

Abstract
Background
Motor neuron disorders involving upper and lower neurons are a genetically and clinically heterogenous group of rare neuromuscular disorders with overlap among spinal muscular atrophies (SMAs) and amyotrophic lateral sclerosis (ALS). Classical SMA caused by recessive mutations in SMN1 is one of the most common genetic causes of mortality in infants. It is characterized by degeneration of anterior horn cells in the spinal cord, leading to progressive muscle weakness and atrophy. Non‐SMN1‐related spinal muscular atrophies are caused by variants in a number of genes, including VRK1, encoding the vaccinia‐related kinase 1 (VRK1). VRK1 variants have been segregated with motor neuron diseases including SMA phenotypes or hereditary complex motor and sensory axonal neuropathy (HMSN), with or without pontocerebellar hypoplasia or microcephaly.
Results
Here, we report an association of a novel homozygous splice variant in VRK1 (c.1159 + 1G>A) with childhood‐onset SMA or juvenile lower motor disease with brisk tendon reflexes without pontocerebellar hypoplasia and normal intellectual ability in a family with five affected individuals. We show that the VRK1 splice variant in patients causes decreased splicing efficiency and a mRNA frameshift that escapes the nonsense‐mediated decay machinery and results in a premature termination codon.
Conclusions
Our findings unveil the impact of the variant on the VRK1 transcript and further support the implication of VRK1 in the pathogenesis of lower motor neuron diseases.
Introduction
Hereditary lower motor neuron diseases are a genetically and clinically heterogenous group of rare neuromuscular disorders within the spectrum spinal muscular atrophy/hereditary motor neuropathy. They are characterized by degeneration of anterior horn cells in the spinal cord, leading to progressive muscle weakness and atrophy.1 Clinical course varies depending on the type of SMA. SMA is most often caused by recessive mutations in the survival motor neuron gene (SMN1) and it is one of the commonest genetic causes of mortality in infants.2 There are, however, a significant number of SMA cases with heterogenous non‐SMN1‐related causes. Non‐SMN1‐related SMAs, including nonproximal SMA, bulbar palsy SMA, spinobulbar muscular atrophy, and variant infantile SMA characterized by SMA features along with additional symptoms and signs or atypical phenotypes.3 Pontocerebellar hypoplasia type 1A (SMA‐PCH) (MIM#607596), is a rare infantile disorder combining SMA with severe congenital microcephaly, mental retardation, and cerebellar signs.4, 5 Variants in VRK1, encoding the vaccinia‐related kinase 1 (VRK1) which is widely expressed in human tissues and has a vital role in embryonic cortical neuronal proliferation,6 have been reported in patients with SMA‐PCH or hereditary complex motor and sensory axonal neuropathy (HMSN) with microcephaly.7, 8, 9 However, the appearance of pontocerebellar hypoplasia or microcephaly has not been a consistent phenotype associated with VRK1 variants. Juvenile‐onset distal hereditary motor neuropathy (dHMN),10 adult‐onset distal SMA,11, 12 and adult‐onset motor neuron disease 13 without pontocerebellar hypoplasia or microcephaly have been associated with VRK1 variants.
Here we report a homozygous splice variant in VRK1 in a family, including five affected individuals with childhood‐onset SMA or distal motor neuropathy with brisk tendon reflexes without pontocerebellar hypoplasia and with normal intellectual ability. The variant causes decreased splicing efficiency and a mRNA frameshift that leads to a premature termination codon.
Materials and Methods
Ethical approval
The study was approved by the ethical standards of the relevant institutional review board, the Ethics Review Committee in the Gothenburg Region (Dn1: 842‐14) and the Human Research Ethics Committee of the University of Western Australia. Informed consent was obtained from all patients, parents, and family members included in this study after appropriate genetic counseling. Blood samples were obtained from patients and their family members.
