

Abstract
Drug development for rare diseases, classified as diseases with a prevalence of <200 000 patients, is limited by high cost of research and low target population. Owing to a lack of representative disease models, research has been challenging for orphan drugs. Human-on-a-chip (HoaC) technology, which models human tissues in interconnected in vitro microfluidic devices, has the potential to lower the cost of preclinical studies and increase the rate of drug approval by introducing human phenotypic models early in the drug discovery process. Advances in HoaC technology can drive a new approach to rare disease research and orphan drug development.
Teaser:
After more than three decades of the Orphan Drug Act, drug development for rare diseases remains a challenge. Human-on-a-chip holds the promise to drive orphan drug development to the next level.
Keywords: Drug development, human-on-a-chip, rare diseases, orphan drug development, PB-PK-PD modeling
Introduction
Rare, or orphan diseases, defined in the USA as diseases that affect <200 000 individuals at a given time, remain an underrepresented area of medical and pharmaceutical research. Although the incidence of any single disease is low, there are >7000 established rare diseases worldwide, and it is estimated that 25 million people suffer a rare disease in the USA alone [1,2]. Because of the expensive and rigorous testing required before a new drug enters the market, most drug development has been historically limited to drugs targeting diseases with high incidence and prevalence. The Orphan Drug Act (ODA), which was passed in the USA in 1983, provides incentives for research into ‘orphan’ diseases that would otherwise not recover costs of development, most of which are rare diseases. This legislation was a landmark for drug development for rare diseases, and many consider the ODA one of the most successful US legislative actions in recent history [1]. From 1983 to 2004, 248 orphan drugs were approved in the USA, a significant increase compared with the ten drugs approved before 1983. Although medical and social issues caused by rare diseases have been increasingly recognized by the public and pharmaceutical industry over the past two decades, almost all rare diseases still lack a cure or effective treatment strategy [3].
Rare disease drug development is hampered by the current issues affecting the pharmaceutical industry at large, namely the high cost of bringing new therapeutic agents to market combined with low success rate of regulatory approval (~9.6% for drugs in 2016) [4]. This is especially true for rare diseases, because many do not have enough patients to perform statistically significant clinical trials and thus have a limited return on investment. The low success rate of drug approval is largely the result of poor predictive power for drug efficacy and toxicity of current preclinical in vitro and in vivo models. Animal models are important tools for drug discovery and development but, in many cases, do not adequately model drug responses observed in the human body [5]. The use of animal models has shown poor predictive power for human response to drugs owing to cross-species discrepancies (i.e., differences in drug metabolism) and increasing ethical concerns surrounding animals in research drives a need for improved models [5,6]. Most in vitro cell culture assays lack complexity and do not model many physiological processes including shear stress and tissue–tissue or organ–organ communication [7].
In the past few decades, advances in nanotechnology, microfabrication and cell biology techniques have enabled the development of microphysiological systems (MPS), also called ‘organ-on-a-chip’ models or, when multiple organs are included on the same platform, ‘body-on-a-chip’ (BoaC) or ‘human-on-a-chip’ (HoaC) models. These platforms, when combined with induced pluripotent stem cell (iPSC) technology, enable the development of patient-specific phenotypic models of rare diseases where disease-specific cells can be differentiated from iPSCs in a cost-effective and disease-relevant approach. These technical advances enabled the ‘Microphysiological Systems for Drug Efficacy and Toxicity Testing Program’ initiatives by the NIH and DARPA in 2012 and have encouraged the maturation and commercialization of this technology [8]. This review discusses the applications of human-based, multi-organ, in vitro models for improved drug discovery and the importance of pharmacokinetics/pharmacodynamics (PK/PD) models and how this can be applied to development of orphan or rare disease drugs.
Advanced in vitro models for drug discovery
The limitations of using animal models to study human diseases and develop therapeutics have been previously established [9]; all animal models exhibit genetic and physiological differences from humans regarding basal metabolism, drug PK/PD, immune system function and lifespan [9]. All of these factors influence the effectiveness of animal models for drug discovery and disease modeling. Further, genetically identical lines of laboratory animals, although ideal for research purposes where limiting variability is crucial, do not account for the complexity and variability observed between humans, especially in the case of rare human diseases and disorders [10].
In vitro human-based models for drug discovery typically involve a combination of plate-based assays with computational modeling or in silico analysis; this strategy has been effective in identifying novel drug combinations and new applications of established drugs in combinatorial drug discovery. Advances in miniaturization and robotics have enabled development of assays based on 96- to 1536-well plates, which have the advantage of being automated and high throughput. However, although these assays predict cell-specific drug response, they are typically not physiological – for example, many liver models use isolated microsomes rather than hepatocytes, leading to a bias toward oxidation and an incomplete enzyme–cofactor spectrum. Computational modeling has led to advances in drug treatment, especially for complex, multifactorial diseases like cancer, metabolic syndrome and autism. This approach relies on existing in vivo experimental data to predict drug responses; therefore, applications are limited for rare diseases, which often lack preclinical and clinical data [11]. Additionally, identification of side-effects is difficult because the specific phenotype of an off-target response is not easily predicted and varies based on genetic profile [11]. To address the technical and ethical limitations of animal or human models for research purposes, there has been increased focus on developing phenotypic human models instead of target-based models to study disease.
HoaC technology integrates cell culture with BioMEMs engineering, surface chemistry and mechatronics, enabling phenotypic modeling of rudimentary organ physiology in a microfluidic device. These devices often include cells grown on sophisticated electronic devices like microelectrode arrays (MEAs) and mechanical systems such as microcantilever arrays that facilitate noninvasive functional measurements of disease or drug effects using these hybrid 3D devices. With the potential to integrate multiple tissues or organ system surrogates, HoaC models provide a unique platform for measuring drug response and toxicity, and for studying the influence of a disease state on the other organ modules in a controlled environment.
Current microfluidic systems range in sophistication from models of a single organ (organ-on-a-chip) to larger-scale combinations of multiple organs or tissues connected through microfluidics in one device. Single-organ models have been developed for most organs, including white adipose tissue [12,13], heart [14], liver [15], skeletal muscle [16], lung [17,18], gastrointestinal (GI) tract [19], kidney [20], reproductive organ [21], central nervous system (CNS) [22], peripheral nervous system (PNS) [23], skin [24] and bone [25]. Developing a microfluidic device to replicate a human organ enables sophistication in modeling beyond that of cells in standard tissue culture; an organ-on-a-chip model can provide more physiologically relevant architecture, including 3D systems that can better replicate in vivo physiology [26], or can provide the flow and shear stress integral to the maturation and physiology of an organ. To enable high-throughput testing or drug screening, models composed of tens or hundreds of systems that can be run simultaneously have been developed [27,28].
Multi-organ HoaC models integrate two or more tissues or cell types to model the relationship between organs, including paracrine and endocrine signaling and determination of the effects of tissue metabolites on other tissues in shared medium [29,30]; these systems mimic the in vivo effects of organ-specific cytokines on the body, or of drug metabolites produced by the liver in other organs [31]. Modules can be connected by blood-memetic medium in either bidirectional or controlled unidirectional flow [32,33]. HoaC models mimicking barrier tissue physiology have also been developed; for example, models of the GI tract, lung epithelium and blood–brain barrier (BBB) have been well characterized [34–37]. This also enables modeling of specific biological processes, such as tumor invasion [25,38] and skin penetration [39,40].
As more tissue and organ modules are established in vitro, there is increasing potential for complex, multi-process HoaC models, mimicking the complex interactions that occur between organ systems and potentially enable whole-body analysis in a single integrated system, the goal of HoaC technology. Toward that end, the Shuler Lab at Cornell University published a 13-organ recirculating system that maintained viability of all cell types for up to 7 days [41]. Additionally, ten connected organ module systems have been included in the PhysioMimix system [42].
The integration of patient-specific iPSCs also enables modeling of diseases utilizing this system [43]. This is especially applicable for developing disease-specific phenotypic models for orphan diseases where it might not be possible or cost effective to establish animal models for the disease. With increasing sophistication in tissue scaffolds, device design and fabrication, and the application of state-of-the-art molecular techniques, HoaC technology shows promise for fine-tuned, personalized medicine in rare diseases and the establishment of more-effective drug development programs while minimizing the risks involved with traditional research on animals or humans.
