

Abstract
Clonal microbial populations are inherently heterogeneous, and this diversification is often considered as an adaptation strategy. In clinical infections, phenotypic diversity is found to be associated with drug tolerance, which in turn could evolve into genetic resistance. Mycobacterium tuberculosis, which ranks among the top ten causes of mortality with high incidence of drug‐resistant infections, exhibits considerable phenotypic diversity. In this study, we quantitatively analyze the cellular dynamics of DNA damage responses in mycobacteria using microfluidics and live‐cell fluorescence imaging. We show that individual cells growing under optimal conditions experience sporadic DNA‐damaging events manifested by RecA expression pulses. Single‐cell responses to these events occur as transient pulses of fluorescence expression, which are dependent on the gene–network structure but are triggered by extrinsic signals. We demonstrate that preexisting subpopulations, with discrete levels of DNA damage response, are associated with differential susceptibility to fluoroquinolones. Our findings reveal that the extent of DNA integrity prior to drug exposure impacts the drug activity against mycobacteria, with conceivable therapeutic implications.
Keywords: DNA damage, drug persistence, microfluidic microscopy, phenotypic variation, tuberculosis
Subject Categories: DNA Replication, Repair & Recombination; Microbiology, Virology & Host Pathogen Interaction; Systems & Computational Biology
Introduction
Microbes are subject to a continuous flux of stimuli, to which they react through diversification processes that ultimately ensure their fitness and survival as a population. In addition to genetic changes, diversification can promptly arise from transient changes in gene expression that produce more than one stable phenotype (multistability), with enhanced adaptive potential (Veening et al, 2008). Changes in gene expression are not only actively orchestrated in response to a stimulus, via intricate feedback‐based networks (Smits et al, 2006; Alon, 2007), but are also passively subject to random molecular fluctuations, which occur at the subcellular scale irrespective of the stimulus (Kærn et al, 2005). The heterogeneity in isogenic populations arising from the ensemble of these processes is implicated in important clinical processes, such as persistent infections and drug tolerance (Balaban et al, 2004; Ackermann, 2015; Dhar et al, 2016).
To ensure long‐term survival and preserve its genetic integrity against the onslaught of mutagenic stresses in the extreme host environment, Mycobacterium tuberculosis is equipped with an arsenal of transcription regulators (Flentie et al, 2016), stress responses (Gengenbacher & Kaufmann, 2012; Sala et al, 2014), and DNA repair systems (Singh, 2017). Double‐strand DNA breaks are fatal if not properly repaired. To resolve such lesions, mycobacteria can employ RecA that binds single‐stranded DNA (ssDNA), generating nucleoprotein filaments, and catalyzes homologous recombination with intact duplex DNA. Additionally, RecA is a cornerstone of the global response to DNA damage, known as the SOS response, which is highly conserved (Baharoglu & Mazel, 2014).
In this study, we focus on the single‐cell dynamics of RecA expression in mycobacteria, whose SOS response is mediated not only by RecA‐dependent inactivation of the global SOS‐response repressor LexA (Baharoglu & Mazel, 2014), with induction of about thirty genes (Davis et al, 2002), but also via a LexA‐independent mechanism that regulates about twenty genes (Gamulin et al, 2004). At present, the interplay between the two mechanisms is still unclear, although they act in concert on some genes, presumably to fine‐tune the cell response to DNA damage (Singh, 2017). Previously, it was shown that recA is dually regulated by both a distal promoter (P2), containing the consensus Cheo box for the SOS‐repressor LexA (Davis et al, 2002), and by a proximal promoter (P1), containing a pseudo‐palindromic sequence that is bound in vitro by the positive regulator ClgR, and whose deletion impairs RecA levels (Gopaul et al, 2003; Russo et al, 2009; Wang et al, 2011). Recently, a second regulator has also been proposed as an alternate P1 activator (Müller et al, 2018). Induction of recA is further influenced by LexA self‐cleavage, which is presumably induced by nucleoprotein filaments formed by RecA on damaged ssDNA. The latter regulation mode represents a typical double‐negative feedback resulting in a final positive loop (Fig 1A; Hasty et al, 2002; Smits et al, 2006; Alon, 2007). Hence, we hypothesized that the recA regulatory circuit met all minimal requirements for bistability. By using fluorescent reporter strains of recA expression, microfluidic cell cultures, and time‐lapse microscopy, we confirmed the coexistence of at least two phenotypes in clonal populations under optimal growth conditions. We observed both dim and bright subpopulations, resulting from sporadic pulses of RecA fluorescence with different levels of intensity, probably due to spontaneous DNA damage. We also found that P2‐regulation was required for pulsing and for RecA induction in the presence of genotoxic agents, while P1‐regulation was associated with the amplitude and frequency of pulses.
Figure 1. M. smegmatis P2P1recA‐GFP des expression is pulsatile under normal growth conditions.