Clinical evaluation
Medical history, physical examination, and imaging were performed as part of routine clinical workup. For the three VRK1 mutation‐positive patients available for study (Fig. 1), extensive clinical follow‐up was performed. The clinical evaluation was performed by experienced neurologists. Cranial MRI scans were performed in all affected individuals and were evaluated by a neuroradiologist. Neurophysiological studies including sensory and motor neurography and concentric needle electromyography were carried out in the affected individuals.
Figure 1.

Pedigree of the family and clinical features. Pedigree and recessive inheritance of VRK1. In the pedigree, squares represent males; circles, females; open symbols, unaffected family members; and slash, deceased. The affected individuals are represented with shaded symbol. −/− indicates WT, −/+ indicates heterozygous presence of the variant and +/+ indicates homozygous appearance of the variant.
Genetic analysis
Prior to this study, the affected individuals (IV:10, IV:11, and V:1) were prescreened for deletion of exon 7 in SMN1. Whole‐exome sequencing was performed on DNA from two affected (IV:10 and V:1) and one unaffected (IV:5) individuals in the family, as previously described.14 Bidirectional Sanger sequencing was performed in the patients and their unaffected parents and siblings.
Transcript analysis
To analyze the impact of the c.1159 + 1G>A variant in splicing efficiency of VRK1 exon 12 in the affected individuals (IV:10, IV:11, and V:1), PCR was performed on cDNA extracted from isolated mRNA from blood with primer pairs covering part of exon 9 through exon 13, spanning the exon 12‐13 junction, and 3’ untranslated region (UTR) (701bp).
Data availability
The data supporting the findings of this study are available within the article or from the corresponding author on request.
Results
Clinical characteristics of patients
We report three sisters (Cases IV:3, IV:10, and IV:11), a cousin (Case IV:1) and a 13‐year‐old second cousin (Case V:1) from a Caucasian family with consanguinity (Fig. 1). They presented with progressive muscle weakness and atrophy. Family history was positive with neurological disorders and occurrence of death in two cases (IV:1 and IV:3).
Case IV:3, a female, was the first child of an apparently healthy first cousin couple (Fig. 1). She presented with similar clinical features as her younger sisters (Cases IV:10 and IV:11), described below, and she died at the age of 32 because of respiratory difficulty.
Case IV:10 is a 33‐year‐old female, the second affected child of this family (Fig. 1). Pregnancy and delivery were unremarkable. She achieved normal motor milestones in childhood. At 12 years of age, she presented with progressive distal muscle weakness of the lower limb that progressed to severely involve proximal and distal muscles of four limbs and she became wheelchair bound at age 25 years. She had completely normal mental activity. Neurological examination revealed global areflexia. The sensory exam was normal. Eye movements were normal. She had scoliosis, bilateral equinovarus, and distal joint hyperlaxity. She had widespread fasciculations. There was no bulbar involvement.
Cardiac examination was normal. At 33 years of age, brain magnetic resonance imaging (MRI) was normal (Fig. 2A).
Figure 2.

Sagittal T1‐weighted MRI of brain. Axial and sagittal Fluid‐attenuated inversion recovery (FLAIR) brain MRI images of Cases (A) IV:10, (B) IV:11, and (C) V:1 reveal normal cerebellum and indicate no abnormality.
Sensory nerve conduction parameters were normal. Absent compound muscle action potential (CMAP) in the lower limbs and very low amplitude in upper limbs were observed. The electromyography (EMG) evaluation indicated chronic neurogenic changes with high‐amplitude motor unit action potentials (MUAP) in all tested muscles. Together, results of neurophysiological examination were consistent with predominantly distal SMA.
