

ABSTRACT
Background
Parkinson's disease (PD) is a common neurodegenerative disorder with both sporadic occurrence and Mendelian heredity, as it is true for autosomal recessive parkin‐related PD (PARK‐parkin). Parkin‐related PD is characterized by early onset, slow progression, frequent lower limb dystonia, and a robust response to levodopa. Clinicians are increasingly confronted with heterozygous PD patients mimicking dominant inheritance. Nevertheless, the exact clinical implications of heterozygosity are not fully understood.
Cases
We present an illustrative PARK‐parkin family with 2 affected sisters (compound heterozygous) and their father (heterozygous). One sister expresses the classical phenotype, whereas the other has isolated jerky tremor. The father has left‐sided action tremor of the hand with some dystonic posturing without clear bradykinesia and normal DaTSCAN.
Conclusion
This case series illustrates the phenotypic variability in parkin‐related PD with 1 classical phenotype and 1 patient with isolated jerky tremor. Unilateral hand tremor of the heterozygous father could mislead genetic testing by mimicking dominant inheritance.
Keywords: parkin, heterozygosity, jerky tremor, Parkinson, paroxysmal exercise‐induced dystonia
Parkinson's disease (PD) is the second‐most common neurodegenerative disorder with an estimated worldwide prevalence of 50 to 200 cases per 100,000 inhabitants and increasing incidence at higher ages. The vast majority of cases belongs to the sporadic form without evident cause, yet partly driven by genetic risk factors. Only about 5% to 10% of PD patients have a monogenic cause. Parkin‐related PD (PARK‐parkin) is the most frequent, accounting for 50% of all autosomal recessive and for 15% of sporadic PD cases with early onset (<40 years).1 Initially, a distinctive phenotype for parkin‐related PD has been proposed with slow disease progression, leg tremor, foot dystonia, brisk reflexes, normosmia, behavioral disturbances, and a robust response to levodopa and deep brain stimulation.1, 2 However, it has been shown that many of these features depend more on age of onset (early onset) than on the underlying genetic cause.3 Because of the vast phenotypic variability of early‐onset PD, accurate diagnosis and appropriate treatment are often grossly delayed.4 An intriguing aspect, which can be observed in some families with PARK‐parkin, is a “pseudodominant” inheritance. The role of heterozygosity in a putative recessive gene such as parkin is still a matter of ongoing discussion.5
With the aim to show and expand the spectrum of PARK‐parkin, we report a family with 3 daughters, 2 of them carrying compound‐heterozygous parkin mutations yet with very different clinical presentations, and their father who is the carrier of a heterozygous exon deletion in the same gene (Fig. 1).
Figure 1.
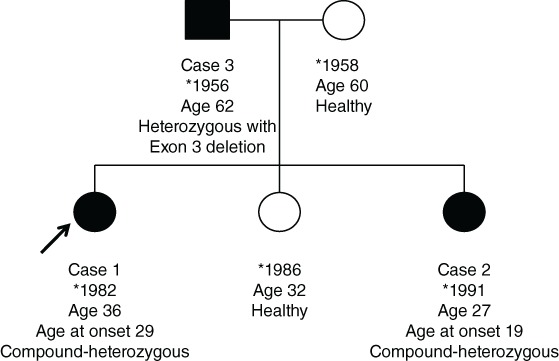
Family pedigree showing the siblings of the index patient and their parents with relevant biographical information.