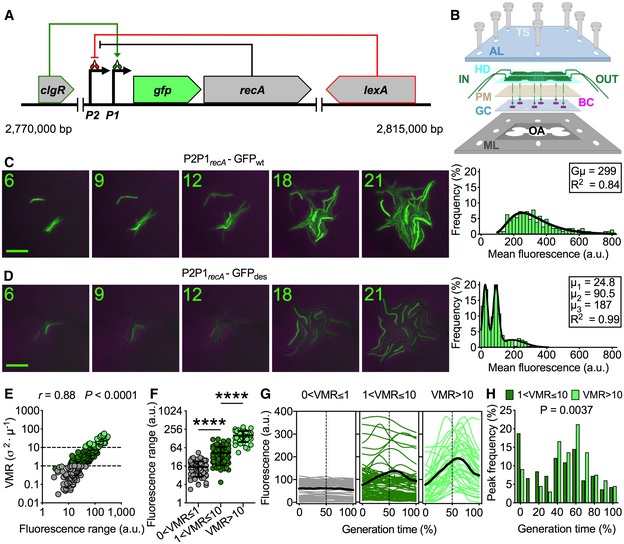
-
ASchematic of recA transcriptional reporter, with gfp inserted between the two promoters and the recA gene, and recA regulatory network. LexA represses the P2 promoter. ClgR activates the P1 promoter. In the presence of ssDNA, RecA promotes the self‐cleavage of LexA dimers, which induces all genes regulated by LexA, including recA.
-
BPerspective drawing of the microfluidic system used for time‐lapse microscopy. The top transparent acrylic layer (AL) and the bottom perforated metal layer (ML) support the system assembly by means of eight thumbscrews (TS). In the middle from above, the hexa‐device (HD) made of silicone elastomer micropatterned with two serpentine channels, connected to inlet and outlet channels (IN and OUT) for solutions to be perfused by means of a dual syringe pump. The solution (green) crosses the permeable membrane (PM), on which bacterial cells (BC) are seeded in superimposition with the six widest areas of the serpentine, and covered by a glass coverslip (GC), to enable monolayer growth. Bacteria are observed through the lens of an inverted microscope that can travel across the entire open area (OA) of the ML.
-
C, DRepresentative time‐lapse image series of exponentially growing M. smegmatis P2P1recA‐GFPwt (C) and P2P1recA‐GFPdes (D) reporters. Phase‐contrast and green fluorescence are merged. Numbers represent hours. Scale bar, 10 μm. See also Movie EV1. Histograms showing the distributions of single‐cell fluorescence (n = 204) averaged over the lifetime of the cell (right panels). Black line indicates fitting of the data with a Lognormal function (C) or with the sum of three Gaussians (D). Insets: P2P1recA‐GFPwt population's geometric mean (Gμ) calculated by computing the logarithm of the single‐cell fluorescence values, the mean of the logarithms and its antilog, P2P1recA‐GFPdes subpopulations’ means (μ), and R 2 values. The two distributions show that the destabilized GFPdes is more suitable to monitor the dynamics of recA expression and to detect subpopulations, which are masked by using the stable GFPwt variant.
-
ESpearman's correlation between single‐cell P2P1recA‐GFPdes fluorescence range and VMR of fluorescence over the lifetime of the cell (n = 204). The data shown are from three independent experiments. Dashed lines separate different levels of fluorescence dispersion over the lifetimes of single cells. Colors denote cells belonging to underdispersed (gray), dispersed (dark green), and highly dispersed (light green) subpopulations.
-
FP2P1recA‐GFPdes fluorescence range over the lifetime of the cell of subpopulations segregated by fluorescence VMR (40 < n < 112). The data shown are from three independent experiments. Black lines indicate means ± SD. Asterisks denote significant difference by Kruskal–Wallis and Dunn's multiple comparison test: ****P < 0.0001.
-
GSingle‐cell time traces of P2P1recA‐GFPdes fluorescence expressed as a percent of generation time of subpopulations segregated by fluorescence VMR. The average fluorescence (solid black lines) and half of the generation time (dashed lines) are indicated. Population statistics (n = 204): 38% (0 < VMR ≤ 1); 44% (1 < VMR ≤ 10); 18% (VMR > 10). The data shown are from three independent experiments.
-
HHistogram showing the distribution of the fluorescence peak time expressed as a percent of generation time. Dark green bars indicate moderately pulsing cells (n = 165). Light green bars indicate highly pulsing cells (n = 66). Significance by Mann–Whitney U‐test. The data shown are from three independent experiments. Peaks mainly occur between 40% and 70% of the generation time.
Interestingly, we found that pulsing subpopulations were more susceptible to fluoroquinolone treatment compared to non‐pulsing subpopulations, which were more tolerant. Most drug‐tolerant cells, which survived the first drug treatment, died at the second drug treatment, whereas only a limited fraction of those cells developed drug resistance and survived during the second drug treatment. In conclusion, here we show, for the first time in a priority clinical pathogen, that preexisting phenotypic variation in DNA damage response is associated with differential activity of fluoroquinolones. Our findings may prove useful not only for a better understanding of the drug's mode of action, but also for the conception of faster and more effective combination therapeutics.
