

ABSTRACT
Clinical History
A 57‐year old woman presented with left hand pain, periodic leg movement during sleep, gradual onset of stiffness, clumsiness, and falls. Neurological examination showed: generalized rigidity and bradykinesia. There was left hand dystonic posturing and ideomotor apraxia, as well as mirror movements of upper limbs and stimulus‐sensitive myoclonus. The patient had a high‐pitched voice and hypophonia (Video S1).
Discussion
Experts discuss localization and the syndromic diagnosis and predict the underlying pathology. The pathological diagnosis is then provided and clinical learning points are considered.
Keywords: corticobasal syndrome, multisystem atrophy, alpha‐synuclein, parkinsonism, myoclonus, cytoplasmic inclusions
Case Presentation
A 57‐year‐old woman presented to her general practitioner with tiredness and sensation of “something jumping in her back.” Within a month, she developed dull non‐neuropathic pain in the left arm unresponsive to Tramadol 50 mg twice a day orally. There was no history of trauma. Within a year, she developed left hand clumsiness when handling small objects. She had a background history of scleroderma, L5/S1 laminectomy, right corneal graft, and a partial bowel resection for rectal prolapse. There was no family history of neurological problems. She was a nonsmoker and consumed alcohol occasionally.
Before the video, the patient fell and fractured her left humerus, severely limiting left arm movement.
Examination revealed normal saccadic and pursuit eye movements, reduced left arm swing; left arm rigidity and dystonic posturing. Fingers of the left hand were extended at the interphalangeal joints and flexed at metacarpo‐phalangeal joints (Video S1). The left hand was cold with dusky discolouration without edema. There was a decreased amplitude and frequency of left heel and toe tapping. Vibration, joint position, and cortical sensation, including two‐point discrimination, were normal. Plantar responses were flexor. Abnormal cerebellar signs were not elicited. The “pull test” was negative. There were no other focal neurological signs present.
Over the next 2 years, the patient developed stiffness in all limbs and intermittent discoloration/coldness of the left hand. The patient had vivid dreams and occasionally cried out during sleep. Periodic leg movements occurred in sleep. Anxiety, forgetfulness, cognitive slowing, and emotional lability developed. At this point, the patient developed “a sweet tooth” and “had sweets impulsively.” Anosmia, stridor, and hallucinations were not present nor were there any autonomic symptoms, such as: postural hypotension and bowel or bladder dysfunction. She was gradually commenced on: levodopa 750 mg a day, pramipexole 1.5 mg three times daily, gabapentin 100 mg twice a day, and amantadine 100 mg daily without any improvement in her symptoms.
At age 58, she developed generalized rigidity, bradykinesia (left more than right), and left hand dystonic posturing with stimulus‐sensitive myoclonus (Video S1). Additionally, there was an ideomotor apraxia in the left hand with mirror movements of the hands. Hypophonia was accompanied by a high‐pitched voice (Video S1). There was no spasticity.
She scored 12/18 on the frontal assessment battery test (points lost: 1 on similarities, 2 on lexical fluency, and 3 on go‐no go task).
A clinical diagnosis of probable corticobasal syndrome (CBS) was made. Initially, idiopathic Parkinson's disease (PD), MSA, or Creutzfeldt‐Jakob disease (CJD; In view of corneal graft with a progressive neurological condition and myoclonus) were all considered. However, the duration of the disease and clinical and radiological features favored CBS. Death occurred at age 64 years, 7 years after the initial presentation.
Laboratory Investigations and Imaging Studies
Full blood count, serum electrolytes, renal function, liver function, coagulation studies, lipid profile, and glucose were normal. MRI brain showed subtle atrophy of the right putamen with mild global brain atrophy and multiple subcortical hyperintensities (Fig. 1). MRI of the cervical spine and left shoulder was normal (Fig. 1). Nerve conduction study of the left hand showed a mild median nerve neuropathy. Cerebrospinal fluid (CSF) analysis was normal and oligoclonal bands were not present. CSF beta A4, Tau, and alpha‐synuclein were not measured in the CSF. A 18FDG PET/CT scan of the brain (Fig. 2) showed moderate reduction of the tracer uptake in the right caudate, in the anterior and posterior right putamen, and decreased activity in the left cerebellum (Fig. 2). A sleep study showed increased periodic leg movements during rapid eye movement (REM) sleep, consistent with REM sleep behavior disorder (RBD).
Figure 1.