Case IV:11, a female aged 29 years, is the third affected offspring of this family. She was born after an uncomplicated full‐term pregnancy and delivery. She acquired normal motor milestones. At 6 years of age, she started to develop a progressive muscle weakness of the lower limbs that eventually progressed to involve the four limbs and she became wheelchair bound at the age of 18 years old. She had respiratory distress and respiratory insufficiency from 24 years of age. Similar to her older sister (Case IV:10), she showed normal intellectual development. Brain MRI was normal at age 29 years (Fig. 2B). Neurological examination revealed absent DTRs in all extremities. Sensory exam was normal and the Babinski sign was absent. She had normal eye movements. She had scoliosis, severe lordosis, equinovarus, and distal joint hyperlaxity. She had urinary incontinence from the age of 20 years. Nerve conduction velocity (NCV) and EMG, revealed absence of response in peroneal and tibial nerves and left ulnar nerve and a decreased amplitude in both median nerves and right ulnar nerve. The sensory nerve conduction study was normal. The EMG revealed chronic denervation in all tested muscles. The clinical and electrodiagnostic findings suggested distal SMA.
The affected sisters had six other siblings with normal motor and intellectual development by history.
Case IV:1 was a male and the only child of an apparently healthy first cousin couple of this family (Fig. 1). He died at the age of 17 because of respiratory difficulty.
Case V:1 is a 13‐year‐old male, the only child of a healthy second cousin couple of this family. Unlike his cousins, individuals IV:10 and IV:11, his motor developmental was delayed. He sat at 12 months of age and was assisted walking at 14 months of age. The first clinical examination at 6 years of age, revealed distal muscle weakness of the lower limbs, that later, spread to proximal and distal muscles of four limbs. His mother described episodic fever, mostly at nights, followed by worsening of muscle functions. He became wheelchair bound at the age of 13. He attended regular school and showed normal intellectual development. Brain MRI was normal at age 13 years (Fig. 2C). Neurological examination at 13 years of age revealed hyperactive deep tendon reflexes (DTRs). Sensory exam was normal. He had normal eye movements. Similar to his cousins (IV:10 and IV:11), he had skeletal deformity including scoliosis, lordosis, and equinovarus. The DTRs were brisk in all extremities. The cranial nerves were intact. Fasciculations were not observed. The motor conduction study revealed absence of response in right ulnar, both peroneal nerves and left tibial nerve. The sensory nerve conduction studies were normal. The EMG showed chronic denervation in all tested muscles including left quadriceps, left tibialis anterior, and left first dorsal interosseous. He was diagnosed with motor neuron disease with brisk tendon reflexes mimicking juvenile ALS.
Genetic findings
Data from whole‐exome sequencing on DNA from individuals IV:10, IV:5, and V:1 were analyzed through the use of the Ingenuity Variant Analysis (IVA) software (Qiagen, Hilden Germany). The filtering strategy initially concentrated on homozygous coding variants in known neurogenetic disease genes. A novel homozygous G to A change affecting a highly conserved nucleotide of the 5’ splice junction of intron 12 (c.1159 + 1G>A) of VRK1 (NM_003384.2) was identified in individuals IV:10 and V:1 (Fig. S1A and Fig. 3A). The variant was identified in heterozygous state in individual IV:5.
Figure 3.