Results
Mycobacterium smegmatis P2P1recA‐GFPdes reporter pulses in normal growth conditions
We generated two fluorescent reporters of recA transcription in M. smegmatis, by alternately inserting the gene encoding the wild‐type green fluorescent protein (GFPwt) or a destabilized variant (GFPdes) into the native recA locus (Fig 1A, Appendix Fig S1A, and see Materials and Methods; Gopaul et al, 2003; Wang et al, 2011). In exponential growth phase, the half‐life of GFPwt was longer than 12 h (about 4 generation times) and largely dilution‐dependent, whereas the half‐life of GFPdes was 49.6 ± 14.4 min (about one‐third of the generation time) and largely degradation‐dependent (Appendix Fig S1B). This was consistent with our previous study in M. tuberculosis and suggests that the rate of GFPdes degradation is species‐specific and scales with the growth rate (Manina et al, 2015). To probe the single‐cell spatiotemporal dynamics of recA, we built a microfluidic system that was suitable for multipoint microscopy imaging, stemming from the principle of multi‐layer assembly (Dhar & Manina, 2015). This “hexa‐device” was conceived to monitor the growth of microcolonies from six independent strains in the same environmental conditions for long periods of time (Fig 1B and Appendix Fig S1C). We imaged exponentially growing cells under constant perfusion of fresh 7H9 medium (Fig 1C and D, and Movie EV1) and found that the reporter strains had no growth defects compared to the wild‐type (WT) strain (Appendix Fig S1D). Both strains exhibited transient pulses of fluorescence, consistent with a stress‐response function and reminiscent of results obtained with a fluorescent RecA reporter strain of Escherichia coli (Smits et al, 2006; Kamensek et al, 2010). Pulsing RecA fluorescence resulted in progressive increment of fluorescence across the population for stable GFPwt, and in marked cell‐to‐cell variation for short‐lived GFPdes, corroborating that short‐lived markers are better suited to monitor the dynamic activity of promoters (Cameron & Collins, 2014). The P2P1recA‐GFPwt reporter produced a fairly homogeneous population that fitted a lognormal distribution (Fig 1C). Conversely, the P2P1recA‐GFPdes reporter produced a highly heterogeneous population comprising three subpopulations with discrete average fluorescence levels (Fig 1D).
To systematically distinguish between the three subpopulations, we measured the single‐cell index of dispersion or noise strength (Raser & O'Shea, 2004; see Materials and Methods), expressed as the variance to the mean ratio (VMR) of fluorescence from birth to division, and defined three subsets: underdispersed (0 < VMR ≤ 1), dispersed (1 < VMR ≤ 10), and highly dispersed (VMR > 10). The VMR positively correlated with the fluorescence range, expressed as the difference between maximum and minimum fluorescence values during the lifetime of the cell, and the three resulting subpopulations were significantly different from each other (Fig 1E and F). VMR‐based segregation revealed subsets of non‐pulsing (38%), moderately pulsing (44%), and highly pulsing (18%) cells (Fig 1G), which had similar sizes but different growth rates (Appendix Fig S1E and F), as the pulsing categories grew more slowly. Interestingly, the fluorescence peaks were predominantly located between 40 and 70% of the generation time, and moderately pulsing cells often peaked at birth (Fig 1H), implying that RecA‐pulsing events can last for more than one generation time.
We also examined the hereditary profile of P2P1recA‐GFPdes, by tracing the fluorescence intensity of single progenitors over four generations (Appendix Fig S1G and H). By reconstructing the single‐cell lineages and tracking the pole age between each related cell pair, we found moderate positive correlations between mothers and older daughter cells, which was less pronounced for younger daughters (Appendix Fig S1I). However, this moderate correlation was not due to a hereditary pattern whereby if a mother cell experiences a pulsing event the daughter cell is also more likely to experience a new pulsing event, but rather to the fact that a pulsing event originating in the mother cell can occasionally span two consecutive cell cycles and resolve in the daughter cell (Fig 1H). Furthermore, we found no correlation between other related cell pairs, nor between random cells pairs used as controls (Appendix Fig S1J–N), collectively implying that recA‐pulsing behavior is not heritable. Asymmetric cell division is a major driver of phenotypic heterogeneity in mycobacteria and is associated with cell age (Appendix Fig S10, Kieser & Rubin, 2014). Here, we did not observe any significant correlation of P2P1recA‐GFPdes pulsing with cell age (Appendix Fig S1P–R), nor did we observe that RecA pulses are heritable within cell lineages, leading us to hypothesize that these events may be stochastic.
Genetic locus architecture impacts recA pulsing in individual M. smegmatis cells
Bacterial phenotypic variation is greatly influenced by feedback‐based regulation of genetic networks (Smits et al, 2006). The mycobacterial recA locus has a relatively complex structure (Fig 1A), which we hypothesized accounts for the pulsing pattern of expression. Bulk studies reported that the recA P2 promoter is subject to LexA negative regulation and plausibly to RecA‐mediated cleavage of LexA, resulting in self‐induction. Additionally, one or more possible transcriptional activators might control the P1 promoter (Davis et al, 2002; Gopaul et al, 2003; Rand et al, 2003; Gamulin et al, 2004; Wang et al, 2011; Smollett et al, 2012; Müller et al, 2018). We probed the transcriptional regulation of recA at the single‐cell level to uncover the molecular bases of its pulsing pattern of expression by constructing a panel of fluorescent reporter strains (Fig 2A, Appendix Fig S2A, and Movie EV2) derived from the P2P1recA‐GFPdes background, here abridged as P2P1wt (see Materials and Methods for strains construction). We deleted MSMEG_2694, encoding the putative positive regulator ClgR (Wang et al, 2011), here referred to as the ΔClgR strain. We generated a red fluorescent reporter of the distal P2 promoter activity, here referred to as P2wtΔP1, by inserting the gene coding for a destabilized red fluorescent protein (mCherry des) downstream of the P2 promoter (Appendix Fig S2A and B) while deleting the proximal P1 promoter and the gfp marker downstream of P1. Lastly, we mutated the Cheo box (Davis et al, 2002) in the P2wtΔP1 background to prevent LexA binding (Fig 2A and Appendix Fig S2C), here referred to as P2mutΔP1. All reporter strains were similar to the WT in relation to growth and drug susceptibility and, following exposure to mitomycin C (MMC), a DNA‐alkylating agent conventionally used to induce double‐strand DNA breaks, we observed an induction of the reference genes, as expected (Appendix Fig S2D–F).
