 45 and methane monooxygenase,46, 47 enzymes that catalyze a similarly large array of oxygenation chemistry. In support of this model, it was demonstrated that addition of H2O2 to NDO or BZDO along with a substrate resulted in formation of the cis-dihydroxylated product.48, 49 Also, a side-on-bound peroxy adduct was trapped and characterized structurally by X-ray crystallography in NDO and spectroscopically in BZDO.49, 50 In the native reaction cycle, it was proposed that O2 binding to the mononuclear Fe(II) resulted in rapid transfer of an electron from the Rieske cluster to yield the two-electron reduced oxygen intermediate which could react with substrate.40
Scheme 2.

Alternative Reaction Mechanisms Proposed for Rieske Dioxygenases
Recently, the mechanism shown in Scheme 2, path A was shown not to operate as proposed in the cis-dihydrodiol forming reaction of BZDO. It was observed that the rate constant for electron transfer from the Rieske cluster is strongly dependent on the nature of the substrate.35, 51 In general, aromatic substrates with more electron withdrawing substituents cause slower electron transfer between the metal centers. This observation implied that the initial reaction of an activated O2 species with substrate occurs prior to electron transfer from the Rieske cluster (Scheme 2, path B). In other words, the activated iron-oxygen reactive species is only one electron reduced, most logically, an Fe(III)-superoxo moiety. Generally, such species only react readily with easily oxidized or activated substrates,52–54 but in the case of the RO, rapid electron transfer from the Rieske cluster after initial attack may greatly amplify the potency of the intermediate.51, 55
The reactivity of the putative Fe(III)-superoxo moiety in cis-dihydrodiol formation does not rule out the possible efficacy of a two-electron reduced oxygen species in other types of RO reactions. Indeed, reactions of model complexes known to form a high-valent species equivalent to that which would result from cleavage of an Fe(III)-peroxo O-O bond in the RO active site have been shown to carry out oxygenase reactions.56, 57 The tight electronic connection between the Fe(II) and Rieske cluster sites makes it quite reasonable that Fe(III)-superoxo, Fe(III)-peroxo, and HO-Fe(V)-oxo could each form in the RO active site.
One approach to probing the mechanistic uniformity or diversity of ROs, is to apply the same mechanistic probes developed for the cis-dihydrodiol forming dioxygenases to a RO that exclusively catalyzes another type of chemistry. Here we examine the reactivity of the RO salicylate 5-hydroxylase (S5H) that catalyzes conversion of salicylate (2-hydroxybenzoate) to gentisate (2,5-dihydroxybenzoate) in Ralstonia sp. strain U2 (Scheme 3). Salicylate occurs at a pathway branch point during bacterial aromatic catabolism.58 Onward metabolism occurs via alternative pathways: either conversion to gentisate by S5H or to catechol by either a Rieske oxygenase59 or a flavoenzyme.60
Scheme 3:

Reaction of Salicylate 5-Hydroxylase
Past studies have shown that S5H is a three component system: Rieske oxygenase (S5HH, 209 kD, α3β3 hydroxylase), reductase (S5HR, 37 kD monomer), and Rieske ferredoxin (S5HF, 12.5 kD monomer).58, 61–63 The components have the same complement of metal centers and/or cofactors as described above for the Rieske dioxygenases. S5H can catalyze monooxygenase reactions for a variety of aromatic substrates, yielding only phenol adducts rather that cis-dihydrodiols. The common aromatic substrate type with different reaction outcomes, allows a useful comparison with the cis-dihydrodiol-forming ROs. In the current study, transient kinetic studies of salicylate and O2 binding as well as Rieske cluster electron transfer are used together with product analysis and an alternative substrate reaction to show that S5H shares many of the mechanistic properties of the cis-dihydrodiol forming dioxygenases up at least up to the point of O-O bond cleavage. Moreover, novel chromophoric properties of the product complex allow several tenets of the regulatory mechanism of S5H to be compared with those of the cis-dihydrodiol-forming dioxygenases. The studies reveal that at least two distinct classes of ROs employ a similar mechanism of O2 activation that differs fundamentally from those of cyt. P450, MMO, and α-keto glutarate-linked oxygenases.
EXPERIMENTAL PROCEDURES
General Methods.
Standard procedures are described in the Supporting Information. Water used in all experiments was purified with a Millipore Super-Q system. All commercial reagents were purchased from standard vendors and used without further purification. Gases were purchased from Matheson. 17O2 gas (50 % 17O2 enriched) was obtained from Monsanto (DOE Mound site) and 18O2 gas (98 atom %) was purchased from Icon Isotopes (Dexter, MI). Unless noted, all enzymatic reactions were conducted in 200 mM HEPES, 100 mM NaCl, 5 % glycerol, pH 8 buffer. The concentration of S5HH is given in terms of the αβ protomer concentration. Iron quantification was carried out as previously described.40
Electronic absorption spectra were recorded using a Hewlett-Packard 8453 diode array spectrophotometer. EPR spectra were recorded using a Bruker ELEXSYS E-500 spectrometer equipped with an Oxford ESR-910 liquid helium cryostat. EPR spectra were analyzed as detailed in the Supporting Information.
Cloning of S5HR, S5HF and S5HH.
Ralstonia sp. strain U2 genes for S5HR (nagAa), S5HF (nagAb) and S5HH (nagGH) were cloned from pWWF6 (Table 1).58, 64 Cloning oligonucleotides used for PCR amplification are shown in Table 1. Restriction sites are bolded and underlined. Following digest with the appropriate restriction enzymes, constructs were ligated into similarly cut over-expression plasmids. The resulting constructs were verified by sequencing at the University of Minnesota Genomics Center. E. coli strains DH5α and BL21(DE3) were used for cloning and over-expression, respectively.
Table 1.
Cloning Oligonucleotides and Plasmids Used to Produce S5H Proteins
| Cloning oligonucleotides | Sequence (restriction sites bolded and underlined) |
|---|---|
| 7nt-NdeI-nagAa-FOR | AGAGTCACATATGATGGAACTGGTAGTAGAACCCCTCAATTTGC |
| 7nt-nagAa-SpeI-REV | AGAGTCAACTAGTTCAGACGCCGCTGGGATAGAACG |
| 7nt-NdeI-nagAb-FOR | AGAGTCACATATGATGACTCAGAACTGGATTGATGCAGC |
| 7nt-nagAb-SpeI-REV | AGAGTCAACTAGTTTACTCCATTTTGAGCATCACGCG |
| NdeI nagG for2 | TGCATATGATGAGTGAACCCCAACGATTAAAACCCGTGTTTCCCC |
| HindIII nagH rev2 | TCAAGCTTTCAGATTGGATAGATCACAGAGTTGGGGATCATTTCGC |
| Plasmids | Description |
| pWWF6 | pUC18 containing Ralstonia sp. strain U2 nag genes58, 64 |
| pACYC-isc | pACYC-Duet containing part of the E. coli isc operon /P15A origin / chloramphenicol resistant65 |
| p11 | pET15b-based plasmid with N-terminal His6-tag and TEV protease site/ ColE1 origin / ampicillin resistanta |
| pET28b(+) | pET plasmid with N-terminal His6-tag and thrombin cleavage site/ pBR322 origin / kanamycin resistantb |
| pS5HR | p11 plasmid containing Ralstonia sp. strain U2 nag genes nahAa |
| pS5HF | p11 plasmid containing Ralstonia sp. strain U2 nag genes nahAb |
| pS5HH | pET28 plasmid containing Ralstonia sp. strain U2 nag genes nagGH |
DNASU Plasmid Repository
Novagen, Madison, USA
Heterologous Expression of S5HR, S5HF, and S5HH.
S5HR and S5HF were over-expressed using the following procedure. E. coli BL21(DE3) were co-transformed with pACYC-isc and either pS5HR or pS5HF. Cultures were grown in LB media with carbenicillin (50 μg/ml) and chloramphenicol (34 μg/ml). A single colony was transferred to 5 ml media, incubated for 7 h (37 °C, 250 rpm), then 20 μl was used to inoculate 500 ml media which was grown overnight (28 °C, 140 rpm). The overnight culture was used to inoculate media (700 ml) containing (NH4)2Fe(SO4)2·6H2O (25 μM). Baffled 2-L flasks were used and 1 drop of Antifoam 204 (Sigma-Aldrich) was added. Cultures were incubated at 30 °C, 160 rpm. When OD600 ~ 0.5 was reached, the culture was cooled to 16 °C, then IPTG (285 μM) was added. After 16 h, cells were harvest by centrifugation, and the cell paste was stored at −80 °C.
S5HH was over-expressed as follows. E. coli BL21(DE3) was co-transformed with pS5HH and pACYC-isc. Liquid starter cultures were grown using LB media with kanamycin (50 μg/ml) and chloramphenicol (34 μg/ml). A single colony was transferred to 5 ml media, incubated for 6 h (37 °C, 250 rpm) then 20 μl was used to inoculate 500 ml media which was grown overnight (28 °C, 160 rpm). The overnight culture was used to inoculate TB-glycerol media (1 L) containing kanamycin (50 μg/ml), chloramphenicol (34 μg/ml) and (NH4)2Fe(SO4)2·6H2O (32 μM). Baffled 2-L flasks were used and 1 drop of Antifoam 204 (Sigma-Aldrich) was added. Cultures were incubated at 30 °C, 200 rpm. When OD600 ~ 0.5 was reached, (NH4)2Fe(SO4)2·6H2O (32 μM) was added, the culture cooled to 16 °C, and IPTG (400 μM) added. After 16 h, cells were harvest by centrifugation, and the cell paste was stored at −80 °C.
Purification of S5HR, S5HF, and S5HH.