Cell sources
In recent years, advances in cell culture resources and stem cell technologies have expanded options for the cellular components utilized in HoaC systems. Whereas some in vitro models use animal cells, human-based models enable more-accurate and improved drug response prediction [44]. Primary human cells are now largely available for many cell types, along with specialized supplies, nutrients and growth factors needed to maintain them, many of them in serum-free formulations for minimizing variability introduced by animal serum. However, in the case of highly differentiated and non-proliferative cells such as neurons or cardiomyocytes, obtaining primary cells can be difficult and expensive. Additionally, any primary cells have a potential for contamination with mycoplasma, viruses or other adventitious agents that can interfere with in vitro viability and physiology and can be difficult to obtain in the case of rare diseases. Primary human cells often have genotypic and phenotypic instability leading to the loss of complex differentiation [45], and cell culture can drive change or loss of function, especially over many passages. To address these concerns, the commercialization of iPSC technology provides an opportunity to culture patient-specific cells that would otherwise be difficult to obtain in the large quantities necessary for research purposes.
More importantly, the development of iPSC technology has enabled the generation of specialized cells without the technical or ethical concerns of obtaining human stem cells directly. The protocol, first established in 2007, enabled the generation of embryonic stem-cell-like cells from mouse somatic cells using four growth factors: Oct3/4, Sox2, c-Myc and Klf4 [46]. The technique was quickly applied to adult human somatic cells [47,48], and has since expanded to a variety of accessible human cells applied to the fields of disease modeling, biological research and clinical regenerative medicine. The combination of iPSC technology and genome editing with CRISPR/Cas9 has made available unique cell options, in which genetic mutations can be induced or removed to model diseases in vitro [49]. The technique has been used to create iPSC lines carrying mutations for rare diseases including cystic fibrosis [50], Tay–Sachs disease [51], familial dysautonomia [52], spinal muscular dystrophy [53] and Barth syndrome [54], among others.
Flow methods
Compared with traditional in vitro cell culture, in which cells are grown in static systems – flasks, plates and dishes – modern microphysiological models support medium flow and recirculation throughout the device, providing greater physiological relevance. Few biological systems are static; blood pressure drives movement of endogenous and exogenous molecules and cells throughout the body, subjecting tissues to varying degrees of shear stress and pressure necessary for proper maturation and physiology [55]. These mechanical forces are crucial for the development, maturation and survival of many tissues including lung, BBB, liver, kidney and blood vessels [56–59]. HoaC systems have been developed to mimic this physiology using gravity or microfluidic pumps and valves. Further automation of device function through the integration of electronic sensors and controllers to monitor medium pH, regulate medium handling and control value function make high-content screening feasible [60]. The ability of HoaC systems to mimic this important physiological process has been established for organs including the endothelium [61] and kidney nephrons [62].
In microphysiological models, fluid movement, shear stress and perfusion flow can be tuned to match the corresponding biological condition [63]. Flow can be finely controlled using a pump-based system, which can be used to drive a specific pressure or to study effect of flow dynamics on cells where pressure gradient is crucial for physiology, for example glomerular filtration in the kidney [64,65]. However, pump systems can be large, difficult to maintain, contain significant dead volumes and the added equipment can increase risk of contamination. Alternatively, pumpless systems enable replication of physiological shear stress in compact systems driven by gravity, surface tension or osmosis, potentially with the use of an external power source such as a rocker [32,66]. Recent advances in chip geometry support unidirectional flow in gravity-driven microfluidic models [63,67].
Architecture
The scaffolds and extracellular matrix (ECM) materials used in HoaC construction are integral for replicating a physiological environment and maintaining in vitro function. Optimal physiological relevance is achieved through systems that replicate the 3D architecture or function of biological systems, through cellular structure, scaffold or flow. Allowing cells to contact ECM, and potentially other cells, results in a healthier cell phenotype, improved differentiation and more-representative cell behavior [68]. In vitro models have been designed using decellularized scaffolds, including a recellularized rat liver [69] and heart [70]. Hydrogels, polymer networks that hold up to 99% water by weight, are effective for modeling soft tissue environments; tissue hydrogel scaffolds can be composed of ECM and cellular adhesion molecules and dosed with growth factors to support cell growth and movement to mimic functional tissues. In conjunction with inkjet, microextrusion or laser-assisted bioprinting, hydrogels have been used for the construction of 3D tissues in vitro [71]. This has been applied for modeling of the intestinal epithelium [72], fibroblast-driven wound healing [73] and tumor angiogenesis [38]. ECM modulation has applications for modeling dysfunctional interaction between cell membranes and the ECM in rare diseases including myasthenia gravis, scleroderma, Marfan syndrome and Ehlers–Danlos syndrome, where extracellular architecture is damaged. However, 2D models provide the advantage of simplicity and have been shown to perform as well as or better than 3D models for tissue maturation and longevity in some cases and, when integrated with BioMEMs devices to form hybrid 3D devices, can be more readily integrated into multi-organ systems (Figure 1) [32].
Figure 1.
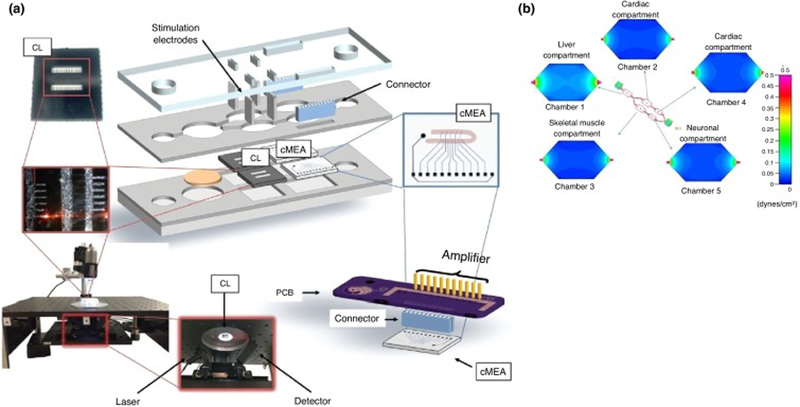
Multi-organ microphysiological system consisting of four different human organ modules (4-organ system): liver, heart, skeletal muscle and neurons. (a) Schematic for noninvasive technology to monitor cellular function in the 4-organ system, (b) shear stress distribution in each compartment of the system. Adapted, with permission, from [32,143].
Physiologically based pharmacokinetics and pharmacodynamics mathematical modeling
PK is the segment of pharmacology that studies the time-course of the ADME of a drug that results from the administration of a drug. PD is the area of pharmacology that studies the time-course of the effect of drug in the body [74,75]. Simply, PK studies are ‘what the body does to a drug’ and PD studies are ‘what a drug does to the body’ [76]. One important tool used to guide the design of HoaC and drug development is mathematical modeling, such as PK and/or PD models. PK models can be divided into compartmental and noncompartmental models. Compartmental models describe PK parameters through nonlinear regression analysis and describe the body as a finite number of interconnected, well-mixed and kinetically homogeneous compartments [75,77]. The degree of complexity varies between compartmental models; the simplest is a one-compartment model, which represents the body as a single uniform compartment but, to provide insight into drug mechanism, a more physiologically relevant model is required (Figure 2). This is accomplished though physiologically based pharmacokinetics (PBPK) models, where organs are represented as separate compartments connected with a hypothetical blood flow [75]. PBPK models are used to predict the concentration profiles for the parent compound and its metabolites associated with compound dosing [31].
Figure 2.
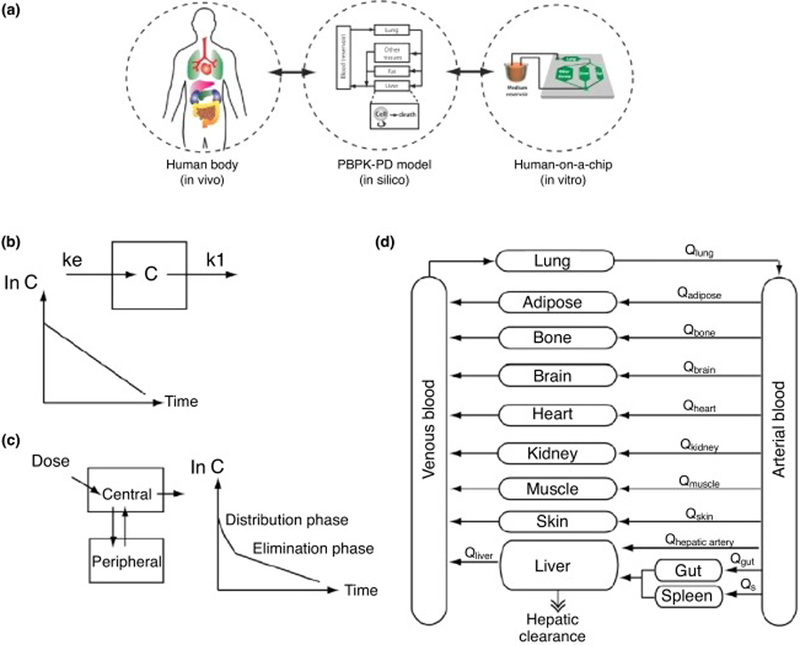
Physiologically based pharmacokinetic (PBPK) and pharmacodynamic (PD) mathematical modeling schematic. (a) Concept of PBPK model as a mathematical representation of the human body, (b) one-compartment model, (c) two-compartment model, (d) PBPK ‘whole-human’ model. Adapted, with permission, from [75,144].