Case Series
Case 1
This 32‐year‐old woman with a 3‐year history of tremor affecting both hands was referred to our center for evaluation of deep brain stimulation. The referring neurologist had made the diagnosis of essential tremor. On clinical examination, there was symmetrical jerky action tremor of both hands in the absence of bradykinesia or rigidity. Further detailed neurological examination was unremarkable. Medication included propranolol 120 mg three times a day as prescribed by the referring physician and oral contraception. Because of the jerkiness of the tremor, further investigations were performed. A diagnostic work‐up including brain magnetic resonance imaging and metabolic testing for Wilson's disease and Manganese storage disorders was unremarkable. Because jerky tremor can often be observed in dystonic syndromes, genetic testing for the most frequent dystonia genes (TOR1A, THAP1, CIZ1, GNAL, and ANO3) was performed, yet without pathological result. After having seen her younger sister (case 2) with subtle parkinsonism and paroxysmal exercise‐induced dystonia (PED), a single‐dose levodopa challenge test was performed with 200‐mg levodopa but without any improvement in symptoms (Unified Parkinson's Disease Rating Scale part III remained 6). By then the patient was pregnant and further testing, including single‐photon emission computed tomography imaging, had to be restricted to case 2. Following the respective results of case 2, further specific genetic testing for parkin gene mutations showed compound heterozygosity identical to case 2. Despite the initial negative results of the levodopa challenge test, a treatment trial with pramipexole 1.5 mg once a day (qd) was initiated and supplemented with levodopa 50 mg 4 times a day, which led to a complete reversal of her tremor. She gave birth to 2 healthy children. During the pregnancies, a monotherapy with levodopa 50 mg 4 times a day was maintained. Following 4 years of stable disease without remarkable side effects, levodopa had to be increased to 100 mg 4 times a day, and pramipexole was substituted for rasagiline 1mg qd.
Case 2
At initial presentation, the 23‐year‐old sister of case 1 reported a 4‐year history of progressive gait difficulties. The referring neurologist had already performed brain magnetic resonance imaging, which was normal. On clinical examination, subtle left‐sided tremor with bradykinesia and left‐sided PED with foot dystonia while walking were evident. The diagnostic work‐up included metabolic testing for Wilson's disease with 24‐hour urine sampling and blood tests for free copper and ceruloplasmin and manganese storage disease, which were unremarkable. A single‐dose levodopa challenge test with 200 mg levodopa left the Unified Parkinson's Disease Rating Scale part III unchanged (raise from 5 to 6 points). Following the proposed diagnostic algorithm for PED by Erro and colleagues,6 DaTSCAN® revealed a severe bilateral presynaptic dopaminergic deficit, which triggered genetic panel diagnostic testing for autosomal‐recessive PD (ATP13A2, DNAJC6, FBXO7, PARK2, PARK7, PINK1, PLA2G6, SYN1). A pathogenic compound‐heterozygous mutation in the parkin gene (point mutation c.714C>G; p.C238W, deletion in exon 3) was detected.
Treatment with levodopa (50 mg three times a day) was initiated and supplemented with pramiprexole (1.5 mg qd). As a result of an insufficient treatment response of PED, levodopa had to be increased up to levodopa 600 mg/d (200 mg three times a day) and pramipexole 3 mg qd. With this regime, symptom control was satisfying only for a short period of time and soon was complicated by wearing‐off phenomena with inner restlessness and impulse‐control disorder with nocturnal binge eating. Pramipexole was tapered down and switched to rasagiline 1 mg per day. Following botulinum neurotoxin type A injections into her left lower leg and foot muscles (Table 1), levodopa could be tapered down to 50 mg to 100 mg/d on demand while maintaining satisfying control of the PED. During the course of 3 years, dystonia spread to the other foot and changed from exercise induced to permanent.
Table 1.
Current botulinum neurotoxin treatment regime
| Location | Dose |
|---|---|
| M. tibialis posterior sinister | 75 IU |
| M. tibialis posterior dexter | 75 IU |
| M. flexor digitorum longus sinister | 30 IU |
| M. flexor digitorum longus dexter | 20 IU |
| M. flexor hallucis longus sinister | 40 IU |
| M. flexor hallucis longus dexter | 20 IU |
| M. flexor hallucis brevis sinister | 30 IU |
| M. flexor hallucis brevis dexter | 20 IU |
| M. flexor digitorum brevis sinister | 40 IU |
| M. flexor digitorum brevis dexter | 20 IU |
IU, international unit; M., Musculus.
Case 3
The 62‐year‐old father of the aforementioned sisters reported a slowly progressive tremor of the left arm without restrictions in daily activities, apart from occasional spilling since the age of 45. On clinical examination, there was action tremor of his left hand with some dystonic posturing but without clear‐cut evidence of parkinsonism and an otherwise normal neurologic examination. Focused diagnostic work‐up showed a carrier status of the known heterozygous deletion within exon 3 of the parkin gene. The DaTSCAN was semiquantitatively (putamen/occipital cortex ratio: left 2.73, right 2.78) and visually normal. No treatment was initiated.
