

Abstract
The direct, site-selective alkylation of unactivated C(sp3)–H bonds in organic substrates is a long-standing goal in synthetic chemistry. General approaches to the activation of strong C–H bonds include radical-mediated processes involving highly reactive intermediates, such as heteroatom-centered radicals. Herein, we describe a catalytic, intermolecular C–H alkylation that circumvents such reactive species via a new elementary step for C–H cleavage involving multisite-proton-coupled electron transfer (multisite-PCET). Mechanistic studies indicate that the reaction is catalyzed by a non-covalent complex formed between an iridium(III) photocatalyst and a monobasic phosphate base. The C–H alkylation proceeds efficiently using diverse hydrocarbons and complex molecules as the limiting reagent and represents a new approach to the catalytic functionalization of unactivated C(sp3)–H bonds.
Graphical Abstract

INTRODUCTION
Intermolecular functionalizations of unactivated C–H bonds hold significant promise in streamlining the synthesis and late-stage derivatization of complex molecules. In recent years, substantial progress has been made in the development of catalytic methods for C–H bond functionalization involving hydrogen atom transfer (HAT) processes, which involve the transfer of both a proton and an electron from a substrate C–H σ-bond to an abstracting atom X•, forming a new X–H σ-bond.1 While HAT processes have a long history in synthetic chemistry2, it has been shown recently that many transformations thought to occur through HAT may actually proceed through a proton-coupled electron transfer (PCET) mechanism, wherein the proton and electron transfer to different orbitals in the product.3 A notable example is C–H bond homolysis mediated by cytochrome P450, in which the proton from the C–H bond travels to the basic Fe(IV) oxo group, while the electron is transferred to an orthogonal ligand-based orbital with considerable porphyrin radical cation character.4
Multisite-PCET mechanisms are also possible, wherein the proton and electron travel to two distinct molecular acceptors.4 While multisite-PCET has been utilized in a variety of synthetically useful transformations,5 this mechanism typically requires pre-equilibrium hydrogen-bonding between the substrate and the proton acceptor prior to reaction with the oxidant (Figure 1A). Accordingly, multisite-PCET processes are generally selective for polar protic functional groups that can participate in H-bonding (e.g., amides and alcohols) while the poor H-bond donor character of aliphatic C–H bonds renders them unreactive in these mechanisms.6
Figure 1.

Mechanisms for homolytic activation of C–H bonds. A) Selectivity differences of HAT reagents in HAT mechanisms versus base/oxidant pairs in multi-site-PCET mechanisms. B) Experimental proof of principle for multisite-PCET of C–H bonds – intramolecular C–H abstraction in fluorenyl benzoate. C) Mechanism for intermolecular C–H bond multisite-PCET via a non-covalent iridium photocatalyst and phosphate base complex.
However, recent work has begun to challenge these assumptions, and demonstrate the feasibility of multisite-PCET activation of aliphatic C–H bonds. For example, theoretical studies of the oxidation of n-butane to maleic anhydride on a vanadium-phosphorus-oxide (VPO) surface have indicated that covalently linked oxidant/base constructs are capable of direct activation of C–H bonds via multisite-PCET.7 A well-characterized experimental example was recently reported by Mayer and coworkers describing the reaction of a covalently linked fluorenyl-benzoate substrate/base construct with exogenous oxidants (Figure 1B).8 Together these insights present an intriguing framework for developing novel catalysts for synthetic applications of multisite-PCET activation of aliphatic C–H bonds by decreasing the molecularity of the abstraction. A possible way to achieve a decrease in molecularity is through non-covalent association of the oxidant, base, or substrate, which would represent a unique strategy for intermolecular C–H cleavage. Herein, we report such a system involving a non-covalent H-bonded complex of an excited-state Ir(III)-polypyridyl center and a monobasic phosphate which catalyzes an efficient C–H alkylation via the intermolecular, homolytic activation of aliphatic C–H bonds via multisite-PCET (Figure 1C).