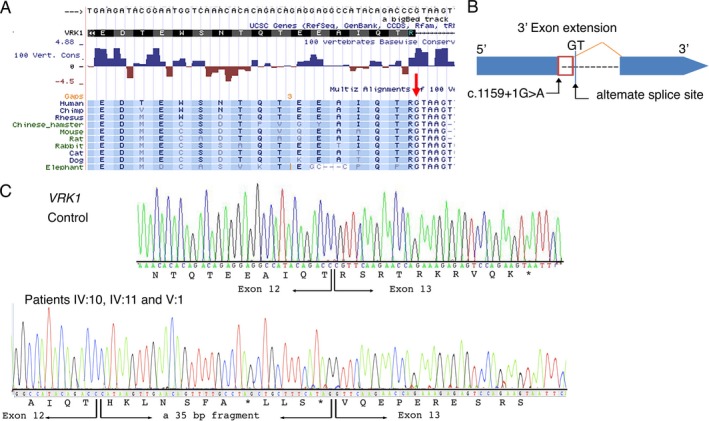
Genetic findings. (A) Species conservation of amino acids and the highly conserved GT nucleotides (red arrow) of the 5’ splice junction of intron 12 of VRK1 (shaded). (B) In silico prediction analysis (MaxEntScan) indicated the likely use of the nearby alternative splice site located 35 bases (red box) from the exon/intron boundary on the 3’ side upstream of exon 13. (C) Sanger sequencing of reverse transcriptase PCR on whole‐blood tissue of the patients (IV:10, IV:11, and V:1) revealed the presence of VRK1 messenger RNA variant in the patients. A larger fragment in the patients indicated a amplicon covering exon 12 and the entire exon 13 with the insertion of a 35 bp fragment from the exon/intron boundary on the 3’ side upstream of exon 13 and a mRNA frameshift and subsequently to a premature termination codon.
The VRK1 variant was validated by PCR and bidirectional Sanger sequencing analysis in individuals IV:10, IV:5, and V:1 (Fig. S1B). The variant is not present on any allele in the Genome Aggregation Database (gnomAD, ExAC or 1000 Genome Project Database (1000G)). In silico combined annotation‐dependent depletion (CADD) analysis of the variant revealed high‐deleterious scores: 25.200. In silico analysis predicted the VRK1 variant to be disease causing, presumably identified as potentially affecting splicing (MutationTaster, http://mutationtaster.org/). In addition, in silico prediction with the Human Splicing Finder version 3.0 and MaxEntScan suggested that the G to A nucleotide changes the highly conserved GU end in the 5’ splice donor site of intron 12 of VRK1 and would result in a decreased RNA splicing efficiency of exon 12 by 90% (from 9.09 to 0.91). In silico transcript prediction analysis (MaxEntScan) further indicated that the variant would result in the use of a nearby cryptic alternative splice site located 35 bases from the exon/intron boundary on the 3’ side. This would cause a mRNA frameshift and subsequently lead to a premature termination codon (Fig. 3B).
The appearance of the VRK1 variant was examined in all available family members by Sanger sequencing analysis, which confirmed segregation of the homozygous variant with the disease phenotype. The asymptomatic parents and siblings were either heterozygous or wild‐type for the VRK1 variant (Fig. S1B).
Transcript analysis
Owing to the location of the variant, at the last exon‐exon junction, the nonsense‐mediated decay (NMD) machinery is not expected to be engaged to initiate degradation of the mutant messenger RNA, and yield a stable mRNA that directs the synthesis of C‐terminally truncated polypeptides.15, 16 To determine the actual effect of the homozygous c.1159 + 1G>A variant on VRK1 gene expression and transcript levels, analysis of the VRK1 mRNA was performed by PCR on complementary DNA (cDNA) extracted from blood samples from the available individuals in the family. The RT‐PCR analysis indicated detectable expression levels of VRK1 transcript in the affected individuals IV:10, IV:11, and V:1. While amplification of the cDNA fragment, covering exon 9 through to the 3’UTR, resulted in a single amplicon of 701bp in controls, a larger fragment (736 bp) was detectable in the three affected individuals. Sanger sequencing of the cDNA fragment in the patients indicated a full‐length amplicon covering exon 12 and the entire exon 13, but with the insertion of a 35 bp fragment from the exon/intron boundary on the 3’ side upstream of exon 13 (Fig. 3C). This indicates an inefficient RNA splicing of exon 12 leading to a frameshift, but synthesis of a stable mRNA which escapes the NMD machinery. Subsequently, this would lead to a premature termination codon, p.Arg387Hisfs*7 (Fig. 3C).
We were, however, not able to determine whether the VRK1 transcript results in production of a stable truncated VRK1 protein or an unstable protein, due to lack of a specific VRK1 antibody suitable for immune blot analysis.