S5HR or S5HF cell paste (38 g) was suspended in imidazole-free 50 mM HEPES, 300 mM NaCl, 10 % glycerol, 1 mM DTT, pH 8 buffer (120 ml) containing DNAseI (5 mg) and lysozyme (10 mg) and then stirred (4 °C, 20 min). The cell suspension was lysed by sonication using a Branson sonifier with 3/4” tip installed. A duty cycle of 90 % and maximum output were used. The solution, in a steel beaker, was cooled to −1 °C using a saturated calcium chloride-ice bath prior to sonication. The beaker remained in the ice bath throughout the procedure. Each sonication cycle was stopped when the solution’s temperature warmed to 10 °C whereupon sonication was ceased until the solution re-cooled to −1 °C. Sonication was performed for a total of 6 min. All subsequent procedures were performed at 4 °C. The lysate was centrifuged at 75,600 RCFmax for 1 h. The cell-free extract was applied to a 25 ml Roche cOmplete His-Tag purification resin (Sigma-Aldrich) equilibrated with imidazole-free 50 mM HEPES, 300 mM NaCl, 10 % glycerol, 1 mM DTT, pH 8 buffer. The column was washed with 10 mM imidazole-containing buffer followed by elution with 250 mM imidazole-containing buffer. Fractions were screened via UV-visible spectroscopy and SDS-PAGE. Pooled protein fractions were concentrated using an Amicon pressure-driven device (S5HR, 10 kDa cut-off; S5HF, 3 kDa cut off). Note that in a previous study the authors reported that S5HF is a dimer.63 Using non-denaturing gel chromatography on a Superdex 75 Increase 10/300 column installed on a Bio-Rad DuoLogic system, we find that S5HF runs as a monomer versus five protein standards of 1350 to 670,000 Da, BioRad 1511901.
For S5HR, buffer exchange and removal of imidazole was achieved by applying the concentrated protein solution (25 ml) to a Sephadex G-25 medium column (GE Healthcare) (250 ml) equilibrated with 50 mM HEPES, 100 mM NaCl, 5 % glycerol, 1 mM DTT, pH 8 buffer. The red-brown fractions were collected and concentrated using a centrifugal concentration device (S5HR, 10 kDa cut off). The resulting S5HR protein was aliquoted, flash-frozen in liquid nitrogen and stored at −80 °C. In the case of the S5HF protein, buffer exchange and removal of imidazole was achieved by dialysis in 50 mM HEPES, 100 mM NaCl, 5 % glycerol, 1 mM DTT, pH 8 buffer using a Slide-a-lyser dialysis cassette (3.5 kDa cut-off) (Thermo-Fisher). The resulting S5HF protein was aliquoted, flash-frozen in liquid nitrogen and stored at −80 °C.
S5HH cell paste (25 g) was suspended in imidazole-free 50 mM HEPES, 300 mM NaCl, 10 % glycerol, 1 mM DTT, pH 8 buffer (100 ml) containing DNAseI (5 mg) and lysozyme (10 mg) and then stirred (4 °C, 20 min). The cell suspension was lysed via sonication as described above. All subsequent procedures were performed at 4 °C. The lysate was centrifuged at 75,600 RCFmax for 1 h. The cell-free extract was applied to a 25 ml Roche cOmplete His-Tag purification resin equilibrated with 5 mM imidazole-containing 50 mM HEPES, 300 mM NaCl, 10 % glycerol, 1 mM DTT, pH 8 buffer. The column was washed with 20 mM imidazole-containing buffer followed by elution with 350 mM imidazole-containing buffer. Fractions were screened via UV-visible spectroscopy and SDS-PAGE. Pooled S5HH protein fractions were concentrated using an Amicon pressure-driven device (30 kDa cut-off). Buffer exchange and removal of imidazole was achieved by applying the concentrated protein solution (25 ml) to a Sephadex G-25 medium column (250 ml) equilibrated with 50 mM HEPES, 100 mM NaCl, 5 % glycerol, 1 mM DTT, pH 8 buffer. The red-brown fractions were collected and concentrated using a centrifugal concentration device (30 kDa cut off). The resulting S5HH protein (220 mg, 890 μM) was aliquoted, flash-frozen in liquid nitrogen and stored at −80 °C. At the conclusion of this purification method, S5HH contains a ferrous mononuclear iron in partial (55 – 65 %) occupancy and an approximately stoichiometric oxidized Rieske cluster per αβ protomer.
S5H protein concentrations were determined via UV absorption. Extinction coefficients were calculated from the molar protein concentration as determined using a modified Lowry method (BioRad DC Protein Assay) and the protein solution’s UV absorption (S5HR ε273 = 43,350 M−1cm−1; S5HF ε276 nm = 18,260 M−1cm−1; S5HH ε280 nm = 127,740 M−1cm−1).
Tagless S5HH.
Tagless S5HH was cloned, heterologously expressed, and purified as described in Supporting Information. The kinetics of steady state turnover were evaluated as described for the His-tagged S5HH and found to be unchanged.
Steady-state Reactions.
Steady-state S5H measurements were made using an Applied Photophysics SX.18MV spectrophotometer under aerobic conditions. Activity assays were performed by recording initial velocity absorbance changes at 340 nm upon rapid mixing of S5HH, S5HR, S5HF with salicylate and NADH. The UV-visible spectra of S5HH reaction cycle intermediates were recorded during steady-state continuous turnover using a diode array detector upon mixing S5HH, S5HR, and S5HF with salicylate and NADH. The reactant concentrations and other conditions for the assay and steady-state turnover experiments are given in the captions of the appropriate figures.
Stopped-Flow Analysis of Single-turnover Reactions.
Anaerobic, reduced S5HH was mixed with oxygen-saturated buffer containing substrate using an Applied Photophysics SX.18MV spectrophotometer at 4 °C. Either single-wavelength measurements at 453 nm and 700 nm or complete UV-visible spectra using a diode array detector were recorded. Methods for the preparation of anaerobic solutions and analysis of stopped-flow data are detailed in the Supporting Information.
Rapid Freeze Quench (RFQ) and Rapid Chemical Quench (RCQ).
Reduced S5HH was rapidly reacted with oxygen-saturated buffer containing salicylate using an Update 715 ram syringe controller to mix and dispense the reactants after a series of delays using calibrated tubing of different lengths (10 – 500 ms delay) or sequential syringe pushes at specific intervals (500 ms – 5 s delay). The reacted solution was either rapidly frozen on counter-rotating aluminum wheels partially submerged in liquid nitrogen to produce a frozen powder (RFQ)66 or quenched by dispensing into rapidly stirring 10 % trifluoroacetic acid (RCQ).51 For reaction times > 5 s, single-turnover reactions were performed in sealed vials followed by manual quench. After transfer into an EPR tube, RFQ samples were quenched via freezing upon rapid immersion in methanol-dry ice, and RCQ samples were quenched by pipetting into rapidly stirring 10 % trifluoroacetic acid (TFA). RCQ single-turnover samples were centrifuged to remove precipitated enzyme. Then, 1 M HEPES, pH 8 was added to the quenched sample (1:1 with TFA volume). The resulting solutions were analyzed using either HPLC or LC-MS and authentic product standards as described in the Supporting Information.
RESULTS
Enzyme Preparation and Characterization.
The genes for the His-tagged S5HH, S5HR, and S5HF components of S5H from Ralstonia sp. strain U2 (nagGH, nagAa, and nagAb, respectively) were cloned and expressed, and the proteins purified as described in Experimental Procedures and Table 1. Although the protein components exhibited physical properties (quaternary structures, molecular weights, metal and cofactor content) similar to those previously reported,63 the specific activity of the current preparation was increased nearly 30-fold at 23 °C (kcat = 1.86 ± 0.35 s−1 vs. 0.065 s−1; KM salicylate = 2.9 ± 0.4 μM vs. 102 μM) (Figure 1). At 4 °C, kcat is decreased to 0.64 ± 0.09 s−1, and this value is used below for comparison with transient kinetic data. The improvement in activity was achieved by new purification procedures, determination of the optimal ratio of components, and use of protein and substrate concentrations appropriate for the determined kcat and Km values (see Supporting Information for a discussion of the latter point). The UV-visible spectra of oxidized and reduced S5HH (Figure 1, inset) and the EPR spectrum of reduced S5HH (compare Figure 2A and B) are due to the Rieske Fe-S cluster and were found to be typical of the oxygenase component of all Rieske oxygenases characterized to date.13, 35, 40
Figure 1.

Steady-state and optical parameters of S5H. A buffered solution of S5HR (0.09 μM), S5HF (1.84 μM), and S5HH (0.23 μM), was reacted with a solution of NADH (742 μM) and salicylate (1–50 μM) using a stopped-flow spectrophotometer (2 mm pathlength, concentrations after mixing). Solutions were prepared in air-saturated 50 mM HEPES, 20 mM NaCl, 5 % glycerol, pH 8.0 buffer (~250 μM O2) at 23 °C. The slope of the initial portion of the time course was determined and converted into an initial velocity using the extinction of NADH at 340 nm, pH 8 corrected for the contribution of the gentisate product at this wavelength and pH (difference ε = 4300 mM−1 cm−1). The solid line is a fit to a hyperbolic expression. Inset: UV-visible spectra of oxidized S5HH (black) (178 μM) and reduced S5HH (red) (178 μM, 5.5 mM sodium dithionite) in standard buffer at 23 °C. Spectra were recorded under anaerobic conditions. The calculated Δε700 is 198 M−1cm−1, a value used in data analysis below.
Figure 2.

EPR spectra of S5HH. (A) As-isolated (mononuclear iron reduced, Rieske cluster oxidized) S5HH (470 μM). (B) S5HH (470 μM) stoichiometrically reduced with sodium dithionite and methyl viologen (40 μM). The * indicates a small overlapping derivative signal from reduced methyl viologen, indicating full reduction of S5HH. (C) In a single-turnover reaction, reduced S5HH (470 μM) with salicylate (7.96 mM) was reacted with oxygen (720 μM) in standard buffer at 4 °C and frozen after 30 s (concentrations after mixing). Instrument conditions: microwave power, 200 μW; modulation amplitude, 10 G; microwave frequency, 9.64 GHz; temperature, the g = 4.3 and g = 9.68 signals were detected at 2 K (black) and the Rieske cluster signal (g = 2.01, 1.91, 1.45) was detected at 20 K (red).
S5HH was also expressed and purified without the His-tag by traditional anion exchange and hydrophobic interaction chromatographies (see Supporting Information Experimental Procedures, and Table S1). The resulting enzyme was devoid of mononuclear iron and consequently inactive. However, the metal center could be reconstituted to the same level as the His-tagged S5HH (~60 %) by addition of ferrous ion under anaerobic conditions. The specific activity of this preparation was comparable to that of the His-tagged enzyme and the transient kinetics of electron transfer during the reaction cycle were unchanged. Consequently, the His-tagged S5HH was used for all experiments described here, because it could be obtained in the quantities required for the single-turnover transient kinetic and spectroscopic studies related below.
Product Formation During a Single Turnover.