PD models can describe the effect of a drug as a linear function of concentration, where effect will increase with higher concentrations of the drug, or as a nonlinear function where a maximum effect will be defined (Emax model). PD and PK models can be combined to create a PK/PD model to analyze the time-dependent changes of the physiological effect of a specific dose of the drug. In this modeling approach, the physiological outcome (PD) is predicted based on the PK profile associated with a certain dose of a drug. For example, Sung and collaborators created a PBPK model and a PD model separately to describe the ADME and efficacy of a chemotherapeutic agent (5-fluorouracil), and then combined the two models to create a ‘PK/PD model-on-a-chip’, which demonstrated significant changes in cancer cell viability between static and dynamic drug concentrations [78]. A more extensive description of PBPK and PD models is reviewed elsewhere [75].
PBPK and PD models can be especially useful in the development of drugs for rare diseases by enhancing the number of applications in clinical pharmacology particularly for specific populations [79,80]. However, these models have only recently been applied pharmaceutically; in 2014, the FDA released the ‘Strategic Plan for Accelerating the Development of Therapies for Pediatric Rare Diseases’, which recognized the importance of a PBPK approach by using it as a strategy to inform the design and conduct of PK/PD studies and clinical trials for investigational drugs in pediatric rare disease populations (http://www.fda.gov).
Disease-on-a-chip
As previously described, HoaC technology can be leveraged to build physiologically relevant human tissue models with a dynamic microenvironment and complex intercellular interactions. HoaC models can be developed using healthy tissues with normal physiology or it can mimic a disease state in one or more tissues in the same system. This design is often described as ‘disease-on-a-chip’ (DoaC). DoaC models can be used to investigate drug toxicity as well as efficacy, in the study of progression and treatment of specific diseases.
One advantage of DoaC models is the ability to incorporate patient-derived iPSCs. This allows doctors to develop patient-specific treatment strategies by identifying therapies that are most effective and least toxic in individual cases. For example, individuals vary widely in their liver cytochrome P450 (CYP) enzyme activity levels – the major enzyme pathways involved in drug metabolism [81]. These differences can lead to substantial interindividual variability in rates of drug activation or elimination, and consequently have a significant impact on patient tolerance of a particular drug treatment protocol. Understanding these inter-patient differences before treatment through the use of patient-specific HoaC systems could greatly improve the efficacy and minimize the toxicity of a drug treatment protocol. This kind of precision medicine can be a powerful tool for understanding complex diseases, such as cancer [82]. The next section of this review will focus on discussing HoaC systems developed specifically for rare diseases.
Human-on-a-chip and rare diseases
Rare diseases represent a wide spectrum of disorders that can affect almost any tissue, organ system or biological process in the body. HoaC technology is capable of representing a disease state where other models are limited or unavailable; human-based multi-organ models have flexibility relating to which organ modules, cell types and microfluidics to include, so platforms can be engineered to recapitulate highly specific disease states. In rare diseases with a genetic component, iPSC technology can be used that includes relevant mutations or patient-specific cells to understand their effect on relevant tissues. Because this technology has only recently begun to be applied to rare disease research, this section will discuss recent developments and potential applications of the technology.
Autoimmune models
Autoimmune diseases occur when the adaptive immune system no longer tolerates self-antigens and instead mounts an immune response against them that leads to impaired function. There are at least 80 defined autoimmune disorders, including type I diabetes, rheumatoid arthritis, celiac disease, inflammatory bowel disease, Graves’ disease, myasthenia gravis and systemic lupus erythematosus, as well as many others defined as rare diseases [83]. Autoimmune diseases arise when the body fails to differentiate self from non-self; as normal physiological processes inactivate or destroy lymphocytes that self-react, in pathological autoimmunity, protective mechanisms fail to prevent an inflammatory response [83]. Despite affecting >23.5 million Americans the cause of autoimmune disorders is poorly understood, and diseases are typically managed rather than cured [83]. Almost any organ can be targeted but, overall, autoimmune diseases share similar effects and mechanisms. Autoimmune diseases disproportionally effect women – 78% of those affected are female, especially during childbearing years, suggesting a hormonal component [84], and occur more frequently in certain ethnic groups, suggesting a genetic component. Risk factors include previous viral infections and some vitamin deficiencies. However, there is no single cause and it has been proposed that these diseases occur as a complex combination of genetic and environmental factors [85]. Because of the difficulty in isolating any one factor in vivo, HoaC models are a compelling platform for research into the causes and mechanisms of autoimmune diseases.
Microphysiological models with recirculating medium can model the cellular and soluble components of the immune system and how they interact with tissues. HoaC models can be engineered to replicate the 3D structure of relevant tissues, and can house or contain circulating immune cells, including patient-derived leukocytes [86]. Systems can also be infused with antibodies or inflammatory molecules such as complement, cytokines or chemokines for investigation into the role of specific immune biomolecules or can use purified blood fractions to represent a disease state. Although no comprehensive autoimmune system model has been established to date, a number of immune system HoaC models have been established [87], including models for the thymus [88], lymph node [86] and hematopoietic bone marrow in microfluidic [89] and 3D [90] platforms, as well as for leukocyte intra- and extra-vasation, specifically neutrophil trafficking [91] and transendothelial migration [92,93]. In vitro models have also been established for modeling autoimmune diseases, including a 3D cartilage model for rheumatoid arthritis [94] and a colon crypt model with potential for studying inflammatory bowel disease [95].
There is a strong genetic component to autoimmunity, but this link is not fully established. Although there are monogenic autoimmune disorders, and some major histocompatibility complex (MHC) mutations are strongly established as disease components, mutations do not always have established effects and are generally not predictive of disease [96]. This makes HoaC technology, where iPSC and CRISPR technology can model mutations in vitro, a useful platform for studying the genetic role in autoimmune responses. Because autoimmune diseases vary significantly in severity between individuals, the potential to model combinations of genetic, environmental and cellular components makes HoaC platforms uniquely able to determine the most relevant factors in disease progression for specific patients. For rare autoimmune diseases, where incidence is too low to gather significant data on affected individuals and shared risk factors, HoaC systems can potentially be engineered with modules representing a patient’s own somatic and immune cells to determine disease cause and to identify or test relevant therapeutics as a personalized medicine application.
Neuromuscular junction models
The neuromuscular junction (NMJ) is a tripartite synapse formed between motoneurons, skeletal muscle and Schwann cells in the PNS [97]. Rare neuromuscular diseases comprise a range of acquired and inherited disorders targeting motoneurons, skeletal muscle or the NMJ – specifically, acute and chronic autoimmune peripheral neuropathies including Guillian–Barré syndrome and myasthenia gravis, respectively. Other peripheral neuropathies include amyotrophic lateral sclerosis (ALS) and the muscular dystrophies. Currently, there are no approved cures for neuromuscular diseases; treatments focus on symptom management [98]. Contributing to the absence of effective treatment options is the heterogeneity of neuromuscular diseases and a lack of appropriate models to study. For example, the muscular dystrophies are a group of >30 genetic diseases affecting ~200 000 people annually in the USA and characterized by progressive muscle weakness and atrophy of muscle tissue, irrespective of nerve degeneration [99]. Duchenne’s muscular dystrophy (DMD), the most common MD, has been extensively studied using mouse models [100]. DMD is caused by a mutation to the dystrophin gene, located on the X chromosome, encoding the dystrophin protein, which plays an integral part in linking the actin cytoskeleton of the muscle cell to the ECM via the dystrophin–glycoprotein complex [101]. The most common mouse model, the mdx mouse, contains a premature stop codon in exon 23 that leads to a loss of full-length dystrophin [102]. These mice exhibit a milder phenotype compared with human DMD patients, living ~80% as long as control mice, significantly longer proportionally to diseased human patients [103]. These mice exhibit muscle fiber necrosis and inflammatory cell infiltration beginning ~3 weeks of age. However, after the 4-week period of extensive necrosis and elevated serum creatine kinase, the damage begins to slow, and only mild necrosis is present for the remainder of the mouse’s lifespan. This pathological phenotype is significantly milder than that observed in DMD patients [104]. The milder mouse phenotype has been attributed to a compensatory upregulation in utrophin expression [104]. At least five additional mdx mouse models have been generated, all with similar phenotypes [100]. Recently, a human-based NMJ-on-a-chip model composed of iPSC-derived motoneurons and primary skeletal muscle myotubes lasting up to 21 days was developed [105]. The system uses a PDMS barrier with microtunnels cast from a microfabricated silicon wafer. The barrier-partitioned motoneurons and myotubes are connected via tunnels where axons migrate through and innervate the myotubes forming functional NMJs. The NMJ system measures the functional transmission of signals from motoneurons to skeletal muscle via neuromuscular junctions, with direct measurements of the muscle function, and can be used for drug testing (Figure 3). Treatment of the NMJ-on-a-chip devices with neurotoxins or myotoxins resulted in dose-dependent neuromuscular dysfunction, indicating the sensitivity of the system for drug discovery research aimed at neuromuscular physiology [105]. Utilizing motoneurons derived from diseased-patient iPSCs, such a system could be used to investigate the dysfunctional neuromuscular signaling in rare diseases like ALS, as well as to screen novel compounds for efficacy in treating the disease. Further, iPSC-derived myoblasts, which have been differentiated by several groups, could be used in the system to study muscle-centric rare diseases such as the muscular dystrophies using mutant-iPSCs [106]. HoaC models of functional neuromuscular junctions will be useful tools for investigating neuromuscular degenerative diseases owing to their ability to sensitively determine the acute and chronic effects of compounds aimed at restoring neuromuscular function.
