

ABSTRACT
Background
The pathological hallmark in MSA is oligodendrocytic glial cytoplasmic inclusions (GCIs) containing α‐synuclein, in addition to neuronal loss and astrogliosis especially involving the striatonigral and olivopontocerebellar systems. Rarely, TAR DNA‐binding protein of 43 kDa (TDP‐43), a component of ubiquitinated inclusions observed mainly in amyotrophic lateral sclerosis and frontotemporal lobar degeneration has been demonstrated in cases of MSA and, more recently, was shown to colocalize with α‐synuclein pathology in GCIs in 2 patients.
Methods
A 66‐year‐old woman presented with a syndrome characterized by spasticity, dysautonomia, bulbar dysfunction, and parkinsonism. Symptoms progressed until her death at age 74. Neuropathological evaluation was performed at the New York Brain Bank at Columbia University.
Results
On gross examination, there was striking severe volume loss of the left striatum compared to mild involvement of the right striatum. Microscopically, neuronal loss and gliosis of the putamen and globus pallidus were severe on the left side, in contrast to mild involvement on the right side. Immunohistochemistry for α‐synuclein revealed widespread GCIs. The sections subjected to TDP‐43 antibodies showed a few GCIs with definite nucleocytoplasmic translocation of the labeling within the lenticular nucleus and within the paracentral cortex.
Conclusions
This report adds to the evidence that TDP‐43 and α‐synuclein colocalize in GCIs. Whether this coexistence contributes to the pathogenesis of a subset of MSA patients or is an age‐related process is not known. More cases with these peculiar pathological hallmarks might help determine whether TDP‐43 contributes to neurodegeneration in a subset of patients with MSA.
Keywords: multiple system atrophy, parkinsonism, TDP‐43, α‐synuclein
MSA is a sporadic neurodegenerative disease characterized by various combinations of progressive autonomic failure, cerebellar ataxia, parkinsonism, and pyramidal features, with generally poor response to levodopa.1 The pathological hallmark in MSA is oligodendrocytic glial cytoplasmic inclusions (GCIs) containing α‐synuclein, in addition to neuronal loss and astrogliosis involving mainly the striatonigral and olivopontocerebellar systems.2 Indeed, GCIs are found throughout the brain with a predilection for the basal ganglia and other motor areas, including both the cerebral cortex and corticospinal tract. In addition, the corticocerebellar systems and supraspinal autonomic systems are also affected in this disease.3 Density of GCIs correlates to the extent of neuronal loss and symptom duration4; however, at the ultimate stage of the degenerative process, the most vulnerable areas display features of status spongiosus with which the density of GCIs gradually decreases.
TAR DNA‐binding protein of 43 kDa (TDP‐43) has been found to be a disease‐defining pathological feature in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD), accumulating in neurons and glia.5 It has also been reported in other neurodegenerative diseases, including Alzheimer's disease (AD) and Lewy body dementia.6, 7 It is not clear whether TDP‐43 has pathological significance in these diseases or is a secondary phenomenon attributable to the aging process, given that it can also be observed in cognitively normal individuals.8 Rarely, TDP‐43 pathology has been reported in cases of MSA and, more recently, was shown to colocalize with α‐synuclein pathology in GCIs in 2 patients, suggesting a possible direct interrelation between the two pathologies.9, 10 We report on a third patient with MSA with colocalization of TDP‐43 and α‐synuclein in GCIs.
Case Report
A 66‐year‐old white woman of Jewish ancestry developed episodes of severe vertigo, followed by gait imbalance, hypophonia, micrographia, urinary urgency/incontinence, and mental fogginess. She had a personal history of mild asthma and depression and a strong family history of cancer, but no history of Parkinson's disease or other parkinsonisms. On initial evaluation, she was found to have subtle hypomimia, truncal rigidity, and brisk reflexes, but otherwise had an intact neurological exam.