Stoichiometrically reduced S5HH rapidly oxidizes in the presence of O2 and salicylate to yield the product gentisate (Figures S1 and S2). Both the metal centers were oxidized during the reaction (formation of mononuclear Fe(III) at g = 9.68 and 4.3 and loss of reduced Rieske cluster at g = 2.01, 1.91, 1.45) (Figure 2C), showing that only two electrons are required for product formation, as we have reported for Rieske dioxygenases in past studies.35, 40 The source of the oxygen in the product is O2 as shown by LC-MS analysis of a single turnover of S5HH in an 18O2 atmosphere (Figure S3) and by EPR studies described below.
Kinetics of Rieske Cluster Oxidation.
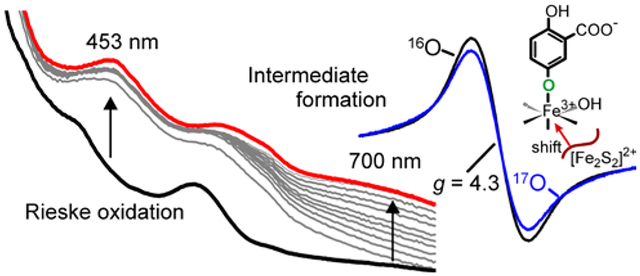
The time course for oxidation of the Rieske cluster during a single-turnover reaction of stoichiometrically reduced S5HH with large excesses of O2 and salicylate (pseudo-first order conditions) can be monitored using a stopped-flow spectrophotometer with UV-visible detection. Diode array spectra show that a large fraction of the Rieske cluster monitored in the 350 to 600 nm region is rapidly oxidized (Figure 3A) and then a species absorbing near 700 nm is formed more slowly, maximizing at 3 s. Over the next 1000 s, and the 700 nm species disappears slowly while spectral species in the 350 to 600 nm region grow in with an apparent isosbestic point at 575 nm, as shown in Figure 3B.
Figure 3.

Stopped-flow diode array spectra of a single-turnover reaction. Fully reduced S5HH (50 μM) with salicylate (1 mM) was reacted with O2 (900 μM) and salicylate (1 mM) (concentrations after mixing). (A) Period from 0 to 3 s. (B) Period from 3 s to 1000 s. Inset: Spectrum of the 700 nm intermediate determined by subtracting the spectral contribution of the Rieske cluster from 3 s spectrum. Conditions: 50 mM HEPES, 100 mM NaCl, 5% glycerol, pH 8, 4 °C.
The known spectra of the oxidized and reduced Rieske cluster (Figure 1, inset) allow the correct summation to be made to simulate the contribution of the Rieske cluster to the spectrum of the reaction mixture at 3 s (95% oxidized, 5% reduced, see below and Supporting Information). Subtraction of this pure Rieske cluster contribution from the composite spectrum reveals the spectrum of the new 700 nm species (Figure 3B, inset).
The kinetic parameters of the single-turnover reaction can be quantified using single wavelength detection (Figure 4). Under pseudo-first order conditions, the time courses can each be fit by a summation of exponential terms (Figure 4, white dashed curves, see Experimental Procedures). The reciprocal relaxation times (1/τ values) for the exponential phases at 453 nm (primarily from the Rieske cluster) and 700 nm (from the new intermediate) and their amplitude values are shown in Table 2. The data indicate two important aspects of the reaction with salicylate. First, the requirement for multiple exponential phases to fit the time course shows that there are multiple physical reaction steps in the process. The number of steps in the physical reaction is at least as great as the number of phases in the multi-exponential simulation, but the steps could be occurring either sequentially or in parallel. If all of the steps are irreversible, as seems likely (see below), the 1/τ values represent rate constants for the steps (but not the order of the steps). The second important aspect of the reaction is that at least some different steps in the overall reaction are reported at the two wavelengths, otherwise phases with the same 1/τ values would be observed in the fits. We have noted in the past for other ROs that the change in the UV-visible spectrum in the 350 to 600 nm region can be accounted for by oxidation of the Rieske cluster without the need to invoke significant contributions from other factors such as conformational changes.51 The nearly flat baseline in Figure 3B, inset, shows that the 700 nm intermediate does not contribute directly to the spectrum in this region. Since the oxidation of the Rieske cluster is a one-step event, it is not possible for a single electron transfer to generate multiple phases in the fit. Thus, the faster observed phases at 453 nm probably represent parallel steps during Rieske oxidation. However, the isosbestic point in the decay of the 700 nm species without a significant change in spectral maxima or lineshape below 600 nm opens the possibility that a process unrelated to S5HH Rieske oxidation contributes to the spectrum in the 350 – 600 nm region during the time period after formation of the Fe(III)-product complex. This possibility is discussed further in the Supporting Information. The data confirm the much larger rate constant for the Rieske cluster oxidation reaction in comparison to the formation and decay rate constants of the intermediate absorbing at 700 nm.
Figure 4.

Single-turnover time course of fully reduced S5HH (30 μM) reacted with salicylate (2.9 mM) and O2 (900 μM) at (top) long and (bottom) short times (concentrations after mixing) in standard buffer at 4 °C. The reaction was monitored at 453 nm (blue) and 700 nm (red). The fits to summed exponential equations are shown as white dashed lines over the data and the residuals are shown as insets in the bottom panel. The kinetic parameters from the fits (n = 5) are shown in Table 2. In the absence of salicylate, the S5HH oxidizes very slowly with a rate constant k of ≤0.0144 s−1.
Table 2.
Multiple Summed Exponential Fits for the Time Course of S5HH Single Turnover Reactions
| 453 nm | 700 nm | |||
|---|---|---|---|---|
| Salicylate | 1/τ, s−1 | Amplitude (abs) [%]a | 1/τ, s−1 | Amplitude (abs)a |
| 1/τ1 | 72 ± 5 | −0.042 ± 0.0035 [63] | 20.9 ± 1.1 | −0.044 ± 0.0035 |
| 1/τ2 | 3.5 ± 0.6 | −0.006 ± 0.0005 [9.3] | 0.48 ± 0.03 | −0.002 ± 0.0004 |
| 1/τ3 | 0.11 ± 0.01 | −0.007 ± 0.0004 [11.0] | 0.0014 ± 0.0002 | +0.041 ± 0.0035 |
| 1/τ4 | 0.003 ± 0.0005 | −0.011 ± 0.0005 [16.5] | ||
| Benzoate | ||||
| 1/τ1 | 4.1 ± 0.2 | −0.035 ± 0.003 [58] | 3.07 ± 0.25 | −0.032 ± 0.003 |
| 1/τ2 | 0.47 ± 0.05 | −0.012 ± 0.001 [19] | 0.32 ± 0.03 | −0.005 ± 0.0005 |
| 1/τ3 | 0.055 ± 0.006 | −0.008 ± 0.001 [12.8] | 0.002 ± 0.0002 | +0.026 ± 0.002 |
| 1/τ4 | 0.005 ± 0.0005 | −0.006 ± 0.0005 [9.9] | ||
| No substrate | ||||
| 1/τ1 | 0.144 ± 0.012 | −0.030 ± 0.002 | Not observed | Not observed |
| 1/τ2 | 0.065 ± 0.005 | −0.019 ± 0.001 | Not observed | Not observed |
A negative amplitude appears as an increase in absorbance during the time course
Kinetics of Product Formation.
The time course of product formation in the active site (as opposed to product release from the active site) can be monitored using single-turnover rapid chemical quench (RCQ). Fully-reduced S5HH was rapidly mixed with salicylate and O2, and then the reaction was quenched in TFA after a series of calibrated delay times (see Supporting Information Experimental Procedures). The product was quantified by HPLC versus an authentic standard. As shown in Figure 5, the formation of product was complete within 40 ms, meaning that the reaction step leading to product formation must have a rate constant much greater than 35 s−1. The only 1/τ for the fit to the reaction time course monitored at 453 nm (Table 2) that exceeds 35 s−1 is the fastest phase of the Rieske cluster oxidation. The nature of the product forming reaction, i.e. O-O bond breaking and addition of one oxygen to an aromatic ring, suggests that the reaction steps are irreversible. Thus, it is likely that the product is formed in only one of the parallel steps implied by the multiexponential fit to the time course at 453 nm, and that the rate constant for this step is given by the 1/τ for the fastest phase of the fit. The remaining steps in the time course are discussed in Supporting Information.
Figure 5.

HPLC analysis of RCQ samples from a single-turnover reaction of S5HH. Fully reduced S5HH (256 μM), salicylate (900 μM) and internal standard caffeic acid (50 μM) were reacted with O2 (900 μM) (concentrations after mixing) in standard buffer at 4 °C. The product formation at 4 °C is largely complete before the fastest chemical quench possible (~10 ms) so an accurate rate constant could not be obtained. The dashed blue line is an exponential curve with k = 200 s−1 meant to guide the eye. Inset: EPR spectra of rapid freeze quench samples from the single-turnover reaction at the times shown. Instrument conditions: microwave power, 200 μW; modulation amplitude, 10 G; microwave frequency, 9. 644 GHz; temperature, 2 K.
The proposal that product is formed in the fastest step of Rieske cluster reoxidation can be tested by comparing the absolute yield of product to the relative amplitude of the fastest phase. The yield of gentisate in the single-turnover reaction was be determined from the HPLC data to be approximately 50 % relative to the concentration of S5HH; 55–65 % of the mononuclear iron sites were occupied in the preparations used for the experiment. Accordingly, the amplitude of the fastest kinetic phase is 63 % of the overall amplitude change for the reaction, as shown in Table 2. The correlation of yield, phase amplitude, and mononuclear site occupancy suggests that the reaction is strongly coupled at the occupied sites, as we have noted previously for the Rieske dioxygenases.35, 40, 51
Although a fast phase associated with product formation can readily be identified in the data monitored at 453 nm, the same is not true at 700 nm where all of the 1/τ values are slower than 35 s−1. This observation suggests that the intermediate detected at 700 nm forms after product is generated, a conclusion compatible with the wavelength-dependent kinetics noted above.
The single-turnover reaction was also monitored using RFQ techniques (Figure 5, inset). The g = 4.3 EPR species forms rapidly, but on a slower time scale than product formation (~25% complete when product is fully formed). The similar time scales for formation of the g = 4.3 signal and 700 nm species suggest a common origin which is explored below. After incubation for 1 h at 4 °C, the sharp g = 4.3 resonance disappears and is replaced by a broad resonance typical of the oxidized mononuclear iron without a substrate or product ligand.
