

Abstract
Diffusion basis spectrum imaging (DBSI) combines discrete anisotropic diffusion tensors and the spectrum of isotropic diffusion tensors to model the underlying multiple sclerosis (MS) pathologies. We used clinical MS subtypes as a surrogate of underlying pathologies to assess DBSI as a biomarker of pathology in 55 individuals with MS. Restricted isotropic fraction (reflecting cellularity) and fiber fraction (representing apparent axonal density) were the most important DBSI metrics to classify MS using brain white matter lesions. These DBSI metrics outperformed lesion volume. When analyzing the normal‐appearing corpus callosum, the most significant DBSI metrics were fiber fraction, radial diffusivity (reflecting myelination), and nonrestricted isotropic fraction (representing edema). This study provides preliminary evidence supporting the ability of DBSI as a potential noninvasive biomarker of MS neuropathology.
Introduction
Multiple sclerosis (MS) is a disease with profound heterogeneity clinically, radiologically, and pathologically. Current clinical subtypes of MS do not fully capture its pathological heterogeneity. Identifying pathologically meaningful subtypes of MS are crucial in tailoring immunotherapies and moving toward a personalized medicine approach in the care of people with MS (PwMS).1
Diffusion basis spectrum imaging (DBSI) models diffusion‐weighted MRI signals as a combination of discrete anisotropic diffusion tensors (reflecting the axon and myelin integrity of fibers), and a spectrum of isotropic diffusion tensors (detecting inflammation and tissue loss surrounding axons).2 The accuracy of DBSI in capturing and quantifying white matter (WM) pathologies has been shown in tissue phantoms, spinal cord, and optic nerves of mice with experimental allergic encephalomyelitis, the corpus callosum (CC) of mice with cuprizone‐induced demyelination, autopsied human MS spinal cord, and in biopsied human brain tissue.3, 4, 5, 6, 7
Here, we assessed the ability of DBSI to provide pathologically meaningful differentiation of MS subtypes in living PwMS.
Methods
This cross‐sectional study included 55 PwMS (according to 2010 Revised McDonald Criteria)8 from John L. Trotter MS Center at Washington University in St. Louis. MS clinical subtypes were confirmed by two MS neurologists independently and included relapsing‐remitting MS (RRMS), secondary‐progressive MS (SPMS), and primary‐progressive MS (PPMS). We used the consensus definition of SPMS which indicates that “in most clinical contexts, SPMS is diagnosed retrospectively by a history of gradual worsening after an initial relapsing disease course, with or without acute exacerbations during the progressive course”. 9 Patients underwent detailed clinical assessment, and structural and diffusion imaging. Diffusion data were collected using a 3 Tesla TIM Trio (Siemens) scanner with a 32‐channel head coil at 2 × 2 × 2 mm3 resolution in the axial plane with repetition time/echo time = 10,000/120 msec, and employing a 99‐direction diffusion‐weighting scheme (maximum b‐value = 1500 sec/mm2).
Voxel‐wise DBSI metrics were calculated for two sets of regions of interest (ROIs) in each PwMS, one consisting of all brain WM lesions, and the other ROI was the normal‐appearing CC (excluding lesions in the CC). Normal‐appearing WM often harbors pathology in MS, and the CC was chosen for study due to its large size and ease of identification, axonal coherence, and predilection to be affected in MS. WM lesion ROIs for the whole brain were created on fluid‐attenuated inversion recovery (FLAIR) images by automatic segmentation using the Lesion Prediction Algorithm implemented in Lesion Segmentation Tool of Statistical Parametric Mapping (SPM) toolbox in MATLAB.10 To create the ROIs for normal‐appearing CC, we first traced CC on high‐resolution magnetization‐prepared rapid acquisition with gradient echo (MP‐RAGE) images using semiautomatic thresholding in Amira 6.0.1. These CC traces were eroded by one voxel to reduce partial volume effects. SPM‐detected lesions within CC were excluded to obtain normal‐appearing CC ROIs.