Initial serum and cerebrospinal fluid laboratory investigation included basic labs and infectious, autoimmune, and paraneoplastic workup, which were unremarkable. Motor nerve evaluation with transcranial magnetic stimulation showed markedly prolonged central conduction times to both legs, worse on the left. MRI brain showed a mild degree of diffuse atrophy with minimal secondary ventricular dilatation. MRI thoracic and cervical spine showed degenerative changes without abnormal signal in the cord. MR spectroscopy did not show abnormal lactate peak. Electromyography/nerve conduction study was normal with no evidence of focal/large fiber neuropathy, plexopathy, radiculopathy, or myopathy. The patient was given a diagnosis of primary lateral sclerosis (PLS) by age 68 after initial evaluation and workup.
Her symptoms eventually worsened over the course of the following 3 years, to include poor hand dexterity, bradykinesia, and leg spasticity and weakness causing wheelchair dependence. She continued to exhibit worsening signs of upper motor neuron (UMN) dysfunction. On evaluation by a movement disorders specialist, she was found to have significant parkinsonism, worse in the legs and on the left. DaTscan revealed symmetrically reduced dopaminergic uptake in the striatum with more decrease in the putamina compared to the caudate nuclei (Fig. 1). Fluorodeoxyglucose‐PET scan showed reduced uptake in the medial and periopercular portions of the frontal lobe, inferobasal temporal cortex, and basal ganglia, more pronounced on the left side. Additionally, radiotracer uptake was bilaterally reduced in the primary motor region. This pattern suggested an atypical parkinsonian syndrome, and the involvement of the primary motor cortices was concerning for tauopathy‐related PLS.
Figure 1.
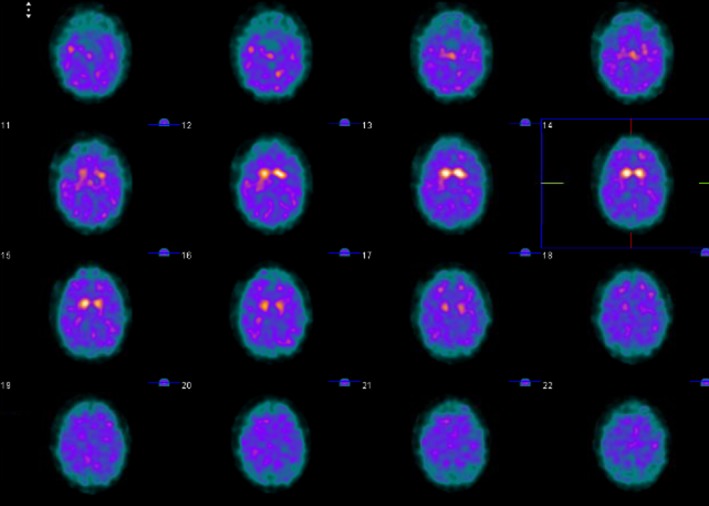
DaTscan shows symmetrically reduced dopaminergic uptake in the striatum with more decrease in the putamina compared to the caudate nuclei.
Her gait initially improved on carbidopa/l‐dopa 300 mg daily; however, she lost this mild benefit after 2 months of treatment. Testing for C9orf72 repeat expansion was negative, and whole‐exome sequencing was unrevealing for any pathogenical mutations. Over the next 2 years, bulbar dysfunction, dysautonomia (constipation, urinary retention, and dizziness), severe neuropathic pain, and anxiety/depression gradually occurred. Through the course of her illness, she did not exhibit signs of cognitive decline. Montreal Cognitive Assessment at ages 69 and 72 were 25/30 and 26/30, respectively. Toward the end of her life, she suffered from opioid hyperalgesia syndrome and was transitioned to hospice care. She passed away at age 74, 8 years after symptom onset.