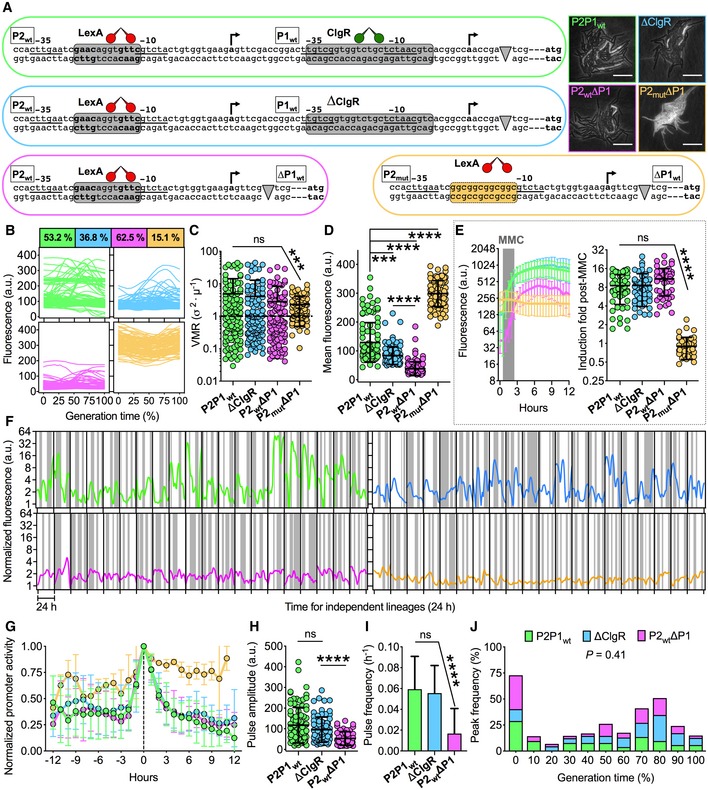
Figure 2. Genetic architecture of recA influences pulsing in individual M. smegmatis cells.
-
ASchematics of the recA regulatory region and of genetically modified variants. The −10 and −35 regions of the two promoters are underlined. The transcriptional start sites are indicated by arrows. The consensus recognized by LexA (red) and ClgR (green) is shown in gray (wt) and orange (mutated) boxes. Gray arrowheads represent the insertion of fluorescent markers. The RecA translation initiation codon is bolded. Genomic loci (left and bottom) and representative time‐lapse images (right) of the four fluorescent reporters in exponential phase are color‐coded. Phase‐contrast and fluorescence images are merged. Scale bar, 10 μm. See also Movie EV2.
-
B–DColor‐coded single‐cell time traces of the four reporters expressed as percent of the generation time (B). Population coefficients of variation of fluorescence (%) are in color‐coded boxes. Single‐cell VMR of fluorescence (C) and fluorescence averaged over the lifetime of the cell (D) in exponential phase. Black lines indicate means ± SD. Asterisks denote significance by Kruskal–Wallis and Dunn's multiple comparison test: ns, not significant; ***P = 0.0002; ****P < 0.0001. The data shown are from two independent experiments (n = 105 cells per strain). Single‐cell fluorescence decreases but variation does not change in strains devoid of positive regulation. Single‐cell fluorescence increases and variation decreases in the strain devoid of negative regulation.
-
EColor‐coded time traces of fluorescence (left panel) averaged for 50 individual cells per strain (mean ± SD) treated with MMC (0.5 μg/ml) for 2 h (gray shading). Fluorescence induction during 6 h from MMC exposure (right panel). Asterisks denote significance by Kruskal–Wallis and Dunn's multiple comparison test: ns, not significant; ****P < 0.0001. The data shown are from two independent experiments.
-
FSingle‐cell analysis of independent lineages separated by black vertical lines. Color‐coded traces represent single‐cell fluorescence of random descendants monitored in fresh 7H9 medium for 24 h (n = 20). White and gray vertical bands represent cell divisions. The data shown are from two independent experiments.
-
GAligned pulse profiles averaged for each reporter strain (color‐coded as in A). Error bars are SD (4 < n < 37). The data shown are from two independent experiments. Pulses are abrogated in the strain devoid of negative regulation.
-
H, IAnalysis of pulse characteristics. Black lines indicate means and SD (52 < n < 88). Significance by Kruskal–Wallis and Dunn's multiple comparison test: ns, not significant; ****P < 0.0001. The data shown are from two independent experiments.
-
JHistogram showing the distribution of the fluorescence peak time of individual cells expressed as a percent of the generation time (52 < n < 88). Different strains are color‐coded. Statistical analysis by one‐way ANOVA and Holm‐Sidak's multiple comparisons test. Pulses are weaker and fewer in the strain devoid of positive regulation but are not displaced relative to the generation time.