The mother and her third daughter were found to be healthy and without clinical signs of tremor or parkinsonism. Genetic testing was not performed because testing was declined. Nevertheless, the mother must be a carrier of the aforementioned complementary point mutation because the same mutation was found in both daughters and not in the father.
Discussion
The accurate diagnosis of PARK‐parkin is often delayed because of the young age of onset and the variable phenotypic presentation.4 Therefore, we describe an illustrative family to show the broad phenotypic spectrum and diagnostic obstacles and clues of PARK‐parkin. The spectrum in this family ranges from isolated jerky action tremor of the hands described in case 1 to paroxysmal exercise‐induced foot dystonia in case 2 and mild action tremor of 1 hand in the heterozygous father.
Case 1 was referred with the diagnosis of essential tremor. Although this diagnosis is consistent with the diagnostic criteria for essential tremor, the jerkiness of her tremor gave rise to doubts about the diagnosis. The diagnostic spectrum of a jerky tremor can be summarized as follows: dystonic tremor often appears jerky or even with superimposed subcortical myoclonus as, for example, described for DYT‐ANO3 (anoctamin‐3).7 Myoclonus‐dystonia, because of mutations in the ε‐sarcoglycan gene, can also cause jerky tremor as a result of superimposed subcortical myoclonus, but in contrast to our case, it typically affects the cranio‐cervical region presenting with dystonia and lightening jerks. Another entity causing jerky (myoclonic) tremor is “familial cortical myoclonic tremor with epilepsy.” Tremor of this entity usually comes along with epilepsy and cortical myoclonus. Finally, but important, jerky tremor can be observed in atypical parkinsonian syndromes such as multiple system atrophy or cortico‐basal syndrome. A jerky tremor of the hand as the sole initial presentation of PARK‐parkin is important to recognize to prevent inappropriate or delayed therapy. In case 1, the clue to the accurate diagnosis was the phenotype of her sister (case 2) presenting with parkinsonism and PED of 1 foot.
Case 2, with a rather classical phenotype of PARK‐parkin, greatly fostered the diagnostic work‐up of case 1 and thus emphasizes the importance of a thorough family history and detailed clinical examination of the relatives. A peculiarity shared by both cases is their unresponsiveness to the single‐shot levodopa (200 mg) challenge test. In idiopathic PD, false‐negative results of the single‐shot levodopa challenge test are seen in up to 40%, whereas no data on the levodopa challenge test is available for PARK‐parkin.8
The clinical and single‐photon emission computed tomography findings of case 3 fuel the discussion on the significance of parkin heterozygosity. One hypothesis suggests that heterozygous carriers are at a higher risk of developing subtle parkinsonism, also showing nigrostriatal dysfunction on positron emission tomography studies.9 This could be explained by haplo‐insufficiency resulting in loss of function of the E3 ubiquitin‐protein ligase coded by the parkin gene.9 Data show that dosage mutations as present in case 3 might have a greater impact than point mutations.10 Clinically, case 3 does not fulfill the diagnostic criteria for parkinsonism because there is no clear‐cut decrement of frequency or amplitude on repetitive movements. There are subtle signs of dystonia, and the unilateral action tremor shown on the video best matches a dystonic tremor. Whether his dystonic tremor is caused by the parkin mutation or just coincidence has yet to be determined. Currently, the normal and symmetrical DaTSCAN does not answer this question. As a subset of patients with this type of tremor and initially normal DaTSCAN show an abnormal DaTSCAN on follow‐up, clinical and single‐photon emission computed tomography imaging follow‐up might answer this question in the future.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Qualitative Data Analysis: A. Execution, B. Review and Critique; (3) Instrument Design: A. Design and Structure, B. Content development, C. Review and Critique; (4) Manuscript Preparation: A. Writing of the first draft, B. Review and Critique
R.S.: 1A, 1B, 1C, 2A
J.W.: 1B, 1C
G.K.: 1A, 1B, 2B
Disclosures
Ethical Compliance Statement
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The authors confirm that neither an approval of an institutional review board nor a patient consent were required for this work.