Pathology
The fresh brain (1,206.5 g) was divided by a sagittal cut through the corpus callosum. The left half (561.1 g) was processed fresh and banked for research; however, in view of the extreme atrophy of the striatum, two blocks (one including the striatum, the other the thalamus with STN) were harvested and immersed in formalin for microscopic evaluation and for comparison with the contralateral regions. The right half of the brain was immersed in formalin (615.0 g) for thorough neuropathological evaluation.
On gross examination of the coronal sections, the outstanding finding was in the neostriatum, which showed severe atrophy on the left compared to the mild atrophy on the right side (Fig. 2). The cut surfaces of the left neostriatum were velvety and tan discolored, especially involving the lateral edge of the head of the caudate nucleus and dorsal lateral edge of the adjacent putamen. This discoloration prevailed in the coronal section passing through the amygdala. The caudal level of the putamen was severely atrophic and the globus pallidus was markedly smaller than normal. There was mild atrophy in the frontal and parietal cortices and white matter. The claustrum and hypothalamus were mildly atrophic. The hippocampal formation and amygdala were normal. There was preservation of the remaining structures.
Figure 2.
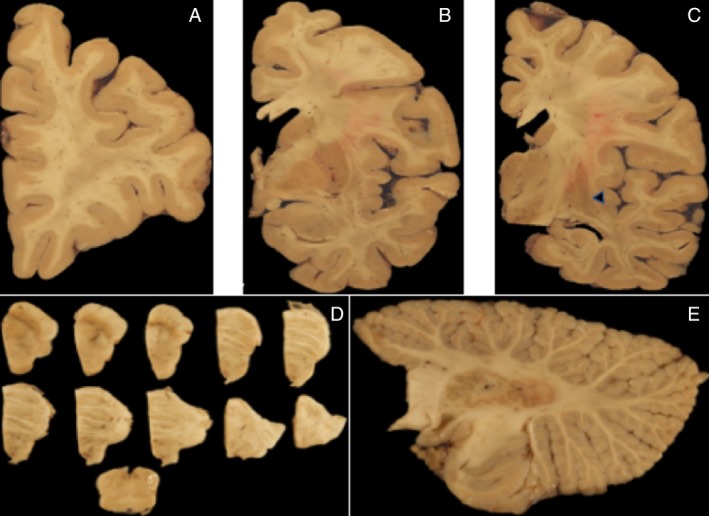
(A–C) Coronal sections of the cerebrum depicting discoloration of the caudal portion of the ventral putamen (black head arrow). (D) Transversal sections of the brainstem showing the SNpc with moderate pallor. (E) Sagittal cerebellar slice passing through the dentate nucleus with minimal changes in the album cerebri.
Microscopically, the brunt of the changes was in the left lenticular nucleus (Fig. 3A,B). There was severe neuronal loss and gliosis of the putamen and globus pallidus (Fig. 3C,D). In contrast, on the right side, these changes were mild. The lenticular sections subjected to α‐synuclein antibodies (A‐Syn, 1:40; Leica Microsystems, Wetzlar, Germany) revealed widespread GCIs. Furthermore, GCI density in cortical areas showed a gradient, markedly increased within the paracentral and cingulate corticosubcortical regions and to a lesser extent in the prefrontal and occipital cortices (Fig. 4A,B). In the hippocampal formation, α‐synuclein–labeled GCIs were rare within the alveus, marked within the fimbria (which harbored the brunt of the synucleinopathic burden), and very rare within the CA1 or within the subiculum. The entorhinal cortex, parahippocampal, and occipitotemporal gyri were spared. α‐synuclein burden was also located in the amygdala, basal ganglia, hypothalamus (marked), thalamus, midbrain, pons, medulla, and white matter. The sections subjected to TDP‐43 antibodies (TDP43, 1:500; Proteintech Group Inc., Rosemont, IL) showed scattered GCIs with definite nucleocytoplasmic translocation of the labeling, especially within lenticular nucleus and within the paracentral cortex (Figs. 4C,D and 5), but not within the prefrontal cortex GCIs. Other sections subjected to TDP‐43 antibodies did not detect nucleocytoplasmic translocation of the labeling. Rare AT8‐labeled neurons and neuropil threads were noted within the rostral ventral neostriatum, including the nucleus accumbens. AT8‐labeled neurons were scattered within the temporal pole, entorhinal cortex, parahippocampal and occipitotemporal gyri, and to a lesser extent within the insular cortex and amygdala. There was no detectable neuronal loss in the corticospinal tract, and the density of Betz was normal.