RESULTS AND DISCUSSION
These studies were prompted by an unexpected observation in the course of developing an intermolecular C–H alkylation involving multisite-PCET. Our initial approach sought to access tuned heteroatom-centered radicals from suitable N–H precursors via the multisite-PCET catalytic strategy outlined in Figure 1A, followed by subsequent intermolecular HAT involving hydrocarbon substrates. Our efforts began with the C–H alkylation of tetrahydrofuran with 1,1-bis(phenylsulfonyl)ethylene as the alkene partner. This alkene was selected owing to its high synthetic versatility, allowing for facile elaboration to a diverse range of products and simple oxidative or reductive removal of the phenylsulfonyl groups.9 Preliminary screening identified N-tBu-p-methoxybenzene-sulfonamide as a putative sulfonamidyl radical precursor, delivering the C–H alkylation product in 75% yield (Table 1, entry 1). Surprisingly, we discovered that removing the sulfonamide had little effect on the reaction outcome, and further optimization increased the reaction yield to 92% in the absence of sulfonamide and with the substrate as the limiting reagent (Table 1, entries 2 & 3). Additional control reactions indicated that all of the other reaction components were crucial to reactivity. These results clearly indicated that an alternative pathway (i.e., not involving nitrogen-centered radicals) was involved in the C–H activation step, and that a simple system comprised of the Ir(III) catalyst and phosphate base efficiently catalyzed the C–H alkylation.10
Table 1.
Development of conditions for C–H alkylation.
 | ||
|---|---|---|
| entry | variation of conditions above | yield (%)a |
| 1 | 1.0 equiv of sulfonamide, 5 mol% Ir(III) catalyst | 75 |
| 2 | no sulfonamide, 5 mol% Ir(III) catalyst | 66 |
| 3 | no sulfonamide | 92 |
Determined by 1H NMR spectroscopy of the crude reaction mixture using an internal standard.
Following this surprising observation, we first evaluated the scope of the Ir(III)-phosphate catalyzed C–H alkylation, with the hydrocarbon substrate used as the limiting reagent in all cases (Figure 2). Alkylations of simple cyclic hydrocarbons (6-7) and adamantyl derivatives (8-10) proceeded in moderate to good yields (45-80%). The functionalizations of substrates 11-15 highlight the ability to alkylate at benzylic sites in good yield. The alkylation of substrate 15 containing both methyl and methine C–H sites occurred exclusively at the methine site, demonstrating selectivity for the weaker C–H bond; this selectivity is consistent with either a HAT or PCET mechanism.11 The allylic C–H of cyclohexene was also functionalized in moderate yield (16). The alkylations of the α-heteroatomic C–H sites of several common heterocycles (17-19) were also successful, and the reaction of 2,3-dihydrobenzofuran 20 selectively delivered the benzylic C–H alkylation product in good yield (67%). While the 1,1-bis(phenylsulfonyl)ethylene coupling partner is highly versatile, we have also examined the C–H alkylation of tetrahydrofuran (1 equiv) using alternative alkenes (Figure 2). The use of methyl-2-phenylacrylate, methyl vinyl ketone, and phenyl vinyl ketone were all competent reaction partners, delivering the respective C–H alkylation products in good yield (47-60%).
Figure 2.

Catalytic intermolecular C–H alkylation using an iridium-phosphate photoredox system. Yields are of isolated product. aYields are determined by 1H NMR spectroscopy of crude reaction mixtures using an internal standard.
We next sought to determine the efficiency of the catalytic C–H alkylation using increasingly complex substrates (Figure 3). The alkylation of the N-phthalimide protected derivative of memantine proceeded solely at the tertiary C–H site to provide product 21 in 40% yield. The functionalization of the terpenoid (–)-ambroxide delivered alkylation product 22 in excellent yield (88%). Ibuprofen methyl ester reacted exclusively at the more electron-rich benzylic C–H site to provide 23 in 83% yield. Notably, prior C–H functionalization of this substrate via HAT of an oxygen-centered radical generated through phosphate oxidation proceeded with poor regioselectivity, suggesting that different mechanisms might be operative.12 The observed selectivity is consistent with selectivity for the weakest C–H bond, with the benzylic C–H bond BDFE ∼85 kcal/mol and the tertiary C–H bond BDFE ∼93 kcal/mol.13 Alkylation of a bioactive pyrimidine substrate provided 24 with excellent yield (84%) and regioselectivity (>20:1).
Figure 3.

Catalytic C–H alkylation of complex substrates. Yields are of isolated product.
