

Abstract
Spinal muscular atrophy (SMA) is a neuromuscular disorder leading to paralysis and death. Recent evidence shows increased susceptibility to dyslipidemia and liver steatosis in patients. Here, we provide evidence that low fat diets nearly double survival in Smn2B/− mice, a model for SMA, independent of changes in SMN levels, liver steatosis, or enhanced hepatic functions. Liver damage and ketone levels were reduced, implying a lower reliance on fatty acid oxidation. This preclinical proof of concept study provides grounds for controlled clinical investigation of dietary needs and offers evidence to inform nutritional guidelines specific to SMA.
Introduction
Spinal muscular atrophy (SMA) is a devastating neurological disease affecting patients of all ages.1 Commonly affecting young infants, SMA results in motor neuron loss leading to generalized weakness and paralysis.1 The genetic basis of SMA is deletion or mutation in the ubiquitously expressed Survival motor neuron 1 (SMN1) gene,2 which encodes a protein (SMN) involved in housekeeping functions, including RNA metabolism and splicing.3
Over time, defects in multiple non‐neuronal cell types have been identified in SMA.4, 5 Metabolic defects were suspected even prior to the identification of the SMA disease gene.6 In early clinical studies, the presence of dicarboxylic aciduria, reduced beta‐oxidation capacity, and a case report of fatty liver highlighted the potential deficits in fatty acid metabolism.6, 7, 8 Recently, we identified the increased susceptibility of SMA patients and preclinical models to develop dyslipidemia and fatty liver.9 More specifically, 13% of a SMA patient cohort showed alterations in more than three common measures of dyslipidemia (total cholesterol, low‐density lipoprotein (LDL), high‐density lipoprotein (HDL), triglycerides (TGs), and non‐HDL cholesterol), which is a greater prevalence than in the normal population.10, 11 Moreover, about one‐third of patients' autopsies showed liver steatosis (commonly known as fatty liver), a feature that less than 0.7% of 2‐ to 4‐year‐old normal children harbor.12 In accordance with these clinical findings, all Smn 2 B/− mice, a commonly used model of SMA that lives about 25 days of age, developed a nonalcoholic fatty liver phenotype and dyslipidemia.9 These findings raise concerns for additional comorbidities such as metabolic syndrome, cardiovascular disease, and stroke in a subset of SMA patients as they age.
Here, we show that low fat diets were sufficient to double lifespan in Smn2B/− mice, without any reduction in the increased hepatic TG content, which suggests that there was no significant change in the fatty liver pathology. Liver function, such as protein output, also showed no amelioration. Some improvements were identified in liver damage markers, and in glucagon‐like peptide 1 (GLP‐1) and ketone levels, which trended toward normal in low fat and high‐sucrose diet (HSD). Altogether, this work highlights the need for additional controlled nutritional preclinical and clinical research. Nutritional guidelines specific to SMA should then be further developed from these evidence‐based studies.
Materials and Methods
Mouse models
The Smn2B/− (C57BL/6J background)13 mice were housed at the University of Ottawa and cared for according to the Canadian Council on Animal Care. Tissues were collected ad libitum between 9 and 11 am to limit the effect of the circadian rhythm.
Diet modulation
Dams were provided excess normal chow (NC) (Teklad Global 18% Protein Rodent Diet), high‐fat diet (HFD) (Research Diets D12492), low‐fat diet (LFD) (Research Diets D12450J), or high‐sucrose/low‐fat diet (HSD) (Research Diets D12450B) at the bottom of the cage 2 weeks after putting the breeding pair together, which was changed every 2 days. Following weaning, mice were provided with the diets until endpoint. At least three males and three females were included in each group to account for sex differences.
Gene expression studies
RNA extraction, cDNA reverse transcription, and qPCR were performed as previously published.9 Primers are as follows: Ywhaz forward 5’ AAG ACA GCA CGC TAA TAA TGC 3’, reverse 5’ TTG GAA GGC CGG TTA ATT TTC 3’, hprt1 forward 5’ CCC AGC GTC GTG ATT AGT GAT G 3’, reverse 5’ TTC AGT CCT GTC CAT AAT CAG TC 3’, and plin2 forward and reverse primer were ordered from primePCR (Biorad ‐ qMmuCIP0033479). Results were normalized with two genes identified as appropriate stable internal reference given M value below 0.5 and coefficient of variance below 0.25.