Discussion
Due to advances in genomic testing, the list of underlying genetic causes of many forms of hereditary motor neuron disease will continue to grow.17 Here, we report a homozygous splice variant in VRK1 and the presence of neurological conditions in a family, including five individuals and occurrence of death in two cases. The affected individuals presented with progressive muscle weakness and atrophy and clinical examination suggested childhood‐onset SMA or juvenile motor neuron disease with hyperreflexia without pontocerebellar hypoplasia or microcephaly and normal intellectual ability.
The ubiquitously expressed VRK1 is a serine/threonine protein kinase that phosphorylates p53 and transcription factor cAMP response element‐binding (CREB) protein. It is essential for cell cycle regulation, histone modification, chromatin organization, and nuclear envelope formation.18, 19, 20 In addition, it has a vital role in embryonic cortical neuronal proliferation and migration through amyloid‐ß precursor protein (APP) and has increased expression in proliferating cells.6
Variants in VRK1 have been segregated with motor neuron diseases including SMA phenotype, adult‐onset motor neuron disease, HMSN, and juvenile‐onset dHMN with or without pontocerebellar hypoplasia or microcephaly 7, 8, 9, 10, 11, 12, 13 (Table S1 and Fig. 4). A homozygous nonsense mutation in VRK1 (p.R358X) has been described to cause an infantile onset SMA‐PCH phenotype associated with progressive microcephaly of prenatal onset, mental deficiency, significant ataxia, and death in infancy or childhood in a consanguineous family of Ashkenazi Jewish origin.7 The same recessive nonsense mutation was reported in a patient of Ashkenazi Jewish origin with a HMSN accompanied by severe nonprogressive microcephaly and cerebral dysgenesis of prenatal onset.8 In addition, homozygous missense mutation in VRK1 (p.R133C) has been associated with SMA‐PCH in a family with four affected siblings 9 and compound heterozygous missense mutations (p.R89Q and p.V236M) have been identified in two siblings with HMSN and severe nonprogressive microcephaly.8 Furthermore, compound heterozygous missense mutations (p.G135R and p.L195V) have been identified in a patient with juvenile motor neuron disease and microcephaly.11 Segregation of VRK1 variants in patients with SMA phenotype or motor neuron disease without pontocerebellar hypoplasia or microcephaly has also been reported. Compound heterozygosity for the same nonsense mutation and a novel missense mutation (p.R358X and p.H119R) in two siblings of Ashkenazi Jewish ancestry and homozygosity for a novel nonsense mutation (p.W375X) in two Chinese siblings with adult‐onset distal SMA and a Chinese patient with Juvenile‐onset dHMN without pontocerebellar hypoplasia have been reported.10, 11, 12 Furthermore, compound heterozygous missense mutations (p.H119R and p.R321C) have been identified in a patient with adult‐onset motor neuron disease without pontocerebellar hypoplasia or microcephaly 13 (Fig. 4 and Table S1).
Figure 4.

Distribution of VRK1 variants in previously reported patients and currently described recessive variant. Schematic representation of the VRK1 transcript and VRK1 protein, including the catalytic domain of the ATP‐binding site, active site of Serine/Threonine protein kinase, activation loop, and nuclear localization signal. Previously reported homozygous (p.R133C, p.R358X, and p.W375X) and compound heterozygous (p.R89Q and p.V236M; p.H119R and p.R321C; p.H119R and p.R358X; and p.G135R and p.L195V) variants (black color) and the currently homozygous c.1159 + 1G>A (blue color) situated at the highly conserved 5’ splice junction of intron 12 are indicated. To date the genetic null variants are C‐terminal, the missense variants more N‐terminal.