Our initial mechanistic hypothesis was that the C–H abstraction step proceeded via HAT involving an oxygen-centered radical derived from single-electron oxidation of the phosphate base by the excited state of the Ir photocatalyst. Oxygen-centered radicals of this type were recently demonstrated to be competent H-atom abstractors in work from Alexanian and Nicewicz, and such a pathway was also proposed in a C–H cyanation by Kanai and coworkers.12,14 Yet upon further examination, thermochemical concerns caused us to doubt this proposal. Specifically, electron transfer from the dibutyl phosphate base (Ep/2(P−/P•) > 2 V vs. SCE) to the excited state of the Ir(III) catalyst 1 (Ep/2(IrIII*/IrII) = 1.68 V vs. SCE) is significantly endergonic and unlikely to be kinetically accessible within the lifetime of the photocatalyst 3MLCT excited state (240 ns). Moreover, C–H alkylations using the much less oxidizing photocatalysts [Ir(dF(CF3)ppy)2(bpy)]PF6 and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, are still viable (though less efficient); in these cases the proposed electron transfer event is endergonic by more than 600 mV and 800 mV respectively (vide infra).
We have performed a series of mechanistic experiments to uncover the details of the C–H functionalization described above. These studies provide a body of evidence that is consistent with a novel reaction mechanism for the functionalization of unactivated C–H bonds involving an unprecedented, intermolecular multisite-PCET between the C–H bond of the organic substrate and a non-covalent complex between the Ir excited state and phosphate base. The complexation of the Ir catalyst and phosphate base serves to decrease the molecularity of this elementary step, facilitating a concerted transfer event. The details of these studies are described below.
We began by studying the association of the phosphate base and iridium photocatalyst. Titration of the base into a solution containing the Ir(III) catalyst leads to downfield shifts in the 1H NMR spectrum of all protons of the bipyridine ligand, with an especially pronounced change associated with the 3,3’-protons (∼1.5 ppm) (Figure 4). This association was also observed in the solid state; the crystal structure of the Ir(III) catalyst and diphenyl phosphate base confirmed the affinity of the oxygen atom of the phosphate for the 3,3’-position of the bipyridyl ligand (Figure 4). Using both 1H NMR and UV/Vis titrations, the equilibrium binding constant of the ion pairing of the Ir(III) catalyst and dibutyl phosphate base was calculated to be (7.8 ± 0.6) x 103 M−1. A Job plot was indicative of the formation of a one-to-one ion paired complex. We would note that phosphate association is conserved across a range of structurally similar cationic Ir(III) photocatalysts that vary in their substituents on the 4 and 5 positions of the bipyridine ligand (Figure 5A).
Figure 4.

1H NMR spectra of Ir photocatalyst upon phosphate base titration, exhibiting downfield shifting of the resonance signal corresponding to C3–H of the bipyridine ligand; X-Ray crystal structure of the iridium-phosphate complex rendered using CYL-View20; determination of binding stoichiometry.
Figure 5.

A) Reaction yields with different iridium catalysts and their binding Keq with phosphate. B) Iridium-fluoride complex and the effect of adding TBAF on the reaction rate, demonstrating that the addition of a competitive binder has an inverse first order concentration dependence.
In order to demonstrate the essential coordination of phosphate to the photocatalyst for C–H alkylation, we prepared photocatalysts with the H-bonding 3,3’-positions on the bipyridyl ligand substituted with either fluorine atoms or methyl esters (Figure 5A). Reactions involving these substituted photocatalysts did not yield any appreciable amount of C–H alkylation product. In order to discount the possibility that the substitution of the bipyridine altered the excited-state redox properties of the complex in a deleterious manner, we performed a previously reported PCET alkene hydrosulfonamidation using these catalysts.15 Product was observed in good yield, an outcome inconsistent with poor photocatalyst activity.
Competition binding studies with fluoride anion also supported a crucial role for the Ir-phosphate complex (Figure 5B). Specifically, we found that fluoride anion (from a 1 M solution of tetrabutylammonium fluoride (TBAF) in THF) associates at the same position on the bipyridyl ligand as the phosphate base with a similar equilibrium binding constant. A Job plot generated from the 1H-NMR shift in the Ir(III) bipyridyl 3,3’-protons versus fluoride concentration also revealed a one-to-one binding stoichiometry. Most importantly, in the alkylation of THF with α-phenyl acrylate, the addition of increasing concentrations of TBAF resulted in decreasing product yield, showing an inverse first-order concentration dependence. For comparison, addition of TBAF to a standard hydrosulfonamidation did not show any decrease in reaction efficiency. This is consistent with the phosphate base-Ir photocatalyst association being necessary for C–H bond activation to occur, but not required for N–H PCET. Hydrogen bonding functionality (e.g., amides) can competitively bind with the dibutyl phosphate and can serve as substrates for multisite-PCET. Consistent with this view, the addition of exogenous amide to the reaction results in a decreased rate.