Dependence of the Single-turnover Kinetics on Salicylate and O2 Concentrations.
The observations that the product forming reaction requires two electrons and that the product is formed coincident with the oxidation of the Rieske cluster in the single-turnover reaction suggest that the kinetic time course monitored at 453 nm is rate-limited by some step between substrate binding and electron transfer between the metal centers. The actual electron transfer through bonds over 12 Å distance as found in NDO is likely to occur with a rate constant many orders of magnitude greater than those reported here,67 so an earlier step must be rate limiting. Figure 6 shows the salicylate and O2 dependencies of the fastest 1/τ of the single-turnover reaction monitored at 453 nm. For both substrates, the 453 nm data fit well to a hyperbolic function approaching the same maximal value. The hyperbolic shapes of the concentration dependent plots suggest that both salicylate and O2 bind rapidly and reversibly in an initial first step and then a slower step(s) follows, the rate constant for which maximizes as the enzyme substrate complex saturates (see Supporting Information Experimental Procedures).51, 68 If so, then the apparent Kd values reflect the binding affinity of the varied substrates. The value observed for salicylate (~ 380 μM) is much greater than the Km value for the overall reaction (~2.9 μM, Figure 1). This means that the Km is dominated by slow steps occurring after the product forming step. In contrast, the maximum 1/τ values observed in Figure 6 are very large and comparable to 1/τ for the fast phase of the Rieske cluster oxidation reaction at 4 °C (Table 2). Thus, the rate-limiting reaction in the product forming segment of the catalytic cycle occurs after substrates bind, but before the ultrafast electron transfer from the Rieske cluster. In contrast, the slow, rate-limiting step of the overall reaction (kcat = 0.64 s−1, 4 °C) occurs after product is formed in the active site and may be related to some aspect of the product release process.
Figure 6.

Dependence of the rate constant for Rieske cluster reoxidation on salicylate or O2 concentration. (Black) Salicylate was reacted with fully reduced S5HH (38 μM) and O2 (900 μM). (Red) O2 was reacted with fully reduced S5HH (50 μM) and salicylate (2 mM). Reactants were mixed using a stopped-flow spectrophotometer in standard buffer at 4 °C and the reaction time course monitored at 453 nm (concentrations after mixing). Rate constants were determined from multiple summed exponential fits to the data at 453 nm (average of 2–5 time courses) and the fastest 1/τ is plotted. The error at each point is approximately ±10 %. Little or no concentration dependence was observed for the reaction monitored at 700 nm.
It is significant that there is little or no salicylate or O2 concentration dependence observed for the time courses observed at 700 nm. This finding shows that an irreversible step such as O-O bond cleavage or phenolic product formation occurs between the addition of substrates and the formation of the new intermediate at 700 nm. Such a step would uncouple the 1/τ values of the slower processes monitored at 700 nm from the second order rate constants of the earlier fast steps. Thus, a reasonable hypothesis is that the product complex is being monitored at 700 nm. The alternative proposal that the chromophore derives from product released into solution can be ruled out by the known spectral properties of gentisate, λmax = 320 nm, where it is masked by the intense absorbance of the Rieske cluster. Although the X-ray crystal structure of a naphthalene dioxygenase product complex has been reported50, and we have suggested that EPR spectra found at the end of single turnover and peroxide shunt reactions of NDO and BZDO are characteristic of a product complex,35, 40, 49 it has not previously been possible to monitor the kinetics and optical spectral properties of the product complex of any Rieske oxygenase.
Nature of the Product Complex.
The nature of the g = 4.3 species was probed directly by conducting a single-turnover reaction in the presence of 17O2, which places 17O (nuclear spin I = 5/2) in the 5-position of the aromatic ring of the substrate. As shown in Figure 7, spectral line broadening from transferred nuclear hyperfine interactions is observed on this very narrow resonance (19 gauss peak to trough). This observation directly reveals three aspects of the reaction: (i) it shows unequivocally that S5H is an oxygenase, (ii) it demonstrates that the g = 4.3 species is an Fe(III)-product complex, and (iii) it shows that the product binds through the 5-position hydroxyl group. The formation of such a product phenolate bond with the Fe(III) would account for the wavelength, intensity, and rate of formation of the 700 nm species (see Discussion), so we propose that the g = 4.3 EPR signal and the 700 nm optical feature have the same origin. Quantification of the g = 4.3 species using protocatechuate 3,4-dioxygenase, a well-characterized standard with a similar EPR spectrum,69 shows that at least 45 % of the S5HH subunits have formed the complex (Supporting Information Experimental Procedures).
Figure 7.

EPR spectra of a 40 s sample of a single-turnover reaction of stoichiometrically reduced S5HH (472 μM) reacted with 16O2 (black, 720 μM) or 17O2 (blue, 676 μM, 50 % 17O2) and salicylate (8.88 mM) in standard buffer at 4 °C (concentrations after mixing). EPR spectra were normalized to the double integrated peak area of the g = 4.3 signal. Instrument conditions: microwave power, 200 μW; temperature, 2 K; modulation amplitude, 3 G; microwave frequency, 9.64 GHz.
Relevance of the Mononuclear Iron-Product Complex to the Reaction Cycle.
The 700 nm/g = 4.3 intermediate persists for several minutes after a single turnover (Figures 2–5). Since the turnover number for the enzyme in steady-state catalysis is much faster (kcat = 0.64 s−1 at 4 °C), the 700 nm/g = 4.3 intermediate is either not part of the reaction cycle or there is a mechanism to accelerate dissociation of the product complex during sustained turnover. In order to determine whether this species actually occurs in the reaction cycle, the reaction of S5HH, S5HR, and S5HF with excess salicylate, O2, and NADH was monitored at 453 nm and 700 nm at 4 °C using a stopped-flow spectrophotometer (Figure 8). Oxygen was limiting in the experiment, so that after a period of steady-state turnover, O2 was exhausted and the enzyme passed through a final cycle before returning to a fully reduced state. At 453 nm, the Rieske cluster optical absorbance rapidly decreased to a steady-state level of 40% reduced, while at 700 nm an increase in absorbance was observed consistent with formation of the 700 nm species. The Rieske cluster has a weak absorbance at 700 nm, and while this decreases during steady-state catalysis (due to S5HH reduction by NADH via S5HR and S5HF), it still contributes to the observed absorbance (Figure 1, inset). The extent of its contribution at 700 nm was estimated based on the fraction of reduced Rieske cluster measured at 453 nm during steady-state and the known extinction coefficients for the cluster at 700 nm (Δε700 = 198 M−1cm−1, See Figure 1). As shown in Figure 8, inset about 60 % of the absorbance at 700 nm during steady-state is due to the 700 nm intermediate, so it is clearly formed during steady-state turnover. Upon exhaustion of the O2, both the 453 nm and 700 nm absorbances decreased with the same rate constant of roughly 1 s−1, a value which is similar to the kcat for the reaction at 4 °C. The formation of the chromophoric Fe(III)-product complex during rapid turnover of the enzyme shows that it is an integral part of the reaction cycle which must dissociate much faster under multiple turnover conditions than during single turnover.
Figure 8.

Optical changes at 453 nm (black) and 700 nm (red) during the S5H steady-state reaction. Oxidized S5HH (34 μM), S5HR (10.4 μM), S5HF (9 μM) and O2 (1.8 mM) were reacted with salicylate (8.5 mM), NADH (2.12 mM) and O2 (1.8 mM) in standard buffer at 4 °C using a stopped-flow spectrophotometer (concentrations after mixing). Inset: The 700 nm time course (red) is composed of contributions from the 700 nm species and the Rieske cluster (simulated in blue). The Rieske cluster contribution is derived from the relative amounts of oxidized and reduced cluster determined from the 453 nm steady-state absorbance and the known extinction coefficients for the cluster at 700 nm (Figure 1, inset).
Formation of the 700 nm Product Complex with an Alternative Substrate.
The occurrence of the Fe(III)-phenolate complex in the S5H reaction cycle was further examined using benzoate, an alternative substrate that is turned over at 57% (at 23 °C) or 50% (at 4 °C) of the maximal rates of salicylate at the same temperatures. Product extraction and analysis by HPLC and LC-MS versus an authentic standard showed that benzoate is readily converted to 3-OH benzoate (Figures S1 and S2). Thus, the hydroxyl function is added in the same position relative to the carboxylate substituent as in the native reaction. Single-turnover stopped-flow reactions of fully reduced S5HH, benzoate, and O2 show the formation of an intermediate in the 700 nm region, so the same type of Fe(III)-product complex appears to form as seen for the salicylate reaction. However, the rate constant for the Rieske cluster oxidation and product formation derived from the fastest 1/τ of the internal electron transfer reaction between metal centers is much slower (k = 4.1 s−1 vs 72 s−1), showing that the chemical nature of the substrate greatly affects the reaction before transfer of the second electron required for product formation. The formation of the 700 nm intermediate is also slower (k = 3.1 s−1 vs 21 s−1), but by a different relative amount, showing that the electron transfer and Fe(III)-product complex forming reactions are not directly correlated. The similar values for the Rieske cluster oxidation and 700 nm formation rate constants with benzoate may also mean that the reaction is completely rate-limited by the reactions leading to product, and the formation of the following 700 nm intermediate could potentially occur with a larger rate constant similar to that observed during salicylate turnover. The decay of the 700 nm species generated by single turnover of benzoate was only slightly affected (Figure 9, inset, Table 2).
Figure 9.

Single-turnover reaction of fully reduced S5HH (30 μM) reacted with benzoate (2.35 mM) and O2 (900 μM) at short and (inset) long times (concentrations after mixing) in standard buffer, 4 °C. The reaction was monitored using a stopped-flow spectrophotometer at 453 nm (blue) and 700 nm (red). Both time courses are an average of 5 replicates. The fits to summed exponential equations are shown as white dashed lines over the data. The kinetic parameters from the fits are shown in Table 2. Inset: Time course of the reaction monitored at 700 nm over 500 s showing the decay of the 700 nm intermediate.