Figure 3.
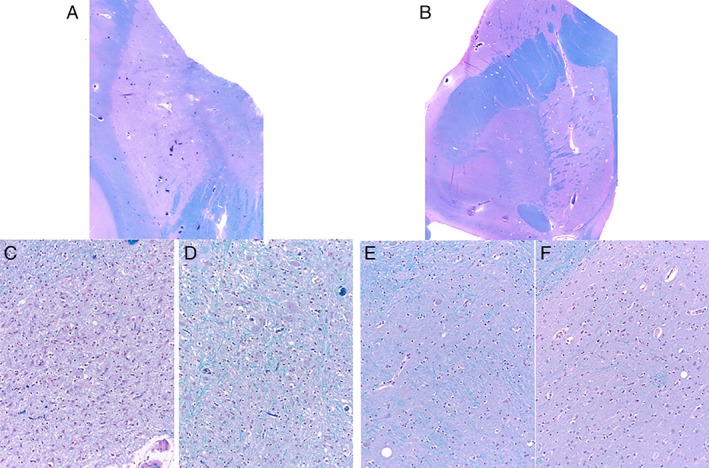
Severe atrophy of the left lenticular nucleus (A) compared to the right lenticular nucleus (B). Severe neuronal loss and reactive gliosis involving the left putamen (C), and left globus pallidus (D) compared to the relatively spared right globus pallidus (E) and to the right putamen (F). Luxol Fast Blue counterstained with hematoxylin and eosin. Original magnification: (A) and (B), ×1; (C), (D), (E), and (F), ×200.
Figure 4.
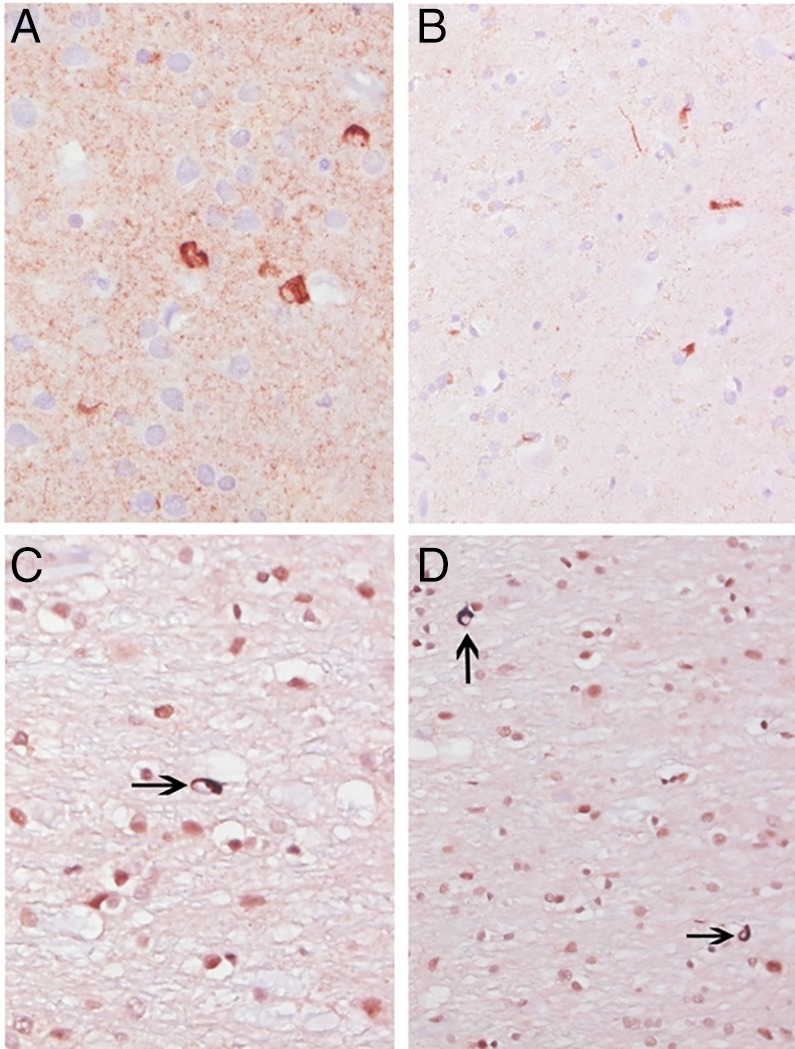