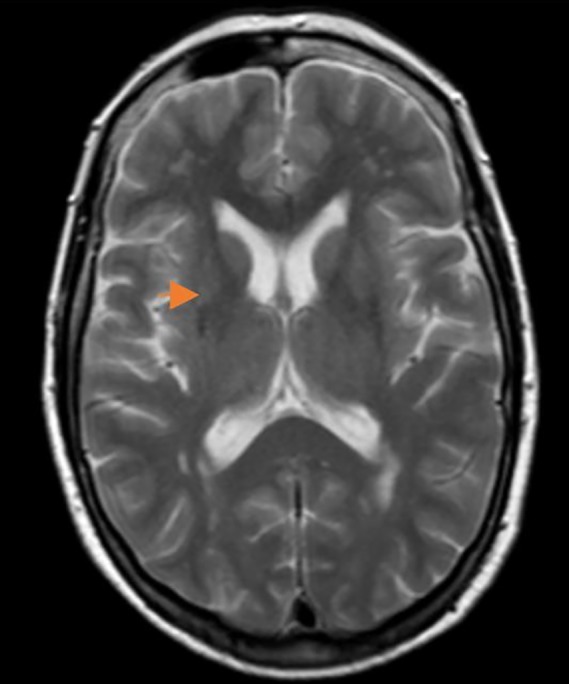
MRI brain axial T2 images show subtle atrophy of the right putamen (orange arrow) with mild global atrophy with multiple subcortical hyperintensities, no putaminal slit sign.
Figure 2.
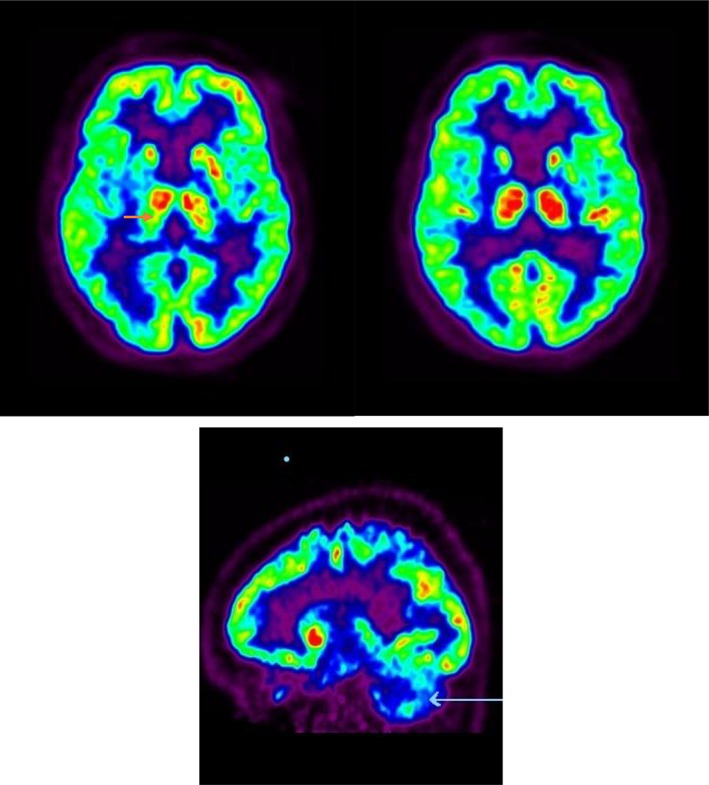
18FDG PET scan of the brain shows moderate reduction in right putaminal uptake (orange arrow) with decreased activity in the left cerebellum (white arrow). No structural abnormality of the basal ganglia. The “light green” images are a comparison with the normal database; red = greater than normal; blue is >3 SDs below normal. SDs, standard deviations.