Figure 3.
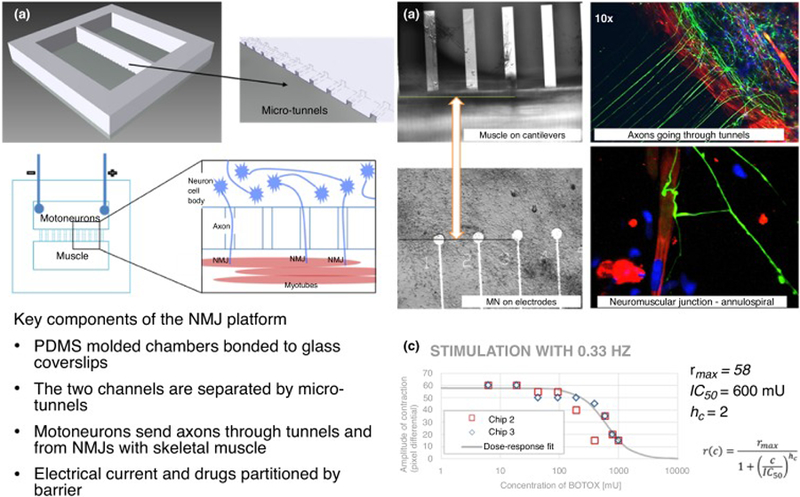
Neuromuscular junction (NMJ) platform. A microtunnel-based system (a) allows the neurons and skeletal muscle to remain in distinct compartments while allowing axons to pass to the muscle side and innervate the myotubes; microfabricated bioMEMS enable direct electrical stimulation of motoneurons and direct measurement of myotube contraction (lower panel). (b) Phase contrast images of myotubes on cantilevers (upper left) and motoneurons on microelectrode array (MEA) electrodes (lower left), immunocytochemistry indicating axons (green) growing through tunnels and forming NMJs with myotubes (red) (right panels). (c) Effect of drugs on the NMJ can be tested in the system, producing a dose–response curve, in this case using BOTOX as the NMJ blocking toxin. As the concentration of BOTOX on the muscle-side increases the amplitude of the myotube contraction decreases. IC50 = 600 mU.
Cancer models
A rare cancer, as defined by the National Cancer Institute, has an incidence of <150 new cases per million per year (15 per 100 000 per year). This corresponds to ~40 000 new cases per year in the USA. Rare cancers affect most major body regions and organ systems including head and neck, digestive, reproductive, respiratory, urogenital, nervous and endocrine systems. Rare cancers also include rare histological variants and molecular subtypes of common cancers, which can significantly influence progression, treatment and prognosis [107]. For example, pleomorphic lobular carcinoma is an extremely rare, aggressive variant of lobular carcinoma with a poor prognosis [108]. Rare cancers are difficult to study owing to their low incidence rate and, consequently, their etiology and progression can be poorly understood making identifying a timely, highly effective therapeutic strategy difficult. HoaC systems are an attractive model for studying cancer, and rare cancers specifically, because of their modularity, scalability and ability to mimic important aspects of tumor biology including the tumor microenvironment. Specifically, the tumor microenvironment, including the ECM, blood vessels, signaling molecules and inflammatory cells, plays an important part in malignancy and metastasis potential of a cancer [109]. For example, breast, prostate and lung cancer have all been linked to a high clinical risk of metastasis [110]. The multistep process involves tumor separation from the primary site, tumor intravasation, tumor extravasation and, finally, colonization of the secondary tissue. Microfluidic HoaC models can be engineered to recreate aspects of the tumor microenvironment using a range of techniques in micropatterning of ECM, microfluidic channel fabrication, 2D or 3D cell and scaffold printing, among others [111,112]. Additionally, systems can be created that reproduce the cellular architecture necessary to study tumor metastasis. Recently, a microfluidic system was used to study circulating tumor cell (CTC) metastasis into bone and liver [113]. The study also demonstrated inhibition of lung metastasis using the drug candidate AMD3100. In another study, a microfluidic 3D model was used to analyze the specificity of breast cancer metastases to bone. The system consisted of osteodifferentiated bone-marrow-derived stem cells, endothelial cells and breast cancer cells and quantified the extravasation and proliferation of cancer cells over 5 days [113]. Systems have also been developed to study tumor intravasation. For example, Zervantonakis et al. demonstrated the utility of a 3D microfluidic model to monitor and quantify tumor intravasation in real-time [114]. Cancer HoaC models can recreate important aspects of tumor physiology including tumor architecture and metastasis. Utilizing HoaC technology to model rare cancers could facilitate investigations into their underlying pathophysiology enabling the development of more-effective therapeutic strategies.
Blood–brain barrier systems
The BBB is the highly regulated microvascular network that serves as a border between the CNS, including the brain, spinal cord and cerebrospinal fluid, and the blood. It comprises highly restrictive capillaries composed of continuous, nonfenestrated endothelial cells and lined with pericytes. The neurovascular unit (NVU) includes ECM molecules and astrocytes, which ensheath and connect neurons and blood vessels through polarized processes that regulate the diffusion and transport of water, molecules and ions for brain homeostasis, playing a part in neurodegeneration, aging and drug delivery to the CNS [115]. BBB dysfunction is relevant to a number of neurodegenerative disorders, including Alzheimer’s disease, multiple sclerosis (MS), stroke and epilepsy, and is a major driving force in other rare or monogenic disorders including microcephaly, Allan-Herndon-Dudley syndrome and Alexander disease [116]. However, difficulties in studying the BBB in vivo and a lack of models for rare diseases make it difficult to elucidate the precise role of the BBB in neurologic conditions.
HoaC systems, where the cellular and extracellular components of the BBB can be readily modeled, represent an ideal platform for determining the role of BBB disfunction in disease progression and treatment. The BBB has been extensively modeled using HoaC systems [117,118] characterized using TEER, tight junction staining, passive diffusion of dextrans through the membrane, active transport of molecules and the ability to block immune cell invasion [119]. Models range from microfluidic devices with representative shear stress on endothelial cells to co-cultures of endothelial cells, pericytes and astrocytes. Three-dimensional microfluidic models have been constructed to provide relevant architecture [119,120] and models have been applied for drug screening [121] and HTS [36]. Furthermore, HoaC systems can be used to study diseases where the precise role of the BBB is unknown, or where BBB dysfunction is suspected to play a part; multiorgan systems, which can include a BBB module, used to study CNS rare diseases, would be useful to understand the role of the BBB in disease progression or mechanisms. For example, Huntington’s disease is a rare CNS disorder where cerebrovascular changes occur, but BBB dysfunction specifically has not been thoroughly investigated [122]. Similarly, BBB breakdown is a component of MS and ALS [123,124]. Multi-organ systems where the BBB is a component can address specific aspects of dysfunction, including whether BBB disfunction is causative or consequential, and can be useful in determining treatment strategies.
A major challenge in research, diagnosis and treatment of neurological and neurodegenerative disorders is symptom overlap between diseases and cumulative disease effects, making a specific disease difficult or impossible to distinguish. In particular, in the case of rare diseases or diseases with a genetic component, individual genotypes can exacerbate disorders, or lead to varying progression and severity between individuals. Identification of many forms of diseases such as dementia, and concurrent incidence of multiple disorders, has driven research into more-precise diagnostic tools [125,126]; however, neurodegeneration exists as a continuum and not all disorders can be discretely classified [127]. In combination with iPSC technology, HoaC systems with a BBB component could be useful for research into treatment strategies. The ability to address permutations of mutant and healthy BBBs alongside other diseased CNS components would facilitate investigations into the causative role the BBB has in rare disease progression. For more individualized treatment, HoaC technology could be used to reconstruct the exact disease state of a patient, including any mutations that would affect the BBB and CNS, enabling personalized research or testing of therapeutics.