Development of pontocerebellar hypoplasia or microcephaly in motor neuron disease patients associated with VRK1 has not been a frequent finding in reported cases, with seven patients so far,7, 8, 9, 11 and it is not a clinical feature in the family reported here. It thus remains unknown whether patients with VRK1‐associated motor neuron disease have a predisposition to pontocerebellar hypoplasia or microcephaly, given the few patients with VRK1 mutation that have been described so far and the lack of functional study associated with the reported variants. There is further no clear correlation between the location of the VRK1 variant and the associated clinical features (Fig. 4, Tables [Link], [Link] and [Link], [Link]). In order to understand the pathological basis of the VRK1‐associated diseases, a greater understanding of the roles of the VRK1 and its subdomains in normal function is needed.
Consistent with the in silico analysis prediction, results from our transcript assay in the patients revealed the production of a larger VRK1 transcript with insertion of a 35 bp fragment from the exon/intron boundary that escapes the NMD machinery, leading to a mRNA frameshift and subsequently to a premature termination codon. To the best of our knowledge, this is the first study enlightening the impact of a VRK1 variant on the transcript production in the patients. Nevertheless, the downstream consequences of the VRK1 variant on the VRK1 production remain unknown. Further studies are required to investigate the regulation of VRK1 and the role of the VRK1 protein in neuronal function to understand which aspect of the VRK1 protein function are affected to lead to motor neuron diseases with or without pontocerebellar hypoplasia or microcephaly and intellectual disability/central nervous system involvement.
Web Resources
The following databases were used in this study: The Exome Variant Server: NHLBI Exome Sequencing Project (ESP), Seattle, WA; URL: http://evs.gs.washington.edu/EVS/. 1000 Genome Project Database: http://browser.1000genomes.org/index.html. Human Background Variant DataBase: http://neotek.scilifelab.se/hbvdb/. Genome Aggregation Database (GnomAD): http://gnomad.broadinstitute.org/. Greater Middle East (GME) Variome web: http://igm.ucsd.edu/gme/index.php. Ensembl genome browser: http://www.ensembl.org/. MutationTaster: http://mutationtaster.org/. MaxEntScan: http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html.
Consent to Publish
The parents in this study provided written informed consent to publish their family trees, and family data, including photographs.
Conflict of Interests
The authors declare that they have no competing interests.
Supporting information
Figure S1. Whole exome and Sanger sequencing. (A) The plus strand of the genome around the VRK1 variant (red arrow) demonstrates the presence of the c.1159 + 1G>A variant in Case IV:10. (B) Sanger sequence analysis of the genomic DNA demonstrates the segregation of the VRK1 c.1159 + 1G>A variant (red arrow) in the family.
Table S1. Clinical features of the patients with VRK1 variants.
Table S2. VRK1 genotype/phenotype correlation.
Acknowledgments
We thank the family members who provided samples and clinical information for this study.
Funding Information
The study was supported by grants from the European Union’s Seventh Framework Programme for research, technological development, and demonstration under grant agreement no. 608473 (HT) and the Swedish Research Council (HT) and the Australian National Health and Medical Research Council Principal Research Fellowship APP1117510. The funders had no role in the design of the study and collection, analysis, decision to publish, interpretation of data, or preparation of the manuscript.
Funding Statement
This work was funded by Vetenskapsrådet grant ; European Union’s Seventh Framework Programme for research, technological development, and demonstration grant 608473; Australian National Health grant ; Medical Research Council Principal Research Fellowship grant APP1117510.