Immunoblotting
Western blots were performed as previously described.9 Results were normalized to total protein.
Lipid quantification and blood chemistry
Tissue lipid analysis was performed at Vanderbilt Mouse Metabolic Phenotyping Center.9 All blood collected in this study was sampled randomly (i.e., no fasting period) between 9 and 11 am. Alanine aminotransferase (ALT), total protein, and non‐esterified fatty acids (NEFA) were assessed at Comparative Clinical Pathology Services, LLC., Columbia, Missouri. Glucagon and GLP‐1 quantification were outsourced to Eve Technologies Corp. (Calgary, Alberta).9 Ketone and glucose were analyzed with Freestyle Precision Neo Blood Glucose and Ketone monitoring system.
Statistics
Data are presented as the mean ± standard error of the mean. One‐way ANOVA analysis was used with Tukey as posttest. Survival curves were compared using a Mantel‐Cox test. Statistical analysis was performed in GraphPad Prism. Grubbs test (alpha = 0.05) was performed to identify outliers, which were removed from the analysis. Significance was set at P ≤ 0.05 for *, P ≤ 0.01 for **, P ≤ 0.001 for ***, and P ≤ 0.0001 for ****. N number for each experiment is as indicated in the figure legends.
Data and materials availability
Raw data can be provided upon request.
Results
low‐fat diet leads to increased survival without a correction of hepatic triglyceride or liver function
We have previously shown that the Smn2B/− mice develop fatty liver, with over 25‐fold more TGs in their liver than control mice.9 At this time, it is unclear whether this is contributing to disease severity and whether it could be modulated through pharmacological or dietary means. Dietary intake plays an important part in liver lipid accumulation.14, 15 Additionally, many families find subjective clinical benefit for SMA children in adhering to a low‐fat diet called the “amino acid diet” (http://www.aadietinfo.com). As such, we tested three different diets: HFD, LFD, and HSD, which were compared to standard NC (Fig. 1). Given the short lifespan of Smn 2 B/− mice, diets were administered to the dams until the pups could freely feed on their own. It has previously been shown that milk of rat dams fed HFD had higher fat content than those on normal chow by 21 days postpartum, while their offspring showed increased weights, change in body composition, and metabolic abnormalities.16 Surprisingly, introduction of LFD and HSD doubled life expectancy of Smn2B/− mice in comparison to normal chow (median lifespan of 38 and 39 vs. 21 days, respectively) (Fig 2A). Note, both LFD and HSD have similar fat and carbohydrate proportion as energy source but differ significantly in the type of carbohydrate they provide (Fig. 1). HFD (median lifespan of 22 days) did not lead to improvement (Fig. 2A). Interestingly, the weight of the mice, SMN levels, or hepatic fat content (TG levels and plin2 mRNA levels17) were only marginally changed regardless of diet (Fig. 2B–E). Hyperglucagonemia was previously observed in the Smn2B/− mice.18 While plasma glucagon levels remain constant (Fig. 2F), a reduction of GLP‐1 in the HSD group, a resultant protein from the proglucagon produced in the intestines, was observed (Fig. 2G). We have previously noticed sustained low glucose9 and high circulating NEFA, a product from peripheral lipolysis of white adipose tissue (data not shown). We next speculated that LFD and HSD may simply restore appropriate proportions of energy substrate for usage, by reducing circulating fat and increasing glucose. Indeed, we noted a modest increase in plasma glucose levels but sustained NEFA in Smn2B/− mice on the HSD regimen (Fig. 2H,J). More importantly, plasma ketones were reduced in both LFD and HSD cohorts, implying reduced beta‐oxidation and a lower reliance on fatty acids as an energy source (Fig. 2I). Interestingly, liver damage was diminished in Smn2B/− fed LFD and HSD diets in comparison to Smn2B/− mice on normal chow, as assessed by plasma ALT levels (Fig. 2K). However, low hepatic function remained as total protein production (Fig. 2L) and Igf1 mRNA production (data not shown) were unchanged amongst the different Smn2B/− diet cohorts. Altogether, diet modulation can significantly improve the lifespan of Smn2B/− mice, likely by altering mitochondrial bioenergetic substrate utilization, without influence on hepatic fat accumulation.