For analysis, we applied recursive partitioning,11 a nonparametric decision tree‐based regression and classification method, to use DBSI metrics to classify PwMS. Recursive partitioning involves constructing a decision tree by dividing a dataset into subsets according to descriptors, or rules that discriminate between different subsets while trying to maximize the homogeneity within subsequent subsets. In other words, as the splits occur, the nodes become more homogeneous. The median values for the following DBSI‐derived metrics were included in the recursive partitioning:
-
Anisotropic components:
Radial diffusivity (representing myelin integrity of residual axons)
Axial diffusivity (representing residual axon integrity)
Fiber fraction (representing apparent axonal density)
-
Isotropic components:
Restricted fraction (reflecting cellularity)
Nonrestricted fraction (reflecting extra‐axonal environment associated with tissue loss, inflammation, and cerebrospinal fluid partial volume).
We also included T2‐weighted lesion volume (standardized using median absolute deviation) in the recursive partitioning of WM lesion ROIs.
The Institutional Review Board at Washington University approved the study.
Results
Of the 55 subjects in the study (22 RRMS, 16 SPMS, and 17 PPMS), females comprised a larger proportion among all subtypes as expected (Table 1). RRMS patients were on average approximately 14 and 11 years younger than SPMS and PPMS patients, respectively. Despite a shorter disease duration, PPMS subjects had similar median expanded disability status scale (EDSS) score (6) to those with SPMS.
Table 1.
Demographic and clinical characteristics of 55 patients with MS included in the study.
| Characteristics |
RRMS (n = 22) |
SPMS (n = 16) |
PPMS (n = 17) |
|---|---|---|---|
| Sex, n (%) | |||
| Female | 15 (68) | 13 (81) | 12 (71) |
| Male | 7 (32) | 3 (19) | 5 (29) |
| Age (years), mean ± SD | 43.0 ± 10.7 | 56.7 ± 7.5 | 54.1 ± 8.2 |
| Disease duration from symptoms onset (years), mean ± SD | 10.4 ± 8.4 | 27.2 ± 11.8 | 12.4 ± 5.9 |
| Expanded disability status scale score, median (range) | 3 (1.5–6) | 6 (2.5–6.5) | 6 (3–7.5) |
| T2 lesion volume (ml), median (interquartile range) | 9.5 (4.4–17.9) | 20.2 (6.1–32.7) | 8.0 (5.2–14.2) |
| DBSI‐derived metrics in brain white matter lesions | |||
| Fiber fraction, median (interquartile range) | 0.40 (0.38–0.42) | 0.39 (0.37–0.47) | 0.37 (0.32–0.41) |
| Isotropic restricted fraction, median (interquartile range) | 0.07 (0.07–0.08) | 0.06 (0.04–0.07) | 0.07 (0.05–0.08) |
| Isotropic nonrestricted fraction, median (interquartile range) | 0.43 (0.40–0.47) | 0.46 (0.39–0.50) | 0.48 (0.43–0.53) |
| Radial diffusivity (μm2/msec), median (interquartile range) | 0.70 (0.65–0.74) | 0.68 (0.64–0.71) | 0.70 (0.66–0.74) |
| Axial diffusivity (μm2/msec), median (interquartile range) | 1.84 (1.74–1.97) | 1.92 (1.83–2.02) | 1.99 (1.79–2.11) |
| DBSI‐derived metrics in normal‐appearing corpus callosum | |||
| Fiber fraction, median (interquartile range) | 0.61 (0.58–0.66) | 0.60 (0.54–0.68) | 0.61 (0.58–0.66) |
| Isotropic restricted fraction, median (interquartile range) | 0.19 (0.18–0.21) | 0.16 (0.12–0.19) | 0.20 (0.17–0.23) |
| Isotropic nonrestricted fraction, median (interquartile range) | 0.04 (0.00–0.10) | 0.04 (0.02–0.15) | 0.05 (0.03–0.07) |
| Radial diffusivity (μm2/msec), median (interquartile range) | 0.31 (0.20–0.35) | 0.30 (0.20–0.34) | 0.33 (0.29–0.38) |
| Axial diffusivity (μm2/msec), median (interquartile range) | 2.25 (2.12–2.36) | 2.13 (2.02–2.24) | 2.11 (2.06–2.25) |
RRMS, relapsing‐remitting multiple sclerosis; SPMS, secondary‐progressive multiple sclerosis; PPMS, primary‐progressive multiple sclerosis; DBSI, diffusion basis spectrum imaging.