DISCUSSION
It has been shown here that the S5H monooxygenase shares many of the mechanistic features we have previously reported for cis-dihydrodiol-forming Rieske dioxygenases.35, 40, 48, 49, 51 Specifically, (i) the substrate and O2 binding reactions to the fully reduced S5HH are faster than the downstream reactions, (ii) the rate-limiting step in the product-forming half of the reaction cycle appears to occur after substrate and O2 bind, but before an electron is transferred from the Rieske cluster, and (iii) the product release phase of the reaction is accelerated by re-reduction of the enzyme. Despite these similarities between Rieske mono- and dioxygenase reactions, not even a minor amount of a cis-dihydrodiol product was detected by HPLC after turnover of salicylate or benzoate by S5HH. The ability described here to directly observe the formation and decay of the S5HH product complex through its unique optical and EPR signatures allows this part of the reaction cycle to be characterized in more detail than previously possible for a Rieske oxygenase. The insight gained regarding the mechanism and regulation of the Rieske monooxygenase reaction are discussed here.
Rate-Limiting Step in Product Formation.
The hyperbolic dependence on salicylate or O2 concentration of the rate constant for electron transfer from the Rieske cluster to the mononuclear iron (Figure 6) shows that the product forming step is not rate limited by these two substrate binding reactions at the high concentrations of substrates used here. Similarly, electron transfer between the metal centers is not rate limiting. This conclusion follows from the facts that: (i) the redox potential of the Rieske cluster is much lower than that of the mononuclear iron (most Rieske oxygenases are isolated with the Rieske cluster oxidized and the mononuclear site >95 % reduced, see for example Figure 2A,35, 40, 70 and (ii) the electron transfer is expected to occur with a rate constant at least five orders of magnitude faster than any other rate constant reported here in the short, through-bond pathway between the metal centers51, 55, 67 Thus, the electron transfer must be rate-limited by a previous step. The cleavage of the O-O bond also does not appear to be rate-limiting because this reaction typically occurs from a two electron reduced state,71 which only occurs after electron transfer. Indeed, we show here that product formation, and by extension the required O-O bond cleavage, occurs at a high rate, comparable to that of observed electron transfer in S5HH (Figure 5). Consequently, it is likely that the rate-limiting step is attack of a one-electron-reduced O2 on the aromatic substrate, a conclusion we also reached for the cis-dihydrodiol-forming Rieske dioxygenases.51, 55
Nature of the Species Absorbing at 700 nm.
The species absorbing at 700 nm forms rapidly during S5HH single-turnover reactions and also persists throughout the extended steady-state occurring during multiple turnover reactions catalyzed by S5H (Figures 3, 4 and 8). Nevertheless, analyses of optical and RCQ transient kinetic data show that the 700 nm species is formed more slowly than either the electron transfer or product formation reactions (Figure 5). Insight into the nature of the 700 nm species derives from the very sharp EPR species at g = 4.3, which forms in single-turnover RFQ experiments on approximately the same time scale as the 700 nm species. The observed broadening of the EPR signal by nuclear hyperfine interactions from 17O inserted during the reaction with 17O2 to form gentisate shows directly that the sharp g = 4.3 signal arises from complex of Fe(III) with gentisate through the C5 hydroxyl function (Figure 7). Similar broadenings observed for 17O-enriched protocatechuate binding to protocatechuate 3,4-dioxygenase (3,4-PCD) and the nitrosyl complex of protocatechuate 4,5-dioxygenase were used to demonstrate direct substrate coordination to the active site iron of those enzymes.72–74 The EPR resonances at g = 4.3 and 9.68 and their temperature dependence identify the iron as high-spin Fe(III) in a maximally rhombic electronic environment with a small zero field splitting (S = 5/2, E/D = 0.333, D = 0.8 ± 0.2 cm−1, P1/2 = 2.8 ± 0.1 mW see Supporting Information and Figure S4A and B). A single, strong, charge-donating phenolate ligand in an otherwise typical ligand field of histidine, aspartate, and water would cause such a distortion. A phenolate ligand would also be consistent with the characteristic optical spectrum of the 700 nm species as discussed below.
The optical and EPR characteristics of Fe(III)-phenolate and catecholate complexes have been studied extensively in both synthetic models and enzymes such as catechol 1,2 dioxygenase or 3,4-PCD.69, 75–77 For example, the two tyrosinate ligands of the active site Fe(III) of 3,4-PCD result in ligand-to-metal-charge-transfer (LMCT) spectra in the visible range at 485 and 568 nm (ε ~ 2 mM−1 cm−1 at each wavelength) as well as a sharp EPR resonance at g = 4.3 with maximal E/D and a small D value (S = 5/2, E/D = 0.333, D = 1.6 cm−1),69, 77, 78 similar to that seen for the S5HH intermediate. Catecholic substrate complexes of 3,4-PCD as well as synthetic model complexes with either catecholate or phenolate ligands also exhibit long wavelength LMCT bands in the 500 −700 nm region with extinction coefficients between 1.0 and 4 mM−1 cm−1.75, 79–82 The extinction coefficient of the 700 nm species reported here can be estimated to be ~2 mM−1 cm−1 from the absorbance data presented in Figure 3B, inset and the concentration of mononuclear iron (~63 % of the protein concentration based on the amount of Rieske cluster oxidized in the fast phase). This relatively large extinction coefficient supports its assignment as a LMCT transition.79 Together, the optical and EPR characteristics argue strongly for the assignment of the 700 nm species as the product complex of the oxidized mononuclear iron. Attempts to force product to rebind to the active site Fe(III) to regenerate the 700 nm species were unsuccessful. This was not unexpected because we have found that when the metal centers for NDO and BZDO are oxidized, substrates do not easily bind, products do not rapidly dissociate, and products cannot be forced to rebind even at high concentrations35, 40, 49.
Dependence of Product Release on Rieske Cluster Oxidation State.
The 700 nm/g = 4.3 intermediate is remarkably long lived following a single turnover of S5HH, but disappears with a rate constant comparable to the S5H turnover number following exhaustion of the O2 and re-reduction of the enzyme in a multiple turnover experiment (Figure 8). The interpretation of this loss of the spectral characteristics of the putative product complex is complicated by the possible reduction of the mononuclear iron by the NADH/S5HR and S5HF components. Both the EPR signal and the LMCT band would be lost when the mononuclear iron is reduced even if the product were still bound. However, turnover of the enzyme system requires release of the product with a rate constant at least as large as the overall turnover number. This observation means that the 700 nm absorbance and g = 4.3 EPR signal must be lost with rate constants at least 300 times greater when a reductant is available for the Rieske cluster than during a single-turnover experiment.
Model for S5H Rieske Monooxygenase Mechanism.
Our studies of the mechanism of BZDO using fluorinated substrates together with computational analysis suggest that Fe(III)-superoxo is the attacking species in the cis-dihydrodiol-forming dioxygenases (see Scheme 2, path B).51, 55 While this substrate radical-forming reaction is not energetically favorable when considered alone, it occurs because a proton-coupled-electron-transfer (PCET) reaction involving the Rieske cluster follows immediately. The result is O-O bond cleavage and formation of an intermediate substrate epoxide, which is proposed to form a bond with the mononuclear Fe(III). This stabilizing bond promotes breakdown of the epoxide to yield an intermediate arenium cation, which is then quenched by the second atom of oxygen from O2 to form the cis-dihydrodiol.
Despite the difference in catalytic outcome, it is possible to propose a similar mechanism for aromatic hydroxylation by the Rieske monooxygenase S5H as illustrated in Scheme 4. All of the steps through attack of the Fe(III)-superoxo on the aromatic substrate are unchanged in accord with the very similar kinetics of organic substrate and O2 binding. Also, the nearly 20-fold decrease in the electron transfer rate constant when using benzoate as the substrate in place of salicylate (Figure 9) confirms the participation of the substrate in the reaction that ultimately allows electron transfer from the Rieske cluster. It is worth noting that benzoate lacks the electron supplying para-substituent to the C5 position of the substrate that would promote electrophilic attack of the Fe(III)-superoxo. Following electron transfer from the Rieske cluster, the mechanism of the di- and monooxygenases may continue along the same track to form an intermediate epoxide, or they may diverge such that O-O bond cleavage occurs directly to form product gentisate rather than an epoxide. This alternative reaction would be favored by protonation of the O2-derived oxygen during bond cleavage and by concurrent ring re-aromatization. Progress of the reaction in this manner would avoid formation of an arenium cation as a site for transfer of the second oxygen, thereby precluding cis-dihydrodiol formation.
Scheme 4.

Proposed Mechanism of S5H Catalysis
Alternative Mechanisms for Monooxygenation.
Alternative mechanisms involving high-valent iron reactive species should also be considered. One such mechanism involves formation of an Fe(IV)-oxo by O-O bond cleavage of the O2 bridging to the substrate prior to electron transfer (Scheme 5A). This mechanism is reminiscent of the reaction between Fe(II), O2, and bound tetrahydrobiopterin in tyrosine hydroxylase that forms hydroxylated tetrahydrobiopterin and the Fe(IV)-oxo species that ultimately effects hydroxylation of the aromatic substrate.83 However, in the S5H case, the high-valent, high-potential species would simply be quenched by electron transfer from the low potential Rieske cluster. In another alternative mechanism, an HO-Fe(V)-oxo reactive species might be formed by O-O bond cleavage after electron transfer, but before attack on the substrate (Scheme 5B).30, 40, 84 This O-O bond cleavage might also occur by a water-assisted mechanism using the water bound in the coordination site adjacent to the peroxide, as demonstrated in model complex studies.85 The high-valent species could then react with substrate by any of several routes involving electron abstraction and oxygen rebound with the likely formation of an arene epoxide and/or cation intermediate. This mechanism is equivalent to that computed for the peroxide shunt reaction of BZDO,55 except that the incorporation of the second atom of oxygen from O2 (to form a dihydrodiol) would have to be prevented in some manner in S5H. Both of these alternative mechanisms are more reminiscent of the traditional electrophilic aromatic substitution reactions proposed for other oxygenases and small molecule reactions.83, 86, 87 However, both mechanisms seem unlikely for S5H because the former would result in hydroxylation without electron transfer, while the latter could result in electron transfer and oxygen activation without involvement of the substrate, neither of which is observed. Also, we have not been able to observe peroxide shunt chemistry for S5H in contrast to the cis-dihydrodiol-forming NDO and BZDO enzymes.48, 49
Scheme 5.