GCIs: (A) second layer of the primary motor cortex; (B), (C), and (D) within the adjacent white matter; (A) and (B) alpha‐synuclein; (C) and (D) TDP‐43. Original magnification: (A), (B), and (D): ×400; (C): ×630.
Figure 5.
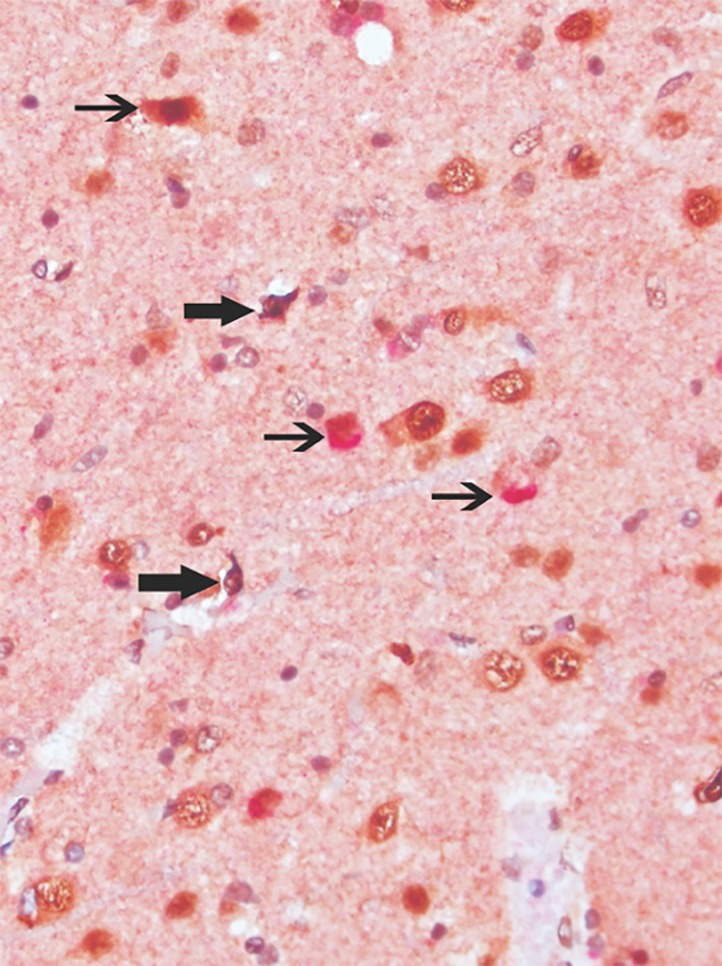
Aberrant nucleocytoplasmic labeling with TDP‐43 antibodies (dark brown chromogene; bold arrows) and colocation of glial cells with cytoplasmic labeling with antibodies directed against alpha‐synuclein (red chromogene; thin arrows). Sixth layer of primary motor cortex. Original magnification: ×400.