Liver models
The addition of a liver component to a HoaC system is useful for studying the metabolism of drugs and the differences in toxicity between parent compound and metabolites. HoaC systems can be beneficial to understand rare liver diseases such as alpha-1 antitrypsin deficiency and Alagille syndrome. Owing to the difficulties of in vitro culture of isolated hepatocyte (i.e., in vitro cell degeneration) Li and colleagues developed a method to form small hepatocyte aggregates on collagen micro-islands using primary rat hepatocytes. These micropatterned hepatocytes enabled cell survival with normal functionality (albumin production and induction of CYP450 drug metabolism enzymes) [128]. In 2016, Bhise et al. created a liver-on-a-chip device for toxicity testing by combining bioprinting of hepatic spheroid-laden hydrogel constructs and bioreactors using human hepatocytes. This system was able to maintain hepatocyte functionality over a 4-week period [129]. Recently, a liver model was integrated into a 4-organ system and maintained function for 4 weeks [31].
Cardiac models
Rare cardiac diseases can also be addressed by HoaC technology. Relevant heart-on-a-chip devices can potentially assess real-time contractile force, speed and frequency of cardiomyocytes. In 2008, Kim et al. developed a methodology to assess cardiomyocyte contractility placing them in a 3D microenvironment using cantilevers [130]. Sidorov and co-workers developed a wire platform that allows mechanical and electrical characterization of 3D cardiac tissue constructs that can be valuable to study cardiac diseases and for drug development [131]. Stancescu et al. developed a human stem-cell-derived cardiomyocyte platform that was able to model electrical readouts and force in the same system (Figure 4) [132]. These technologies can be assembled into microdevices for the development of a heart-on-a-chip system. Wang et al. developed an interesting heart-on-a-chip device that proved to be useful to provide new insights into the pathogenesis of Barth syndrome – a rare cardiac disease – using patient-derived iPSCs [54]. This study is one example of how HoaC devices can make a significant impact on understanding rare diseases and on orphan drug development.
Figure 4.
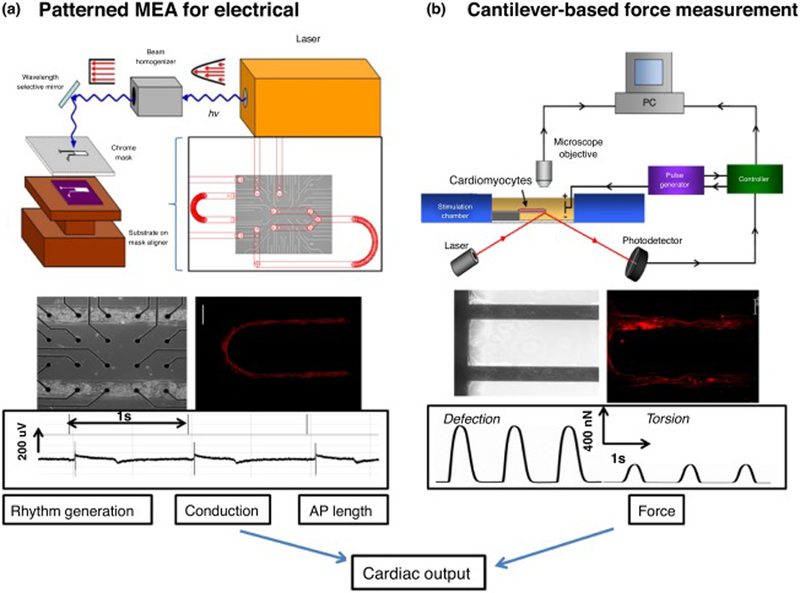
Key elements for a platform for determining cardiac physiology using human cardiomyocytes. Cardiac function was extrapolated from measurement of rhythm generation (frequency and amplitude), conduction velocity, action potential length (QT interval) and force generation of the heart (a) schematic of the system used to pattern SAMs on microelectrode arrays (MEAs) (top). Phase contrast micrograph of patterned human-derived cardiomyocytes on top of substrate-embedded extracellular electrodes. Immunostaining verified that human-derived cells differentiated to cardiomyocytes (middle) and exhibited cardiac rhythm generation as measured by the embedded electrodes (bottom). (b) Diagram of the cantilever-based force measurement system (top) cardiomyocytes integrated into the BioMEMs device and immunocytochemistry indicating cardiac alignment along the cantilever (middle). Example traces of deflection and torsional force with the device after myocyte contractions (bottom) [132].
Other systems and rare diseases
In addition to the models discussed above, HoaC platforms are applicable to other rare diseases, including rare respiratory and kidney diseases. Most rare respiratory diseases involve lung damage; rare lung diseases can affect the lungs exclusively, as part of a systemic disease, or can involve iatrogenic lung disease caused by the treatment of a rare condition. Respiratory diseases include vasculitides of the lung (granulomatosis with polyangiitis), microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis, Behçet’s disease, Takayasu’s arteritis and anti-basement membrane syndrome pulmonary alveolar proteinosis [133]. Lung-on-a-chip models are valuable for studying the biological processes of lung diseases for developing pharmaceutical treatments [134]. In 2010, Huh et al. developed a lung-on-a-chip model that reconstituted multiple physiological functions of a breathing lung. This device was able to reconstitute the microarchitecture of the alveolar–capillary unit by combining 2D cellular models with a BioMEMs device, while maintaining alveolar epithelial cells at an air–liquid interface, enabling analysis of the effect of these forces on different pathological and physiological lung functions [135]. Most recently, other groups have also developed lung-on-a-chip models that reproduced the functionality of the alveolar barrier using human cells [136,137]. Moreover, in 2018, Jain and colleagues created a human pulmonary thrombosis disease model. Bovard et al. developed an integrated lung/liver-on-a-chip device to study acute and chronic toxicity of inhaled compounds [136,138].
The kidney is an important organ to consider in drug development, because drug-induced nephrotoxicity is the main cause of up to 25% of all cases of severe acute renal failure during treatment, making it one of the major factors leading to drug development failure [139]. There are ~150 known rare kidney diseases with an overall incidence of ~60–80 cases per 100 000 in Europe and the USA [140] with two of the most common being focal segmental glomerulosclerosis (FSGS) and atypical hemolytic uremic syndrome (aHUS). There are currently no drugs for the treatment of FSGS, and aHUS has only one drug that is approved for treatment in the USA. Kidney-on-a-chip models could be used for the development of orphan drugs for these diseases; however, these models are still in early development, with most devices using animal cells instead of human cells. Wang et al. constructed a glomerulus-on-a-chip device with isolated rat glomerulus and established an in vitro disease model for diabetic nephropathy induced by high blood glucose [6]. In this model, glomeruli functionality (barrier function and integrity) was maintained for 2 weeks [6]. Another interesting device was developed by Qu and colleagues where they constructed the basic structure of a nephron [139]. This nephron-on-a-chip device was constructed using primary rat glomerular endothelial cells, podocytes, tubular epithelial cells, peritubular endothelial cells, renal blood flow involving plasma proteins and glomerular filtrate flow. In this study, they also showed that this microdevice was able to identify different pathogenesis of cisplatin-and-doxorubicin-induced acute kidney injury [139].
Concluding remarks and future directions
Rare diseases, which often have complex genetic and environmental causes, are difficult and expensive to study owing to a limit in cases for study and a small market for testing and marketing treatments. Even after the ODA, scientific resources and treatments for rare diseases are low and few drugs aimed at treating rare diseases exist. However, rare diseases affect >25 million Americans, and managing rare diseases can be socially and financially damaging for those who suffer from them. A major problem in rare disease research is the lack of appropriate models for studying the diseases and subsequently testing pharmaceuticals. Animal models are expensive and are poor predictors of drug response in humans and, although many isolated in vitro models for rare diseases have been established, they often use animal cells or immortalized or cancer-derived human cells, which show loss of in vivo function or do not model crucial elements of human physiology.