Figure 1A shows the recursive partitioning tree using DBSI metrics for brain WM lesions. The most significant splits were based on DBSI‐derived restricted isotropic fraction and fiber fraction. RRMS subtype was predicted by relatively higher values for both restricted isotropic fraction and fiber fraction. Four final classes (terminal nodes) were identified, with two predicted to contain individuals with PPMS, one predicted to contain RRMS, and one predicted as SPMS. Of the nine subjects predicted to have SPMS, seven also had SPMS as a clinical designation. Figure 1B shows the confusion matrix of the classifier using DBSI metrics of WM lesions versus predefined clinical subtypes. Based on DBSI metrics of WM lesions, 35 PwMS (64%) were predicted to have the same subtype as their predefined clinical subtypes. Notably, WM lesion volume, as well as other DBSI‐derived metrics, did not improve the classification.
Figure 1.
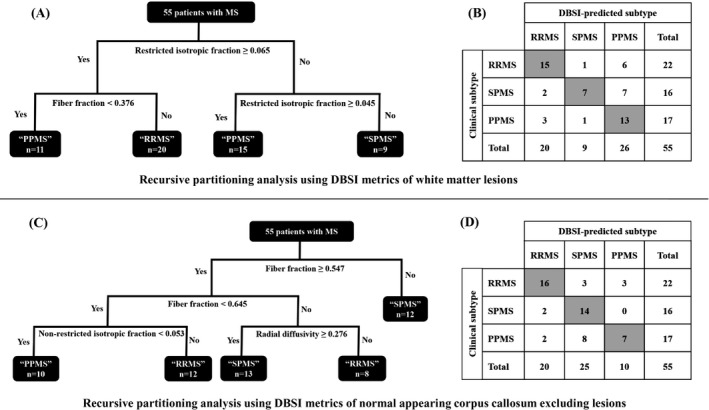
Results of recursive partitioning analysis for 55 people with multiple sclerosis using diffusion basis spectrum imaging (DBSI) metrics of white matter (WM) lesions (A), and normal‐appearing corpus callosum excluding lesions (C), and the corresponding confusion matrices ((B) and (D)). The predictors included in recursive partitioning analysis were DBSI‐derived anisotropic components (radial diffusivity, axial diffusivity, and fiber fraction), isotropic components (restricted fraction, and nonrestricted fraction), and lesion volume (only when analyzing WM lesions). The most significant splits in recursive partitioning of WM lesions were based on restricted fraction and fiber fraction (A). This suggests that cellularity and apparent axon density were the most important characteristics (based on DBSI metrics) of WM lesions for predicting clinical subtypes. Using DBSI metrics of WM lesions, 35 individuals (64%) were predicted to have the same disease subtype as their predefined clinical subtype as shown in the diagonal of the matrix (B). When analyzing normal‐appearing corpus callosum, the most important DBSI‐derived characteristics were fiber fraction followed by nonrestricted isotropic fraction and radial diffusivity (C), with 37 individuals (67%) predicted to have the same disease subtype as their predefined clinical subtypes (D).
Recursive partitioning using DBSI metrics of normal‐appearing CC showed that the most significant (first and second) splits were based on DBSI‐derived fiber fraction (Fig. 1C). At the top split, lower fiber fraction (consistent with lower axon density) predicted a subgroup of PwMS as having SPMS. Lower but important splits were based on nonrestricted fraction and radial diffusivity. Of note, DBSI‐derived radial diffusivity helped predict SPMS versus RRMS. Those with higher radial diffusivity were predicted as having SPMS versus RRMS (consistent with more severe demyelination in SPMS). Five final nodes were identified including two classes predicted as having RRMS, two as having SPMS, and one as having PPMS. DBSI of normal‐appearing CC predicted 37 PwMS (67%) to have the same subtype as their predefined clinical subtypes (Fig. 1D).