Pathological Findings
The right cerebral hemisphere was frozen at –70oC and the left cerebral hemisphere immersion fixed in formalin. Examination of both cerebral hemispheres immediately following autopsy showed mild sulcal widening, particularly of the left parietal lobe. The subcortical white matter appeared normal. There was atrophy of the caudate nucleus, putamen, and globus pallidus. The mesial temporal structures, including amygdale and hippocampi, were normal. The midbrain was artifactually disrupted. The pons showed a normally pigmented locus coeruleus. The medulla together with cerebellar vermis, dentate nucleus, and cerebellar hemispheres were normal. Microscopic re‐examination of the left cerebral hemisphere showed that the cerebral cortex was normally laminated. "Neuritic plaques and neurofibrillary tangle pathology" were not observed. Abnormal cortical vessels were not noted. The subcortical white matter appeared normal. Arteriolosclerosis and hyalinosis of subcortical vessels were evident, but leukoariaosis was not apparent. Immunostaining for Tau, Beta A4, and alpha‐synuclein was negative in the cortex, but alpha‐synuclein–positive glial cytoplasmic inclusions were widespread in the subcortical white matter and were especially numerous in the corpus callosum. The caudate nucleus was normal. However, the putamen, globus pallidus, and thalamus all showed variable gliosis and neuronal loss. The putamen was particularly gliotic. Alpha‐synuclein–positive inclusions were prominent in oligodendroglia and in striatal neurones but were not observed within the hippocampus. The SN showed a decrease in pigmented neurones with increased pigmentary incontinence. The red nucleus was normal. Alpha‐synuclein–positive inclusions and threads were widespread and especially within the glia of the corticopontine fibers and within glia and neurones of the pontine gray matter. In summary, the features were those of gliosis most severely affecting the putamen and globus pallidus, but also involving the thalamus, SN, midbrain, and pontine white matter. Within these areas, a synucleinopathy was confirmed through the presence of immunopositive inclusions predominantly within oligodendroglia, but also within neurones. Moreover, oligodendroglial cytoplasmic inclusions were also noted within subcortical white matter extending into the corpus callosum.
Pathological Summary
Neuropathological examination showed (Fig. 3) gliosis of lentiform nucleus, thalamus, SN, midbrain, and, to a lesser extent, of pontine white matter. There was widespread glial cytoplasmic alpha‐synuclein accumulation in the corticopontine fibers, pontine gray matter and oligodendroglia. Alpha‐synuclein–positive glial cytoplasmic inclusions was also found to be present in the deeper layers of cerebral cortex, corpus callosum, and subcortical white matter. Phosphorylated‐Tau was not present.
Figure 3.
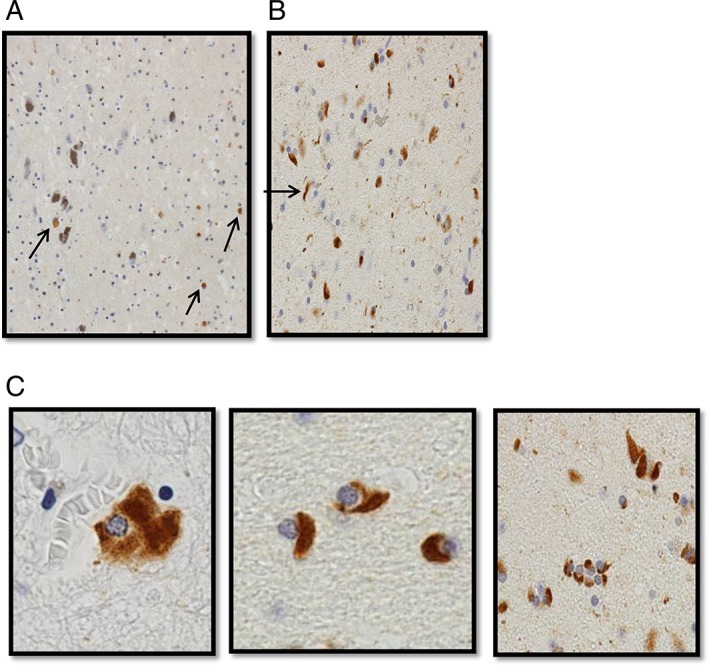
(A) Alpha‐synuclein immunohistochemistry of the SN showing neuronal cytoplasmic synuclein immunoreactivity. (B) Globus pallidus showing alpha‐synuclein–positive threads (arrowed). (C) Oligodendrocyte alpha‐synuclein–positive inclusions.
Pathological diagnosis was MSA.
Learning Points
MSA is a rapidly progressive neurodegenerative disorder involving the central, peripheral, and autonomic nervous systems.1 It usually presents with symmetrical parkinsonism (MSA‐P) or cerebellar ataxia (MSA‐C) with early autonomic failure (sexual and bladder dysfunction, postural hypotension, cold blue hands, constipation, and sweating) and relative sparing of cognition.2
In contrast, CBS is a clinical syndrome and presents with l‐dopa‐unresponsive parkinsonism, apraxia, dystonia, stimulus‐sensitive myoclonus, alien limb phenomenon, and impaired cognition. The clinical syndrome of CBS may be associated with a variety of pathologies, including those noted in corticobasal degeneration (CBD), PSP, stroke, Alzheimer's disease, frontotemporal lobar degeneration, dementia of Lewy body, CJD, C9orf72 hexanucleotide expansions, and argyrophilic grain disease. CBD is characterized pathologically by widespread deposition of abnormally hyperphosphorylated protein tau in the somatosensory, premotor, and supplementary motor cortices, as well as in the brainstem and basal ganglia. CBD and PSP may be difficult to distinguish pathologically, especially late in the disease process. Typically, CBD is distinguished microscopically from PSP by the presence of ballooned or achromatic neurons, astrocytic plaques, and Tau threads/coiled bodies and by the absence of tufted astrocytes and globose tangles.
