

Abstract
NLRP3 inflammasome has recently emerged as an attractive drug target for neurodegenerative disorders. In our continuing studies, a new chemical scaffold was designed as selective inhibitors of NLRP3 inflammasome. Initial characterization of the lead HL16 demonstrated improved however non-selective inhibition on the NLRP3 inflammasome. Structure-activity relationship studies of HL16 identified a new lead, 17 (YQ128), with an IC50 of 0.30 ± 0.01 μM. Further studies from in vitro and in vivo models confirmed its selective inhibition on the NLRP3 inflammaome and its brain penetration. Furthermore, pharmacokinetic studies in rats at 20 mg/kg indicated extensive systemic clearance and tissue distribution, leading to a half-life of 6.6 hours. However, the oral bioavailability is estimated to be only 10%, which may reflect limited GI permeability and possibly high first-pass effects. Collectively, these findings strongly encourage development of more potent analogs with improved pharmacokinetic properties from this new chemical scaffold.
Graphical Abstract
Introduction
Inflammasomes are important components of the innate immunity to initiate immune responses through pattern recognition receptors (PRRs).1 Three major types of PRRs have been identified and characterized to regulate the functions of inflammasomes and this includes the NOD-like receptor (NLR) containing family, the absent in melanoma 2 (AIM2), and retinoic acid-inducible gene I (RIG-I) like receptors (RLRs).2 Upon recognition of the damage associated molecular pattern molecules (DAMPs) released during tissue injury or stress and/or pathogen-associated molecular patterns (PAMPs), inflammasome complexes are assembled to include a sensor component, an adaptor component (the apoptosis-associated speck-like protein containing a caspase recruitment domain---ASC), and an effector component, typically pro-caspase-1, and the substrate component.2,3 Subsequently, pro-caspase-1 will be cleaved and activated to produce pro-inflammatory cytokines interleukin (IL)-1 β and IL-18.2,4 Among the members of the NLR family, the NLRP3 inflammasome plays essential roles in the maturation and production of IL-1β and IL-18.1 Notably, emerging studies have shown dysregulation of this inflammasome and IL-1β in the pathogenesis of many human diseases, such as autoinflammatory disorders, diabetes, and neurodegenerative disorders.5-12 Therefore, NLRP3 inflammasome has attracted extensive interests as a promising target of drug development for pathological conditions in which the dysregulation or overactivation of this inflammasome is evident.
Several small molecule inhibitors have recently been reported to block the NLRP3 inflammasome pathways. This includes MCC950,13-15, Bay 11-7082,16 CY-09,17 Oridonin,18 Tranilast,19 INF39,20 Glyburide,21,22 JC124.23, 24 among which MCC950 has been used in many studies as a pharmacological tool to demonstrate NLRP3 inflammasome as a viable drug target to development therapeutics for human diseases (Figure 1).13, 15 Our research group has recently designed and developed sulfonamide analogs as active NLRP3 inflammasome inhibitors and potential therapeutics for Alzheimer’s disease (AD), multiple sclerosis (MS) and traumatic brain injury (TBI).23-26 Furthermore, our studies suggested that the sulfonamide analogs directly interfere with the formation of the NLRP3 inflammasome complex.24,25 Studies of one of our lead compounds JC124 have shown both in vitro and in vivo activities in animal models of AD23 and TBI,26 thus strongly suggesting further development of analogs based on this chemical scaffold with improved potency and drug like properties. Herein, we report the design and discovery of a series of compounds from a new chemical scaffold. Structure-activity relationship (SAR) studies were conducted to understand the contributions of different structural features of the lead structure and to provide guidance for further structural optimization/refinement of this chemical scaffold.
Figure 1.

Structures of small molecule inhibitors targeting the NLRP3 inflammasome pathway.
Results and Discussion
Design of a new chemical scaffold as NLRP3 inhibitors.
Our SAR studies of JC124 (1, Figure 2) suggested that only limited modifications can be tolerated on the phenyl ring of the benzamide moiety.24 To expand the scope for structural variations and optimization of this lead structure, we designed a new chemical scaffold exemplified by HL16 (2, Figure 2). In this structure, we changed the amide functional group to an appendix position of the structure and this should allow introduction of a variety of substituents to explore the SAR and to optimize the biological activity. Specifically, we incorporated a propargyl substituent on the sulfonamide moiety and an acrylamide moiety based on the results of our chemical probe studies (unpublished data). Biological characterization from murine macrophage J774A.1 cells that release IL-1β upon the activation of NLRP3 inflammasome by lipopolysaccharide (LPS) and adenosine triphosphate (ATP)27 established an IC50 of 1.30 ± 0.23 μM for HL16 (Figure 3A), 2.5-fold increase compared that of JC124. The inhibitory activity of HL16 was also confirmed in mouse peritoneal macrophages (Figure 3B). However, when the selectivity was examined (J774A.1 cells were stimulated with LPS/poly(dA:dT) or LPS/flagellin to activate the NLRC4 and AIM2 inflammasome, respectively), HL16 also significantly inhibited NLRC4 and AIM2 inflammasomes at 10 μM concentration (Figure 3C).
Figure 2.

Designed new chemical scaffold from JC124.
Figure 3.

HL16 inhibits NLRP3 inflammasome. J774A. 1 cells (A) or mouse peritoneal macrophages (B) were primed with LPS (1 μg/mL) for 4.5 h and then treated with indicated concentrations of HL16 when adding ATP (5 mM) stimulation for 30 min. IL-β in the culture media was assayed by ELISA. (C) J774A.1 cells were treated with LPS (1 μg/mL) and HL16 (10 μM) for 1 h. Flagellin (1 μg/mL) was added and allowed to incubate for 6 hr or (Poly(dA:dT)) (4 μg/ml) for 8 hr. The supernatants were collected and levels of IL-1β were measured by ELISA. Data are expressed as mean ± SEM from at least 3 independent experiments with at least triplicates for each experiment. Statisitical analysis by student t-test.
Further testing in mice challenged with LPS also demonstrated that treatment with HL16 (10 mg/kg) led to significant suppression of both IL-1β (Figure 4A) and TNF-α (Figure 4B), while MCC950 (10 mg/kg), the known NLRP3 inhibitor13-15 as a positive control, only inhibited IL-1β, suggesting the nonspecific action of HL16.
Figure 4.
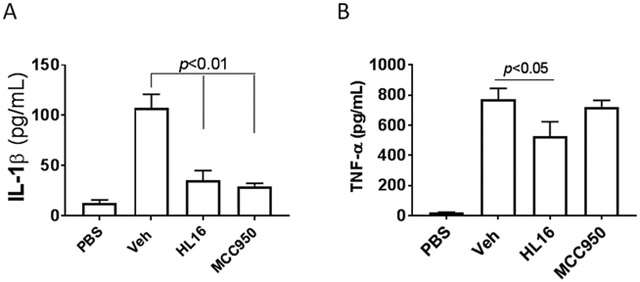
HL16 nonselectively suppressed the production of cytokines in vivo. Serum levels of IL-1β (A) and TNF-α (B) from C57BL/6 mice (n=4 per group) pretreated with HL16 (10 mg/kg) or MCC950 (10 mg/kg) by intraperitoneal injection (i.p.) were measured by ELISA 2.5 h after i.p. injection of LPS (50 mg/kg). Data are expressed as mean ± SD. Statistical analysis by student t-test.
Structural exploration of HL16 by SAR studies.
To explore whether structural modifications of HL16 will improve inhibitory potency and selectivity to NLRP3 inflammasome, a series of analogs were designed and synthesized. As shown in Figure 5, structural modifications were mainly focused on two positions of HL16: the appendix amide and the methoxy group on the benzyl moiety. To evaluate the role of the chlorine substitution, we also designed analogs with a fluorine or bromine at this position. In total, twenty-six analogs were designed and synthesized.
Figure 5.

Chemical structures of the designed analogues of HL16.
The chemical syntheses were achieved by the conditions outlined in Schemes 1 and 2. Briefly, reaction of sulfonyl chloride 29 with propargylamine gave 30, followed by de-protection using methylhydrazine in benzene to provide the amine 31. Aldehyde 32 was reacted with iodomethane or 1-bromopropane to obtain compound 33 or 35, respectively. Reductive amination of 33 with 31 in the presence of NaBH3CN yielded 34, which on coupling reaction with various carboxylic acids in the presence of 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) and 1-hydroxybenzotriazole (HOBt) gave the target compounds 2-6. Alkylation of 34 with allyl bromide or 1-bromopropane afforded 7 and 8, respectively. Similarly, compound 9-25 were synthetized following the conditions outlined in schemes 1 and 2 with 35, 37, or 40 as the starting material.
Scheme 1a.

aReagents and conditions: (a) Propargylamine, TEA, DCM; (b) Methylhydrazine, benzene; (c) Iodomethane, K2CO3, DMF; (d)TEA, Acetic acid, NaCNBH3; (e) various acids, EDCI, HOBt, TEA, DCM; For 7 and 8: Allylbromide or 1-Bromopropane, K2CO3, DMF; (f) 1-Bromopropane, K2CO3, DMF; (g) TEA, Acetic acid, NaCNBH3; (h) various acids, EDCI, HOBt, TEA, DCM. (i) 3-Thiopheneacetic acid, EDCI, HOBt, TEA, DCM.
Scheme 2a.

aReagents and conditions: (a) Bromobutane or 1-Bromopentane or 2-Bromobutane, K2CO3, DMF (b) TEA, Acetic acid, NaCNBH3; (c) 3-Thiopheneacetic acid, EDCI, HOBt, TEA, DCM.
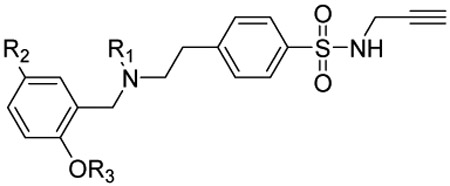
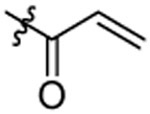
After synthesis, the analogs were tested using the J774A. 1 cell model upon challenge with LPS/ATP to measure their inhibitory potencies on the release of IL-1β by an enzyme-linked immunosorbent assay (ELISA). As shown in Table 1, saturation of the acrylamide to a propanamide as represented by compound 3 retained the inhibitory potency.. Chain extension to a hexanamide as in 4 did not lead to improved inhibitory potency. Notably, replacement with a 2-phenylacetamide lead to ~2-fold increase of inhibitory potency as evidenced by compound 5. Bioisoteric replacement of the phenyl ring of 5 with a thiophene (6) retained its inhibitory potency. To evaluate whether the appendix amide is essential to the biological activity, compounds 7 and 8 were designed and tested. The results of these two analogs suggest that a tertiary amine can still maintain the inhibitory potency. Replacement of the methoxy substituent in the structure of HL16 with a propoxyl one as illustrated by 9 led to a slight improvement of inhibitory potency. Therefore, in the following analogs, we kept this propoxyl substituent and varied the appendix amide to evaluate their effects on the inhibitory potency. Surprisingly, the combination of a 2-phenylacetamide and a propoxyl substituent in compound 10 did not result in increased inhibitory potency compared to 5 or 9. Introduction of a para-methoxy on the phenyl ring of 10 led to reduction of inhibitory potency as seen by compound 11. However, structural extension to a 3-phenylpropanamide in 12 retained the inhibitory potency. Bioisosteric replacement of the phenyl ring of the 2-phenylacetamide with a 2-(13) or 3-pyridine (14), or 3-indole (15) retained the inhibitory potency while replacement with an imidazole-4-yl moiety (16) led to a ~2-fold reduction of inhibitory potency. Notably, replacement of the phenyl ring of the 2-phenylacetamide with a 3-thiophene (17) further increased the inhibitory potency by 2-fold. Change of the 2-phenylacetamide of 10 to a furan-3-carboxamide (18), thiophene-3-carboxamide (19), 1,3-thiazole-4-carboxamide (20), or (2,5-diaza-1-thiazole)-3-carboxamide (21) all slightly increased the inhibitory potency while replacement with (1,2,4-traizole)-3-carboxamide (22) and pyrazine-2-carboxamide (23) led to slight reduction of inhibitory potency. Based on these observations, we further replaced the Cl of compound 17 with a Br (24) or F (25) and this led to reduction of inhibitory potency. We also evaluated how the propoxyl substituent of 17 can be further modified in analogs 26-28. The results demonstrated that increase of chain length or steric hindrance did not improve the inhibitory potency. Although some of the structural modifications represented by these analogs only led to slight improvements of inhibitory potency, overall, the SAR studies identified critical postions to optimize and improve inhibitory potency for the design of next generation analogs.