We used the hexa‐device (Fig 1B) and quantitative time‐lapse imaging to analyze the single‐cell dynamics of each reporter strain in exponential growth phase and upon MMC exposure (Movie EV2), which caused a growth rate reduction and cell elongation (Appendix Fig S2G and H). In exponentially growing microcolonies, the single‐cell fluorescence exhibited marked variation both at the population and single‐cell level, except for the P2mutΔP1 strain, whose fluorescence levels were relatively homogeneous over the cell lifetime and from cell to cell (Fig 2B and C). The mean fluorescence values were significantly lower in ΔClgR cells, consistent with the role of ClgR as a positive regulator (Wang et al, 2011), and further decreased in P2wtΔP1 cells, consistent with a second regulatory mechanism acting on the proximal promoter P1 (Müller et al, 2018). Conversely, P2mutΔP1 cells exhibited significantly higher levels of fluorescence, almost resembling constitutive recA expression, due to loss of LexA binding (Fig 2D). Interestingly, all strains exhibited comparable induction of fluorescence on exposure to MMC, apart from P2mutΔP1 (Fig 2E).
Since RecA pulses were erratic, we sought to track their dynamics over 24 h (about 8 consecutive generations) under constant perfusion of fresh 7H9 medium (Fig 2F). We detected bona fide pulses in all strains except for P2mutΔP1 (Fig 2G), which we excluded from the rest of the analysis. The structure and duration of pulses overlapped in P2P1wt, ΔClgR, and P2wtΔP1 strains (Fig 2G and Appendix Fig S2I). As expected, the baseline of the pulses was progressively lower in the ΔClgR and P2wtΔP1 strains (Appendix Fig S2J), whereas only the absence of the proximal promoter led to a significant reduction in the amplitude and frequency of pulses (Fig 2H and I). Interestingly, the peak positioning relative to the cell lifetime was unchanged in the ΔP1 mutants, indicating that pulsing events per se are the result of a global cellular stimulus that might be associated with the cell cycle (Fig 2J). In sum, while lack of positive regulation affects recA basal expression in ΔClgR and P2wtΔP1 strains, and the intensity and frequency of RecA pulses in the P2wtΔP1 strain, but has no effect on the magnitude of the stress response, lack of negative regulation in the P2mutΔP1 strain abrogates RecA pulsing and induction upon stress exposure.
The DNA damage response is largely driven by extrinsic signals
We asked whether RecA pulses were exclusively dependent on factors intrinsic to the regulatory structure of the recA locus or if they were also triggered by DNA damage.
First, we wanted to monitor the expression of RecA at the translational level, to assess whether it paralleled the transcriptional level of expression from the P2P1recA‐GFPdes reporter strain. We also wanted to check the possible implication of factors related to editing of the native locus in RecA pulses. To these ends, we constructed a dual‐fluorescent reporter strain of recA transcription and translation (Appendix Fig S2B, S3A and B), by duplicating the recA locus into the attB phage attachment site of the P2P1recA‐GFPdes strain (Pena et al, 1996; see Materials and Methods). To compare the P2P1recA‐GFPdes and RecA‐mCherrywt reporters in the recA‐merodiploid strain, we made a correlative analysis of the fluorescence range of individual cells growing inside the hexa‐device (Fig 1B and Movie EV3). We found a significant positive correlation, whether computed along the cell cycle of individual cells or at the subpopulation level (Fig 3A and Appendix Fig S3C–E). The lack of absolute correlation could either be attributed to different positioning of the two recA loci on the chromosome, or to differential stability of the fluorescent proteins. The similarity in the behavior of the two independent recA reporters enabled us to confirm the authenticity of pulses, and to rule out the implication of other factors, such as the fusion of the SsrA degradation tag to GFP (Andersen et al, 1998), or merely the gfp insertion.
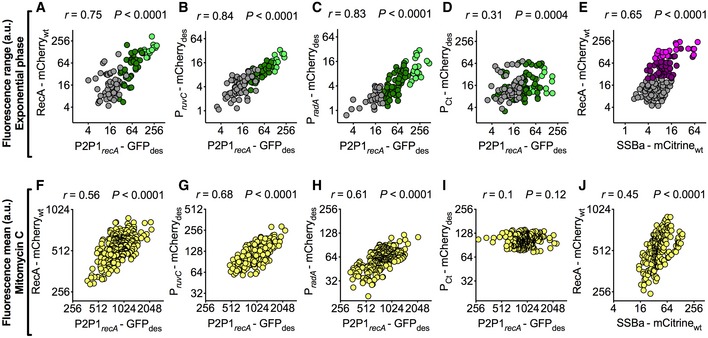
Figure 3. The DNA damage response is largely driven by extrinsic signals.
-
A–ESpearman's correlation between green (A‐D) or yellow (E) fluorescence range and red fluorescence range over the lifetime of the cell (105 < n < 153). The data shown are from two independent experiments. On the x‐axes, P2P1recA‐GFPdes (A–D) or a C‐term translational reporter of SSBa fused to mCitrinewt (E). On the y‐axes, a C‐term translational reporter of RecA fused to mCherrywt (A and E), transcriptional reporters of ruvC (B) and radA (C), and mCherry des expressed from a constitutive control promoter (D). Colors (A–D) indicate subpopulations segregated by VMR of P2P1recA‐GFPdes: gray (0 < VMR ≤ 1), dark green (1 < VMR ≤ 10), and light green (VMR > 10). Colors (E) indicate subpopulations segregated by VMR of RecA‐mCherrywt: gray (0 < VMR ≤ 1), plum (1 < VMR ≤ 10), and magenta (VMR > 10). See also Movie EV3.