Figure 1.
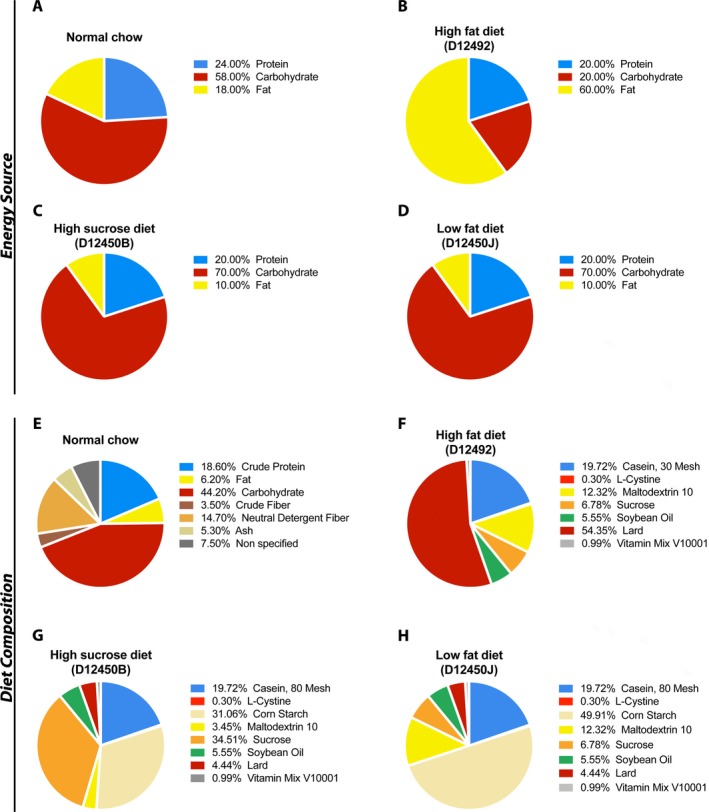
Composition of diets used in this study. Energy sources for normal chow (A), high‐fat diet – Research Diets D12492 (B), high‐sucrose diet – Research Diets D12450B (C), and low‐fat diet – Research Diets D12450J (D) are presented. The diet content is shown for normal chow (E), high‐fat diet – Research Diets D12492 (F), high‐sucrose diet – Research Diets D12450B (G), and low‐fat diet – Research Diets D12450J (H).
Figure 2.
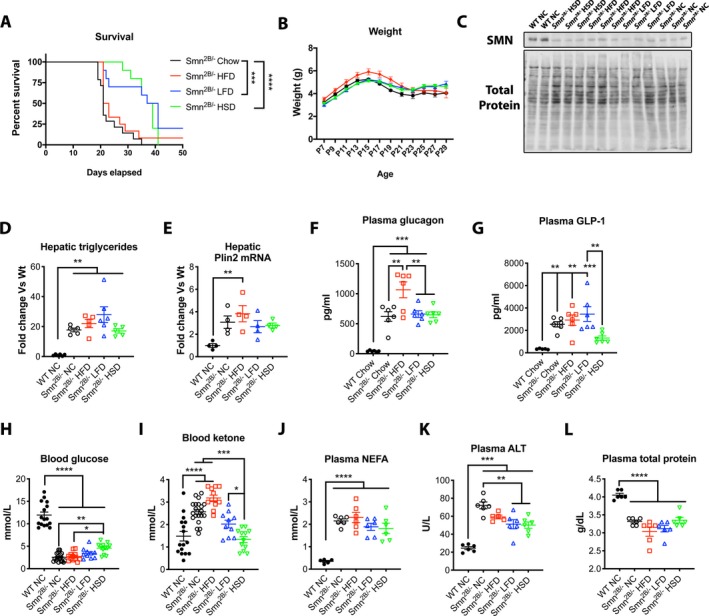
Low fat diets enhance survival by switching cell metabolism away from fat as an energy source. (A) LFD and HSD lead to a median survival of 38 and 39 days for Smn2B/− mice in comparison to 21 and 22 days when fed NC and HFD, respectively. (B–E) Weight, hepatic SMN, hepatic triglycerides, and plin2 mRNA levels were unchanged by the introduction of different diets in Smn2B/− mice. (F) Glucagon was further elevated by HFD diet in Smn2B/− mice while no change was observed by the introduction of other diets. (G) GLP‐1 was considerably diminished upon administration of HSD but not with other diets. (H) Glucose was mostly unchanged by diet modulation, albeit slightly elevated in HSD group. (I) Levels of ketone bodies were enhanced further by introduction of HFD, significantly diminished by LFD diet, while back to normal with the HSD diet in Smn2B/− mice. (J) Non‐esterified fatty acids were unchanged upon diet modulation, albeit slightly reduced in the HSD cohort. (K) Significant reduction in plasma ALT levels was observed in LFD and HSD cohort, supporting reduced hepatocyte damage. (L) Liver function, as shown by total protein output, was not restored in LFD and HSD cohorts. qPCR data were normalized with HPRT1 and Ywhaz in (E) (N value for each experiment is as follows: N = 10–19 for A‐B, 3‐6 for C–G & J–M, 10–20 for H‐I, one‐way ANOVA with Tukey's correction, Grubbs test (alpha – 0.05) for outliers, P ≤ 0.05 for *, P ≤ 0.01 for **, P ≤ 0.001 for *** and P ≤ 0.0001 for ****).
Discussion
Consensus on standards of care in SMA19, 20 repeatedly highlighted the need for more research in SMA metabolism. Despite this, optimal nutritional guidelines for SMA patients are still lacking. Currently, many SMA patients are either malnourished, underfed, or overfed.21, 22 Over the years, many SMA families have adopted the “amino acid diet.” This diet was developed purely based on observations and experiences of caregivers, and consists of reduced fat intake consumption (10–20%) and elemental free amino acid formula amongst other components. Nevertheless, scientific and preclinical evidence on the benefit of this diet is lacking.