In addition to PBPK and PD models, microfluidic HoaC platforms with circulating medium and multiple organ modules can provide the unique advantage of modeling interaction between human tissues by enabling the development of phenotypic models. When studying human drug metabolism, no compound acts in isolation – in addition to affecting the target organ, administration of a drug can result in toxicity and drug metabolites can drive downstream effects or damage. For disease research, models can be used to study disease by driving a change in the system and monitoring response. For drug testing, multi-organ HoaC models can analyze effectiveness of the drug on the target and effects on other tissues, potentially on the same platform. Further, a disease state can alter how drugs are metabolized, and dysfunction in one organ can have a whole-body response. HoaC models are capable of recapitulating drug response when the target organ or other tissues are damaged or missing. HoaC models also enable improved research into combinatorial drug treatment; it is established that the optimal treatment of diseases is often a combination of multiple drugs, strategized so that effectiveness is maximized while toxicity and side-effects are minimized. Potential combinations can be identified through HTS and analysis of existing genetic and clinical data [11]. For rare diseases, HoaC systems can be used as a platform to test new drug combinations, providing data in cases where limited clinical drug data exists; and models can be used to compare treatment strategies for efficacy and toxicity directly in a physiologically relevant model.
Because of their flexibility in construction, HoaC systems are useful tools for studying rare diseases. A DoaC-focused platform can be tailored to represent a specific disease in one or all organ modules, and blood-surrogate medium formulations can mimic signaling molecules sent by other tissues. For a genetic element, as genomic modification and iPSC technologies have improved, options have expanded for modeling of rare diseases in vitro; with the opportunity to drive specific mutations in addition to established diseases, even in combination, research can be conducted on cells of any organ in a diseased state, leading to a more personalized approach to research and medicine. Furthermore, patient-derived iPSCs can potentially be used to study an individual’s disease state, which can include several specific mutations and/or epigenetic modifications. Because one condition can interfere with the response to treatment of another, HoaC modeling of rare diseases can be used to test personalized drug response and side-effects [141,142].
Currently, the applications of HoaC systems have been limited. Although many studies have established microphysiological tissue interactions in vitro, and devices have been constructed containing multiple organs relative to drug metabolism [41,42,143], a complete model for the human body has yet to be developed. As a new technology, areas of modeling relevant to rare disease are still being developed; for example, many rare diseases feature inflammation as a key disease driver, so the addition of an immune system to multi-organ devices would be an important achievement toward the construction of reliable disease models. Nevertheless, over the past decade, HoaC technology has improved greatly. Here, we have shown the latest advances in HoaC systems and its promising potential for rare-disease research and orphan drug development.
Highlights:
Drug development and disease modeling for rare diseases remain challenges
Advances in in vitro modeling has led to development of human-on-a-chip technology
PK and PD mathematical modeling is a powerful tool for orphan drug development
HoaC systems have potential for facilitating rare disease and orphan drug research
Acknowledgments
We would like to acknowledge funding from the National Institutes of Health, grant numbers R01NS050452 and R44AG059511.
Biography

Camilly P. Pires de Mello
Dr Camilly Pires de Mello obtained her PhD in science and biotechnology from the Universidade Federal Fluminense. She is currently a Post-Doctoral Research Associate in the Hybrid Systems Lab, in the NanoScience Technology Center at the University of Central Florida (UCF). Previously, she was a Post-Doctoral Associate at the University of Florida in the College of Medicine’s Institute for Therapeutic Innovation. She has 10 years of experience working with pharmacology, pharmacokinetics/pharmacodynamics, drug discovery, molecular biology, RNA and DNA viruses, preclinical research, cell culture and bioreactors.

James J. Hickman
Prof. James Hickman is the Founding Director of the NanoScience Technology Center and Professor of Nanoscience Technology, Chemistry, Biomolecular Science and EE at UCF and has a PhD from MIT in chemistry. His career has focused on studying the interaction of biological species with modified surfaces, first in industry and later in academia. He is interested in creating hybrid systems for biosensor and biological computation applications and the creation of functional in vitro systems for human-on-a-chip applications. He is also the founder and current Chief Scientist of Hesperos, which is focusing on multi-organ systems for drug discovery and toxicity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cheung RY et al. (2004) Orphan drug policies: implications for the United States, Canada, and developing countries. Health Law J 12, 183–200 [PubMed] [Google Scholar]
- 2.Mullard A (2016) Parsing clinical success rates. Nat. Rev. Drug Discov 15, 447. [DOI] [PubMed] [Google Scholar]
- 3.Schieppati A et al. (2008) Why rare diseases are an important medical and social issue. Lancet 371, 2039–2041 [DOI] [PubMed] [Google Scholar]
- 4.Griggs RC et al. (2009) Clinical research for rare disease: opportunities, challenges, and solutions. Mol. Genet. Metab 96, 20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang YI et al. (2017) Self-contained, low-cost body-on-a-chip systems for drug development. Exp. Biol. Med 242, 1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L et al. (2017) A disease model of diabetic nephropathy in a glomerulus-on-a-chip microdevice. Lab Chip 17, 1749–1760 [DOI] [PubMed] [Google Scholar]
- 7.Esch MB et al. (2014) How multi-organ microdevices can help foster drug development. Adv. Drug Deliv. Rev 69–70, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Low LA and Tagle DA (2017) Microphysiological systems (“organs-on-chips”) for drug efficacy and toxicity testing. Clin. Transl. Sci 10, 237–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bracken MB (2009) Why animal studies are often poor predictors of human reactions to exposure. J. R. Soc. Med 102, 120–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barré-Sinoussi F and Montagutelli X (2015) Animal models are essential to biological research: issues and perspectives. Future Sci. O.A 1, FSO63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun X et al. (2013) High-throughput methods for combinatorial drug discovery. Sci. Transl. Med 5, 205rv1. [DOI] [PubMed] [Google Scholar]
- 12.Daquinag AC et al. (2013) Adipose tissue engineering in three-dimensional levitation tissue culture system based on magnetic nanoparticles. Tissue Eng. Part C Methods 19, 336–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loskill P et al. (2017) WAT-on-a-chip: a physiologically relevant microfluidic system incorporating white adipose tissue. Lab Chip 17, 1645–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menon NV et al. (2018) A tunable microfluidic 3D stenosis model to study leukocyte-endothelial interactions in atherosclerosis. APL Bioeng 2, 016103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feaver RE et al. (2016) Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 1, e90954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agrawal G et al. (2017) Skeletal muscle-on-a-chip: an in vitro model to evaluate tissue formation and injury. Lab Chip 17, 3447–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandorkar P et al. (2017) Fast-track development of an in vitro 3D lung/immune cell model to study Aspergillus infections. Sci. Rep 7, 11644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller AJ and Spence JR (2017) In vitro models to study human lung development, disease and homeostasis. Physiology 32, 246–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esch MB et al. (2016) Modular, pumpless body-on-a-chip platform for the co-culture of GI tract epithelium and primary liver tissue. Lab Chip 16, 2719–2729 [DOI] [PubMed] [Google Scholar]
- 20.Wilmer MJ et al. (2016) Kidney-on-a-chip technology for drug-induced nephrotoxicity screening. Trends Biotechnol 34, 156–170 [DOI] [PubMed] [Google Scholar]
- 21.Xiao S et al. (2017) A microfluidic culture model of the human reproductive tract and 28-day menstrual cycle. Nat. Commun 8, 14584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haring AP et al. (2017) Microphysiological human brain and neural systems-on-a-chip: potential alternatives to small animal models and emerging platforms for drug discovery and personalized medicine. Stem Cell Rev 13, 381–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gribi S et al. (2018) A microfabricated nerve-on-a-chip platform for rapid assessment of neural conduction in explanted peripheral nerve fibers. Nat. Commun 9, 4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wufuer M et al. (2016) Skin-on-a-chip model simulating inflammation, edema and drug-based treatment. Sci. Rep 6, 37471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hao S et al. (2018) A spontaneous 3D bone-on-a-chip for bone metastasis study of breast cancer cells. Small 14, e1702787. [DOI] [PubMed] [Google Scholar]