Alternative Mechanisms Proposed for Rieske Monooxygenase Catalysis
Past studies have been used to propose two additional mechanisms for Rieske dioxygenases which might be adapted to account for S5H monooxygenase chemistry. In one study, it was shown that NDO can catalyze para-hydroxylation of meta-cresol (3-OH toluene) to form methyl-hydroquinone.88 The adaptation of this mechanism, shown in Scheme 5C, might yield the gentisate product of S5H. The author of the NDO study proposed a mechanism involving substrate activation by direct coordination of the substrate hydroxyl substituent to a high-valent iron formed by the mechanism shown in Scheme 5B, thereby creating a site for nucleophilic attack by water at the para-position. Accordingly, the source of the oxygen in the new hydroxyl function was found to be water rather than O2. This finding rules out this mechanism for S5H where the source of the oxygen is O2 (Figures 7 and S3). The final alternative mechanism (Scheme 5D) was based the observation that cis-dihydrodiols formed synthetically or by a Rieske dioxygenase would spontaneously dehydrate to phenol analogs when heated to 110 – 180 °C. An NIH-shift was observed in all cases when using specifically deuterated substrates.89 This result was interpreted to suggest that monooxygenation in Rieske oxygenases could occur through initial dioxygenation to form a cis-dihydrodiol product. In our opinion, this route is unlikely because the authors were able to isolate several of the cis-dihydrodiols, indicating that they were stable on the time scale of enzyme turnover at room temperature; this correlates with our experience. Neither the cis-dihydrodiol product nor the alternative 2,4-dihydroxybenzoate product that might be also formed from cis-dihydrodiol dehydration are observed after S5H turnover of salicylate. Moreover, the native organism harboring S5H has no downstream enzymes to further metabolize products other than gentisate.58, 61 A dehydration mechanism was also deemed unlikely based on the products formed in whole cell assays of S5H reactions with alternative substrates.62
Importance of Active Site Flexibility.
Our past spectroscopic and kinetic studies of Rieske dioxygenases35, 40, 42, 43, 48, 49 as well as X-ray crystallographic studies by others16, 17 have favored a model in which the oxidation state of the Rieske cluster controls the relative proximity of the substrate and the mononuclear iron. Reduction of the Rieske cluster results in separation of the substrate and mononuclear iron so that O2 can bind to the latter. This model provides an explanation for why no binding of O2 occurs before the organic substrate is bound in the active site and the Rieske cluster is reduced.35, 40 Similarly, we find no evidence for reactivity of S5H with O2 for the as-isolated enzyme with an oxidized Rieske cluster. Protein conformational shifts of this type in an enzyme linked to substrate binding and redox state must be reversible, so that as the Rieske cluster changes oxidation state during the catalytic cycle, the substrate/product to iron distance should also shift in concert. The rate of protein conformational shifts, however, may not correlate with the rates of the chemical steps in the cycle.
The mechanism shown in Scheme 4 requires an active site that can accommodate both the ~5 Å bridging superoxo and a significantly shorter Fe(III)-product phenolate bond. This flexibility can be accounted for by the proposed movement of the mononuclear iron in response to the Rieske cluster oxidation state. Separation upon Rieske cluster reduction allows the bridging peroxo to form and then Rieske cluster oxidation reverses the separation to allow formation of the Fe(III)-product phenolate bond. In previous studies, direct coordination of the dihydrodiol product of NDO to the mononuclear iron was observed in the X-ray crystal structure after turnover of naphthalene.50 However, it was unclear whether this species actually occurs in the reaction cycle, or whether it was the result of equilibration to a stable state in the crystal where the product cannot readily depart. In the current study, the Fe(III)-product phenolate complex can be directly observed in solution reactions, and it is shown to be part of the reaction cycle. It is noteworthy that the 700 nm/g = 4.3 intermediate appears more slowly than product formation in the active site based on the RCQ experiments shown in Figure 5. This delay may reflect a difference between the rate of O-O bond cleavage and the rate of conformational change required to bring the product into bonding distance. The link between the Rieske cluster oxidation state and the relative positions of the mononuclear iron and the substrate or product provides a means to regulate the catalytic cycle to ensure efficiency and specificity. The cycle cannot initiate until organic substrate binding has occurred and the Rieske cluster is reduced to permit O2 binding. At the end of the reaction, another substrate cannot replace the product and lock the enzyme in an unreactive, fully oxidized state because reduction of the Rieske cluster and mononuclear iron must first occur to allow release of the product. The effect of a source of electrons on the ability of the enzyme to complete its catalytic cycle is made clear by the greatly accelerated release of the 700 nm/g = 4.3 intermediate under multiple turnover conditions.
CONCLUSION
It is shown here that Rieske aromatic monooxygenase and dioxygenases use similar mechanistic strategies to oxidize substrates at least up to the point of determining the fate of the second oxygen from O2. The reactive species that initiates catalysis in each case appears to be an Fe(III)-superoxo species (or its electronic equivalent) which is made more reactive by the subsequent rapid transfer of an electron from the Rieske cluster. This transfer promotes O-O bond cleavage and drives the product forming reaction to completion. We speculate that conformational shifts in response to the Rieske cluster oxidation state modulate the separation of the mononuclear iron and organic substrate, thereby facilitating the chemistry and enforcing a specific sequence of steps in the reaction cycle. The direct observation of the product complex reported here for S5H and the dramatic dependence of its rate of breakdown on the availability of additional reducing equivalents provide a direct test of this overall regulatory hypothesis. While the Fe(III)-superoxo intermediate appears to be reactive species in S5H, this does not rule out different types of reactive species in other types of Rieske oxygenases. Indeed, the active site is set up to form Fe(III)-(H)peroxo or high-valent iron intermediates, and this may well occur in cases where reaction of a substrate with Fe(III)-superoxo is slow.
Supplementary Material
ACKNOWLEDeGEMENTS
We thank Jung-Kul Lee for the gift of the pACYC-isc plasmid. The pWWF6 plasmid containing nagGH, nagAa, and nagAb was a kind gift from Thomas K. Wood and Tammy Wood. We thank Michael P. Hendrich for help using SpinCount to interpret EPR spectra and Rahul Banerjee for valuable discussions. We thank Joseph Dalluge and Sean Murray for analysis of mass spectrometry data.
Funding
This work was supported by National Institutes of Health Grant GM118030 (to J.D.L.)
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.biochem.9b00292
Additional experimental procedures including the cloning, heterologous expression, and purification of tagless S5H. Procedures for anaerobic techniques and chemical reduction of S5HH, HPLC and LC-MS product analysis, spectroscopy, stopped-flow and EPR measurements and analysis. Also found here are a discussion of special assay considerations and the origin of multiple kinetic phases. Table S1 and Figures S1 to S4 (PDF).
REFERENCES
- [1].Sydor PK, Barry SM, Odulate OM, Barona-Gomez F, Haynes SW, Corre C, Song L, and Challis GL (2011) Regio- and stereodivergent antibiotic oxidative carbocyclizations catalysed by Rieske oxygenase-like enzymes, Nat. Chem 3, 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Serrano-Plana J, Company A, and Costas M (2017) O–O bond activation in Cu- and Fe-based coordination complexes: Breaking it makes the difference, In Adv. Inorg. Chem (van Eldik R, and Hubbard CD, Eds.), pp 63–105, Academic Press. [Google Scholar]
- [3].Capyk JK, D’Angelo I, Strynadka NC, and Eltis LD (2009) Characterization of 3-ketosteroid 9α-hydroxylase, a Rieske oxygenase in the cholesterol degradation pathway of Mycobacterium tuberculosis, J. Biol. Chem 284, 9937–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hauenstein M, Christ B, Das A, Aubry S, and Hortensteiner S (2016) A role for TIC55 as a hydroxylase of phyllobilins, the products of chlorophyll breakdown during plant senescence, Plant Cell 28, 2510–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Berim A, Park JJ, and Gang DR (2014) Unexpected roles for ancient proteins: flavone 8-hydroxylase in sweet basil trichomes is a Rieske-type, PAO-family oxygenase, Plant J 80, 385–395. [DOI] [PubMed] [Google Scholar]
- [6].Rottiers V, Motola DL, Gerisch B, Cummins CL, Nishiwaki K, Mangelsdorf DJ, and Antebi A (2006) Hormonal control of C. elegans dauer formation and life span by a Rieske-like oxygenase, Dev. Cell 10, 473–482. [DOI] [PubMed] [Google Scholar]
- [7].Yoshiyama-Yanagawa T, Enya S, Shimada-Niwa Y, Yaguchi S, Haramoto Y, Matsuya T, Shiomi K, Sasakura Y, Takahashi S, Asashima M, Kataoka H, and Niwa R (2011) The conserved Rieske oxygenase DAF-36/Neverland is a novel cholesterol metabolizing enzyme, J. Biol. Chem 286, 25756–25762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yoshiyama T, Namiki T, Mita K, Kataoka H, and Niwa R (2006) Neverland is an evolutionally conserved Rieske-domain protein that is essential for ecdysone synthesis and insect growth, Development 133, 2565–2574. [DOI] [PubMed] [Google Scholar]