-
F–JPearson's correlation between green (F–I) or yellow (J) fluorescence and red fluorescence, measured 3 h after MMC exposure (203 < n < 314). The data shown are from two independent experiments. On the x‐axes, P2P1recA‐GFPdes (F–I) or a C‐term translational reporter of SSBa fused to mCitrinewt (J). On the y‐axes, a C‐term translational reporter of RecA fused to mCherrywt (F and J), transcriptional reporters of ruvC (G) and radA (H), and mCherry des expressed from a constitutive control promoter (I). See also Movie EV3. Positive correlations between recA and other DNA damage markers both under normal growth conditions, where spontaneous DNA damage events occur at the subpopulation level, and in the presence of an alkylating agent that induces double‐strand breaks in the entire population, validate recA as a reliable proxy for DNA damage in single cells.
Next, we sought other DNA damage response genes whose regulatory sequences contained either the Cheo box or the ClgR operator (Gamulin et al, 2004). We focused on RuvC (MSMEG_2943), a LexA‐regulated crossover junction endodeoxyribonuclease that resolves Holliday junctions in stalled replication forks; RadA (MSMEG_6079), a ClgR‐regulated RecA/Rad51 paralog that catalyzes homologous DNA strand exchange (Inoue et al, 2017); and SSBa (MSMEG_6896), a ssDNA‐binding protein that protects 3′‐single‐stranded tails resulting from double‐strand breaks, interacts with RecA, and has a role in RecA loading onto ssDNA (Reddy et al, 2001; Santi & McKinney, 2015; Hung‐Yi et al, 2017; Singh, 2017). MSMEG_6896 contains a putative ClgR operator sequence in its regulatory region (Gamulin et al, 2004). We constructed a panel of dual transcriptional reporter strains in the P2P1recA‐GFPdes background (Movie EV3), by inserting the unstable mCherry des variant downstream of the native promoter of either ruvC or radA (Appendix Fig S2B, S3F, and G, and see Materials and Methods). The strains showed no growth defects and normal levels of transcription (Appendix Fig S3A and H). By comparing each pair of reporters, we found a robust linear relationship between recA and ruvC expression (Fig 3B), as well as between recA and radA (Fig 3C). As a control, we inserted mCherry des driven by a constitutive promoter into the attB site of the P2P1recA‐GFPdes strain. Surprisingly, we also found a weak positive correlation between P2P1recA‐GFPdes and the control marker (Fig 3D). The latter observation is likely due to the fact both markers were fused to the SsrA destruction tag, which directs the two independent reporter fusion proteins to the same degradation pathway (Andersen et al, 1998). Lastly, we wanted to probe the dynamics of the RecA‐mCherry translational reporter in association with a different DNA damage‐related protein. To this aim, we generated a dual‐translational reporter of RecA‐mCherrywt and SSBa‐mCitrinewt (see Materials and Methods). We also found a moderate positive correlation at the protein level between SSBa‐mCitrinewt and RecA‐mCherrywt (Fig 3E and Movie EV3), implying their coordinated activity in the presence of ssDNA (Reddy et al, 2001; Hung‐Yi et al, 2017; Singh, 2017). Importantly, we also measured positive correlations between all dual‐fluorescent reporter pairs (Fig 3F–H and J, and Movie EV3) following DNA damage caused by MMC exposure, except in the control strain (Fig 3I).
Collectively, these results imply that recA, ruvC, radA, and ssbA have similar single‐cell expression dynamics that are dependent on DNA damage, which is the sole factor shared by all four genes (Reddy et al, 2001; Rand et al, 2003; Gamulin et al, 2004; Nautiyal et al, 2016; Inoue et al, 2017; Singh, 2017), and validate recA as a robust indicator of DNA damage events in mycobacteria.
The single‐cell RecA‐pulsing pattern is predictive of cell fate in M. smegmatis treated with the DNA‐damaging antibiotic ciprofloxacin
Transition to low‐metabolic states and expression of detoxifying enzymes and efflux systems are recognized causes of phenotypic drug tolerance, whose common trait is their inherent stochasticity (Adams et al, 2011; Dhar et al, 2016; Pu et al, 2016; Bergmiller et al, 2017; Defraine et al, 2018). Since RecA pulsing is probably of stochastic origin, we enquired whether pulses could be associated with drug tolerance. We decided to focus on fluoroquinolones, a broad‐spectrum drug class that targets DNA gyrase and is effective against M. tuberculosis (Zumla et al, 2014). We tracked the single‐cell behavior of rapidly growing cells (cultured in fresh medium) and slowly growing cells (cultured in spent medium) during a multistage time‐lapse microscopy assay (Movie EV4). Cells were first cultured in either fresh or spent medium, then exposed to ciprofloxacin (CIP) fourfold above the minimum inhibitory concentration (4X‐MIC) for 6 h, then exposed to CIP‐free fresh medium to assess viability. At the end of the recovery phase, cells were stained with SYTOX dye (SX) to detect membrane permeabilization. At this stage, we were able to clearly recognize two categories, namely cells dead by lysis or SX+, and survivors that were able to resume growth after CIP washout. Lastly, we exposed the survivors a second time to CIP (4X‐MIC), to assess whether they had acquired genetic resistance, which was never the case (Fig 4A–D). Not surprisingly, the RecA‐mCherrywt reporter exhibited a patchy localization pattern (Movie EV4), which conceivably reflects RecA binding to ssDNA. This appearance intensified during exposure to ciprofloxacin and declined after removal of the stress, suggestive of DNA damage resolution. Also, we sporadically visualized RecA‐mCherrywt bundles (Vijayan, 2005; Lesterlin et al, 2014) in a minority of cells in the stage immediately preceding death.