The AD pathological changes in the brain were minimal, and the likelihood of dementia attributable to AD changes were most likely nihil, based on the diagnostic categorization of AD recommended by the National Institute on Aging‐Alzheimer's Association.11
Discussion
This report identified widespread α‐synuclein GCIs with colocalization of TDP‐43 within both the paracentral cortex and lenticular nucleus in a patient with pathologically confirmed MSA‐P. The concomitant occurrence of TDP‐43 and α‐synuclein–labeled inclusions is rare in MSA. Geser et al. localized TDP‐43 in 4 of 29 cases with at least mild pathology, predominantly in subcortical brain areas such as the amygdala, midbrain, or medulla oblongata, and there was almost no neocortical TDP‐43 pathology detected.9 In these cases, TDP‐43 pathology comprised mainly dystrophic cellular processes and perivascular lesions, and there was no clear evidence of TDP‐43 immunoreactivity in GCIs.9 Koga et al. showed TDP‐43 immunoreactivity in 13 MSA cases, predominantly in the medial temporal lobes (specifically amygdala) and, rarely, in subcortical structures and brainstem.10 Morphologically, TDP‐43 most commonly aggregated as subpial astrocytic lesions and neuronal cytoplasmic inclusions.10 In 2 cases, Koga et al. were further able to show colocalization of TDP‐43 and α‐synuclein in GCIs, specifically in the thalamic fasciculus and mammillothalamic tract.10 Our case is the third reported case of concomitant TDP‐43 and α‐synuclein pathology coexisting in GCIs in MSA. It is yet to be determined whether this co‐occurrence is clinically and pathologically significant in the disease process.
Colocalization of TDP‐43 and α‐synuclein in GCIs in MSA suggests a possible interaction between the two pathologies. Interestingly, in our case, TDP‐43 was colocalized with α‐synuclein in the motor cortex, which possibly correlates to the patient's severe UMN dysfunction on clinical exam, despite the lack of clear neurodegeneration of the corticospinal tract on pathology. This lack of neurodegeneration also refutes the initial misdiagnosis of PLS, given that past autopsies of patients who were thought to have PLS or PLS‐spectrum disorders very commonly had degeneration of the corticospinal tract and loss of Betz cells.12 In addition, the widespread α‐synuclein noted in our patient makes PLS unlikely given that this is not a common pathological finding. It is also interesting that the colocalization in our patient was found in the paracentral cortex, but not in the amygdala or hippocampal area, which is in contrast to earlier suggestions that TDP‐43 pathology progresses from medial temporal limbic structures and higher‐order association cortices are affected later in the disease process.9, 13 So far, there is not a clear pattern between the 2 reported cases and our case. More cases of MSA with colocalization of the two pathologies are necessary to determine region‐specific patterns of accumulation and whether TDP‐43 truly has an impact on clinical manifestations.
Although widely accepted as the pathological hallmark observed in ALS and FTD,5 TDP‐43 pathology has been found in other distinct neurodegenerative diseases with dissimilar clinical manifestations, including AD, Lewy body dementia, hippocampal sclerosis, PSP, and corticobasal degeneration, among others.6, 7, 13, 14, 15 TDP‐43 has been found in only 7% to 13% of MSA cases, and the extent and severity in most cases were minimal.9, 10 It has also been shown to occur in up to 40% of elderly individuals with minimal senile‐related neuropathological changes, predominantly in the amygdala and hippocampal area.8, 16 This localization is similar to MSA cases described by Geser et al. and Koga et al., suggesting that TDP‐43 has a propensity to accumulate in the amygdala, regardless of disease process or lack thereof. This is in contrast to the 3 cases of MSA (including ours) where α‐synuclein and TDP‐43 colocalization occurred in the thalamic fasciculus, mammillothalamic tract, and paracentral cortex. TDP‐43–positive cases in normal individuals were more likely to be have higher age of death and unlikely to be younger than 65 years.8, 16 Koga et al. also found that age is an independent risk factor for TDP‐43 pathology in MSA.10 These findings suggest that TDP‐43 pathology in MSA is likely an age‐related finding and not associated with the disease process. Clinically, our patient did not have any significant cognitive impairment that would suggest an effect of TDP‐43 on the disease process. This makes it unlikely that their presence is truly clinically significant.