Discussion
Prior histopathological studies of relapsing and progressive MS central nervous system (CNS) tissues have described pathological heterogeneity.12, 13 However, CNS tissue cannot be accessed easily and safely in living PwMS. A major goal of this study was to use the relative pathological differences among MS clinical subtypes, based on published studies of neuropathology, to investigate the ability of DBSI as a noninvasive imaging method to differentiate components of MS pathology, and to determine which DBSI metrics were most important in this regard.
We previously showed in preclinical models, and autopsied and biopsied human tissue that DBSI restricted isotropic fraction correlated significantly and positively with cellularity, and fiber fraction had a significant positive correlation with axonal density.2, 3, 5, 7 The current study suggests that DBSI may be useful in living individuals with MS. The clinical subtypes of 64% and 67% of PwMS in our study were predicted by recursive partitioning of DBSI‐derived metrics of WM lesions and of normal‐appearing CC, respectively. It is noteworthy that our recursive partitioning was solely based on DBSI metrics and did not include any demographic or clinical metrics such as age, gender, disease duration, and EDSS, all of which may contribute to differentiating MS subtypes clinically.
We found that restricted isotropic fraction and fiber fraction of WM lesions were the most important DBSI metrics when using brain WM lesions to predict MS clinical subtypes. It is noteworthy that DBSI metrics performed better than T2‐weighted lesion volume at discerning clinical subtypes in our study, although prior studies have often reported higher T2 lesion loads to be associated with SPMS versus RRMS. Out of 22 subjects designated as RRMS clinically, 15 were predicted to have RRMS by DBSI metrics, 6 were predicted to have PPMS and only one was predicted to have SPMS. These results suggest that RRMS WM lesions in our subjects were more similar to PPMS than SPMS in terms of axon density and cellularity.
When analyzing DBSI metrics of normal‐appearing CC, the most significant factor to predict MS subtype was fiber fraction, with radial diffusivity and nonrestricted isotropic fraction also of importance. Based on the known pathology of SPMS, it was not unexpected that those predicted to be SPMS had the lowest fiber fractions (representing apparent axonal density) within normal‐appearing CC.14 Compared to RRMS, SPMS was also predicted by greater apparent demyelination based on higher DBSI‐derived radial diffusivity, also not unexpected.15 A higher nonrestricted isotropic fraction in normal‐appearing CC predicted RRMS over PPMS. This is possibly due to more edema in the RRMS group, about 15% of whom had gadolinium‐enhancing brain lesions (albeit not all in the CC). In contrast, only 7% of PPMS and none of the SPMS subjects given gadolinium displayed an enhancing brain lesion.
Our study has some limitations. First, we base our pathological interpretations of these DBSI findings on animal and MS autopsy studies without much pathological confirmation in living humans with MS. However, we recently showed that findings from DBSI metrics were consistent with pathology findings in a biopsied inflammatory demyelinating WM brain lesion.7 While the majority of the MS participants in our study did not have active disease, we cannot completely rule out if active inflammation in some might have confounded the findings with our limited sample size. DBSI of the spinal cord was not a part of the present studies. We are working to overcome technical challenges in applying DBSI to the human spinal cord and hope to include it in future studies. Gray matter lesions are particularly common in progressive forms of MS, but DBSI metrics of gray matter were not included due to the lower image resolution in diffusion‐weighted images. Our study was cross‐sectional. Longitudinal studies, which are underway, will be valuable to evaluate the meaning of DBSI metrics in relation to the pathological heterogeneity of MS over time. It would also be interesting to compare DBSI with other diffusion models. Last but not the least, our findings need to be validated in external independent datasets, including more subjects with concurrent biopsies.
Overall, this study provides preliminary evidence supporting the ability of DBSI as a potential noninvasive biomarker of MS neuropathology. Future studies are needed to evaluate the utility of DBSI as a potential outcome measure in trials of remyelinating and neuroprotective agents.
Author Contributions
A.H.C., S‐K.S., K.T., R.T.N, P.S., and R.S. contributed to the conception and design of the study. A.S., P.S., D.P., A.G., A.H.C., S‐K.S, and R.T.N. contributed to acquisition of the data. K.T. and A.S. contributed to statistical analysis. A.S., K.T., P.S., R.T.N., D.P., R.S., A.H.C., and S‐K.S. contributed to interpretation of the data. A.S. wrote the first draft of the manuscript. A.S., K.T., P.S., R.T.N., D.P., A.G., R.S., A.H.C., and S‐K.S. reviewed the manuscript and provided revisions for intellectual content.