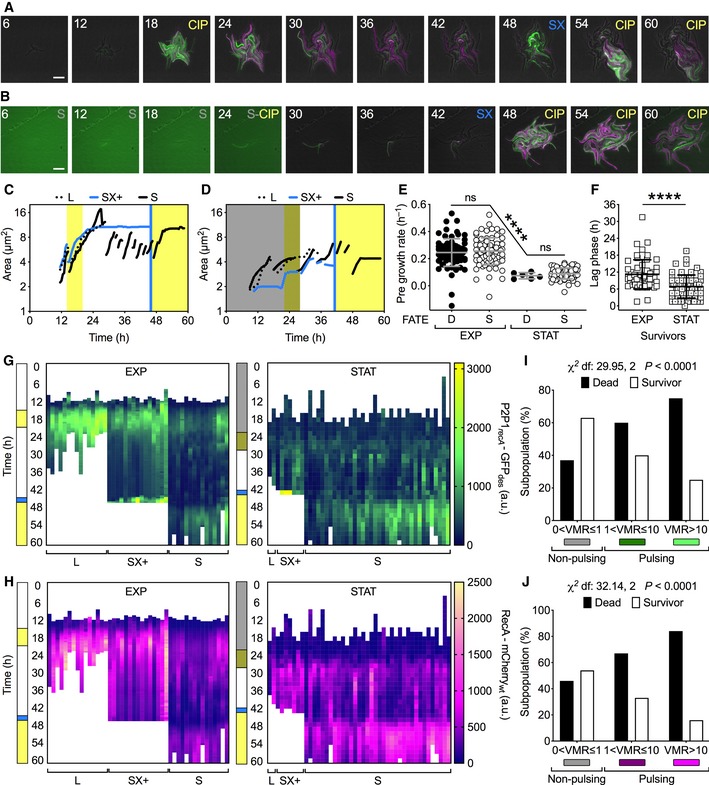
Figure 4. RecA pulsing is predictive of cell fate in M. smegmatis .
-
A, BRepresentative time‐lapse image series of the merodiploid recA reporter strain (P2P1recA‐GFPdes_RecA‐mCherrywt) cultured in fresh (A) or spent (B) flow medium (see Materials and Methods). Fresh 7H9 medium is unlabeled; spent medium (S), ciprofloxacin 1 μg/ml in fresh (CIP) or spent (S‐CIP) medium, and SYTOX 0.5 μM (SX) are labeled. Phase‐contrast (gray), GFPdes (green), and mCherrywt (magenta) fluorescence are merged. Fluorescence images of each channel are scaled to the brightest frame. Light green background in the first 4 frames (B) is due to the autofluorescence of spent medium. Cells were imaged at 30‐min intervals, and numbers represent hours. Scale bar, 10 μm. See also Movie EV4.
-
C, DRepresentative time traces of individual lineages of cells fed with fresh medium (C) or spent medium (D). Different styles are used for lineages with different fates: death and lysis (L); SYTOX‐positive (SX+); and survival (S). Line breaks represent division events in the same lineage, where a single random daughter cell is tracked for clarity. Vertical bars represent fresh (white) or spent (dark shading) 7H9 medium, CIP (yellow), and SX (blue).
-
E, FComparison of single‐cell growth rates prior to CIP exposure for “D” cells that die and “S” cells that survive and regrow after CIP washout (E), and lag time of regrowth (F). Cells were cultured in fresh (EXP) or spent (STAT) flow medium. Gray and black lines indicate mean ± SD. Significance by Kruskal–Wallis and Dunn's multiple comparison test (E) and unpaired Mann–Whitney U‐test (F): ns, not significant; ****P < 0.0001; 6 < n < 84. The data shown are from two independent experiments. Single‐cell fate is independent of the growth rate before exposure to the drug. However, cells cultured in spent medium (STAT) grow at significantly slower rates, survive at higher frequencies, and recover faster after CIP washout compared to cells cultured in fresh medium (EXP).
-
G, HRepresentative GFPdes (G) and mCherrywt (H) fluorescence time traces for single‐cell lineages of cells cultured in fresh medium (EXP) and cells cultured in spent medium (STAT), and having different fates. Vertical bars are color‐coded as in (C, D).
-
I, JSingle‐cell fate as a function of VMR of P2P1recA‐GFPdes (I) and RecA‐mCherrywt (J) fluorescence prior to CIP exposure. Significance by Chi‐square test of independence for cells that die (n = 84) and cells that survive (n = 54). The data shown are from two independent experiments. Cells that do not experience DNA damage prior to drug exposure (RecA‐non‐pulsing cells) survive more than cells that do experience DNA damage (RecA‐pulsing cells).
The fate of single cells was independent of both the growth rate and cell age (Fig 4E and Appendix Fig S4A). Cells cultured in fresh medium produced only 1.5% of survivors, whereas cells cultured in spent medium produced 86% of survivors, which is consistent with reduced CIP efficacy during starvation and stationary phase (Sarathy et al, 2013; Gutierrez et al, 2017). Although cells cultured in spent medium had slower growth rates (Fig 4E) and greater chromosomal condensation, expressed as an increase in standard deviation of DRAQ5 fluorescence (Appendix Fig S4B), they continued to grow and divide even during CIP exposure, whereas cells cultured in fresh medium continued to elongate without dividing until they arrested (Appendix Fig S4C). The recovery time after withdrawal of CIP was longer in cells cultured in fresh medium compared to cells cultured in spent medium (Fig 4F). Survivors, especially from cells cultured in fresh medium, exhibited a disproportionate elongation in the recovery phase until the first division event, after which they progressively recovered a normal size during the following division events (Appendix Fig S4C).