Our patient was initially diagnosed with probable CBS, because of asymmetrical rigidity, bradykinesia, ideomotor apraxia, dystonia, stimulus‐sensitive myoclonus, cognitive decline (Frontal Assessment Battery 12/18), a poor response to l‐dopa, and the lack of significant autonomic dysfunction.3 However, our clinical diagnosis of CBS was not associated with the anticipated tau pathology, but instead a synucleinopathy characteristic of MSA was found at autopsy. In retrospect, we should have reconsidered the diagnosis of MSA if we had heeded the history of possible RBD and the patient's high‐pitched voice.
MSA remains the most commonly misdiagnosed form of parkinsonism often mimicking idiopathic PD. Litvan et al. reported MSA misdiagnosis of 50% of pathologically proven MSA patients among general neurologists, whereas movement disorders neurologists correctly diagnosed 70% of patients.4 Autopsy studies reveal that the antemortem diagnostic accuracy of MSA varies between 29% and 86%. One study showed that 2 of 51 pathologically proven MSA patients were misdiagnosed with CBS.5 Another study revealed that 50% (2 of the 4) pathologically proven MSA patients were clinically misdiagnosed with PSP, 2 as undetermined parkinsonism, and only 1 of the 3 clinically diagnosed CBD had postmortem changes consistent with CBD.6 The other 2 had idiopathic PD and frontotemporal dementia.6 On the other hand, of the 19 autopsy proven CBD patients from the Queen Square Brain Bank only 5 patients were diagnosed as CBS while alive and 8 were diagnosed as having PSP largely because of vertical gaze palsy. In summary, early and reliable diagnosis of atypical parkinsonism is a major clinical challenge especially when they lack their cardinal features.
There are several limitations in current International Parkinson and Movement Disorder Society (MDS) diagnostic criteria for MSA.1 The clinical criteria rely predominantly on autonomic dysfunction to diagnose MSA. However, MSA can progress very slowly and the autonomic dysfunction may only occur late with a latency of up to 11 years after initial symptoms.7 Disease duration from onset to death in MSA is usually 6 to 9 years. Later development of dysautonomia in “atypical presentations of atypical parkinsonian conditions,” as in our patient, makes the diagnosis very difficult. Moreover, the MDS criteria emphasize early‐onset ataxia as a strong predictor of probable MSA‐C, but PSP variants rarely present with predominant cerebellar ataxia.5 Furthermore, cognitive decline is observed early in some MSA patients, although the MDS diagnostic criteria suggest that dementia is an exclusion criterion for MSA.1 However, our patient with pathologically confirmed MSA scored 12/18 in a frontal assessment battery indicating cognitive dysfunction. Future diagnostic criteria will need to address “atypical presentations of atypical parkinsonian conditions” to improve diagnostic accuracy of probable or possible MSA.
MSA is usually not mistaken for CBS, but it can present with features that mimic CBS in keeping with atypical presentations of atypical parkinsonian conditions. In CBS, the myoclonus is usually stimulus sensitive, and asymmetrical and superimposed dystonic posturing is often noted. MSA can also present with spontaneous distal myoclonus, rather than the stimulus‐sensitive myoclonus of CBS. Asymmetry can occur in MSA, but ideomotor apraxia, as observed in our patient, is rare. The other subtle MSA “red flags” in our patient, which in retrospect favored synucleinopathy, were the patient's high‐pitched voice, RBD, and cold discoloration of the peripheries. RBD occurs in 81.9% of patients with MSA, but only in 1% to 2% of patients with CBD.8, 9 Cold hand sign is probably a feature of autonomic dysfunction in MSA, but it is not uncommon to see a cold blue hand in patients with idiopathic PD. In these clinically puzzling cases, MRI findings of olivopontocerebellar atrophy or a hot cross bun or putaminal rim sign may aid the diagnosis. However, in our experience, these changes often occur late in the disease course, and the putaminal rim sign can be a normal finding with 3T MRI, thus undermining its diagnostic value in MSA.10 Thirty‐eight percent of MSA patients can have normal MRI scans early in the disease.5 PET and single‐photon emission computed tomography imaging can help to distinguish PD from atypical parkinsonism, although their role in distinguishing among the subtypes of atypical parkinsonism remains unclear.