Table 1.
Inhibitory potency of the designed analogs on the production of IL-1β by J774A.1 cells upon stimulation with LPS/ATPa.
 |
||||
|---|---|---|---|---|
| Cpd | R1 | R2 | R3 | IC50 (μM)a |
|
2 (HL16) |
 |
Cl | 1.30±0.23 | |
| 3 | 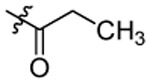 |
Cl | 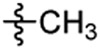 |
1.47±0.20 |
| 4 | 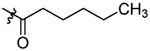 |
Cl | 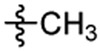 |
1.93±0.11 |
| 5 | 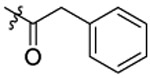 |
Cl | 0.69±0.15 | |
| 6 | 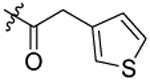 |
Cl | 0.90±0.06 | |
| 7 | 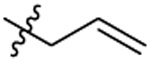 |
Cl | 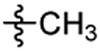 |
1.78±0.21 |
| 8 | 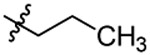 |
Cl | 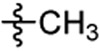 |
1.15±0.12 |
| 9 | 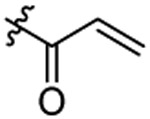 |
Cl | 0.97±0.12 | |
| 10 | 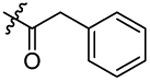 |
Cl | 0.87±02 | |
| 11 | 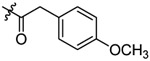 |
Cl | 1.49±0.07 | |
| 12 | 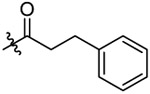 |
Cl | 0.62±0.02 | |
| 13 | 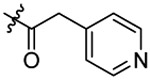 |
Cl | 0.73±0.07 | |
| 14 | 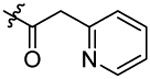 |
Cl | 0.89±0.08 | |
| 15 | 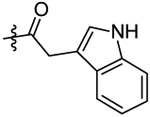 |
Cl | 0.79±0.07 | |
| 16 | 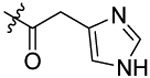 |
Cl | 1.66±0.10 | |
| 17 | 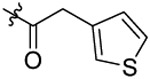 |
Cl | 0.30±0.01 | |
| 18 | 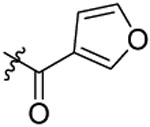 |
Cl | 0.47±0.04 | |
| 19 | 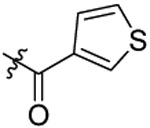 |
Cl | 0.31±0.04 | |
| 20 | 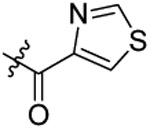 |
Cl | 0.58±0.09 | |
| 21 | 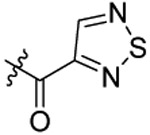 |
Cl | 0.46±0.03 | |
| 22 | 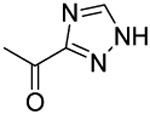 |
Cl | 1.06±0.04 | |
| 23 | 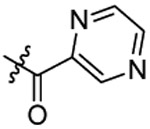 |
Cl | 0.88±0.09 | |
| 24 | 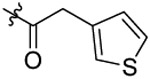 |
Br | 0.91±0.09 | |
| 25 | 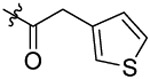 |
F | 0.67±0.07 | |
| 26 | 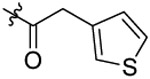 |
Cl | 0.55±0.05 | |
| 27 | 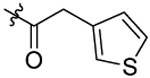 |
Cl | 0.47±0.14 | |
| 28 | 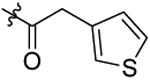 |
Cl | 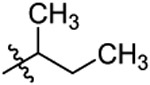 |
0.78±0.19 |
The inhibitory potency was derived from results of at least 3 independent experiments with at least triplicates for each experiment; data are shown as mean ± SEM.
Compound 17 is a selective inhibitor of NLRP3 inflammasome.
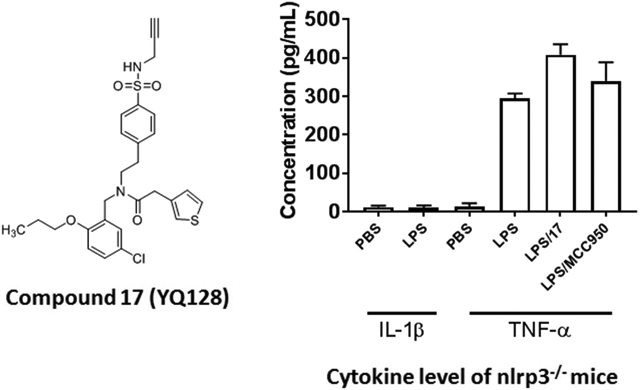
After the establishment of inhibitory potency on IL-1β release in J774A.1 cells for the designed analogs, we selected 17 (YQ128), the most potent analog among this series, for further characterization. Before moving to test its selectivity, we confirmed its inhibitory activity in primary mouse peritoneal macrophages. As shown in Figures 6A, both 17 and MCC950, the known NLRP3 inhibitor as a positive control, dose dependently suppressed the release of IL-1β from peritoneal macrophages upon LPS/ATP challenge with an IC50 of 1.59 ± 0.60 and 0.04 ± 0.0008 μM, respectively. Compound 17 is ~5 times less potent in peritoneal macrophagess than in J774A.1 cells to inhibit the release of IL-1β. This may suggest that J774A.1 are more sensitive to 17 under the current experimental conditions. We next examined the effects of 17 on NLRC4 and AIM2 inflammasomes. As shown in Figure 6B, treatment of J774A.1 cells with 17 under these experimental conditions did not significantly interfere with the production of IL-1β by NLRC4 or AIM2 inflammasome (by student t-test analysis), thus suggesting the specific inhibition of NLRP3 inflammasome by 17. We then tested the in vivo engagement of and selectivity to the NLRP3 inflammasome by 17. C57BL/6 mice (n=4 per group) were pretreated with 17 or MCC950 (positive control) at 10 mg/kg before intraperitoneal injection of LPS, which has been shown to trigger IL-1β production in a NLRP3-dependent manner.28 As shown in Figure 6C, serum level of IL-1β was significantly reduced while no significant inhibition on the TNF-α level (Figure 6D) was observed by the treatment of both compounds at the tested dose, thus strongly suggesting the selective in vivo engagement of NLRP3 inflammasome in the observed effects by 17 and MCC950. Lastly, we confirmed the selective inhibition on NLRP3 inflammasome by 17 in nlrp3−/− mice (n=3 per group). As expected, upon stimulation with LPS, nlrp3 deficiency abolished the production of IL-1β while the production of TNF-α is normal (Figure 6E). Treatment of nlrp3−/− mice with 17 (10 mg/kg) did not produce inhibition on the level of TNF-α and this again confirmed the selective inhibition on the NLRP3 inflammasome by our compound. The results from the selectivity studies serve as an indirect evidence to support the MOA that analogs derived from this chemical scaffold interfere with the NLRP3 inflammasome complex, instead of the upstream priming step by the LPS, which is consistent with our previously reported results.25
Figure 6.

Compound 17 selectively inhibits the NLRP3 inflammasome. (A) Mouse peritoneal macrophages were primed with LPS (1 μg/mL) for 4.5 h and then treated with indicated compounds at indicated concentrations when adding ATP (5 mM) stimulation for 30 min. IL-β in the culture media was assayed by ELISA. (B) J774A. 1 cells were treated with LPS (1 μg/mL) and 17 (10 μM) for 1 h. Flagellin (1 μg/mL) was added and allowed to incubate for 6 hr or (Poly(dA:dT)) (4 μg/ml) for 8 hr. The supernatants were collected and levels of IL-1β were measured by ELISA. Serum levels of IL-1β.(C) and TNF-α (D) from C57BL/6 (n=4 per group) mice pretreated 17 (10 mg/kg) or MCC950 (10 mg/kg) were measured by ELISA 2.5 h after i.p. injection of LPS (50 mg/kg). (E) Serum levels of IL-1β and TNF-α under indicated treatment conditions (both 17 and MCC950 were tested at 10 mg/kg) of nlrp3−/− mice (n=3 per group) were measured by ELISA 2.5 h after i.p. injection of LPS (25 mg/kg). For in vitro assasys, data are expressed as mean ± SEM from at least 3 independent experiments with at least triplicates for each experiment. For in vivo assays, data are expressed as mean ± SD. Statistical analysis by student t-test.
Compound 17 is a BBB penetrant but shows poor oral pharmacokinetic properties.
Since our ultimate goal is to develop small molecule inhibitors of the NLRP3 inflammasome as potential therapeutics for neurodegenerative disorders, especially for AD, effective drug candidates need to cross the blood-brian barrier (BBB) and reach the brain tissue and be suitable for chronic once-daily oral administration. To test whether 17 is a brain penetrant, we first determined its permeability and transport directionality using immortalized human cerebral microvascular endothelial cells hCMEC/D3 as the human BBB model.29 This model expresses functional efflux transporters such as P-glycoprotein which are also expressed at the BBB; it has been widely used as a surrogate for human BBB.29, 30 Apparent permeability (Papp) was calculated using Papp = (dX/dT • Vr )/(A • Co), where dX/dT is the mass of transported compound (X) over time (T), Vr is the volume in the receiver compartment, A is the surface area of the membrane insert, and Co is the initial concentration in the donor compartment.30 We first examined the potential cytotoxicity of 17 on hCMEC/D3 cells to rule out any potential interference with results interpretation, and the results demonstrated that 17 at 20 μM did not show significant toxic effects on these cells (Figure 7A). The apical-to-basolateral (A to B) and basolateral-to-apical (B to A) Papp of 17 was 5.21 ± 0.56 × 10−6 and 1.11 ± 0.12 × 10−6 cm/sec, respectively (Figure 7B). Thus, 17 exhibits an efflux ratio of 0.22, suggesting that 17 is not likely subject to active efflux. We next confirmed the in vivo BBB penetration of 17 in C57BL/6 mice (n=3 per time point) by oral (PO) administration (20 mg/kg, single dose). To accurately quantify the amount of 17 delivered to the brain, we perfused the mice brain to wash out the vascular blood completely prior to collecting and homogenizing brain tissues. Plasma and brain homogenate samples were analyzed by Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS). As shown in Table 2, compound 17 appeared in plasma with the highest concentration at as early as 30 min after PO administration. Brain concentrations of compound 17 after 0.5, 1, and 4 h were 21.3, 20.5, and 6.6 ng/g, respectively, indicating that 17 penetrated into the CNS after oral administration at the tested dose. Furthermore, the brain-to-plasma concertation ratio increases with time. However, after proximate conversion (based on the assumption that 1 g of brain tissue is around 1 mL in volume), the brain concentration of 17 was around 12 nM after 4 h of oral ingestion, 25 fold less than its in vitro IC50 value. Lastly, we evaluated the basic pharmacokinetic (PK) properties of 17 in Sprague-Dawley rats (n=3 and 11 serial plasma samples over a period of 24 h; plasma concentrations were quantified by LC-MS/MS) after a dose of 20 mg/kg via intravenous (IV) and PO administration. The results showed that after IV administration, 17 exhibits extensive extravascular distribution with a large steady-state volume of distribution (Vdss) of 8.5 L/kg and rapid total clearance (CLtot) of 41 mL/min/kg, approaching hepatic (and renal) blood flow with the possibility of extrahepatic clearance mechanisms. This resulted in an intermediate terminal plasma half-life (t1/2) of 6.6 h after IV administration. After oral administration of an ad-hoc suspension using 10% Cremophor EL in PBS, this compound shows delayed gastrointestinal absorption with a tmax and cmax of 12 h and 73 ng/mL, respectively (Fig. 7C); oral bioavailability (Foral) was estimated as 10%. This suggests the possibility of poor GI solubility/permeability and/or high first-pass effects at the tested dose of 20 mg/kg PO.
Figure 7.