Significant nonlinear photoluminescence quenching of the Ir photocatalyst by the phosphate base is observed, which is also consistent with the formation of a less emissive ion pair complex (i.e., iridium-phosphate).16 We next studied the Ir(III) excited-state quenching mechanism by phosphate base using transient absorption spectroscopy–in the absence of substrate–to determine if electron transfer was occurring via the complex. Pulsed-light excitation of a dichloromethane solution containing the Ir(III) catalyst [0.18 mM] and approximately 20 equivalents of phosphate [3.7 mM] produced no Ir(II) signal indicative of electron transfer (purple overlapped signal, Figure 6, right).17 When this quenching was investigated under a much larger excess of phosphate (∼300 equivalents), only a minute signal for Ir(II) was observed (ΔC = 0.25 μM). This indicates that electron transfer between the excited state Ir(III) catalyst and phosphate is unlikely to underlie the observed reactivity and the decrease in photoluminescence is likely a result of the formation of a less emissive ion-paired complex, leading to both steady-state quenching and a much shorter excited-state lifetime.
Figure 6.
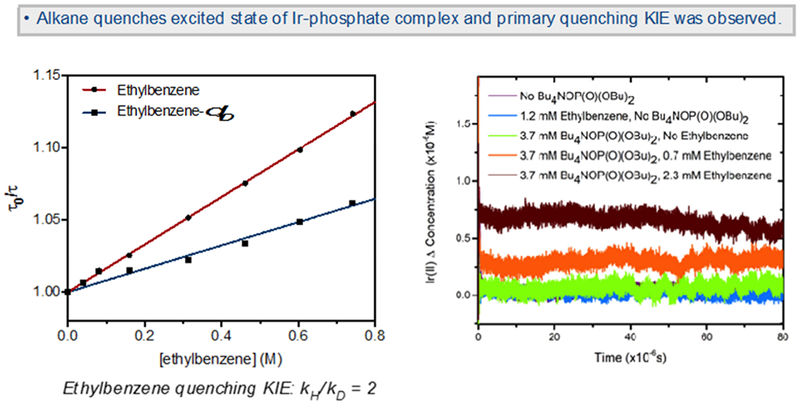
Stern-Volmer plot of time-resolved excited-state quenching of a solution of Ir photocatalyst and phosphate base by ethylbenzene and ethylbenzene-d10, showing a quenching KIE of 2.0 ± 0.2; transient absorption signal at 540 nm (maximum of Ir(II) absorbance-see SI for complete absorption spectrum of Ir(II)), demonstrating an increase in the concentration of Ir(II) as ethylbenzene is added to a solution of the Ir photocatalyst and phosphate base.
We continued by introducing the hydrocarbon substrate to the solutions of Ir(III) catalyst and phosphate base to determine whether electron transfer required the presence of substrate. Consistent with this scenario, as ethylbenzene was added to the Ir-phosphate solution, additional quenching of the Ir(III) excited state was observed by both time-resolved photoluminescence and transient absorption spectroscopy. Remarkably, when ethylbenzene was added to the solution of the Ir(III) photocatalyst containing 20 equiv of phosphate base, a signal for monoreduced Ir(II) was observed by transient absorption spectroscopy, indicating that electron exchange was involved in the quenching event. Additionally, as increasing amounts of ethylbenzene were added, the concentration of Ir(II) in the solution increased proportionally (orange and maroon signals, Figure 6, right). In the absence of phosphate, the addition of ethylbenzene to the Ir(III) catalyst did not result in any Ir(II) formation (green signal, Figure 6, right). This data suggests that both alkane and phosphate base are involved in the electron transfer step. The abstracted Stern-Volmer plot for the ethylbenzene quenching was linear with a quenching rate constant, kq, of 2.3 × 106 M−1s−1(Figure 6, left).18
Finally, to specifically determine whether quenching of the excited-state occurred via C–H bond cleavage, we studied the reaction kinetic isotope effect (KIE) using ethylbenzene-d10. The excited-state quenching KIE (kH/kD) using ethylbenzene was 2.0 ± 0.2, consistent with C–H bond cleavage during the electron transfer event (Figure 6, left). Furthermore, this observation is directly inconsistent with a phosphate oxidation mechanism, in which no quenching KIE would be expected. Additionally, this excited-state quenching KIE was equal to the initial rate KIE observed for the preparative C–H alkylation reaction; the reaction of ethylbenzene with bis(phenylsulfonyl)ethylene proceeded with an initial rate KIE of 1.9 ± 0.3. This also aligns with the KIE measured in competition experiments using either α-phenyl acrylate or phenyl vinyl ketone. The KIE of the C–H alkylation of THF/THF-d8 using α-phenyl acrylate as the olefin acceptor also resulted in a KIE of 2.0 ± 0.1, which did not vary in the presence of added TBAF, suggesting a constant mechanism of cleavage despite a decrease in the overall rate due to competitive fluoride binding.19
We posit that the mechanistic studies outlined above are consistent with a new elementary step for catalytic, intermolecular C–H bond cleavage via multisite-PCET between an excited-state Ir-phosphate complex and hydrocarbon substrate.