- [9].Wollam J, Magomedova L, Magner DB, Shen Y, Rottiers V, Motola DL, Mangelsdorf DJ, Cummins CL, and Antebi A (2011) The Rieske oxygenase DAF-36 functions as a cholesterol 7-desaturase in steroidogenic pathways governing longevity, Aging Cell 10, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schlenzka W, Shaw L, Kelm S, Schmidt CL, Bill E, Trautwein AX, Lottspeich F, and Schauer R (1996) CMP-N-acetylneuraminic acid hydroxylase: the first cytosolic Rieske iron-sulphur protein to be described in Eukarya, FEBS Lett 385, 197–200. [DOI] [PubMed] [Google Scholar]
- [11].Bergfeld DAK, and Varki PA (2014) Cytidine monophospho-N-acetylneuraminic acid hydroxylase (CMAH), In Handbook of Glycosyltransferases and Related Genes (Taniguchi N, Honke K, Fukuda M, Narimatsu H, Yamaguchi Y, and Angata T, Eds.), pp 1559–1580, Springer; Japan. [Google Scholar]
- [12].Gibson DT (1968) Microbial degradation of aromatic compounds, Science 161, 1093–1097. [PubMed] [Google Scholar]
- [13].Barry S, and Challis GL (2013) Mechanism and catalytic diversity of Rieske non-heme iron-dependent oxygenases, ACS Catal 3, 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Perry C, de Los Santos ELC, Alkhalaf LM, and Challis GL (2018) Rieske non-heme iron-dependent oxygenases catalyse diverse reactions in natural product biosynthesis, Nat. Prod. Rep 35, 622–632. [DOI] [PubMed] [Google Scholar]
- [15].Rosche B, Tshisuaka B, Fetzner S, and Lingens F (1995) 2-Oxo-1,2-dihydroquinoline 8-monooxygenase, a two-component enzyme system from Pseudomonas putida 86, J. Biol. Chem 270, 17836–17842. [DOI] [PubMed] [Google Scholar]
- [16].Martins BM, Svetlitchnaia T, and Dobbek H (2005) 2-Oxoquinoline 8-monooxygenase oxygenase component: active site modulation by Rieske-[2Fe-2S] center oxidation/reduction, Structure 13, 817–824. [DOI] [PubMed] [Google Scholar]
- [17].Ashikawa Y, Fujimoto Z, Usami Y, Inoue K, Noguchi H, Yamane H, and Nojiri H (2012) Structural insight into the substrate- and dioxygen-binding manner in the catalytic cycle of Rieske nonheme iron oxygenase system, carbazole 1,9a-dioxygenase, BMC Structural Biology 12, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lukowski AL, Ellinwood DC, Hinze ME, DeLuca RJ, Du Bois J, Hall S, and Narayan ARH (2018) C–H hydroxylation in paralytic shellfish toxin biosynthesis, J. Am. Chem. Soc 140, 11863–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bernhardt FH, Ruf HH, and Staudinger H (1971) Purification of a 4-methoxybenzoate O-demethylase from Pseudomonas putida, Hoppe-Seyler’s Z. Physiol. Chem 352, 1091–1099. [DOI] [PubMed] [Google Scholar]
- [20].Bernhardt FH, Heymann E, and Traylor PS (1978) Chemical and spectral properties of putidamonooxin, the iron-containing and acid-labile-sulfur-containing monooxygenase of a 4-methoxybenzoate O-demethylase from Pseudomonas putida, Eur. J. Biochem 92, 209–223. [DOI] [PubMed] [Google Scholar]
- [21].D’Ordine RL, Rydel TJ, Storek MJ, Sturman EJ, Moshiri F, Bartlett RK, Brown GR, Eilers RJ, Dart C, Qi Y, Flasinski S, and Franklin SJ (2009) Dicamba monooxygenase: Structural insights into a dynamic Rieske oxygenase that catalyzes an exocyclic monooxygenation, J. Mol. Biol 392, 481–497. [DOI] [PubMed] [Google Scholar]
- [22].Jiang W, Wilson MA, and Weeks DP (2013) O-demethylations catalyzed by Rieske nonheme iron monooxygenases involve the difficult oxidation of a saturated C-H bond., ACS Chem. Biol 8, 1687–1691. [DOI] [PubMed] [Google Scholar]
- [23].Lee J, Simurdiak M, and Zhao H (2005) Reconstitution and characterization of aminopyrrolnitrin oxygenase, a Rieske N-oxygenase that catalyzes unusual arylamine oxidation., J. Biol. Chem 280, 36719–36727. [DOI] [PubMed] [Google Scholar]
- [24].Schuster J, Schäfer F, Hübler N, Brandt A, Rosell M, Härtig C, Harms H, Müller RH, and Rohwerder T (2012) Bacterial degradation of tert-amyl alcohol proceeds via hemiterpene 2-methyl-3-buten-2-ol by employing the tertiary alcohol desaturase function of the Rieske nonheme mononuclear iron oxygenase MdpJ., J. Bacteriol 194, 972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Torok DS, Resnick SM, Brand JM, Cruden DL, and Gibson DT (1995) Desaturation and oxygenation of 1,2-dihydronaphthalene by toluene and naphthalene dioxygenase, J. Bacteriol 177, 5799–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen Q, Wang CH, Deng SK, Wu YD, Li Y, Yao L, Jiang JD, Yan X, He J, and Li SP (2014) Novel three-component Rieske non-heme iron oxygenase system catalyzing the N-dealkylation of chloroacetanilide herbicides in sphingomonads DC-6 and DC-2, Appl. Environ. Microbiol 80, 5078–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Withall DM, Haynes SW, and Challis GL (2015) Stereochemistry and mechanism of undecylprodigiosin oxidative carbocyclization to streptorubin B by the Rieske oxygenase RedG, J. Am. Chem. Soc 137, 7889–7897. [DOI] [PubMed] [Google Scholar]
- [28].Najle SR, Nusblat AD, Nudel CB, and Uttaro AD (2013) The sterol-C7 desaturase from the ciliate Tetrahymena thermophila is a Rieske oxygenase, which is highly conserved in animals., Mol. Biol Evol 30, 1630–1643. [DOI] [PubMed] [Google Scholar]
- [29].Higgins TP, Demarco P, and Murrell JC (1997) Purification and molecular characterization of the electron transfer protein of methanesulfonic acid monooxygenase, J. Bacteriol 179, 1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Resnick SM, Lee K, and Gibson DT (1996) Diverse reactions catalyzed by naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816, J. Ind. Microbiol. Biotechnol 17, 438–457. [Google Scholar]
- [31].Gally C, Nestl BM, and Hauer B (2015) Engineering Rieske non-heme iron oxygenases for the asymmetric dihydroxylation of alkenes, Angew. Chem. Int. Ed 54, 12952–12956. [DOI] [PubMed] [Google Scholar]
- [32].Parales RE, Lee K, Resnick SM, Jiang H, Lessner DJ, and Gibson DT (2000) Substrate specificity of naphthalene dioxygenase: effect of specific amino acids at the active site of the enzyme, J. Bacteriol 182, 1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ferraro DJ, Okerlund A, Brown E, and Ramaswamy S (2017) One enzyme, many reactions: structural basis for the various reactions catalyzed by naphthalene 1,2-dioxygenase, IUCrJ 4, 648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ensley BD, Gibson DT, and Laborde AL (1982) Oxidation of naphthalene by a multicomponent enzyme system from Pseudomonas sp. strain NCIB 9816, J. Bacteriol 149, 948–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wolfe MD, Altier DJ, Stubna A, Popescu CV, Münck E, and Lipscomb JD (2002) Benzoate 1,2-dioxygenase from Pseudomonas putida: Single turnover kinetics and regulation of a two-component Rieske dioxygenase, Biochemistry 41, 9611–9626. [DOI] [PubMed] [Google Scholar]
- [36].Batie CJ, LaHaie E, and Ballou DP (1987) Purification and characterization of phthalate oxygenase and phthalate oxygenase reductase from Pseudomonas cepacia, J. Biol. Chem 262, 1510–1518. [PubMed] [Google Scholar]
- [37].Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, and Ramaswamy S (1998) Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase, Structure 6, 571–586. [DOI] [PubMed] [Google Scholar]
- [38].Parales RE, Parales JV, and Gibson DT (1999) Aspartate 205 in the catalytic domain of naphthalene dioxygenase is essential for activity, J. Bacteriol 181, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ashikawa Y, Fujimoto Z, Noguchi H, Habe H, Omori T, Yamane H, and Nojiri H (2005) Crystallization and preliminary X-ray diffraction analysis of the electron-transfer complex between the terminal oxygenase component and ferredoxin in the Rieske non-haem iron oxygenase system carbazole 1,9a-dioxygenase, Acta Crystallogr. F 61, 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wolfe MD, Parales JV, Gibson DT, and Lipscomb JD (2001) Single turnover chemistry and regulation of O2 activation by the oxygenase component of naphthalene 1,2-dioxygenase, J. Biol. Chem 276, 1945–1953. [DOI] [PubMed] [Google Scholar]
- [41].Ohta T, Chakrabarty S, Lipscomb JD, and Solomon EI (2008) Near-IR MCD of the nonheme ferrous active site in naphthalene 1,2-dioxygenase: Correlation to crystallography and structural insight into the mechanism of Rieske dioxygenases, J. Am. Chem. Soc 130, 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yang T-C, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, and Hoffman BM (2003) Modulation of substrate binding to naphthalene 1,2-dioxygenase by Rieske cluster reduction/oxidation, J. Am. Chem. Soc 125, 2034–2035. [DOI] [PubMed] [Google Scholar]
- [43].Yang T-C, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, and Hoffman BM (2003) Substrate binding to NO-ferro-naphthalene 1,2-dioxygenase studied by high-resolution Q-band pulsed 2H-ENDOR spectroscopy, J. Am. Chem. Soc 125, 7056–7066. [DOI] [PubMed] [Google Scholar]
- [44].Groves JT (2006) High-valent iron in chemical and biological oxidations, J. Inorg. Biochem 100, 434–447. [DOI] [PubMed] [Google Scholar]
- [45].Rittle J, and Green MT (2010) Cytochrome P450 Compound I: capture, characterization, and C-H bond activation kinetics, Science 330, 933–937. [DOI] [PubMed] [Google Scholar]
- [46].Lee SK, Nesheim JC, and Lipscomb JD (1993) Transient intermediates of the methane monooxygenase catalytic cycle, J. Biol. Chem 268, 21569–21577. [PubMed] [Google Scholar]
- [47].Lee SK, Fox BG, Froland WA, Lipscomb JD, and Münck E (1993) A transient intermediate of the methane monooxygenase catalytic cycle containing a FeIVFeIV cluster, J. Am. Chem. Soc 115, 6450–6451. [Google Scholar]
- [48].Wolfe MD, and Lipscomb JD (2003) Hydrogen peroxide-coupled cis-diol formation catalyzed by naphthalene 1,2-dioxygenase, J. Biol. Chem 278, 829–835. [DOI] [PubMed] [Google Scholar]