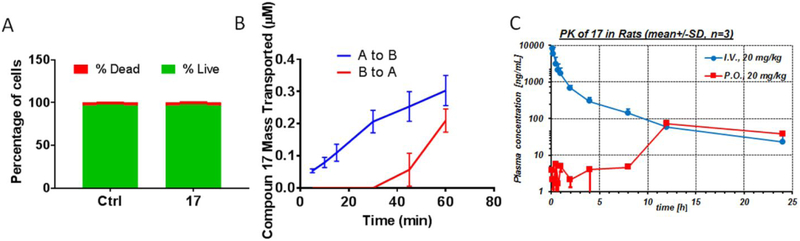
Permeability of 17 in hCMEC/D3 cells and PK properties in rats. (A) hCMEC/D3 cells were treated with 17 (20 μM) for 2 h, then cell viability was measured using the Live/DeadTM kit. (B) hCMEC/D3 cells were plated on transwell filters. Compound 17 (20 μM) was added to either the apical or basolateral side, then samples were analyzed by HPLC to determine flux (A-B: apical-to-basolateral; B-A: basolateral-to-apical) at indicated time points. (C) Spraque-Dawley rats were treated with 17 (20 mg/kg) via IV and PO (n=3 per rout). Plasma samples were collected at indicated time points and analyzed by LC-MS/MS. Data are expressed as mean ± SEM for studies in hCMEC/D3 cells from 3 independent experiments with at least triplicates for each experiement, as mean ± SD for studies in rats.
Table 2.
BBB penetration of 17 (PO, 20 mg/kg) in mice (n=3, mean ± SD) at various time points
| 0.5 h | 1 h | 4 h | |
|---|---|---|---|
| Brain (ng/g) | 21.25 ± 16.90 | 20.48 ± 4.19 | 6.58 ± 11.39 |
| Plasma (ng/mL) | 277.33 ± 182.15 | 57.60 ± 55.65 | 8.00 ± 10.51 |
| Brain-to-Plasma ratio | 0.077 | 0.36 | 0.82 |
Conclusions
In our continuing efforts to develop small molecule inhibitors of the NLRP3 inflammasome, a new chemical scaffold represented by HL16 was designed to allow more scope for structural modifications. Although HL16 exhibited improved inhibitory potency on IL-1β production from J774A.1 cells compared to our previously reported inhibitor, it suffered the loss of selectivity to the NLRP3 inflammasome. Further SAR studies of HL16 established that the 2-OCH3 can be modified to improve inhibitory potency. The acrylamide domain of HL16 can tolerate strucutral modifications and a heteroaromatic acetamide tend to provide analogs with improved potency. As a result of this SAR study, one new lead compound, 17, was identified with a more than 4-fold increased inhibitory potency compared to HL16. Biological characterization in murine peritoneal macrophages and J774A.1 cells confirmed its inhibitory potency and selectivity to the NLRP3 inflammasome. More importantly, studies in LPS-challenged mice, a mouse model in which the release of IL-1β is NLRP3 inflammasome dependent, demonstrated that 17 after a single dose of 10 mg/kg signifiantly and selectively suppressed the production of IL-1β, but not TNF-α, thus supporting its in vivo engagement of NLRP3 inflammasome. In addition, results from studies in nlrp3−/− mice echoed its selective engagement of NLRP3 inflammsome since no inhibitory activity on the production of TNF-α was observed. Studies from both in vitro and in vivo models supported that 17 can cross the BBB to reach the CNS and is not likely subject to efflux transport. However, while PK studies in rats suggested adequate systemic characteristics with a plasma t1/2 of 6.6 hours, oral bioavailability was quite low (10%), likely due to poor GI solubility and possibly high first-pass effects. Collectively, these findings strongly encourage further development of new analogs based on this chemical scaffold with improved PK properties as inhibitors of the NLRP3 inflammasome and explore their potential therapeutic applications.
Experimental Section
Chemistry.
Reagents and solvents were obtained from commercial suppliers and used as received unless otherwise indicated. All reactions were carried out under inert atmosphere (N2) unless otherwise noted. Reactions were monitored by thin-layer chromatography (TLC) (precoated silica gel 60 F254 plates, EMD Chemicals) and visualized with UV light or by treatment with Phosphomolybdic acid (PMA). Flash chromatography was performed on silica gel (200-300 mesh, Fisher Scientific) using solvents as indicated. 1HNMR and 13C NMR spectra were routinely recorded on Bruker ARX 400 spectrometer. The NMR solvent used was CDCl3 or DMSO-d6 as indicated. Tetramethylsilane (TMS) was used as internal standard. The purity of target compounds was determined by HPLC using Varian 100-5 C18 250 × 4.6 mm column with UV detection (230 nm) (60% acetonitrile/40% H2O/0.1 % trifluoroacetic acid (TFA) and 80% methanol/19.9% H2O/0.1% TFA, two solvent systems) to be ≥ 95%.
Method C. N-(5-chloro-2-methoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)acryl-amide (2).
Acrylic acid (0.03 g, 0.4 mmol) and HOBt (0.068 g, 0.5 mmol) were dissolved in anhydrous DCM (20 mL) under ice bath followed by the addition of EDCI (0.096 g, 0.5 mmol). The mixture was stirred at r.t. for 30 min. Compound 34 (0.20 g, 0.5 mmol) and TEA (0.07 mL) in DCM (30 mL) were added directly. The reaction was stirred at r.t. for 3 h. Then DCM solution was washed with 1N HCl, saturated NaHCO3 and brine for 3 times, dried over anhydrous Na2SO4 and evaporated under vacuum. The crude product was purified by chromatography using EtOAc–hexane (1:1) as the mobile phase, to obtain 2 as a white solid (0.088 g, yield: 47%).1H-NMR (400 MHz CDCl3): δ 2.03-2.04 (m, 1H), 2.84-2.91 (m, 2H), 3.51-3.58 (m, 2H), 3.74-3.76 (m, 5H), 4.37-4.55 (m, 2H), 4.89-4.91 (m, 1H), 5.60-5.64 (m, 1H), 6.31-6.45 (m, 2H), 6.74 (d, J=8.68 Hz, 1H), 6.92 (d, J= 2.52 Hz, 1H), 7.09-7.22 (m, 2H), 7.27 (d, J=8.24 Hz, 1H), 7.72-7.77 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 167.1, 156.0, 144.8, 138.3, 129.5, 129.2, 128.7, 128.3, 127.8, 127.6, 127.3, 126.6, 125.9, 111.5, 77.9, 73.1, 55.8, 48.7, 33.8, 32.9, 29.7. HRMS (AP-ESI) m/z calcd for C22H23ClN2O4S [M + Na]+ 469.0965, found 469.1003.
N-(5-chloro-2-methoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)propion-amide(3).
Compound 3 was prepared from propanoic acid and 34 following the procedure of Method C in 55% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.93-0.98 (m, 3H), 2.24-2.36 (m, 2H), 2.82 (t, J=7.44 Hz, 1H), 2.95 (t, J=7.52 Hz, 1H), 3.03-3.05 (m, 1H), 3.49 (t, J=7.36 Hz, 1H), 3.54 (t, J=7.48 Hz, 1H), 3.64-3.66 (m, 2H), 3.82 (d, J=5.12 Hz, 3H), 4.44 (d, J=8.32 Hz, 2H), 6.98-7.07 (m, 2H), 7.27-7.36 (m, 1H), 7.39 (d, J=8.16 Hz, 1H), 7.47 (d, J=8.20 Hz, 1H), 7.70-7.74 (m, 2H), 8.05-8.10 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.1, 155.7, 144.3, 138.3, 129.5, 129.3, 128.1, 127.5, 127.0 126.8, 126.5, 124.2, 112.6, 79.3, 74.6, 55.7, 48.5, 46.9, 34.1, 31.9, 25.4, 9.4. HRMS (AP-ESI) m/z calcd for C22H25ClN2O4S [M + Na]+ 471.1121, found 471.1156.
N-(5-chloro-2-methoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)hexan-amide (4).
Compound 4 was prepared from hexanoic acid acid and 34 following the procedure of Method C in 62% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.82-0.88 (m, 3H), 1.18-1.29 (m, 4H), 1.43-1.48 (m, 2H), 2.26 (t, J=7.32 Hz, 2H), 2.83 (t, J=7.68 Hz, 1H), 2.94 (t, J=7.32 Hz, 1H), 3.03-3.05 (m, 1H), 3.50 (t, J=7.40 Hz, 1H), 3.56 (t, J=7.28 Hz, 1H), 3.64-3.66 (m, 2H), 3.82 (d, J=3.92 Hz, 3H), 4.44 (d, J=7.40 Hz, 2H), 6.99 (dd, J12.68 Hz, J2=9.48 Hz, 1H), 7.01-7.07 (m, 1H), 7.27-7.37 (m, 1H), 7.40 (d, J=8.32 Hz, 1H), 7.46 (d, J=8.36 Hz, 1H), 7.70-7.75 (m, 2H), 8.05-8.10 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 172.4, 155.7, 144.3, 138.4, 129.6, 129.3, 128.1, 127.6, 126.9, 126.8, 124.2 112.6, 79.3, 74.6, 55.7, 48.5, 46.9, 34.1, 31.9, 31.6, 30.9, 24.5, 22.0, 13.9. HRMS (AP-ESI) m/z calcd for C25H31C1N2O4S [M + Na]+ 513.1591, found 513.1599.
N-(5-chloro-2-methoxybenzyl)-2-phenyl-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)aceamide (5).
Compound 5 was prepared from 2-phenyl acetic acid and 34 following the procedure of Method C in 69% yield. 1H-NMR (400 MHz DMSO-d6): δ 2.83 (t, J=7.20 Hz, 1H), 2.89 (t, J=7.56 Hz, 1H), 3.02-3.03 (m, 1H), 3.49 (t, J=7.00 Hz, 1H), 3.58 (t, J=7.00 Hz, 1H), 3.64-3.69 (m, 4H), 3.82 (d, J=2.72 Hz, 3H), 4.47 (d, J=8.88 Hz, 2H), 6.99 (dd, J1=2.64 Hz, J2=16.64 Hz, 1H), 7.07 (dd, J1=8.80 Hz, J2=17.36 Hz, 1H), 7.13-7.36 (m, 7H), 7.45 (d, J=8.40 Hz, 1H), 7.69 (d, J=8.28 Hz, 1H), 7.76 (d, J=8.83 Hz, 1H), 8.05-8.11 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 155.8, 144.1, 138.5, 135.7, 129.6, 129.3, 129.0, 128.9, 128.3, 127.8, 127.6, 127.2, 126.9, 124.3, 112.7, 79.3, 74.6, 55.8, 48.9, 46.7, 42.8, 34.1, 31.9. HRMS (AP-ESI) m/z calcd for C27H27C1N2O4S [M + Na]+ 533.1278, found 533.1285.
N-(5-chloro-2-methoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-2-(thiophen-3-yl)acetamide (6).
Compound 6 was prepared from thiophene-3-yl-acetic acid and 34 following the procedure of Method C in 57% yield. 1H-NMR (400 MHz DMSO-d6): δ 2.79-2.89 (m, 2H), 3.02-3.04 (m, 1H), 3.47 (t, J=7.56 Hz, 1H), 3.59 (t, J=7.40 Hz, 1H), 3.64-3.69 (m, 4H), 3.81 (s, 3H), 4.46 (d, J=4.68 Hz, 2H), 6.93-7.07 (m, 3H), 7.25-7.29 (m, 3H), 7.44-7.50 (m, 2H), 7.70 (d, J=8.32 Hz, 1H), 7.75 (d, J=8.32 Hz, 1H), 8.05-8.10 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.3, 155.7 144.1, 138.5, 135.5, 129.6, 129.2, 128.6, 128.2, 127.7, 127.2, 127.0, 126.8, 125.9, 124.3, 112.7, 79.3, 74.6, 55.8, 48.9, 46.7, 34.6, 34.3, 31.9. HRMS (AP-ESI) m/z calcd for C25H25C1N2O4S2 [M + Na]+ 539.0842, found 539.0866.
Method D. 4-(2-(allyl(5-chloro-2-methoxybenzyl)amino)ethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (7).
A mixture of 34 (0.2 g, 0.5 mmol) and K2CO3 (0.14 g, 1.0 mmol) in DMF was stirred at room temperature. Then allyl bromide (0.06 g, 0.5 mmol) was added and the mixture was stirred at room temperature for 6 h. After removing DMF under reduced pressure, the residue was taken up to DCM. The DCM solution was washed with water for 3 times, dried over anhydrous Na2SO4, filtered and evaporated. The crude product was purified by chromatography using EtOAc–Hexane (1:1) as the mobile phase, to obtain 7 as a white solid (0.058 g, yield: 27%). 1HNMR (400 MHz DMSO-d6): δ 2.78-2.84 (m, 4H), 3.06 (t, J=2.48 Hz, 1H), 3.69 (s, 2H), 3.76-3.80 (m, 5H), 4.03 (d, J=2.52 Hz, 2H), 5.19-5.28 (m, 2H), 5.66-5.76 (m, 1H), 6.98 (d, J=8.76 Hz, 1H), 7.26 (dd, J1=2.72 Hz, J2=8.68 Hz, 1H), 7.31 (d, J=2.76 Hz, 1H), 7.46 (d, J=8.32 Hz, 2H), 7.76 (d, J=8.36 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 155.7, 146.3, 136.0, 132.2, 130.6, 129.4, 128.1, 127.3, 127.1, 119.4, 112.1, 76.9, 76.2, 55.6, 49.7, 48.9, 46.6, 36.0, 35.4. HRMS (AP-ESI) m/z calcd for C22H25C1N2O3S [M + H]+ 433.1347, found 433.1338.