The reaction mechanism commences with the ground state association of the Ir(III) photoredox catalyst and phosphate base (Figure 7). Upon excitation, a concerted PCET occurs involving the C–H bond of the substrate and the Ir(III)*-phosphate complex, generating a carbon-centered radical, an Ir(II) species, and the protonated phosphate base. This Ir(III)-phosphate complex has an effective BDFE of 103 kcal/mol, indicating that the abstraction of the substrate C–H bonds is thermodynamically favorable. This high BDFE represents an exceptionally strong abstracting agent, notably stronger than cytochrome P450s (BDFEs ∼95 kcal/mol).21 The carbon-centered radical next adds to the electron-poor alkene to form the C–C bond, and electron transfer from Ir(II) [Ep/2(IrII/IrIII) = –0.69 V vs. SCE] then regenerates the Ir(III) photocatalyst. Protonation of the resulting anion by the conjugate acid of the phosphate then closes the catalytic cycle, delivering the C–H alkylation product and phosphate base.
Figure 7.

Proposed reaction mechanism for the catalytic C–H alkylation involving an intermolecular multisite-PCET via an iridium-phosphate complex.
CONCLUSIONS
The catalytic intermolecular alkylation described herein proceeds under mild conditions, and constitutes a unique, general approach to C–H alkylation. The transformation efficiently functionalizes a diverse set of small molecules, which are used as limiting reagent in all cases. The unique mechanism of C–H cleavage involved obviates the requirement of highly reactive heteroatom-centered radicals which are typically required in related functionalizations involving photoredox catalysis.11,14,22 Extensive mechanistic studies are consistent with intermolecular multisite-PCET activation of a C–H bond via a non-covalent iridium-phosphate complex–the first example of such a multisite-PCET activation in the absence of covalent attachment of reacting functionality. We anticipate that this fundamental step for intermolecular C–H functionalization will unlock a range of new, catalytic approaches to achieve the site-selective functionalization of hydrocarbons.
Supplementary Material
ACKNOWLEDGMENT
Steady-State and time-resolved absorption and photoluminescence experiments were performed using instrumentation in the Alliance for Molecular PhotoElectrode Design for Solar Fuels (AMPED) EFRC, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences BES, under Award DE-SC0001011. We thank the UNC Department of Chemistry Mass Spectrometry Core Laboratory for assistance with MS analysis. We thank Anthony Metrano and Phillip Jeffrey for obtaining the crystal structure of the the Ir-phosphate complex. This work was supported by Award R01 GM 120163 (EJA) and Award R01 GM 113105 (RRK) from the National Institute of General Medical Sciences.