The clinical presentation of our patient was unusual and consistent with the recently described atypical presentations of atypical parkinsonism.11 Batla et al. reported on 5 patients with asymmetric presentation of MSA mimicking CBS with pathology in 1 patient.12 Moreover, Aoki et al. described 4 MSA patients who had a CBS type presentation (similar to our patient).13 MSA presents with variable phenotypes and often mimics PD, PD dementia (PDD), diffuse Lewy body disease (DLBD), or PSP in the early stages. Ideomotor and ideational apraxia can occur in MSA, but it is never a presenting sign, and it is exceptional for MSA to be associated with all the major features of CBS.
Conclusion
In hindsight, the patient's high‐pitched hypophonic voice (Video S1) and history suggestive of RBD should have led us to an earlier diagnosis of MSA. Finally, the autopsy findings in this patient re‐emphasize the critical role of brain banking in neurodegeneration to confirm or refute the clinical diagnosis and can provide vital tissue for research.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
V.P.: 3A
D.A.O.: 3A
S.B.: 3A
A.C.: 3B
M.O.’C.: 3B
M.F.: 1C, 3B
T.L.: 3A, 3B
Disclosures
Ethical Compliance Statement
The authors confirm that the approval of an institutional review board was not required for this work. Informed consent was obtained for this article. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest
Funding source for the study was received from Health Research Board Ireland, Michael J Fox Foundation. The authors report no conflicts of interest.
Financial Disclosures for previous 12 months
SB received a travel grant from the Merck Serono limited, UK to attend dystonia update meeting in Londont. LT was on the scientific advisory Board for An2H up to 2018.
Supporting information
Appendix S1: Supporting information
Video S1. Dystonic posturing, ideomotor apraxia, and stimulus‐sensitive myoclonus of the left hand with high‐pitched, hypophonic voice.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jellinger KA, Wenning GK. Multiple system atrophy: pathogenic mechanisms and biomarkers. J Neural Transm (Vienna) 2016;123:555–572. [DOI] [PubMed] [Google Scholar]
- 3. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Litvan I, Goetz CG, Jankovic J, et al. What is the accuracy of the clinical diagnosis of multiple system atrophy? A clinicopathologic study. Arch Neurol 1997;54:937–944. [DOI] [PubMed] [Google Scholar]
- 5. Koga S, Aoki N, Uitti RJ, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology 2015;85:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hughes AJ, Daniel SE, Ben‐Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 2002;125(Pt 4):861–870. [DOI] [PubMed] [Google Scholar]
- 7. Ling H, Asi YT, Petrovic IN, et al. Minimal change multiple system atrophy: an aggressive variant? Mov Disord 2015;30:960–967. [DOI] [PubMed] [Google Scholar]
- 8. Munhoz RP, Teive HA. REM sleep behaviour disorder: how useful is it for the differential diagnosis of parkinsonism? Clin Neurol Neurosurg 2014;127:71–74. [DOI] [PubMed] [Google Scholar]
- 9. Cooper AD, Josephs KA. Photophobia, visual hallucinations, and REM sleep behavior disorder in progressive supranuclear palsy and corticobasal degeneration: a prospective study. Parkinsonism Relat Disord 2009;15:59–61. [DOI] [PubMed] [Google Scholar]
- 10. Lee WH, Lee CC, Shyu WC, Chong PN, Lin SZ. Hyperintense putaminal rim sign is not a hallmark of multiple system atrophy at 3T. AJNR Am J Neuroradiol 2005;26:2238–2242. [PMC free article] [PubMed] [Google Scholar]
- 11. Stamelou M, Quinn NP, Bhatia KP. “Atypical” atypical parkinsonism: new genetic conditions presenting with features of progressive supranuclear palsy, corticobasal degeneration, or multiple system atrophy—a diagnostic guide. Mov Disord 2013;28:1184–1199. [DOI] [PubMed] [Google Scholar]
- 12. Batla A, Stamelou M, Mensikova K, et al. Markedly asymmetric presentation in multiple system atrophy. Parkinsonism Relat Disord 2013;19:901–905. [DOI] [PubMed] [Google Scholar]
- 13. Aoki N, Boyer PJ, Lund C, et al. Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with alpha‐synuclein. Acta Neuropathol 2015;130:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information
Video S1. Dystonic posturing, ideomotor apraxia, and stimulus‐sensitive myoclonus of the left hand with high‐pitched, hypophonic voice.