4-(2-((5-chloro-2-methoxybenzyl)(propyl)amino)ethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (8).
Compound 8 was prepared from 1-bromopropane and 34 following the procedure of Method D in 21% yield. 21% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.86 (t, J=7.36 Hz, 3H), 1.48-1.57 (m, 2H), 2.78-2.83 (m, 4H), 3.04-3.09 (m, 3H), 3.68 (s, 2H), 3.76 (s, 3H), 4.09 (d, J=2.48 Hz, 2H), 6.98 (d, J=8.72 Hz, 1H), 7.26 (dd, J1=2.76 Hz, J2=8.68 Hz, 1H), 7.31 (d, J=2.76 Hz, 1H), 7.45 (d, J=8.36 Hz, 2H), 7.73 (d, J=8.32 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 155.7, 146.1, 130.6, 129.3, 128.1, 127.2, 127.1, 123.9, 112.1, 77.5, 75.9, 55.6, 49.7, 48.2, 46.6, 36.2, 35.4, 20.4 10.9. HRMS (AP-ESI) m/z calcd for C22H27C1N2O3S [M + H]+ 435.1503, found 435.1457.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)acryl-amide (9).
compound 9 was synthetized from acrylic acid and compound 36 following the procedure of Method C in 58% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.95-1.00 (m, 3H), 1.70-1.76 (m, 2H), 2.86 (t, J=7.64 Hz, 1H), 2.93 (t, J=7.68 Hz, 1H), 3.02-3.04 (m, 1H), 3.54-3.69 (m, 4H), 3.98 (t, J=6.44 Hz, 2H), 4.44-4.53 (m, 2H), 5.63-5.69 (m, 1H), 6.09-6.20 (m, 1H), 6.65-6.76 (m, 1H), 6.99-7.10 (m, 2H), 7.26-7.46 (m, 3H), 7.73 (d, J=8.16 Hz, 2H), 8.04-8.06 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 165.6, 155.2, 144.1, 138.4, 129.6, 129.2, 128.3, 127.9, 127.8, 127.3, 126.8, 123.9, 113.4, 79.3, 74.6, 69.6, 48.4, 47.2, 34.7, 31.9, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C24H27C1N2O4S [M + Na]+ 497.1277, found 497.1248.
N-(5-chloro-2-propoxybenzyl)-2-phenyl-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)acetamide (10).
Compound 10 was prepared from 2-phenylacetic acid and 36 following the procedure of Method C in 49% yield as a white solid. 1HNMR. (400 MHz DMSO-d6): δ 0.93-0.99 (m, 3H), 1.70-1.73 (m, 2H), 2.80 (t, J=7.52 Hz, 1H), 2.87 (t, J=7.44 Hz, 1H), 3.01-3.02 (m, 1H), 3.49 (m, 1H), 3.60 (t, J=7.56 Hz, 1H), 3.64-3.68 (m, 4H), 3.92-3.98 (m, 2H), 4.46 (t, J=14.96 Hz, 2H), 6.98-7.05 (m, 2H), 7.13-7.33 (m, 7H), 7.42 (d, J=8.16 Hz, 1H), 7.68 (d, J=8.32 Hz, 1H), 7.74 (d, J=8.32 Hz, 1H), 8.05-8.08 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.5, 155.2, 144.1 138.5, 135.7, 129.5, 129.2, 129.1, 128.9, 128.3, 127.9, 127.6, 127.3, 126.8, 126.4, 124.1, 113.4 79.3, 74.6, 69.5, 48.8, 46.6, 42.9, 34.1, 31.9, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C29H31C1N2O4S [M + Na]+ 561.1591, found 561.1595.
N-(5-chloro-2-propoxybenzyl)-2-(4-methoxyphenyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)acetamide (11).
Compound 11 was prepared from 2-(4-methoxy)-phenylacetic acid and 36 following the procedure of Method C in 51% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.95-1.00 (m, 3H), 1.71-1.76 (m, 2H), 2.85 (t, J=7.52 Hz, 1H), 2.97 (t, J=7.40 Hz, 1H), 3.02-3.03 (m, 1H), 3.51 (t, J=6.96 Hz, 1H), 3.57-3.67 (m, 5H), 3.72-3.77 (m, 3H), 3.94-3.99 (m, 2H), 4.45 (d, J=4.00 Hz, 2H), 6.87-7.10 (m, 5H), 7.20-7.31 (m, 2H), 7.37 (d, J=8.20 Hz, 1H), 7.45 (d, J=8.24 Hz, 1H), 7.71 (d, J=8.32 Hz, 1H), 7.77 (d, J=8.28 Hz, 1H), 8.03-8.09 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.7, 156.7, 155.0, 144.2, 138.5, 130.6, 130.4, 129.5, 129.2, 128.1, 127.9, 127.5, 127.0, 126.8, 124.3, 123.9, 120.2, 113.3, 110.6, 79.3, 74.6, 69.6, 55.4, 49.0, 47.1, 34.3, 31.9, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C30H33C1N2O5S [M + Na]+ 591.1696, found 591.1656.
N-(5-chloro-2-propoxybenzyl)-3-phenyl-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)propanamide (12).
Compound 12 was prepared from 3-phenylpropanoic acid and 36 following the procedure of Method C in 55% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.90-0.99 (m, 3H), 1.64-1.75 (m, 2H), 2.57-2.64 (m, 2H), 2.76-2.88 (m, 4H), 3.01-3.05 (m, 1H), 3.49 (t, J=7.40 Hz, 1H), 3.55 (t, J=7.32 Hz, 1H), 3.63-3.66 (m, 2H), 3.91-3.96 (m, 2H), 4.38-4.44 (m, 2H), 6.99-7.04 (m, 2H), 7.16-7.32 (m, 6H), 7.35 (d, J=8.36 Hz, 1H), 7.40 (d, J=8.32 Hz, 1H), 7.69-7.73 (m, 2H), 8.05-8.10 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 171.6, 155.1, 144.2, 141.3 138.4, 129.5, 129.2, 128.3, 128.2, 128.1, 127.7, 127.4, 127.3, 126.8, 125.8, 124.0, 113.4, 79.3 74.6, 69.6, 48.3, 46.9, 34.1, 33.4, 31.9, 30.7, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C30H33C1N2O4S [M + Na]+ 575.1747, found 575.1750.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-2-(pyri-din-4-yl)acetamide (13).
Compound 13 was prepared from 2-(pyridine-4-yl)acetic acid and 36 following the procedure of Method C in 41% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.99-1.05 (m, 3H), 1.75-1.81 (m, 2H), 2.88 (t, J=7.52 Hz, 1H), 3.02 (t, J=7.40 Hz, 1H), 3.08 (t, J=2.48 Hz, 1H), 3.52-3.56 (m, 1H), 3.66-3.71 (m, 3H), 3.78-3.83 (m, 2H), 3.99-4.05 (m, 2H), 4.53 (s, 2H), 7.05-7.15 (m, 2H), 7.20-7.23 (m, 2H), 7.29-7.33 (m, 1H), 7.40 (d, J=8.16 Hz, 1H), 7.53 (d, J=8.32 Hz, 1H), 7.76 (d, J=8.24 Hz, 1H), 7.82 (d, J=8.28 Hz, 1H), 8.11-8.14 (m, 1H), 8.51-8.54 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 169.4, 156.6, 155.2, 149.3, 144.8, 144.0, 138.6, 129.6, 129.2, 128.4, 127.7, 127.0, 126.8, 124.9, 124.6, 124.1, 113.4, 79.3, 74.6, 69.6, 48.7, 47.5, 46.7, 34.3, 31.9, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C28H30C1N3O4S [M + Na]+ 562.1543, found 562.1579.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-2-(pyri-din-2-yl)acetamide (14).
Compound 14 was prepared from 2-(pyridine-2-yl)acetic acid and 36 following the procedure of Method C in 43% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.94-1.00 (m, 3H), 1.69-1.77 (m, 2H), 2.81 (t, J=7.48 Hz, 1H), 2.96 (t, J=7.40 Hz, 1H), 3.08 (t, J=2.52 Hz, 1H), 3.49 (t, J=7.40 Hz, 1H), 3.63-3.67 (m, 3H), 3.87 (d, J=9.08 Hz, 2H), 3.93-3.97 (m, 2H), 4.46-4.52 (m, 2H), 6.97-7.34 (m, 6H), 7.46 (d, J=8.40 Hz, 1H), 7.66-7.78 (m, 3H), 8.04-8.09 (m, 1H), 8.54-8.56 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 169.9, 156.2, 155.1, 148.9, 144.1, 138.5, 136.6, 129.5, 129.2, 128.2, 127.8, 127.4, 127.3, 126.9, 124.1, 123.9, 121.9, 113.4, 79.3, 74.6, 69.6, 49.2, 46.7, 42.3, 34.3, 31.9, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C28H30C1N3O4S [M + Na]+ 562.1543, found 562.1566.
N-(5-chloro-2-propoxybenzyl)-2-(1H-indol-3-yl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)-phenethyl)acetamide (15).
Compound 15 was prepared from 2-(indole-3-yl)acetic acid and 36 following the procedure of Method C in 25% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.92-0.99 (m, 3H), 1.69-1.73 (m, 2H), 2.74-2.80 (m, 2H), 2.99-3.02 (m, 1H), 3.48 (t, J=7.56 Hz, 1H), 3.61 (t, J=7.51 Hz, 1H), 3.63-3.66 (m, 2H), 3.71-3.76 (m, 2H), 3.92-3.97 (m, 2H), 4.46-4.48 (m, 2H), 6.96-7.32 (m, 8H), 7.37 (d, J=8.12 Hz, 1H), 7.52 (t, J=7.60 Hz, 1H), 7.65 (d, J=8.28 Hz, 1H), 7.72 (d, J=8.28 Hz, 1H), 7.96-8.08 (m, 1H), 10.90-10.95 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 171.1, 155.2, 144.2, 138.4, 136.1, 129.4, 129.2, 128.1, 127.5, 127.4, 127.1, 127.0, 126.8, 124.1, 123.5, 121.1, 118.5, 113.4, 111.4, 108.1, 79.3, 74.5, 69.6, 49.1, 46.9, 34.2, 33.0, 31.9, 21.9, 10.4. HRMS (AP-ESI) m/z calcd for C31H32C1N3O4S [M + Na]+ 600.1699, found 600.1695.
N-(5-chloro-2-propoxybenzyl)-2-(1H-imidazol-4-yl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)acetamide (16).
Compound 16 was prepared from 2-(imidazole-4-yl)acetic acid and 36 following the procedure of Method C in 35% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.94-1.00 (m, 3H), 1.70-1.78 (m, 2H), 2.75-2.79 (m, 2H), 3.02-3.04 (m, 1H), 3.38-3.45 (m, 1H), 3.56-3.72 (m, 5H), 3.93-3.98 (m, 2H), 4.43-4.53 (m, 2H), 6.83-6.87 (m, 1H), 7.03-7.11 (m, 2H), 7.24 (dd, J12.68 Hz, J2=8.72 Hz 1H), 7.30-7.32 (m, 1H), 7.45 (d, J=8.36 Hz, 1H), 7.54-7.57 (m, 1H), 7.68 (d, J=8.36 Hz, 1H), 7.74 (d, J=8.36 Hz, 1H), 8.05-8.08 (m, 1H), 11.09 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.2, 155.0, 144.2, 138.4, 134.8, 129.5, 129.2, 128.2, 127.9, 127.4, 127.3, 126.8, 124.1, 113.4, 79.3, 74.6, 69.5, 49.2, 46.8, 34.2, 32.9, 31.9, 21.9, 10.5. HRMS (APESI) m/z calcd for C26H29C1N4O4S [M + Na]+ 551.1495, found 551.1471.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-2-(thio-phen-3-yl)acetamide (17).