- [49].Neibergall MB, Stubna A, Mekmouche Y, Münck E, and Lipscomb JD (2007) Hydrogen peroxide dependent cis-dihydroxylation of benzoate by fully oxidized benzoate 1,2-dioxygenase, Biochemistry 46, 8004–8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, and Ramaswamy S (2003) Crystal structure of naphthalene dioxygenase: Side-on binding of dioxygen to iron, Science 299, 1039–1042. [DOI] [PubMed] [Google Scholar]
- [51].Rivard BS, Rogers MS, Marell DJ, Neibergall MB, Chakrabarty S, Cramer CJ, and Lipscomb JD (2015) Rate-determining attack on substrate precedes Rieske cluster oxidation during cis-dihydroxylation by benzoate dioxygenase, Biochemistry 54, 4652–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chiang C-W, Kleespies ST, Stout HD, Meier KK, Li P-Y, Bominaar EL, Que L, Münck E, and Lee W-Z (2014) Characterization of a paramagnetic mononuclear nonheme iron-superoxo complex, J. Am. Chem. Soc 136, 10846–10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hong S, Sutherlin KD, Park J, Kwon E, Siegler MA, Solomon EI, and Nam W (2014) Crystallographic and spectroscopic characterization and reactivities of a mononuclear non-haem iron(III)-superoxo complex, Nat. Commun 5, 5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chung LW, Li X, Hirao H, and Morokuma K (2011) Comparative reactivity of ferric-superoxo and ferryl-oxo species in heme and non-heme complexes, J. Am. Chem. Soc 133, 20076–20079. [DOI] [PubMed] [Google Scholar]
- [55].Sutherlin KD, Rivard BS, Bottger LH, Liu LV, Rogers MS, Srnec M, Park K, Yoda Y, Kitao S, Kobayashi Y, Saito M, Seto M, Hu M, Zhao J, Lipscomb JD, and Solomon EI (2018) NRVS studies of the peroxide shunt intermediate in a Rieske dioxygenase and its relation to the native Fe(II) O2 reaction, J. Am. Chem. Soc 140, 5544–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Oloo WN, Banerjee R, Lipscomb JD, and Que L Jr. (2017) Equilibrating (L)Fe(III)-OOAc and (L)Fe(V)(O) species in hydrocarbon oxidations by bio-inspired nonheme iron catalysts using H2O2 and AcOH, J. Am. Chem. Soc 139, 17313–17326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chen K, Costas M, Kim J, Tipton AK, and Que L Jr. (2002) Olefin cis-dihydroxylation versus epoxidation by non-heme iron catalysts: two faces of an FeIII-OOH coin, J. Am. Chem. Soc 124, 3026–3035. [DOI] [PubMed] [Google Scholar]
- [58].Fuenmayor SL, Wild M, Boyes AL, and Williams PA (1998) A gene cluster encoding steps in conversion of naphthalene to gentisate in Pseudomonas sp. strain U2, J. Bacteriol 180, 2522–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jouanneau Y, Micoud J, and Meyer C (2007) Purification and characterization of a three-component salicylate 1-hydroxylase from Sphingomonas sp. strain CHY-1, Appl. Environ. Microbiol 73, 7515–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yamamoto S, Katagiri M, Maeno H, and Hayaishi O (1965) Salicylate hydroxylase, a monooxygenase requiring flavin adenine dinucleotide: I. Purification and general properties, J. Biol. Chem 240, 3408–3413. [PubMed] [Google Scholar]
- [61].Zhou N-Y, Fuenmayor SL, and Williams PA (2001) nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism, J. Bacteriol 183, 700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhou N-Y, Al-Dulayymi J, Baird MS, and Williams PA (2002) Salicylate 5-hydroxylase from Ralstonia sp. strain U2: a monooxygenase with close relationships to and shared electron transport proteins with naphthalene dioxygenase, J. Bacteriol 184, 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Fang T, and Zhou N-Y (2014) Purification and characterization of salicylate 5-hydroxylase, a three-component monooxygenase from Ralstonia sp. strain U2., Appl. Microbiol. Biotechnol 98, 671–679. [DOI] [PubMed] [Google Scholar]
- [64].Keenan BG, Leungsakul T, Smets BF, Mori M. a., Henderson DE, and Wood TK (2005) Protein engineering of the archetypal nitroarene dioxygenase of Ralstonia sp. strain U2 for activity on aminonitrotoluenes and dinitrotoluenes through alpha-subunit residues leucine 225, phenylalanine 350, and glycine 407, J. Bacteriol 187, 3302–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tiwari MK, Lee JK, Moon HJ, and Zhao H (2011) Further biochemical studies on aminopyrrolnitrin oxygenase (PrnD), Bioorg Med Chem Lett 21, 2873–2876. [DOI] [PubMed] [Google Scholar]
- [66].Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, and Lipscomb JD (2010) Trapping and spectroscopic characterization of an FeIII-superoxo intermediate from a nonheme mononuclear iron-containing enzyme, Proc. Natl. Acad. Sci. USA 107, 16788–16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gray HB, and Winkler JR (1996) Electron transfer in proteins, Ann. Rev. Biochem 65, 537–561. [DOI] [PubMed] [Google Scholar]
- [68].Whittaker JW, and Lipscomb JD (1984) Transition state analogs for protocatechuate 3,4-dioxygenase. Spectroscopic and kinetic studies of the binding reactions of ketonized substrate analogs, J. Biol. Chem 259, 4476–4486. [PubMed] [Google Scholar]
- [69].Que L Jr., Lipscomb JD, Zimmermann R, Münck E, Orme-Johnson NR, and Orme-Johnson WH (1976) Mössbauer and EPR spectroscopy of protocatechuate 3,4-dioxygenase from Pseudomonas aeruginosa, Biochim. Biophys. Acta 452, 320–334. [DOI] [PubMed] [Google Scholar]
- [70].Coulter ED, Moon N, Batie CJ, Dunham WR, and Ballou DP (1999) Electron paramagnetic resonance measurements of the ferrous mononuclear site of phthalate dioxygenase substituted with alternate divalent metal ions: direct evidence for ligation of two histidines in the copper(II)-reconstituted protein, Biochemistry 38, 11062–11072. [DOI] [PubMed] [Google Scholar]
- [71].Solomon EI, Goudarzi S, and Sutherlin KD (2016) O2 activation by non-heme iron enzymes, Biochemistry 55, 6363–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Orville AM, and Lipscomb JD (1993) Simultaneous binding of nitric oxide and isotopically labeled substrates or inhibitors by reduced protocatechuate 3,4-dioxygenase, J. Biol. Chem 268, 8596–8607. [PubMed] [Google Scholar]
- [73].Arciero DM, and Lipscomb JD (1986) Binding of 17O-labeled substrate and inhibitors to protocatechuate 4,5-dioxygenase-nitrosyl complex. Evidence for direct substrate binding to the active site Fe2+ of extradiol dioxygenases, J. Biol. Chem 261, 2170–2178. [PubMed] [Google Scholar]
- [74].Orville AM, and Lipscomb JD (1989) Binding of isotopically labeled substrates, inhibitors, and cyanide by protocatechuate 3,4-dioxygenase, J. Biol. Chem 264, 8791–8801. [PubMed] [Google Scholar]
- [75].Cox DD, Benkovic SJ, Bloom LM, Bradley FC, Nelson MJ, Que L Jr., and Wallick DE (1988) Catecholate LMCT bands as probes for the active sites of nonheme iron oxygenases, J. Am. Chem. Soc 110, 2026–2032. [Google Scholar]
- [76].Carrano CJ, Carrano MW, Sharma K, Backes G, and Sanders-Loehr J (1990) Resonance Raman spectra of high- and low-spin ferric phenolates. Models for dioxygenases and nitrile hydratase, Inorg. Chem 29, 1865–1870. [Google Scholar]
- [77].Whittaker JW, Lipscomb JD, Kent TA, and Münck E (1984) Brevibacterium fuscum protocatechuate 3,4-dioxygenase. Purification, crystallization, and characterization, J. Biol. Chem 259, 4466–4475. [PubMed] [Google Scholar]
- [78].Davis MI, Orville AM, Neese F, Zaleski JM, Lipscomb JD, and Solomon EI (2002) Spectroscopic and electronic structure studies of protocatechuate 3,4-dioxygenase: Nature of tyrosinate-Fe(III) bonds and their contribution to reactivity, J. Am. Chem. Soc 124, 602–614. [DOI] [PubMed] [Google Scholar]
- [79].Pau MYM, Davis MI, Orville AM, Lipscomb JD, and Solomon EI (2007) Spectroscopic and electronic structure study of the enzyme-substrate complex of intradiol dioxygenases: substrate activation by a high-spin ferric non-heme iron site, J. Am. Chem. Soc 129, 1944–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Valley MP, Brown CK, Burk DL, Vetting MW, Ohlendorf DH, and Lipscomb JD (2005) Roles of the equatorial tyrosyl iron ligand of protocatechuate 3,4-dioxygenase in catalysis, Biochemistry 44, 11024–11039. [DOI] [PubMed] [Google Scholar]
- [81].Pyrz JW, Roe AL, Stern LJ, and Que L Jr. (1985) Model studies of iron-tyrosinate proteins, J. Am. Chem. Soc 107, 614–620. [Google Scholar]
- [82].Makhlynets OV, Das P, Taktak S, Flook M, Mas-Balleste R, Rybak-Akimova EV, and Que L Jr. (2009) Iron-promoted ortho- and/or ipso-hydroxylation of benzoic acids with H2O2, Chem. Eur. J 15, 13171–13180. [DOI] [PubMed] [Google Scholar]
- [83].Eser BE, Barr EW, Frantom PA, Saleh L, Bollinger JM Jr., Krebs C, and Fitzpatrick PF (2007) Direct spectroscopic evidence for a high-spin Fe(IV) intermediate in tyrosine hydroxylase, J. Am. Chem. Soc 129, 11334–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bugg TDH, and Ramaswamy S (2008) Non-heme iron-dependent dioxygenases: unravelling catalytic mechanisms for complex enzymatic oxidations, Curr. Opin. Chem. Biol 12, 134–140. [DOI] [PubMed] [Google Scholar]
- [85].Oloo WN, Fielding AJ, and Que L Jr. (2013) Rate-determining water-assisted O-O bond cleavage of an Fe(III)-OOH intermediate in a bio-inspired nonheme iron-catalyzed oxidation, J. Am. Chem. Soc 135, 6438–6441. [DOI] [PubMed] [Google Scholar]
- [86].Olah GA (1971) Aromatic substitution. XXVIII. Mechanism of electrophilic aromatic substitutions, Acc. Chem. Res 4, 240–248. [Google Scholar]
- [87].Galabov B, Nalbantova D, Schleyer P. v. R., and Schaefer HF (2016) Electrophilic aromatic substitution: New insights into an old class of reactions, Acc. Chem. Res 49, 1191–1199. [DOI] [PubMed] [Google Scholar]
- [88].Lee K (2006) p-Hydroxylation reactions catalyzed by naphthalene dioxygenase, FEMS Microbiol. Lett 255, 316–320. [DOI] [PubMed] [Google Scholar]
- [89].Barr SA, Bowers N, Boyd DR, Sharma ND, Hamilton L, Austin R, McMordie S, and Dalton H (1998) The potential role of cis-dihydrodiol intermediates in bacterial aromatic hydroxylation and the NIH shift, Perkin Trans 1, 3443–3451. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.