In our study, it was intriguing that low‐fat/low‐sucrose diet or low‐fat/HSD led to doubling of the lifespan in a mouse model of SMA. Interestingly, it was previously speculated that a high‐carbohydrate/low‐fat diet would provide energy substrates that do not depend on fatty acid oxidation, keep the level of free fatty acids under control, and diminish production of dicarboxylic acid in the circulation – factors thought to have toxic potential to SMA patients.7, 23 This is consistent with some aspects of the “amino acid” diet. A LFD/high‐carbohydrate diet was tested in 13 SMA patients in the 1990s.24 While the authors claimed a beneficial outcome, there was controversy about the classification scheme used, given the lack of genetic diagnostic tools.7 Hence, the interpretation of the results is difficult7 and the study has not since been reproduced. One previous study concluded that higher fat content may confer protective benefits.25 However, the diets used consisted of two different chows with many differences amongst them, did not control exclusively for the fat content, consisted of only a 5% fat difference, and showed marginal difference in survival (3 days).25 In our study, the use of “research diets” minimized the effect of other substances while maximizing the differences in fat intake or sucrose, especially fat content. Nevertheless, it is unknown whether the full extent of the nutritional content of each diet was carried in the milk of the dams. On another note, it was interesting to note that hepatic TGs did not change significantly upon diet modulation. However, it is thought that dietary lipids only contribute to 15% of lipid accumulation in the liver in nonalcoholic fatty liver disease.26 As such, it appears that diet modulation simply decreases fatty substrate availability coming directly from dietary intake, diminishing the load on fatty acid oxidation, as shown by normalization of ketone bodies. While we were able to identify a clear difference between high‐ and low fat diets (LFD and HSD vs. HFD), there is a more subtle difference apparent between the effects of LFD and HSD. This may be attributed to the different composition of carbohydrate in the respective diets, even though the carbohydrate provided the same equivalence of energy. HSD contains sucrose as its major carbohydrate while the LFD major carbohydrate was cornstarch. This difference may impact the ease of use of the sugar and/or the underlying defects in the pancreas18 and the propensity for low blood sugar in the Smn2B/− mice and patients.9 Overall, this study suggests that supplementation of nonfatty substrates for energy use is a key beneficial determinant of survival. Our findings strongly suggest that clinical nutritional guidelines need to be established from evidence‐based research to provide better care for SMA patients.