Compound 17 was prepared from 2-(thiophene-3-yl)acetic acid and 36 following the procedure of Method C in 61% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.93-0.99 (m, 3H), 1.69-1.74 (m, 2H), 2.80 (t, J=7.58 Hz, 1H), 2.88 (t, J=7.48 Hz, 1H), 3.01-3.03 (m, 1H), 3.46 (t, J=7.48 Hz, 1H), 3.60 (t, J=7.68 Hz, 1H), 3.63-3.71 (m, 4H), 3.92-3.97 (m, 2H), 4.48 (d, J=8.28 Hz, 2H), 6.93-7.04 (m, 3H), 7.18-7.33 (m, 3H), 7.43 (d, J=7.88 Hz, 1H), 7.46-7.49 (m, 1H), 7.68 (d, J=7.88 Hz, 1H), 7.74 (d, J=7.88 Hz, 1H), 8.04-8.09 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.2, 155.2, 144.1, 138.5, 135.5, 129.5, 129.2, 128.6, 128.3, 127.9, 127.4, 126.8, 125.9, 124.1, 122.4, 113.4, 79.3, 74.6, 69.5, 48.9, 46.6, 34.8, 34.3, 31.9, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C27H29C1N2O4S2 [M + Na]+ 567.1155, found 567.1177.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)furan-3-carboxamide (18).
Compound 18 was prepared from 2-(furan-3-yl)acetic acid and 36 following the procedure of Method C in 53% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.91-0.98 (m, 3H), 1.67-1.72 (m, 2H), 2.86-2.92 (m, 2H), 3.03 (t, J=2.52 Hz, 1H), 3.58-3.66 (m, 4H), 3.95-4.04 (m, 2H), 4.59 (s, 2H), 6.53-6.62 (m, 1H), 7.05 (d, J=8.84 Hz, 1H), 7.12-7.18 (m, 1H), 7.32-7.40 (m, 3H), 7.69-7.72 (m, 3H), 7.89-7.99 (m, 1H), 8.08 (t, J=5.92 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.2, 155.2, 150.7, 145.9, 143.4, 138.4, 129.3, 128.4, 128.2, 127.9, 127.3, 126.8, 124.0, 120.9, 113.4, 110.2, 79.3, 74.5, 69.6, 50.0, 47.4, 34.2, 31.9, 21.9, 10.4. HRMS (AP-ESI) m/z calcd for C26H27C1N2O5S [M + Na]+ 537.1227, found 537.1219.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)thio-phene-3-carboxamide (19).
Compound 19 was prepared from 3-thiophene carboxylic acid and 36 following the procedure of Method C in 48% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.86-0.99 (m, 3H), 1.63-1.74 (m, 2H), 2.91-3.02 (m, 3H), 3.59 (t, J=7.44 Hz, 2H), 3.65 (s, 2H), 3.91-3.98 (m, 2H), 4.47-4.64 (m, 2H), 7.03-7.33 (m, 5H), 7.42 (s, 1H), 7.59-7.71 (m, 4H), 8.07 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 166.5, 155.2, 144.0, 138.4, 136.4, 129.3, 128.1, 127.5, 126.9, 126.8, 126.6, 124.0, 113.4, 79.3, 74.5, 69.6, 49.8, 47.8, 34.1, 31.9, 21.9, 10.4. HRMS (AP-ESI) m/z calcd for C26H27C1N2O4S2 [M + Na]+ 553.1000, found 553.1026.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)thiazole-4-carboxamide (20).
Compound 20 was prepared from 4-thiazole carboxylic acid and 36 following the procedure of Method C in 44% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.85-1.02 (m, 3H), 1.58-1.79 (m, 2H), 2.93-3.04 (m, 3H), 3.58-3.67 (m, 3H), 3.81-3.90 (m, 2H), 4.01 (t, J=6.32 Hz, 1H), 4.66-4.81 (m, 2H), 6.98-7.31 (m, 4H), 7.42 (d, J=7.92 Hz, 1H), 7.67 (d, J=7.92 Hz, 1H), 7.74 (d, J=7.92 Hz, 1H), 8.04-8.21 (m, 2H)), 9.13-9.24 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 163.6, 155.3, 153.9, 150.9, 144.1, 138.3, 129.2, 128.2, 127.9, 127.7, 127.6, 126.8, 124.9, 123.9, 113.3, 79.4, 74.6, 69.6, 49.7, 47.0, 34.6, 31.9, 22.0, 10.5. HRMS (AP-ESI) m/z calcd for C25H26C1N3O4S2 [M + Na]+ 554.0951, found 554.0970.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-1,2,5-thiadiazole-3-carboxamide (21).
Compound 21 was prepared from 3-1,2,5-thiadiazole carboxylic acid and 36 following the procedure of Method C in 40% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.80-1.01 (m, 3H), 1.52-1.78 (m, 2H), 2.92-3.05 (m, 3H), 3.64-3.69 (m, 3H), 3.82-3.88 (m, 2H), 3.98-4.00 (m, 1H), 4.72-4.74 (m, 2H), 6.98-7.08 (m, 1H), 7.22-7.34 (m, 3H), 7.43 (d, J=8.24 Hz, 1H), 7.66 (d, J=8.32 Hz, 1H), 7.74 (d, J=8.20 Hz, 1H), 8.06-8.09 (m, 1H)), 8.82-9.10 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 161.6, 157.0, 155.4, 152.5, 143.8, 138.5, 129.3, 128.6, 128.2, 126.9, 126.7, 123.9, 113.5, 79.4, 74.5, 69.7, 49.3, 47.1, 34.2, 31.9, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C24H25C1N4O4S2 [M + Na]+ 555.0904, found 555.0917.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-1H-1,2,4-triazole-3-carboxamide (22).
Compound 22 was prepared from 3-1,2,4-triazole carboxylic acid and 36 following the procedure of Method C in 36% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.85-1.01 (m, 3H), 1.58-1.77 (m, 2H), 2.89-3.04 (m, 3H), 3.58-3.67 (m, 3H), 3.89 (t, J=6.40 Hz, 1H), 3.97-4.17 (m, 2H), 4.65 (s, 2H), 6.98-7.06 (m, 1H), 7.21-7.41 (m, 4H), 7.69 (d, J=7.92 Hz, 1H), 7.74 (d, J=8.28 Hz, 1H), 8.03-8.08 (m, 1H), 8.65-8.72 (m, 1H), 14.42-14.89 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 162.3, 155.3, 152.7, 143.8, 143.4, 138.4, 129.2, 128.2, 128.1, 127.3, 127.1, 126.8, 123.9, 113.4, 79.3, 74.5, 69.7, 49.5, 46.6, 34.6, 31.9, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C24H26C1N5O4S [M + Na]+ 538.1292, found 538.1315.
N-(5-chloro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)pyrazine-2-carboxamide (23).
Compound 23 was prepared from 2-1,4-diazine carboxylic acid and 36 following the procedure of Method C in 38% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.81-1.03 (m, 3H), 1.54-1.79 (m, 2H), 2.92-3.04 (m, 3H), 3.63-3.69 (m, 4H), 3.84-4.03 (m, 2H), 4.57-4.71 (m, 2H), 6.96-7.08 (m, 1H), 7.20-7.34 (m, 3H), 7.45 (d, J=8.24 Hz, 1H), 7.65 (d, J=8.28 Hz, 1H), 7.75 (d, J=8.24 Hz, 1H), 8.08 (t, J=5.92 Hz, 1H), 8.52-8.82 (m, 3H). 13C NMR (100 MHz, DMSO-d6): δ 166.5, 155.3, 149.4, 145.5, 144.4, 143.2, 142.8, 138.5, 129.3, 128.5, 128.1, 127.9, 127.1, 126.8, 126.7, 123.9, 113.5, 79.5, 74.6, 69.7, 49.5, 47.1, 34.1, 31.9, 22.0, 10.5. HRMS (AP-ESI) m/z calcd for C26H27C1N4O4S [M + Na]+ 549.1339, found 549.1340.
N-(5-bromo-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-2-(thio-phen-3-yl)acetamide (24).
Compound 24 was synthetized from 39a ( 0.26 g, 0.55 mmol) and 3-thiopheneacetic acid (0.07 g, 0.5 mmol) following the procedure of Method C in 47% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.94-0.99 (m, 3H), 1.69-1.75 (m, 2H), 2.80 (t, J=7.72 Hz, 1H), 2.87 (t, J=7.48 Hz, 1H), 3.04 (q, J=2.60 Hz, 1H), 3.47 (t, J=7.72 Hz, 1H), 3.61 (t, J=7.48 Hz, 1H), 3.64-3.72 (m, 4H), 3.93-3.98 (m, 2H), 4.46 (d, J=7.64 Hz, 2H), 6.94-7.00 (m, 2H), 7.13 (dd, J1=2.56 Hz, J2=6.64 Hz, 1H), 7.18-7.25 (m, 1H), 7.32 (d, J=8.36 Hz, 1H), 7.36-7.50 (m, 3H), 7.69 (d, J=8.36 Hz, 1H), 7.75 (d, J=8.32 Hz, 1H), 8.05-8.11 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.2, 155.7, 144.1, 138.5, 135.5, 131.3, 130.3, 129.5, 129.2, 128.6, 128.5, 127.6, 126.8, 125.9, 122.3, 113.9, 111.8, 79.3, 74.6, 69.6, 48.9, 46.6, 34.8, 34.4, 31.9, 21.9, 10.5. HRMS (AP-ESI) m/z calcd for C27H29BrN2O4S2 [M + Na]+ 611.0649, found 611.0637.
N-(5-fluoro-2-propoxybenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-2-(thio-phen-3-yl)acetamide (25).
Compound 25 was prepared from 3-thipheneacetic acid and 39b following the procedure of Method C in 37% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.94-1.00 (m, 3H), 1.68-1.76 (m, 2H), 2.82 (t, J=7.08 Hz, 1H), 2.89 (t, J=7.52 Hz, 1H), 3.03 (q, J=2.60 Hz, 1H), 3.49 (t, J=6.60 Hz, 1H), 3.62 (t, J=7.28 Hz, 1H), 3.64-3.67 (m, 2H), 3.71 (d, J=3.76 Hz, 2H), 3.91-3.96 (m, 2H), 4.47 (d, J=13.52 Hz, 2H), 6.79-6.85 (m, 1H), 6.94-7.12 (m, 3H), 7.25 (dd, J1=1.12 Hz, J2=21.88 Hz, 1H), 7.33 (d, J=8.20 Hz, 1H), 7.44 (d, J=8.24 Hz, 1H), 7.46-7.49 (m, 1H), 7.69 (d, J=8.32 Hz, 1H), 7.75 (d, J=8.32 Hz, 1H), 8.04-8.10 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.2, 155.0, 152.6, 144.2, 138.5, 135.6, 129.5, 129.2, 128.8, 128.5, 127.8, 126.8, 125.8, 122.4, 114.6, 113.9, 112.9, 79.3, 74.6, 69.8, 48.9, 46.7, 34.8, 34.2, 31.9, 22.1, 10.5. HRMS (AP-ESI) m/z calcd for C27H29FN2O4S2 [M + Na]+ 551.1450, found 551.1433.
N-(2-butoxy-5-chlorobenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-2-(thio-phen-3-yl)acetamide (26).
Compound 26 was synthetized from 41a (0.26 g, 0.55 mmol) and 3-thiopheneacetic acid (0.07 g, 0.5 mmol) following the procedure of Method C in 49% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.89-0.94 (m, 3H), 1.38-1.48 (m, 2H), 1.65-1.72 (m, 2H), 2.81 (t, J=7.56 Hz, 1H), 2.88 (t, J=7.56 Hz, 1H), 3.04 (q, J=2.52 Hz, 1H), 3.47 (t, J=7.44 Hz, 1H), 3.61 (t, J=7.48 Hz, 1H), 3.64-3.66 (m, 2H), 3.71 (d, J=7.22 Hz, 2H), 3.97-4.02 (m, 2H), 4.45 (d, J=6.84 Hz, 2H), 6.93-7.06 (m, 3H), 7.18-7.33 (m, 3H), 7.44 (d, J=8.36 Hz, 1H), 7.47-7.49 (m, 1H), 7.69 (d, J=8.36 Hz, 1H), 7.75 (d, J=8.32 Hz, 1H), 8.04-8.10 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.2, 155.2, 144.1, 138.5, 135.5, 129.5, 129.2, 128.6, 128.5, 127.9, 127.6, 127.4, 126.8, 125.9, 124.1, 122.3, 113.4, 79.3, 74.6, 67.8, 48.9, 46.6, 34.8, 34.3, 31.9, 30.7, 18.7, 13.7. HRMS (AP-ESI) m/z calcd for C28H31C1N2O4S2 [M + Na]+ 581.1311, found 581.1314.