Conflict of Interest
Afsaneh Shirani is funded through a clinician scientist development award from the National Multiple Sclerosis Society (USA), and a clinical research training scholarship from the American Academy of Neurology. Peng Sun: nothing to disclose. Kathryn Trinkaus: nothing to disclose. Dana Perantie: nothing to disclose. Ajit George: nothing to disclose. Robert Naismith has received honoraria for consulting for Alkermes, Biogen, Celgene, Novartis, TG Therapeutics; and for speaking for EMD Serono, Genzyme, Genentech, and Novartis. Robert Schmidt: nothing to disclose. Sheng‐Kwei Song is currently funded by NIH U01EY025500, R01NS047592, P01NS059560, and NMSS RG‐1701‐26617. Anne Cross was funded in part by the Manny & Rosalyn Rosenthal – Dr. John L Trotter MS Center Chair in Neuroimmunology of Barnes‐Jewish Hospital Foundation. She has received honoraria for consulting for Biogen, Celgene, EMD‐Serono, Genzyme, Genentech, Novartis, and TG Therapeutics. Washington University may receive royalty income based on a technology licensed by Washington University to DxGPS LLC. That technology is evaluated in this research.
Acknowledgment
The research reported in this article was funded by a grant from the U.S. National Institutes of Health (P01 NS059560, PI: A.H.C.).
Funding information
The research reported in this article was funded by a grant from the U.S. National Institutes of Health (P01 NS059560, PI: A.H.C.).
Funding Statement
This work was funded by National Institutes of Health grant P01 NS059560.
References
- 1. De Jager PL. Identifying patient subtypes in multiple sclerosis and tailoring immunotherapy: challenges for the future. Ther Adv Neurol Disord 2009;2:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cross AH, Song SK. A new imaging modality to non‐invasively assess multiple sclerosis pathology. J Neuroimmunol 2017;304:81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Y, Sun P, Wang Q, et al. Differentiation and quantification of inflammation, demyelination and axon injury or loss in multiple sclerosis. Brain 2015;138:1223–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin TH, Chiang CW, Perez‐Torres CJ, et al. Diffusion MRI quantifies early axonal loss in the presence of nerve swelling. J Neuroinflammation 2017;14:78 10.1186/s12974-017-0852-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y, Wang Q, Haldar JP, et al. Quantification of increased cellularity during inflammatory demyelination. Brain 2011;134:3587–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang X, Cusick MF, Wang Y, et al. Diffusion basis spectrum imaging detects and distinguishes coexisting subclinical inflammation, demyelination and axonal injury in experimental autoimmune encephalomyelitis mice. NMR Biomed 2014;27:843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shirani A, Sun P, Schmidt RE, et al. Histopathological correlation of diffusion basis spectrum imaging metrics of a biopsy‐proven inflammatory demyelinating brain lesion: A brief report. Mult Scler 2018; doi: 10.1177/1352458518786072. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lublin FD, Reingold SC, Cohen JA, etc. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Egger C, Opfer R, Wang C, et al. MRI FLAIR lesion segmentation in multiple sclerosis: Does automated segmentation hold up with manual annotation? Neuroimage Clin 2017;13:264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Strobl C, Malley J, Tutz G. An introduction to recursive partitioning: rationale, application and characteristics of classification and regression trees, bagging and random forests. Psychol Methods 2009;14:323–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005;128(Pt 11):2705–2712. [DOI] [PubMed] [Google Scholar]
- 13. Revesz T, Kidd D, Thompson AJ, et al. A comparison of the pathology of primary and secondary progressive multiple sclerosis. Brain 1994;117(Pt 4):759–765. [DOI] [PubMed] [Google Scholar]
- 14. Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci 2003;206:165–1671. [DOI] [PubMed] [Google Scholar]
- 15. Patrikios P, Stadelmann C, Kutzelnigg A, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006;129(Pt 12):3165–3172. [DOI] [PubMed] [Google Scholar]