Author Contribution
MOD designed study, performed experiments, main contributor for Figure 2. Analyzed data and created all figures. Wrote the manuscript. AT performed and provided support for experiments in Figure 2. LC provided support for experiments in Figure 2. AB provided support for experiments. RK designed study and prepared manuscript.
Conflict of Interest
Marc‐Olivier Deguise received honoraria and travel accommodations by Biogen for the SMA Summit 2018 held in Montreal and the SMA academy 2019 held in Toronto, Canada. RK and the Ottawa Hospital Research Institute have a licensing agreement with Biogen for the Smn2B/− mouse model. These COI are outside the scope of this study. All other authors have no competing interests to declare.
Acknowledgments
We extend our gratitude to Sabrina Gibeault, My Tran Trung, and Rebecca Yaworski, and the Animal Care and Veterinary Services staff of the University of Ottawa for their assistance with experiments. RK was supported by Cure SMA/Families of SMA Canada; Muscular Dystrophy Association (USA) (grant number 575466); and Canadian Institutes of Health Research (CIHR) (grant number PJT‐156379). The Vanderbilt Mouse Metabolic Phenotyping Center was supported by NIH grant DK59637. MOD was supported by Frederick Banting and Charles Best CIHR Doctoral Research Award.
Lucia Chehade and Alexandra Tierney authors are contributed equally to the work.
Funding information
RK was supported by Cure SMA/Families of SMA Canada; Muscular Dystrophy Association (USA) (grant number 575466); and Canadian Institutes of Health Research (CIHR) (grant number PJT‐156379). The Vanderbilt Mouse Metabolic Phenotyping Center was supported by National Institutes of Health grant DK59637. MOD was supported by Frederick Banting and Charles Best CIHR Doctoral Research Award.
Funding Statement
This work was funded by Cure SMA grant ; Canadian Institutes of Health Research grant PJT‐156379; Muscular Dystrophy Association grant 575466; National Institutes of Health grant DK59637.
References
- 1. Oskoui M, Darras B, De Vivo D. Spinal Muscular Atrophy: 125 Years Later and on the Verge of a Cure In: Sumner C. J., Paushkin S., Ko C. P. eds. Spinal Muscular Atrophy: Disease Mechanisms and Therapy. 1st edition. pp. 3–19. London: Academic Press, 2017. [Google Scholar]
- 2. Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 1995;80(1):155–65. [DOI] [PubMed] [Google Scholar]
- 3. Tisdale S, Pellizzoni L.RNA‐Processing Dysfunction in Spinal Muscular Atrophy In: Sumner C. J., Paushkin S., Ko C. P.eds. Spinal Muscular Atrophy: Disease Mechanisms and Therapy. 1st ed. pp. 75–97. London: Academic Press, 2017. [Google Scholar]
- 4. Deguise MO, Patitucci TN, Ebert AD, et al. Contributions of Different Cell Types to Spinal Muscular Atrophy Pathogenesis In: C. J. Sumner, S. Paushkin, Ko C. P., eds. Spinal Muscular Atrophy: Disease Mechanisms and Therapy, 1st ed London: Academic Press, 2017:167–190. [Google Scholar]
- 5. Hamilton G, Gillingwater TH. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med 2013;19:40–50. [DOI] [PubMed] [Google Scholar]
- 6. Kelley RI, Sladky JT. Dicarboxylic aciduria in an infant with spinal muscular atrophy. Ann Neurol. 1986;20:734–6. [DOI] [PubMed] [Google Scholar]
- 7. Tein I, Sloane AE, Donner EJ, et al. Fatty acid oxidation abnormalities in childhood‐onset spinal muscular atrophy: primary or secondary defect(s)? Pediatr Neurol. 1995;12:21–30. [DOI] [PubMed] [Google Scholar]
- 8. Crawford TO, Sladky JT, Hurko O, et al. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann Neurol. 1999;45:337–43. [DOI] [PubMed] [Google Scholar]