N-(5-chloro-2-(pentyloxy)benzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-2-(thiophen-3-yl)acetamide (27).
Compound 27 was synthetized from 41b and 3-thiopheneacetic acid following the procedure of Method C in 45% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.86-0.90 (m, 3H), 1.32-1.40 (m, 4H), 1.68-1.75 (m, 2H), 2.80 (t, J=7.68 Hz, 1H), 2.88 (t, J=7.56 Hz, 1H), 3.01-3.03 (m, 1H), 3.48 (t, J=7.52 Hz, 1H), 3.61 (t, J=7.56 Hz, 1H), 3.64-3.71 (m, 4H), 3.97-4.02 (m, 2H), 4.45-4.46 (m, 2H), 6.93-7.06 (m, 3H), 7.23-7.34 (m, 3H), 7.44 (d, J=8.32 Hz, 1H), 7.47-7.50 (m, 1H), 7.69 (d, J=8.28 Hz, 1H), 7.75 (d, J=8.32 Hz, 1H), 8.04-8.10 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.2, 155.1, 144.1, 138.5, 135.5, 129.5, 129.2, 128.6, 128.4, 127.9, 127.6, 127.5, 126.8, 125.9, 124.0, 122.3, 113.4, 79.3, 74.6, 68.1, 48.9, 46.5, 34.8, 34.3, 31.9, 28.3, 27.7, 21.8, 13.9. HRMS (AP-ESI) m/z calcd for C29H33C1N2O4S2 [M + Na]+ 595.1468, found 595.1462.
N-(2-(sec-butoxy)-5-chlorobenzyl)-N-(4-(N-(prop-2-yn-1-yl)sulfamoyl)phenethyl)-2-(thiophen-3-yl)acetamide (28).
Compound 28 was synthetized from 41c and 3-thiopheneacetic acid following the procedure of Method C in 52% yield as a white solid. 1H-NMR (400 MHz DMSO-d6): δ 0.88-0.93 (m, 3H), 1.20-1.22 (m, 3H), 1.54-1.67 (m, 2H), 2.80 (t, J=7.56 Hz, 1H), 2.89 (t, J=7.40 Hz, 1H), 3.01-3.03 (m, 1H), 3.46 (t, J=7.36 Hz, 1H), 3.60 (t, J=7.44 Hz, 1H), 3.64-3.72 (m, 4H), 4.40-4.42 (m, 2H), 6.93-7.08 (m, 3H), 7.18-7.32 (m, 3H), 7.44 (d, J=8.36 Hz, 1H), 7.47-7.49 (m, 1H), 7.69 (d, J=8.32 Hz, 1H), 7.75 (d, J=8.28 Hz, 1H), 8.04-8.10 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.2, 154.3, 144.1, 138.5, 135.5, 129.5, 129.2, 128.7, 128.5, 127.9, 127.8, 127.6, 126.8, 125.9, 123.8, 122.3, 114.6, 79.3, 74.9, 74.6, 48.8, 46.7, 34.9, 34.3, 31.9, 28.5, 18.9, 9.5. HRMS (AP-ESI) m/z calcd for C28H31C1N2O4S2 [M + Na]+ 581.1311, found 581.1308.
4-(2-(1,3-dioxoisoindolin-2-yl)ethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (30).
Propargylamine (1.1 g, 20.0 mmol) and TEA (2.8 mL, 20.0 mmol) were dissolved in anhydrous acetonitrile (100 mL) under ice bath followed by the addition of 29, synthetized following previously reported procedures24 (3.5 g, 10.0 mmol). The mixture was stirred for 4 h at room temperature (r.t.). The solvent was evaporated and the residue was dissolved in dichloromethane (DCM, 20 mL). The DCM solution was washed with water for 3 times, dried over anhydrous Na2SO4 and evaporated under vacuum. The crude product was purified by chromatography using DCM–Methanol (100:1) as the mobile phase, to obtain 30 as a white solid (2.9 g, yield: 78%). 1H-NMR (400 MHz CDCl3): δ 3.04 (t, J=7.44 Hz, 2H), 3.74-3.76 (m, 2H), 3.91 (t, J=7.32 Hz, 2H), 4.67 (s, 1H), 7.31-7.39 (m, 2H), 7.63-7.77 (m, 6H).
4-(2-aminoethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (31).
30 (1.3 g, 3.5 mmol) was dissolved in anhydrous benzene (15 mL) and then methylhydrazine (1.8 mL) was added. The reaction was stirred at r.t. for 12 h. After removing the benzene, the crude product was purified by chromatography using DCM–methanol (100:5) to give 31 as an oil (0.41 g, yield: 49%). 1H-NMR (400 MHz MeOD): δ 2.47 (d, J=2.56 Hz, 1H), 2.84-2.95 (m, 4H), 3.76 (d, J=2.52 Hz, 2H), 7.44 (d, J=8.32 Hz, 2H), 7.83 (d, J=8.36 Hz, 2H).
Method A. 5-chloro-2-methoxybenzaldehyde (33).
A mixture of 5-chloro-2-hydroxy benzaldehyde (0.63 g, 4.0 mmol), iodomethane (0.74 g, 5.2 mmol) and K2CO3 (1.1 g, 8.0 mmol) in dimethylformamide (DMF, 30 mL) was stirred at r.t. for 48 h. DMF was evaporated under reduced pressure and the residue was taken up to DCM (30 mL). The DCM solution was washed with water for 3 times, dried over anhydrous Na2SO4, filtered and evaporated to obtain 33 as a yellow solid and was used in the next step without any further purification. 1H-NMR (400 MHz DMSO-d6): δ 3.94 (s, 3H), 7.31 (d, J=8.96 Hz, 1H), 7.63 (d, J=2.80 Hz, 1H), 7.73 (dd, J12.84 Hz, J2=8.96 Hz, 1H), 10.29 (s, 1H).
Method B. 4-(2-((5-chloro-2-methoxybenzyl)amino)ethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (34).
The solution of compound 33 (1.7 g, 10.0 mmol), 31 (2.6 g, 11.0 mmol) and TEA (1.5 mL, 11.0 mmol) in methanol (50 mL) was stirred at r.t. for 1 h. Then acetic acid (0.6 mL) was added and the mixture was stirred at r.t. for another 1 h. NaCNBH3 (0.87 g, 13.0 mmol) in methanol was added portion wise under ice bath. Then, the reaction was stirred at r.t. for 12 h. After removing the solvents, the residue was taken up to DCM (50 mL). The DCM layer was washed with brine for 3 times, dried over anhydrous Na2SO4 and evaporated under vacuum. The crude produce was purified by chromatography using DCM–methanol (100:1.5) to get 34 as oil (2.4 g, yield: 61%). 1H-NMR (400 MHz CDCl3): δ 2.03 (t, J=2.56 Hz, 1H), 2.85-2.89 (m, 4H), 3.68 (s, 3H), 3.75-3.77 (m, 4H), 6.70 (d, J= 8.56 Hz, 1H), 7.11-7.15 (m, 2H), 7.28 (d, J= 8.36 Hz, 2H), 7.75 (d, J= 8.36 Hz, 2H).
5-chloro-2-propoxybenzaldehyde (35).
Compound 35 was synthetized from 1-Bromopropane (1.3 g, 11.0 mmol) and 5-chloro-2-hydroxy benzaldehyde (1.6 g, 10.0 mmol) following the procedure of Method A in 90% yield. 1H-NMR (400 MHz DMSO-d6): δ 1.03 (t, J=7.40 Hz, 3H), 1.75-1.84 (m, 2H), 4.12 (t, J=6.40 Hz, 2H), 7.29 (d, J=8.92 Hz, 1H), 7.62 (d, J=2.80 Hz, 1H), 7.69 (dd, J12.84 Hz, J2=8.96 Hz, 1H), 10.33 (s, 1H).
4-(2-((5-chloro-2-propoxybenzyl)amino)ethyl)-N-(prop-2-yn-1-yl)benzenesulfon-amide (36).
Compound 36 was synthetized from compound 35 (2.0 g, 10.0 mmol) and compound 31 (2.6 g, 11.0 mmol) following the procedure to Method B in 43% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.97 (t, J=7.68 Hz, 3H), 1.64-1.73 (m, 2H), 2.81-2.84 (m, 4H), 3.05 (t, J=2.52 Hz, 1H), 3.65-3.66 (m, 2H), 3.72 (s, 2H), 3.92 (t, J=6.36 Hz, 2H), 6.97 (d, J=8.72 Hz, 1H), 7.25 (dd, J1=2.76 Hz, J2=8.68 Hz, 1H), 7.33 (d, J=2.72 Hz, 1H), 7.43 (d, J=8.36 Hz, 2H), 7.72 (d, J=8.36 Hz, 2H), 8.05 (s, 1H).
5-bromo-2-propoxybenzaldehyde (38a).
Compound 38a was synthetized from 1-Bromopropane (0.65 g, 5.5 mmol) and 5-bromo-2-hydroxy benzaldehyde (1.0 g, 5.0 mmol) following the procedure of method A in 93% yield. 1H-NMR (400 MHz DMSO-d6): δ 1.03 (t, J=7.40 Hz, 3H), 1.75-1.84 (m, 2H), 4.12 (t, J=6.40 Hz, 2H), 7.24 (d, J=8.92 Hz, 1H), 7.74 (d, J=2.68 Hz, 1H), 7.81 (dd, J1=2.12 Hz, J2=8.96 Hz, 1H), 10.31 (s, 1H).
5-Fluoro-2-propoxybenzaldehyde (38b).
Compound 38b was synthetized from 1-Bromopropane (0.65 g, 5.5 mmol) and 5-fluoro-2-hydroxy benzaldehyde (1.0 g, 5.0 mmol) following the procedure of method A in 93% yield.1H-NMR (400 MHz DMSO-d6): δ 1.03 (t, J=7.40 Hz, 3H), 1.75-1.83 (m, 2H), 4.10 (t, J=6.44 Hz, 2H), 7.29 (dd, J1=4.08 Hz, J2=9.20 Hz, 1H), 7.41 (dd, J1=3.40 Hz, J2=8.52 Hz, 1H), 7.49-7.54 (m, 1H), 10.35 (d, J=3.20 Hz, 1H).
4-(2-((5-bromo-2-propoxybenzyl)amino)ethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (39a).
Compound 39a was synthetized from 38a ( 0.48 g, 2.0 mmol) and 31 (0.51 g, 2.2 mmol) following the procedure of method B in 50% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.97 (t, J=7.36 Hz, 3H), 1.66-1.71 (m, 2H), 2.77-2.82 (m, 4H), 3.04 (t, J=2.52 Hz, 1H), 3.66-3.68 (m, 4H), 3.91 (t, J=6.36 Hz, 2H), 6.91 (d, J=8.76 Hz, 1H), 7.35 (dd, J1=2.60 Hz, J2=8.68 Hz, 1H), 7.40-7.43 (m, 3H), 7.72 (d, J=8.32 Hz, 2H), 8.03 (s, 1H).
4-(2-((5-fluoro-2-propoxybenzyl)amino)ethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (39b).
Compound 39b was synthetized from 38b ( 0.48 g, 2.0 mmol) and 31 (0.51 g, 2.2 mmol) following the procedure of method B in 43% yield. 1H-NMR (400 MHz DMSO-d6): δ 1.03 (t, J=7.32 Hz, 3H), 1.71-1.79 (m, 2H), 2.75-2.97 (m, 4H), 3.08 (t, J=2.52 Hz, 1H), 3.68-3.71 (m, 4H), 3.98 (t, J=6.48 Hz, 2H), 6.96-7.08 (m, 2H), 7.17-7.23 (m, 1H), 7.48 (d, J=8.36 Hz, 2H), 7.78 (d, J=8.32 Hz, 2H), 8.08 (s, 1H).
2-butoxy-5-chlorobenzaldehyde (40a).