Footnotes
Supporting Information
Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Bergman RG C–H Activation. Nature 446, 391–393 (2007). [DOI] [PubMed] [Google Scholar]; (b) Chen MS, White MC A predictably selective C–H oxidation reaction for a complex molecule synthesis. Science 318, 783–787 (2007). [DOI] [PubMed] [Google Scholar]
- (2).Hynes JT; Klinman JP; Limbach HH; Schowen RL Hydrogen-Transfer Reactions (Wiley-VCH, 2007). [Google Scholar]
- (3).Darcy JW; Koronkiewicz B; Parada GA; Mayer JM A continuum of proton-coupled electron transfer reactivity. Acc. Chem. Res 51, 2391–2399 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV)hydroxide pKa and the role of thiolate ligation in C–H bond activation by cytochrome P450. Science 343, 825–829 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature 539, 268–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yayla HG, Wang H, Tarantino KT, Orbe HS, Knowles RR Catalytic ring-opening of cyclic alcohols enabled by PCET activation of strong O−H bonds. J. Am. Chem. Soc 138, 10794–10797 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Warren JJ; Mayer JM Moving protons and electrons in biomimetic systems. Biochem. 54, 1863–1878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cheng M; Goddard III WA The critical role of phosphate in vanadium phosphate oxide for the catalytic activation of functionalization of n-butane to maleic anhydride. J. Am. Chem. Soc 135, 4600–4603 (2013). [DOI] [PubMed] [Google Scholar]
- (8).Markle TF; Darcy JW; Mayer JM A new strategy to efficiently cleave C–H bonds using proton-coupled electron transfer. Sci. Adv 4, 5776–5782 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kamijo S; Takao G; Kamijo K; Tsuno T; Ishiguro K; Murafi T Alkylation of nonacidic C(sp3)–H bonds by photoinduced catalytic Michael-type radical addition. Org. Lett 18, 4912–4915 (2016). [DOI] [PubMed] [Google Scholar]
- (10).During these studies, Kanai reported a C–H cyanation catalyzed by an iridium (III) photocatalyst and a BINOL phosphate base, which is proposed to occur via a different mechanism involving phosphate oxidation and standard HAT by a reactive oxygen-centered radical (14).
- (11).Baciocchi E; D’Acunzo F; Galli C; Lanzalunga O Tertiary: secondary: primary C–H bond relative reactivity in the one-electron oxidation of alkylbenzenes. A tool to distinguish electron transfer from hydrogen atom transfer mechanisms. J. Chem. Soc., Perkin Trans. 2., 133–140 (1996). [Google Scholar]
- (12).Margrey KA; Czaplyski WL; Nicewicz DA; Alexanian EJ A general strategy for aliphatic C–H functionalization enabled by organic photoredox catalysis. J. Am. Chem. Soc 140, 4213–4217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).McMillen DF; Golden DM Hydrogen bond dissociation energies. Ann. Rev. Phys. Chem 33, 493–532 (1982). [Google Scholar]
- (14).Wakaki T; Sakai K; Enomoto T; Kondo M; Masaoka S; Oisaki K; Kanai M C(sp3)—H cyanation promoted by visible-light photoredox/phosphate hybrid catalysis. Chem. Eur. J 24, 8051–8055 (2018). [DOI] [PubMed] [Google Scholar]
- (15).Zhu Q; Graff DE; Knowles RR Intermolecular anti-Markovnikov hydroamination of unactivated alkenes with sulfonamides enabled by proton-coupled electron transfer. J. Am. Chem. Soc 140, 741–747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ward WM; Farnum BH; Siegler M; Meyer GJ Chloride ion-pairing with Ru(II) polypyridyl compounds in dichloromethane. J. Phys. Chem. A 117, 8883–8894 (2013). [DOI] [PubMed] [Google Scholar]
- (17).While the lack of observable photoproducts with 20 equiv. of phosphate and the iridium catalyst is in line with the uphill driving force for electron transfer, the possibility of fast back-electron transfer between the two neutral species, i.e. Ir(II) and (RO)2PO2• cannot be strictly ruled out.
- (18).The small degree of excited state quenching observed in the steady-state experiments is attributed to the low quantum yield of the reaction (0.0025).
- (19).The competition KIE measured from the C–H alkylation using bis(phenylsulfonyl)ethylene was found to be somewhat higher (4.0), so we cannot rule out the presence of an alternate mechanism with this particular alkene partner. One such possibility could be a radical chain mechanism involving the formation of an unusually strong C–H bond in the alkylation product in this case (C–H BDFE ∼99 kcal/mol). We therefore investigated this possibility using diverse radical initiators; in each case, no product was obtained. This indicates that if a chain mechanism proceeds in this case, the initiation step is likely the same–through the proposed multisite-PCET pathway. The low quantum yield of the reaction (ethylbenzene and sulfone trap, 0.0025) is also inconsistent with a chain mechanism.
- (20).CYLview, 1.0b; Legault CY, Université de Sherbrooke, 2009. (http://www.cylview.org)
- (21).Mittra K; Green MT Reduction potentials of PF450 compounds I and II: Insight into the thermodynamics of C–H bond activation. J. Am. Chem. Soc 141, 5504–5510 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Le C; Liang Y; Evans RW; Li X; MacMillan DWC Selective sp3 C–H alkylation via polarity-match based cross-coupling. Nature 547, 79–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ahneman DT; Doyle AG C–H functionalization of amines with aryl halides by nickel-photoredox catalysis. Chem. Sci 7, 7002–7006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.