A limitation in this study is that data were obtained from a single case study, and hence broad conclusions about the role of TDP‐43 pathology in MSA cannot be made. However, this report adds to the evidence that TDP‐43 and α‐synuclein do occur in GCIs. Whether coexistence of the synucleinopathic burden and TDP‐43 findings contributes to the pathogenesis of a subset of MSA patients or is coincidental is not known. More cases with these peculiar pathological hallmarks might help determine whether or not there is a pathogenic interaction between these two proteins, and whether TDP‐43 contributes to neurodegeneration in a subset of patients with MSA.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: N/A; (3) Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
L.N.: 1A, 1B, 1C, 3A, 3B
D.T.: 1B, 3B
N.A.S.: 1A, 3B
S.F.: 1A, 3B
J.P.V.: 1A, 1B, 1C, 3B
E.C.: 1A, 1B, 1C, 3B
Disclosures
Ethical Compliance Statement
Institutional review board approval was not required for this work because this is a case report. Informed patient consent was not necessary for this work because there is no possibility for identification of the patient. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest
Columbia University Parkinson's Brain Bank is funded by the Parkinson's Foundation. The authors report no conflicts of interest.
Financial Disclosures for previous 12 months
The authors declare that there are no disclosures to report.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Wenning GK, Tison F, Ben Shlomo Y, Daniel SE, Quinn NP. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 1997;12:133–147. [DOI] [PubMed] [Google Scholar]
- 2. Ramirez EP, Vonsattel JP. Neuropathologic changes of multiple system atrophy and diffuse Lewy body disease. Semin Neurol 2014;34:210–216. [DOI] [PubMed] [Google Scholar]
- 3. Papp MI, Lantos PL. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain 1994;117:235–243. [DOI] [PubMed] [Google Scholar]
- 4. Ozawa T, Paviour D, Quinn NP, et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain 2004;127:2657–2671. [DOI] [PubMed] [Google Scholar]
- 5. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 6. Lin WL, Dickson DW. Ultrastructural localization of TDP‐43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol 2008;116:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geser F, Martinez‐Lage M, Kwong LK, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP‐43 diseases. J Neurol 2009;256:1205–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uchino A, Takao M, Hatsuta H, et al. Incidence and extent of TDP‐43 accumulation in aging human brain. Acta Neuropathol Commun 2015;3:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geser F, Malunda JA, Hurtig HI, et al. TDP‐43 pathology occurs infrequently in multiple system atrophy. Neuropathol Appl Neurobiol 2011;37:358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koga S, Lin WL, Walton RL, Ross OA, Dickson DW. TDP‐43 pathology in multiple system atrophy: colocalization of TDP‐43 and α‐synuclein in glial cytoplasmic inclusions. Neuropathol Appl Neurobiol 2018;44:707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathologica 2011;123:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singer MA, Statland JM, Wolfe GI, Barohn RJ. Primary lateral sclerosis. Muscle Nerve 2007;35:291–302. [DOI] [PubMed] [Google Scholar]
- 13. Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP‐43 pathology in aging and Alzheimer disease. Ann Neurol 2015;77:942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koga S, Sanchez‐Contreras M, Josephs KA, et al. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov Disord 2017;32:246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kouri N, Oshima K, Takahashi M, et al. Corticobasal degeneration with olivopontocerebellar atrophy and TDP‐43 pathology: an unusual clinicopathologic variant of CBD. Acta Neuropathol 2013;125:741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geser F, Robinson JL, Malunda JA, et al. Pathological 43‐kDa transactivation response DNA‐binding protein in older adults with and without severe mental illness. Arch Neurol 2010;67:1238–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]