Funding Sources and Conflict of Interest
The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for the Previous 12 Months
G.K. reports advisory boards of Bayer and Zambon and research grants from the Swiss Heart Foundation, Swiss Parkinson Association, and Swiss National Foundation. R.S. and J.W. declare that there are no additional disclosures to report.
Supporting information
Video S1. This video shows the pathological findings of (1) case 1 with symmetrical jerky action (postural) tremor of both hands in the absence of both resting tremor and bradykinesia on finger tapping. Normal gait with preserved arm swing on both sides. (2) Case 2 with abnormal dystonic posturing of the left hand while holding outstretched. There is mild postural tremor of the left hand. Normal gait with mildly reduced arm swing on the right side. After walking about 100 m, the left foot shows marked dystonia during walking, improved on walking backward (not shown).
Video S2. This video shows case 3 with left‐sided action tremor (task, posture) and dystonic posturing of the hand. There is no rest tremor. Repetitive movements of the left hand are slightly irregular and less fluent when compared with the right side but without clear cut signs of bradykinesia. Gait is normal with normal arm swing. Postural reflexes are preserved.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Lohmann E, Periquet M, Bonifati V, et al. How much phenotypic variation can be attributed to parkin genotype? Ann Neurol 2003;54:176–185. [DOI] [PubMed] [Google Scholar]
- 2. Khan NL, Graham E, Critchley P, et al. Parkin disease: a phenotypic study of a large case series. Brain 2003;126:1279–1292. [DOI] [PubMed] [Google Scholar]
- 3. Kim HJ, Kim HJ, Lee JY, et al. Phenotype analysis in patients with early onset Parkinson's disease with and without parkin mutations. J Neurol 2011;258:2260–2267. [DOI] [PubMed] [Google Scholar]
- 4. Ruiz‐Lopez M, Freitas ME, Oliveira LM, et al. Diagnostic delay in Parkinson's disease caused by PRKN mutations. Parkinsonism Relat Disord 2019;63:217–220. [DOI] [PubMed] [Google Scholar]
- 5. Klein C, Lohmann‐Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol 2007;6:652–662. [DOI] [PubMed] [Google Scholar]
- 6. Erro R, Stamelou M, Ganos C, et al. The clinical syndrome of paroxysmal exercise‐induced dystonia: diagnostic outcomes and an algorithm. Mov Disord Clin Pract 2014;1:57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stamelou M, Charlesworth G, Cordivari C, et al. The phenotypic spectrum of DYT24 due to ANO3 mutations. Mov Disord 2014;29:928–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schade S, Sixel‐Döring F, Ebentheuer J, Schulz X, Trenkwalder C, Mollenhauer B. Acute levodopa challenge test in patients with de novo Parkinson's disease: data from the DeNoPa Cohort. Mov Disord Clin Pract 2017;4:755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khan NL, Brooks DJ, Pavese N, et al. Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain 2002;125:2248–2256. [DOI] [PubMed] [Google Scholar]
- 10. Pankratz N, Kissell DK, Pauciulo MW, et al. Parkin dosage mutations have greater pathogenicity in familial PD than simple sequence mutations. Neurology 2009;73:279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. This video shows the pathological findings of (1) case 1 with symmetrical jerky action (postural) tremor of both hands in the absence of both resting tremor and bradykinesia on finger tapping. Normal gait with preserved arm swing on both sides. (2) Case 2 with abnormal dystonic posturing of the left hand while holding outstretched. There is mild postural tremor of the left hand. Normal gait with mildly reduced arm swing on the right side. After walking about 100 m, the left foot shows marked dystonia during walking, improved on walking backward (not shown).
Video S2. This video shows case 3 with left‐sided action tremor (task, posture) and dystonic posturing of the hand. There is no rest tremor. Repetitive movements of the left hand are slightly irregular and less fluent when compared with the right side but without clear cut signs of bradykinesia. Gait is normal with normal arm swing. Postural reflexes are preserved.