- 26.Moroni L et al. (2018) Biofabrication strategies for 3D in vitro models and regenerative medicine. Nat. Rev. Mater 3, 21–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chi CW et al. (2016) Microfluidic cell chips for high-throughput drug screening. Bioanalysis 8, 921–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langhans SA (2018) Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol 9, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauer S et al. (2017) Functional coupling of human pancreatic islets and liver spheroids on-a-chip: towards a novel human ex vivo type 2 diabetes model. Sci. Rep 7, 14620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang YS et al. (2017) Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. U. S. A 114, E2293–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oleaga C et al. (2018) Investigation of the effect of hepatic metabolism on off-target cardiotoxicity in a multi-organ human-on-a-chip system. Biomaterials 182, 176–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oleaga C et al. (2019) Long-term electrical and mechanical function monitoring of a human-on-a-chip system. Adv. Funct. Mater 29, 1805792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang YI et al. (2018) Multiorgan microphysiological systems for drug development: strategies, advances, and challenges. Adv. Healthcare Mater 7, 1701000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cordonnier C et al. (2015) Dynamic in vitro models of the human gastrointestinal tract as relevant tools to assess the survival of probiotic strains and their interactions with gut microbiota. Microorganisms 3, 725–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rackley CR and Stripp BR (2012) Building and maintaining the epithelium of the lung. J. Clin. Invest 122, 2724–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wevers NR et al. (2018) A perfused human blood–brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 15, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilhelm I and Krizbai IA (2014) In vitro models of the blood–brain barrier for the study of drug delivery to the brain. Mol. Pharm 11, 1949–1963 [DOI] [PubMed] [Google Scholar]
- 38.Caballero D et al. (2017) Tumour-vessel-on-a-chip models for drug delivery. Lab Chip 17, 3760–3771 [DOI] [PubMed] [Google Scholar]
- 39.Alberti M et al. (2017) Multi-chamber microfluidic platform for high-precision skin permeation testing. Lab Chip 17, 1625–1634 [DOI] [PubMed] [Google Scholar]
- 40.Sriram G et al. (2018) Full-thickness human skin-on-chip with enhanced epidermal morphogenesis and barrier function. Mater. Today 21, 326–340 [Google Scholar]
- 41.Miller PG and Shuler ML (2016) Design and demonstration of a pumpless 14 compartment microphysiological system. Biotechnol. Bioeng 113, 2213–2227 [DOI] [PubMed] [Google Scholar]
- 42.Edington CD et al. (2018) Interconnected microphysiological systems for quantitative biology and pharmacology studies. Sci. Rep 8, 4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu C et al. (2018) Modeling human diseases with induced pluripotent stem cells: from 2D to 3D and beyond. Development 2018, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niu N and Wang L (2015) In vitro human cell line models to predict clinical response to anticancer drugs. Pharmacogenomics 16, 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geraghty RJ et al. (2014) Guidelines for the use of cell lines in biomedical research. Br. J. Cancer 111, 1021–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takahashi K and Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- 47.Takahashi K et al. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 [DOI] [PubMed] [Google Scholar]
- 48.Yu J et al. (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 [DOI] [PubMed] [Google Scholar]
- 49.Shi Y et al. (2017) Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov 16, 115–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleischer A et al. (2018) Generation of two induced pluripotent stem cell (iPSC) lines from p.F508del cystic fibrosis patients. Stem Cell Res 29, 1–5 [DOI] [PubMed] [Google Scholar]
- 51.Liu Z and Zhao R (2016) Generation of HEXA-deficient hiPSCs from fibroblasts of a Tay–Sachs disease patient. Stem Cell Res 17, 289–291 [DOI] [PubMed] [Google Scholar]
- 52.Lee G et al. (2009) Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461, 402–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ebert AD et al. (2009) Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang G et al. (2014) Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stemcell and heart-on-chip technologies. Nat. Med 20, 616–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen G et al. (2013) Matrix mechanics and fluid shear stress control stem cells fate in three dimensional microenvironment. Curr. Stem Cell Res. Ther 8, 313–323 [DOI] [PubMed] [Google Scholar]
- 56.Cucullo L et al. (2011) The role of shear stress in blood–brain barrier endothelial physiology. BMC Neurosci 12, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duan Y et al. (2008) Shear-induced reorganization of renal proximal tubule cell actin cytoskeleton and apical junctional complexes. PNAS 105, 11418–11423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gimbrone MAJ et al. (1999) Special communicationthe critical role of mechanical forces in blood vessel development, physiology and pathology. J. Vasc. Surg 29, 1104–1151 [DOI] [PubMed] [Google Scholar]
- 59.Sato Y et al. (1999) Role of shear stress and immune responses in liver regeneration after a partial hepatectomy. Surg. Today 29, 1–9 [DOI] [PubMed] [Google Scholar]
- 60.Xiong B et al. (2014) Recent developments in microfluidics for cell studies. Adv. Mater 26, 5525–5532 [DOI] [PubMed] [Google Scholar]
- 61.Traub O and Berk BC (1998) Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler. Thromb. Vasc. Biol 18, 677–685 [DOI] [PubMed] [Google Scholar]
- 62.Maggiorani D et al. (2015) Shear stress-induced alteration of epithelial organization in human renal tubular cells. PloS One 10, e0131416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee DW et al. (2019) A microfluidic chip with gravity-induced unidirectional flow for perfusion cell culture. Biotechnol. Prog 35, e2701. [DOI] [PubMed] [Google Scholar]
- 64.Elliott W et al. (2015) In vitro model of physiological and pathological blood flow with application to investigations of vascular cell remodeling. J. Vis. Exp 2015, 53224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peng X et al. (2000) In vitro system to study realistic pulsatile flow and stretch signaling in cultured vascular cells. Am. J. Physiol. Cell Physiol 279, C797–805 [DOI] [PubMed] [Google Scholar]
- 66.Komeya M et al. (2017) Pumpless microfluidic system driven by hydrostatic pressure induces and maintains mouse spermatogenesis in vitro. Sci. Rep 7, 15459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang YI and Shuler ML (2018) UniChip enables long-term recirculating unidirectional perfusion with gravity-driven flow for microphysiological systems. Lab Chip 18, 2563–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tibbitt MW and Anseth KS (2009) Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng 103, 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robertson MJ et al. (2018) Recellularization of rat liver: an in vitro model for assessing human drug metabolism and liver biology. PLoS One 13, e0191892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nguyen DT et al. (2018) Humanizing miniature hearts through 4-flow cannulation perfusion decellularization and recellularization. Sci. Rep 8, 7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yanagawa F et al. (2016) Hydrogel microfabrication technology toward three dimensional tissue engineering. Regen. Ther 3, 45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dosh RH et al. (2017) Use of hydrogel scaffolds to develop an in vitro 3D culture model of human intestinal epithelium. Acta Biomater 62, 128–143 [DOI] [PubMed] [Google Scholar]
- 73.Smithmyer ME et al. (2014) Hydrogel scaffolds as in vitro models to study fibroblast activation in wound healing and disease. Biomater. Sci 2, 634–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meibohm B and Derendorf H (1997) Basic concepts of pharmacokinetic/pharmacodynamic (PK/PD) modelling. Int. J. Clin. Pharmacol. Ther 35, 401–413 [PubMed] [Google Scholar]
- 75.Sung JH et al. (2014) Using physiologically-based pharmacokinetic-guided “body-on-a-chip” systems to predict mammalian response to drug and chemical exposure. Exp. Biol. Med 239, 1225–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robinson DC (1986) Principles of pharmacokinetics. In Topics in Clinical Pharmacology and Therapeutics (Maronde RF, ed.), pp. Springer-Verlag. New York [Google Scholar]
- 77.Fan J and de Lannoy IA (2014) Pharmacokinetics. Biochem. Pharmacol 87, 93–120 [DOI] [PubMed] [Google Scholar]
- 78.Sung JH et al. (2010) A microfludic device for a pharmacokinetic-pharmacodynamic (PK-PD) model on a chip. Lab Chip 10, 446–455 [DOI] [PubMed] [Google Scholar]
- 79.Jones HM et al. (2015) Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin. Pharmacol. Ther 97, 247–262 [DOI] [PubMed] [Google Scholar]
- 80.Sager JE et al. (2015) Phsiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, appiication, and model verification. Drug Metab. Dispos 43, 1823–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tracy TS et al. (2016) Interindividual variability in cytochrome P450-mediated drug metabolism. Drug Metab. Dispos 44, 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hachey SJ and Hughes CCW (2018) Applications of tumor chip technology. Lab Chip 18, 2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang L et al. (2015) Human autoimmune diseases: a comprehensive update. J. Intern. Med 278, 369–395 [DOI] [PubMed] [Google Scholar]
- 84.Fairweather D and Rose NR (2004) Women and autoimmune diseases. Emerg. Infect. Dis 10, 2005–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Watad A et al. (2017) Seasonality and autoimmune diseases: the contribution of the four seasons to the mosaic of autoimmunity. J. Autoimmun 82, 13–30 [DOI] [PubMed] [Google Scholar]