Compound 40a was synthetized from bromobutane (0.6 g, 4.4 mmol) and 5-chloro -2-hydroxy benzaldehyde (0.6 g, 4 mmol) following the procedure of Method A in 88% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.97 (t, J=7.36 Hz, 3H), 1.43-1.52 (m, 2H), 1.73-1.79 (m, 2H), 4.17 (t, J=6.40 Hz, 2H), 7.30 (d, J=8.92 Hz, 1H), 7.62 (d, J=2.80 Hz, 1H), 7.69 (dd, J1=2.80 Hz, J2=8.92 Hz, 1H), 10.31 (s, 1H).
5-chloro-2-(pentyloxy)benzaldehyde (40b).
Compound 40b was synthetized from 1-Bromopentane (0.83 g, 5.5 mmol) and 5-chloro-2-hydroxy benzaldehyde (0.78 g, 5.0 mmol) following the procedure of method A in 89% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.92 (t, J=7.16 Hz, 3H), 1.31-1.47 (m, 4H), 1.75-1.82 (m, 2H), 4.16 (t, J=6.44 Hz, 2H), 7.29 (d, J=8.92 Hz, 1H), 7.62 (d, J=2.80 Hz, 1H), 7.69 (dd, J1=2.84 Hz, J2=8.92 Hz, 1H), 10.32 (s, 1H). 2-(sec-butoxy)-5-chlorobenzaldehyde (40c). Compound 40c was synthetized from 2-Bromobutane (0.75 g, 5.5 mmol) and 5-chloro-2-hydroxy benzaldehyde (0.78 g, 5.0 mmol) following the procedure of method A in 82% yield.1H-NMR (400 MHz DMSO-d6): δ 0.97 (t, J=7.44 Hz, 3H), 1.30 (d, J=6.08 Hz, 3H), 1.58-1.79 (m, 2H), 4.58-4.65 (m, 1H), 7.33 (d, J=8.96 Hz, 1H), 7.61 (d, J=2.84 Hz, 1H), 7.68 (dd, J1=2.88 Hz, J2=8.92 Hz, 1H), 10.31 (s, 1H).
4-(2-((2-butoxy-5-chlorobenzyl)amino)ethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (41a).
Compound 41a was synthetized from 40a ( 0.42 g, 2 mmol) and 31 (0.51 g, 2.2 mmol) following the procedure of Method B in 43% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.94 (t, J=7.36 Hz, 3H), 1.37-1.46 (m, 2H), 1.63-1.69 (m, 2H), 2.76-2.82 (m, 4H), 3.04 (t, J=2.52 Hz, 1H), 3.66-3.69 (m, 4H), 3.96 (t, J=6.36 Hz, 2H), 6.97 (d, J=8.72 Hz, 1H), 7.23 (dd, J1=2.76 Hz, J2=8.68 Hz, 1H), 7.31 (d, J=2.72 Hz, 1H), 7.43 (d, J=6.36 Hz, 2H), 7.72 (d, J=8.36 Hz, 2H), 8.05 (s, 1H).
4-(2-((5-chloro-2-(pentyloxy)benzyl)amino)ethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (41b).
Compound 41b was synthetized from 40b ( 0.45 g, 2.0 mmol) and 31 (0.51 g, 2.2 mmol) following the procedure of method B in 51% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.90 (t, J=7.32 Hz, 3H), 1.28-1.41 (m, 4H), 1.65-1.73 (m, 2H), 2.69-2.99 (m, 4H), 3.01-3.04 (m, 1H), 3.57-3.67 (m, 4H), 3.92-3.99 (m, 2H), 6.95 (d, J=6.52 Hz, 1H), 7.20 (dd, J1=2.76 Hz, J2=6.68 Hz, 1H), 7.32-7.35 (m, 1H), 7.46 (d, J=6.32 Hz, 2H), 7.76 (d, J=8.24 Hz, 2H), 8.08 (s, 1H).
4-(2-((2-(sec-butoxy)-5-chlorobenzyl)amino)ethyl)-N-(prop-2-yn-1-yl)benzenesulfonamide (41c).
Compound 41c was synthetized from 40c ( 0.42 g, 2.0 mmol) and 31 (0.51 g, 2.2 mmol) following the procedure of method B in 46% yield. 1H-NMR (400 MHz DMSO-d6): δ 0.97 (t, J=7.48 Hz, 3H), 1.27 (t, J=6.04 Hz, 3H), 1.58-1.72 (m, 2H), 2.74-2.98 (m, 4H), 3.07-3.09 (m, 1H), 3.62-3.72 (m, 4H), 4.40-4.47 (m, 1H), 7.03 (d, J=8.56 Hz, 1H), 7.25 (dd, J12.84 Hz, J28.56 Hz, 1H), 7.32-7.33 (m, 1H), 7.41 (d, J=6.56 Hz, 2H), 7.79 (d, J=8.32 Hz, 2H), 8.09 (s, 1H).
Biological assays.
Cells:
J774A.1 murine macrophage cells and hCMEC/D3 cells were purchased from American Type Cell Culture (ATCC, Manassas, VA). J774A.1 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin. J774A.1 cells were used under passage 50 for the experiments. hCMEC/D3 cells were cultured using Endothelial Growth mediun-2 (EGM-2) supplemented with a Bullet Kit™. The medium was supplemented with 10 mM HEPES (5mL), 1× penicillin-streptomycin (5 mL), and 1 ng/mL of bFGF (2.5 mL) at 5% CO2, 37°C and 95% relative humidity. This media was termed as growth media. EGM-2 media supplemented with 10 mM HEPES (5 mL), 1× penicillin-streptomycin (5mL), 2.5% FBS, and 1 ng/mL of bFGF (1.25mL) was termed as maintenance media. hCMEC/D3 cells were used between passage 25-35 for the experiments. For cell-based assays, at least 3 independent experiments with at least triplicates for each data point of each experiment were conducted.
IL-1 β assays in J774A.1 cells.
J774A.1 cells were plated into a 96-well plates (1 × 105 cells/well) for 24 h in growth medium. Cells were primed with Escherichia coli 0111 :B4 LPS (Sigma-Aldrich) (final concentration: 1 μg/mL) for 4.5 h. Next, test compounds (0.1, 0.3, 1.0. 3.0, and 10.0 μM) were added for 30 min. ATP (5 mM) was added at the same time when compounds were added to induce NLRP3 inflammasome activation. After 30 min, the supernatants were collected and the level of IL-1β was measured with a mouse IL-1β ELISA kit (DuoSet ELISA, R&D Systems) following the manufacturer’s instructions.
IL-1β assays in mouse peritoneal macrophages.
C57BL/6 mice were injected with 1 mL of 3% thioglycolate into the peritoneal cavity. Peritoneal cells were collected 3 days after injection by flushing the peritoneal cavity with cold PBS. The peritoneal cells were plated in complete RPMI1640 media containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin. Floating cells were removed after 2 h and adherent peritoneal macrophages were similarly treated as described with J774A.1 cell line followed by ELISA analysis for IL-β production.
Inhibition on the NLRC4 and AIM2 inflammasomes assays.
J774A.1 cells were plated into 96-well plates (1 × 105 cells/well) for 24 h in growth medium. Cells were treated with LPS (1 μg/mL) and test compounds for 1 h. Flagellin or poly-deoxyadenylic-deoxythymidylic acid sodium salt (Poly(dA:dT)) was used to induce the formation of the NLRC4 and the AIM2 inflammasomes. Flagellin (Enzo Life Sciences, Farmingdale, NY), isolated from Salmonella typhimurium strain 14028, was added in DMEM (Invitrogen) without fetal bovine serum (FBS) to the plate (1 μg/mL) and allowed to incubate for 6 h. Flagellin cell-transfection was accomplished utilizing the Polyplus transfection kit (PULSin, New York, NY). For AIM2 activation, cells were incubated with Poly(dA:dT) (4 μg/ml) (InvivoGen, San Diego, CA) for 8 h. The supernatants were collected and levels of IL-1β were measured with a mouse IL-1β ELISA kit following the manufacturer’s instructions.
In vitro BBB assay.
hCMEC/D3 cells were used from passage 25 to 35. The in vitro model of BBB was built by seeding 150,000 cells/well on the apical surface of 12-well transwell with 3.0 micron pores.30 The cells were supplemented with growth medium for the initial 48 h period and then with maintenance medium for the next 72 h. Medium was replenished every day until the transport experiments were performed. The apical side of the transwell represents the blood side, while basolateral side represents the brain side. On the fifth day, vectorial transport (A-to-B, B-to-A) of 17 (20 μM, dissolved in DPBS with 0.01% DMSO) was determined by adding compound in one compartment and sampling the appearance of compound in the opposing compartment. Samples were collected at 5, 10, 15, 30, 45, and 60 min. Compound 17 was quantified by HPLC. Apparent permeability (Papp) was then calculated.
Animals:
All animal experiments were conducted under the guidelines of the “Guide for the care and use of laboratory animals” published by National Institutes of Health (revised 2011). C57BL/6 male mice were purchased from the National Cancer Institute (Bethesda, MD). nlrp3−/− male mice on C57BL/6 background and male Sprague-Dawley rats were purchased from the Jackson Laboratory (Bar Harbor, ME).
LPS challenge in vivo and compound treatment
C57BL/6 mice were injected intraperitoneally (i.p.) with 50 mg/kg LPS (Sigma-Aldrich) or PBS one hour after compound (10 mg/kg) or vehicle treatment by i.p. injection. For nlrp3−/− mice, 25 mg/kg of LPS was injected by i.p.. Serum levels of IL-1β and TNF-α were measured by ELISA 2.5 h after LPS injection.
In vivo BBB penetration and PK studies
C57BL/6 mice (n=3 for each time point) were given compound 17 by PO administration (20 mg/kg) or vehicle treatment. Plasma samples (200 – 500 μL) and brain tissues (after saline perfusion) were collected at 0.5, 1, and 4 h time points and stored in −80 °C freezer for later analysis.
Sprague-Dawley rats (200-250 g, n=3) were given compound 17 by IV and PO administration (20 mg/kg, for oral suspension an ad-hoc formulation of 10% Cremophor EL in PBS was used). Plasma samples (~500 μL) were collected at 0.08, 0.17, 0.25, 0.5, 0.75, 1.0, 2.0, 4.0, 8.0, 12.0, and 24.0 h time points through orbital vein and were stored in −80 °C freezer for later analysis.
LC-MS/MS analysis.
Plasma samples were thawed and centrifuged at 3000 rpm (2095 g). 40 μL of each sample was added to a 1.5 mL microcentrifuge tube and 25 μL of internal standard (100 ng/mL glipizide) was added. Samples were mixed by vortexing for 30 seconds. In order to precipitate the proteins, 250 μL of 1% ammonium formate in methanol was added to each sample and mixed by vortex for 2 minutes. Samples were centrifuged at 14000 rpm (10,956 g) for 5 minutes, and the supernatant was transferred to a 1.5 mL microcentrifuge tube with a microfilter tube (0.45 μm) filter insert (Pall Corporation, New York, USA). The remaining supernatant was evaporated under a nitrogen stream at 55 °C. The dry residue was reconstituted with 90:10 methanol:water. Samples were transferred to 96-well plate for analysis by LC-MS/MS. For tissue samples, brain samples were thawed and centrifuged at 3000 rpm (2095 g). To each tissue sample, 100 μL of PBS, 25 μL of internal standard (100 ng/mL glipizide), and 250 μL of 1% ammonium formate in methanol was added. The tissue was homogenized at a gradually increasing speed for 30 seconds using a beadbug mixer (Benchmark Scientific, Atkinson, NH, USA) with 6 cycles, then centrifuged at 14000 rpm (10,956 g) for 5 min. The supernatant was transferred to 1.5 mL microtube with a microfilter tube (0.45 μm) filter insert (Pall Corporation, New York, USA). The remaining supernatant was evaporated under a nitrogen stream at 55 °C. The dry residue was reconstituted with 90:10 methanol:water. Samples were transferred to 96-well plate for analysis by LC-MS/MS. Chromatographic separation was achieved using a Waters Acquity HPLC with a Phenomenex Gemini 2.1×30mm 5-micron column (Phenomenex, Torrance, CA, USA) and a gradient mobile phase (1% formic acid, Mobile Phase A, and acetonitrile, Mobile Phase B). Flow was a constant 0.40 μL/min, with 95% A from 0 to 1.0 minutes. From 1.0 to 3.25 minutes, the composition changed to 95% mobile phase B, and held until 4.25 min. From 4.5 minutes to 5.0 min initial conditions were re-established with 95% 1% formic acid in water. The LC-MS/MS method employed was positive electrospray ionization and the following MRM transitions used were as follows: Compoun 17 546.400 > 155.1 and glipizide 446.2 > 321.200. Results were processed using Analyst 1.5.2, on an AB Sciex 4000 QTrap hybrid linear ion trap tandem mass spectrometer. The linear range for the method was 1-5000 ng/mL, with a linear, 1/x regression method. The detailed chromatographic conditions are as the following:
HPLC Column: Phenomenex Gemini C18 5 um, 30 × 2 mm, (Phenomenex, Torrance, CA, USA) Column Temperature: 40 C
Mobile phase A: 95% 0.5% formic acid 5% acetonitrile
Mobile phase B: 0.5% formic acid in acetonitrile
Flow rate: 0.4 mL/min
Gradient Conditions:
| Time | Events |
|---|---|
| 0.10 | Total Flow 0.400 mL/min |
| 0.10 | Mobile phase A Conc.95 |
| 1.00 | Mobile phase A Conc.95 |
| 2.75 | Mobile phase A Conc.5 |
| 4.00 | Mobile phase A Conc.5 |
| 5.00 | Mobile phase A Conc.95 |
| 6.50 | Stop |
Supplementary Material
Acknowledgements
The work was supported in part by the NIA of the NIH under award number R01AG058673 (SZ), Alzheimer’s Drug Discovery Foundation 20150601 (SZ), Award No. 18-2 from the Commonwealth of Virginia’s Alzheimer’s and Related Disease Research Award Fund administered by the Center on Aging, School of Allied Health Professions, Virginia Commonwealth University (SZ).