- 9. Deguise MO, Baranello G, Mastella C, et al. Abnormal fatty acid metabolism is a core component of spinal muscular atrophy. Ann Clin Transl Neurol. 2019;6:1519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kit BK, Kuklina E, Carroll MD, et al. Prevalence of and trends in dyslipidemia and blood pressure among US children and adolescents, 1999–2012. JAMA Pediatr. 2015;169:272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li J, Motsko SP, Goehring EL Jr, et al. Prevalence of pediatric dyslipidemia: comparison of a population‐based claims database to national surveys. Pharmacoepidemiol Drug Saf 2010;19:1031–40. [DOI] [PubMed] [Google Scholar]
- 12. Schwimmer JB, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics 2006;118:1388–93. [DOI] [PubMed] [Google Scholar]
- 13. Eshraghi M, McFall E, Gibeault S, Kothary R. Effect of genetic background on the phenotype of the Smn2B/‐ mouse model of spinal muscular atrophy. Hum Mol Genet 2016;25:4494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol 2016;11:451–96. [DOI] [PubMed] [Google Scholar]
- 15. Fan JG, Cao HX. Role of diet and nutritional management in non‐alcoholic fatty liver disease. J Gastroenterol Hepatol. 2013;28(Suppl 4):81–87. [DOI] [PubMed] [Google Scholar]
- 16. Sun B, Purcell RH, Terrillion CE, et al. Maternal high‐fat diet during gestation or suckling differentially affects offspring leptin sensitivity and obesity. Diabetes 2012;61:2833–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen E, Tsai TH, Li L, et al. PLIN2 is a Key Regulator of the Unfolded Protein Response and Endoplasmic Reticulum Stress Resolution in Pancreatic beta Cells. Sci Rep 2017;7:40855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bowerman M, Swoboda KJ, Michalski J‐PP, et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol 2012;72:256–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang CH, Finkel RS, Bertini ES, Schroth M. Consensus statement for standard of care in spinal muscular atrophy. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol 2007;22:1027–1049. [DOI] [PubMed] [Google Scholar]
- 20. Finkel RS, Sejersen T, Mercuri E, Group ESWS . 218th ENMC International Workshop: Revisiting the consensus on standards of care in SMA Naarden, The Netherlands, 19–21 February 2016. Neuromuscul Disord 2017;27:596–605. [DOI] [PubMed] [Google Scholar]
- 21. Mehta NM, Newman H, Tarrant S, Graham RJ. Nutritional status and nutrient intake challenges in children with spinal muscular atrophy. Pediatr Neurol 2016;57:80–83. [DOI] [PubMed] [Google Scholar]
- 22. Poruk KE, Davis R, Smart AL, et al. Observational study of caloric and nutrient intake, bone density, and body composition in infants and children with spinal muscular atrophy type I. Neuromuscul Disord 2012;22:966–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bruce AK, Jacobsen E, Dossing H, Kondrup J. Hypoglycaemia in spinal muscular atrophy. Lancet 1995;2:609–610. [DOI] [PubMed] [Google Scholar]
- 24. Harpey JP, Charpentier C, Paturneau‐Jouas M, et al. Secondary metabolic defects in spinal muscular atrophy type II. Lancet 1990;8:629–630. [DOI] [PubMed] [Google Scholar]
- 25. Butchbach ME, Rose FF, Rhoades S, et al. Effect of diet on the survival and phenotype of a mouse model for spinal muscular atrophy. Biochem Biophys Res Comm 2010;391:835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hardwick JP, Osei‐Hyiaman D, Wiland H, et al. PPAR/RXR Regulation of fatty acid metabolism and fatty acid omega‐hydroxylase (CYP4) Isozymes: implications for prevention of lipotoxicity in fatty liver disease. PPAR Res 2009;2009:952734. [DOI] [PMC free article] [PubMed] [Google Scholar]