- 86.Moura Rosa P et al. (2016) The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab Chip 16, 3728–3740 [DOI] [PubMed] [Google Scholar]
- 87.Irimia D and Wang X (2018) Inflammation-on-a-chip: probing the immune system ex vivo. Trends Biotechnol 36, 923–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pinto S et al. (2015) 3D organotypic co-culture model supporting medullary thymic epithelial cell proliferation, differentiation and promiscuous gene expression. J. Vis. Exp 2015, e52614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Torisawa Y-S et al. (2014) Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat. Methods 11, 663. [DOI] [PubMed] [Google Scholar]
- 90.Sieber S et al. (2018) Bone marrow-on-a-chip: long-term culture of human haematopoietic stem cells in a three-dimensional microfluidic environment. J. Tissue Eng. Regen. Med 12, 479–489 [DOI] [PubMed] [Google Scholar]
- 91.Hamza B and Irimia D (2015) Whole blood human neutrophil trafficking in a microfluidic model of infection and inflammation. Lab Chip 15, 2625–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han S et al. (2012) A versatile assay for monitoring in vivo-like transendothelial migration of neutrophils. Lab Chip 12, 3861–3865 [DOI] [PubMed] [Google Scholar]
- 93.Wu X et al. (2017) A versatile microfluidic platform for the study of cellular interactions between endothelial cells and neutrophils. Biochim. Biophys. Acta Gen. Subj 1861, 1122–1130 [DOI] [PubMed] [Google Scholar]
- 94.Peck Y et al. (2010) Establishment of an in vitro three-dimensional model for cartilage damage in rheumatoid arthritis. J. Tissue Eng. Regen. Med 12, e237–249 [DOI] [PubMed] [Google Scholar]
- 95.Wang Y et al. (2010) Microdevice to capture colon crypts for in vitro studies. Lab Chip 10, 1596–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ramos PS et al. (2015) Genetics of autoimmune diseases: insights from population genetics. J. Hum. Genet 60, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bloch-Gallego E (2015) Mechanisms controlling neuromuscular junction stability. Cell. Mol. Life Sci 72, 1029–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li L et al. (2018) Neuromuscular junction formation, aging, and disorders. Annu. Rev. Physiol 80, 159–188 [DOI] [PubMed] [Google Scholar]
- 99.Verschuuren J et al. (2016) Neuromuscular junction disorders. Handb. Clin. Neurol 133, 447–466 [DOI] [PubMed] [Google Scholar]
- 100.Ng R et al. (2012) Animal models of muscular dystrophy. Prog. Mol. Biol. Transl. Sci 105, 83–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Flanigan KM (2014) Duchenne and Becker muscular dystrophies. Neurol. Clin 32, 671–688 [DOI] [PubMed] [Google Scholar]
- 102.Im WB et al. (1996) Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet 5, 1149–1153 [DOI] [PubMed] [Google Scholar]
- 103.Chamberlain JS et al. (2007) Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J 21, 2195–2204 [DOI] [PubMed] [Google Scholar]
- 104.Matsumura K et al. (1992) Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature 360, 588–591 [DOI] [PubMed] [Google Scholar]
- 105.Santhanam N et al. (2018) Stem cell derived phenotypic human neuromuscular junction model for dose response evaluation of therapeutics. Biomaterials 166, 64–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kodaka Y et al. (2017) Skeletal muscle cell induction from pluripotent stem cells. Stem Cell Int 2017, 1376151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Boyd N et al. (2016) Rare cancers: a sea of opportunity. Lancet Oncol 17, e52–61 [DOI] [PubMed] [Google Scholar]
- 108.Ohashi R et al. (2017) Pleomorphic lobular carcinoma of the breast: a comparison of cytopathological features with other lobular carcinoma variants. Cytopathology 28, 122–130 [DOI] [PubMed] [Google Scholar]
- 109.Hui L and Chen Y (2015) Tumor microenvironment: sanctuary of the devil. Cancer Lett 368, 7–13 [DOI] [PubMed] [Google Scholar]
- 110.Gupta GP and Massagué J (2006) Cancer metastasis: building a framework. Cell 127, 679–695 [DOI] [PubMed] [Google Scholar]
- 111.Caballero D et al. (2017) Organ-on-chip models of cancer metastasis for future personalized medicine: from chip to the patient. Biomaterials 149, 98–115 [DOI] [PubMed] [Google Scholar]
- 112.Pavesi A et al. (2015) Controlled electromechanical cell stimulation on-a-chip. Sci. Rep 5, 11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Y et al. (2016) Microfluidic chip for isolation of viable circulating tumor cells of hepatocellular carcinoma for their culture and drug sensitivity assay. Cancer Biol. Ther 17, 1177–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zervantonakis IK et al. (2012) Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. U. S. A 109, 13515–13520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Daneman R and Prat A (2015) The blood–brain barrier. Cold Spring Harb. Perspect. Biol 7, a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhao Z et al. (2015) Establishment and dysfunction of the blood–brain barrier. Cell 163, 1064–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bang S et al. (2017) A low permeability microfluidic blood–brain barrier platform with direct contact between perfusable vascular network and astrocytes. Sci. Rep 7, 8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van der Helm MW et al. (2016) Microfluidic organ-on-chip technology for blood–brain barrier research. Tissue Barriers 4, e1142493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cho H et al. (2015) Three-dimensional blood–brain barrier model for in vitro studies of neurovascular pathology. Sci. Rep 5, 15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang JD et al. (2016) Organization of endothelial cells, pericytes, and astrocytes into a 3D microfluidic in vitro model of the blood–brain barrier. Mol. Pharm 13, 895–906 [DOI] [PubMed] [Google Scholar]
- 121.Wang YI et al. (2017) Microfluidic blood–brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol. Bioeng 114, 184–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Drouin-Ouellet J et al. (2015) Cerebrovascular and blood–brain barrier impairments in Huntington’s disease: potential implications for its pathophysiology. Ann. Neurol 78, 160–177 [DOI] [PubMed] [Google Scholar]
- 123.Brylev LV et al. (2012) Disruption of blood–brain barrier in amyotrophic lateral sclerosis: an update. Neurochemical J 6, 64–70 [Google Scholar]
- 124.Ortiz GG et al. (2014) Role of the blood–brain barrier in multiple sclerosis. Arch. Med. Res 45, 687–697 [DOI] [PubMed] [Google Scholar]
- 125.Agrawal M and Biswas A (2015) Molecular diagnostics of neurodegenerative disorders. Front. Mol. Biosci 2, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Koikkalainen J et al. (2016) Differential diagnosis of neurodegenerative diseases using structural MRI data. NeuroImage Clin 11, 435–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Armstrong RA (2012) Review paper on the ‘classification’ of neurodegenerative disorders: discrete entities, overlap or continuum. Folia Neuropathol 50, 201–218 [DOI] [PubMed] [Google Scholar]
- 128.Li CY et al. (2014) Micropatterned cell-cell interactions enable functional encapsulation of primary hepatocytes in hydrogel microtissues. Tissue Eng. Part A 20, 2200–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bhise NS et al. (2016) A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 8, 014101. [DOI] [PubMed] [Google Scholar]
- 130.Kim J et al. (2008) Quantitative evaluation of cardiomyocyte contractility in a 3d microenvironment. J. Biomech 41, 2396–2401 [DOI] [PubMed] [Google Scholar]
- 131.Sidorov VY et al. (2017) I-wire heart-on-a-chip i: three-dimensional cardiac tissue constructs for physiology and pharmacology. Acta Biomater 48, 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stancescu M et al. (2015) A phenotypic in vitro model for the main determinants of human whole heart function. Biomaterials 60, 20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cordier JF (2011) Orphan lung diseases. In European Respiratory Monograph (Welte T, ed.) (vol. 54), pp. European Respiratory Society [Google Scholar]
- 134.Konar D et al. (2016) Lung-on-a-chip technologies for disease modeling and drug development. Biomed. Eng. Comput. Biol 7 (suppl. 1), 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huh D et al. (2010) Reconstituting organ-level lung functions on a chip. Science 328, 1662–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jain A et al. (2018) Primary human lung alveolus-on-a-chip model of intravascular thrombosis for assessment of therapeutics. Clin. Pharmacol. Ther 103, 332–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stucki AO et al. (2015) A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab Chip 15, 1302–1310 [DOI] [PubMed] [Google Scholar]
- 138.Bovard D et al. (2018) A lung/liver-on-a-chip platform for acute and chronic toxicity studies. Lab Chip 18, 3814–3829 [DOI] [PubMed] [Google Scholar]
- 139.Qu Y et al. (2018) A nephron model for study of drug-induced acute kidney injury and assessment of drug-inducednephrotoxicity. Biomaterials 155, 41–53 [DOI] [PubMed] [Google Scholar]
- 140.Devuyst O et al. (2014) Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet 383, 1844–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Matsa E et al. (2016) Transcriptome profiling of patient-specific human iPSC-cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell 19, 311–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pinto N and Dolan ME (2011) Clinically relevant genetic variations in drug metabolizing enzymes. Curr. Drug Metab 12, 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Oleaga C et al. (2016) Multi-organ toxicity demonstration in a functional human in vitro system composed of four organs. Sci. Rep 6, 20030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sung JH et al. (2013) Microfabricated mammalian organ systems and their integration into models of whole animals and humans. Lab Chip 13, 1201–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]