Abbreviations
- AD
Alzheimer’s disease
- AIM2
absent in melanoma 2
- ASC
apoptosis-associated speck-like protein containing a CARD
- ATP
adenosine triphosphate
- BBB
blood-brian barrier
- CNS
Central nervous system
- DAMPs
damage associated molecular pattern molecules
- DCM
Dichloromethane
- DMEM
Dulbecco’s Modified Eagle Medium
- DMF
N,N-Dimethylformamide
- EDCI
N-(3-(di methylamino)propyl)-N-ethyl-carbodiimide hydrochloride
- EGM
Endothelia growth medium
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- HOBt
hydroxybenzotriazole
- IL-1β
interleukin-1β
- IP
intraperitoneal
- IV
intravenous injection
- LPS
lipopolysaccharide
- LC-MS/MS
Liquid chromatography tandem mass spectrometry
- MOA
mechanisms of action
- NLR
NOD-like receptor
- NLRP3
NOD-like receptor family pyrin-domain-containing 3
- NLRP3Is
NLRP3 inflammasome inhibitors
- NOR
Novel Object Recognition
- PAMPs
pathogen-associated molecular patterns
- PBS
phosphate-buffered saline
- PK
pharmacokinetic
- PMA
phosphomolybdic acid
- PO
per os
- Poly(dA:dT)
poly-deoxyadenylic-deoxythymidylic acid sodium salt
- PRRs
pattern recognition receptors
- RIG-I
retinoic acid-inducible gene I
- SAR
structure activity relationship
- TBI
traumatic brain injury
- TEA
triethylamine
- TFA
trifluoroacetic acid
- TMS
Tetramethylsilane
- TNF-α
Tumor Necrosis Factor-α
Footnotes
Supporting Information
Molecular formula strings (CSV)
References
- (1).Martinon F; Burns K; Tschopp J The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of Proil-Beta. Mol. Cell 2002, 10, 417–426. [DOI] [PubMed] [Google Scholar]
- (2).Schroder K; Tschopp J The Inflammasomes. Cell. 2010, 140, 821–832. [DOI] [PubMed] [Google Scholar]
- (3).Latz E; Xiao TS; Stutz A Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol 2013, 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shao BZ; Xu ZQ; Han BZ; Su DF; Liu C Nlrp3 Inflammasome and Its Inhibitors: A Review. Front. Pharmacol 2015, 6, 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hoffman HM; Mueller JL; Broide DH; Wanderer AA; Kolodner RD Mutation of a New Gene Encoding a Putative Pyrin-Like Protein Causes Familial Cold Autoinflammatory Syndrome and Muckle-Wells Syndrome. Nat. Genet 2001, 29, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yang F; Qin Y; Wang Y; Meng S; Xian H; Che H; Lv J; Li Y; Yu Y; Bai Y; Wang L Metformin Inhibits the Nlrp3 Inflammasome Via Ampk/Mtor-Dependent Effects in Diabetic Cardiomyopathy. Int. J. Biol. Sci 2019, 15, 1010–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).De Nardo D; Latz E Nlrp3 Inflammasomes Link Inflammation and Metabolic Disease. Trends Immunol. 2011, 32, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Choi AJ; Ryter SW Inflammasomes: Molecular Regulation and Implications for Metabolic and Cognitive Diseases. Mol. Cells 2014, 37, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Couturier J; Stancu IC; Schakman O; Pierrot N; Huaux F; Kienlen-Campard P; Dewachter I; Octave JN Activation of Phagocytic Activity in Astrocytes by Reduced Expression of the Inflammasome Component Asc and Its Implication in a Mouse Model of Alzheimer Disease. J. Neuroinflammation 2016, 13, 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Freeman LC; Ting JP The Pathogenic Role of the Inflammasome in Neurodegenerative Diseases. J Neurochem. 2016, 136 Suppl 1, 29–38. [DOI] [PubMed] [Google Scholar]
- (11).Heneka MT; Kummer MP; Stutz A; Delekate A; Schwartz S; Vieira-Saecker A; Griep A; Axt D; Remus A; Tzeng TC; Gelpi E; Halle A; Korte M; Latz E; Golenbock DT Nlrp3 Is Activated in Alzheimer's Disease and Contributes to Pathology in App/Ps1 Mice. Nature. 2013, 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Venegas C; Kumar S; Franklin BS; Dierkes T; Brinkschulte R; Tejera D; Vieira-Saecker A; Schwartz S; Santarelli F; Kummer MP; Griep A; Gelpi E; Beilharz M; Riedel D; Golenbock DT; Geyer M; Walter J; Latz E; Heneka MT Microglia-Derived Asc Specks Cross-Seed Amyloid-Beta in Alzheimer's Disease. Nature. 2017, 552, 355–361. [DOI] [PubMed] [Google Scholar]
- (13).Coll RC; Robertson AA; Chae JJ; Higgins SC; Munoz-Planillo R; Inserra MC; Vetter I; Dungan LS; Monks BG; Stutz A; Croker DE; Butler MS; Haneklaus M; Sutton CE; Nunez G; Latz E; Kastner DL; Mills KH; Masters SL; Schroder K; Cooper MA; O'Neill LA A Small-Molecule Inhibitor of the Nlrp3 Inflammasome for the Treatment of Inflammatory Diseases. Nat. Med 2015, 21, 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tapia-Abellan A; Angosto-Bazarra D; Martinez-Banaclocha H; de Torre-Minguela C; Ceron-Carrasco JP; Perez-Sanchez H; Arostegui JI; Pelegrin P Mcc950 Closes the Active Conformation of Nlrp3 to an Inactive State. Nat. Chem. Biol 2019, 15, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Coll RC; Hill JR; Day CJ; Zamoshnikova A; Boucher D; Massey NL; Chitty JL; Fraser JA; Jennings MP; Robertson AAB; Schroder K Mcc950 Directly Targets the Nlrp3 Atp-Hydrolysis Motif for Inflammasome Inhibition. Nat. Chem. Biol 2019, 15, 556–559. [DOI] [PubMed] [Google Scholar]
- (16).Irrera N; Vaccaro M; Bitto A; Pallio G; Pizzino G; Lentini M; Arcoraci V; Minutoli L; Scuruchi M; Cutroneo G; Anastasi GP; Ettari R; Squadrito F; Altavilla D Bay 11-7082 Inhibits the Nf-Kappab and Nlrp3 Inflammasome Pathways and Protects against Imq-Induced Psoriasis. Clin. Sci (Lond) 2017, 131, 487–498. [DOI] [PubMed] [Google Scholar]
- (17).Jiang H; He H; Chen Y; Huang W; Cheng J; Ye J; Wang A; Tao J; Wang C; Liu Q; Jin T; Jiang W; Deng X; Zhou R Identification of a Selective and Direct Nlrp3 Inhibitor to Treat Inflammatory Disorders. J. Exp. Med 2017, 214, 3219–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).He H; Jiang H; Chen Y; Ye J; Wang A; Wang C; Liu Q; Liang G; Deng X; Jiang W; Zhou R Oridonin Is a Covalent Nlrp3 Inhibitor with Strong Anti-Inflammasome Activity. Nat. Commun 2018, 9, 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Huang Y; Jiang H; Chen Y; Wang X; Yang Y; Tao J; Deng X; Liang G; Zhang H; Jiang W; Zhou R Tranilast Directly Targets Nlrp3 to Treat Inflammasome-Driven Diseases. EMBO Mol. Med 2018, 10, e8689–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cocco M; Pellegrini C; Martinez-Banaclocha H; Giorgis M; Marini E; Costale A; Miglio G; Fornai M; Antonioli L; Lopez-Castejon G; Tapia-Abellan A; Angosto D; Hafner-Bratkovic I; Regazzoni L; Blandizzi C; Pelegrin P; Bertinaria M Development of an Acrylate Derivative Targeting the Nlrp3 Inflammasome for the Treatment of Inflammatory Bowel Disease. J Med. Chem 2017, 60, 3656–3671. [DOI] [PubMed] [Google Scholar]
- (21).Lamkanfi M; Mueller JL; Vitari AC; Misaghi S; Fedorova A; Deshayes K; Lee WP; Hoffman HM; Dixit VM Glyburide Inhibits the Cryopyrin/Nalp3 Inflammasome. J Cell. Biol 2009, 187, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tamura K; Ishikawa G; Yoshie M; Ohneda W; Nakai A; Takeshita T; Tachikawa E Glibenclamide Inhibits Nlrp3 Inflammasome-Mediated Il-1 beta Secretion in Human Trophoblasts. J Pharmacol. Sci 2017, 135, 89–95. [DOI] [PubMed] [Google Scholar]
- (23).Yin J; Zhao F; Chojnacki JE; Fulp J; Klein WL; Zhang S; Zhu X Nlrp3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer's Disease. Mol. Neurobiol 2018, 55, 1977–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fulp J; He L; Toldo S; Jiang Y; Boice A; Guo C; Li X; Rolfe A; Sun D; Abbate A; Wang XY; Zhang S Structural Insights of Benzenesulfonamide Analogues as Nlrp3 Inflammasome Inhibitors: Design, Synthesis, and Biological Characterization. J Med. Chem 2018, 61, 5412–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Guo C; Fulp JW; Jiang Y; Li X; Chojnacki JE; Wu J; Wang XY; Zhang S Development and Characterization of a Hydroxyl-Sulfonamide Analogue, 5-Chloro-N-[2-(4-Hydroxysulfamoyl-Phenyl)-Ethyl]-2-Methoxy-Benzamide, as a Novel Nlrp3 Inflammasome Inhibitor for Potential Treatment of Multiple Sclerosis. ACS Chem. Neurosci 2017, 8, 2194–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kuwar R; Rolfe A; Di L; Xu H; He L; Jiang Y; Zhang S; Sun D A Novel Small Molecular Nlrp3 Inflammasome Inhibitor Alleviates Neuroinflammatory Response Following Traumatic Brain Injury. J. Neuroinflammation 2019, 16, 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Marchetti C; Chojnacki J; Toldo S; Mezzaroma E; Tranchida N; Rose SW; Federici M; Van Tassell BW; Zhang S; Abbate A A Novel Pharmacologic Inhibitor of the Nlrp3 Inflammasome Limits Myocardial Injury after Ischemia-Reperfusion in the Mouse. J Cardiovasc. Pharmacol 2014, 63, 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).He Y; Franchi L; Nunez G Tlr Agonists Stimulate Nlrp3-Dependent Il-1beta Production Independently of the Purinergic P2×7 Receptor in Dendritic Cells and in Vivo. J Immunol. 2013, 190, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Weksler B; Romero IA; Couraud PO The Hcmec/D3 Cell Line as a Model of the Human Blood Brain Barrier. Fluids Barriers CNS. 2013, 10, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Patel S; Leibrand CR; Palasuberniam P; Couraud PO; Weksler B; Jahr FM; McClay JL; Hauser KF; McRae M Effects of HIV-1 Tat and Methamphetamine on Blood-Brain Barrier Integrity and Function in Vitro. Antimicrob. Agents Chemother 2017, 61, e01307–01317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.