References
- 1. Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 1995;80:155–165. [DOI] [PubMed] [Google Scholar]
- 2. Moultrie RR, Kish‐Doto J, Peay H, Lewis MA. A review on spinal muscular atrophy: awareness, knowledge, and attitudes. J Genet Couns 2016;25:892–900. [DOI] [PubMed] [Google Scholar]
- 3. Zerres K, Rudnik‐Schoneborn S. 93rd ENMC international workshop: non‐5q‐spinal muscular atrophies (SMA) ‐ clinical picture (6–8 April 2001, Naarden, The Netherlands). Neuromuscul Disord 2003;13:179–183. [DOI] [PubMed] [Google Scholar]
- 4. Barth PG. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev 1993;15:411–422. [DOI] [PubMed] [Google Scholar]
- 5. Rudnik‐Schoneborn S, Sztriha L, Aithala GR, et al. Extended phenotype of pontocerebellar hypoplasia with infantile spinal muscular atrophy. Am J Med Genet A 2003;117A:10–17. [DOI] [PubMed] [Google Scholar]
- 6. Vinograd‐Byk H, Sapir T, Cantarero L, et al. The spinal muscular atrophy with pontocerebellar hypoplasia gene VRK1 regulates neuronal migration through an amyloid‐beta precursor protein‐dependent mechanism. J Neurosci 2015;35:936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Renbaum P, Kellerman E, Jaron R, et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet 2009;85:281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gonzaga‐Jauregui C, Lotze T, Jamal L, et al. Mutations in VRK1 associated with complex motor and sensory axonal neuropathy plus microcephaly. JAMA Neurol 2013;70:1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011;478:57–63. [DOI] [PubMed] [Google Scholar]
- 10. Feng SY, Li LY, Feng SM, Zou ZY. A novel VRK1 mutation associated with recessive distal hereditary motor neuropathy. Ann Clin Transl Neurol 2019;6:401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stoll M, Teoh H, Lee J, et al. Novel motor phenotypes in patients with VRK1 mutations without pontocerebellar hypoplasia. Neurology 2016;87:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li N, Wang L, Sun X, et al. A novel mutation in VRK1 associated with distal spinal muscular atrophy. J Hum Genet 2019;64:215–219. [DOI] [PubMed] [Google Scholar]
- 13. Nguyen TP, Biliciler S, Wiszniewski W, Sheikh K. Expanding Phenotype of VRK1 Mutations in Motor Neuron Disease. J Clin Neuromuscul Dis 2015;17:69–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kariminejad A, Dahl‐Halvarsson M, Ravenscroft G, et al. TOR1A variants cause a severe arthrogryposis with developmental delay, strabismus and tremor. Brain 2017;140:2851–2859. [DOI] [PubMed] [Google Scholar]
- 15. Nagy E, Maquat LE. A rule for termination‐codon position within intron‐containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 1998;23:198–199. [DOI] [PubMed] [Google Scholar]
- 16. Thermann R, Neu‐Yilik G, Deters A, et al. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J 1998;17:3484–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wee CD, Kong L, Sumner CJ. The genetics of spinal muscular atrophies. Curr Opin Neurol 2010;23:450–458. [DOI] [PubMed] [Google Scholar]
- 18. Vega FM, Sevilla A, Lazo PA. p53 Stabilization and accumulation induced by human vaccinia‐related kinase 1. Mol Cell Biol 2004;24:10366–10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Valour D, Ochala J, Ballay Y, Pousson M. The influence of ageing on the force‐velocity‐power characteristics of human elbow flexor muscles. Exp Gerontol 2003;38:387–395. [DOI] [PubMed] [Google Scholar]
- 20. Salzano M, Sanz‐Garcia M, Monsalve DM, et al. VRK1 chromatin kinase phosphorylates H2AX and is required for foci formation induced by DNA damage. Epigenetics 2015;10:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Whole exome and Sanger sequencing. (A) The plus strand of the genome around the VRK1 variant (red arrow) demonstrates the presence of the c.1159 + 1G>A variant in Case IV:10. (B) Sanger sequence analysis of the genomic DNA demonstrates the segregation of the VRK1 c.1159 + 1G>A variant (red arrow) in the family.
Table S1. Clinical features of the patients with VRK1 variants.
Table S2. VRK1 genotype/phenotype correlation.
Data Availability Statement
The data supporting the findings of this study are available within the article or from the corresponding author on request.