Next, we analyzed the single‐cell fluorescence intensities of both P2P1recA‐GFPdes and RecA‐mCherrywt. At the population level, both reporters were induced during CIP exposure and decreased during the post‐CIP recovery phase with marked cell‐to‐cell variation (Fig 4G and H, Appendix Fig S4D and E). To understand the subpopulation dynamics, we probed whether the pulsing state prior to CIP exposure was related to cell fate. Consistently between the two RecA reporters, we found that pulsing cells were more likely to die and non‐pulsing cells were more likely to survive (Fig 4I and J). We did not observe any increased mortality in pulsing cells in the absence of stress. Cells that ultimately died exhibited more heterogeneous and pronounced fluorescence induction (Appendix Fig S4F and G). These results suggest that the extent of DNA damage prior to CIP exposure has a significant impact on the onset of drug tolerance.
M. tuberculosis RecA‐mCherrywt reporter pulses in normal growth conditions
In addition to the transcriptional regulation mediated by promoters P1 and P2 (Gopaul et al, 2003), the M. tuberculosis RecA protein is subject to post‐translational splicing (Mills et al, 1998). To track the single‐cell dynamics of functional RecA in M. tuberculosis, we fused mCherrywt to the C‐terminus of RecA (Appendix Fig S5A and B), generating a merodiploid recA strain in the background of a previously described green fluorescent reporter of ribosomal RNA (rrn) operon expression (Prrn‐GFPdes) (Manina et al, 2015; see Materials and Methods). We carried out time‐lapse imaging of exponential‐phase bacilli growing inside our hexa‐device (Fig 1B), continuously fed with fresh 7H9 medium (Movie EV5). The dual M. tuberculosis reporter exhibited sporadic pulses of RecA‐mCherrywt fluorescence and marked cell‐to‐cell variation, which was consistent with our findings in M. smegmatis (Fig 1C–H and 3A). In contrast, Prrn‐GFPdes fluorescence was relatively homogeneous over the generation time, reflecting its housekeeping role (Appendix Fig S5C).
We applied the VMR‐segregation method for identifying subpopulations with discrete ranges of RecA‐mCherrywt fluorescence (Fig 5A and B), which enabled us to identify 40% of non‐pulsing (0 < VMR ≤ 1), 44% of moderately pulsing (1 < VMR ≤ 10), and 16% of highly pulsing (VMR > 10) bacilli (Fig 5C). Consistent with our results in M. smegmatis, also in M. tuberculosis the fluorescence peaks mainly occur in the second half of the generation time (Figs 1H and 5D). More precisely, most peaks occur between 80 and 100% of the generation time in M. tuberculosis (Fig 5D), as opposed to M. smegmatis where they mainly occur between 40 and 70% of the generation time (Fig 1H). Additionally, moderately pulsing cells also peaked at birth in both species. Highly pulsing cells were also slightly larger and grew at slower rates (Fig 5E and F), which supported the hypothesis of ongoing DNA damage in this cell subpopulation. Although there was no correlation between Prrn‐GFPdes fluorescence and RecA‐mCherrywt fluorescence in non‐pulsing cells, pulsing cells exhibited a moderate but significant negative correlation, suggesting that exponential‐phase cells with spontaneous DNA damage exhibit a slowdown of metabolic activity (Appendix Fig S5C and D). Moreover, we measured a decrease of the Prrn‐GFPdes output and an increase of the RecA‐mCherrywt output under different host‐mimetic conditions, further validating our dual reporter as a reliable gauge of stress response in single cells (Appendix Fig S5E). Finally, RecA‐mCherrywt pulsing was neither heritable nor age‐dependent, consistent with a probabilistic origin of RecA pulses in M. tuberculosis (Appendix Fig S5F).
Figure 5. M. tuberculosis RecA‐mCherrywt expression dynamics under normal growth conditions.
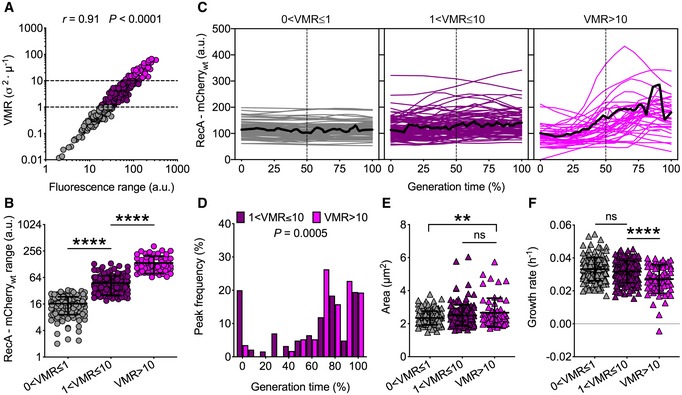
-
ASpearman's correlation between single‐cell RecA‐mCherrywt fluorescence range and fluorescence VMR over the lifetime of the cell (n = 360). The data shown are from three independent experiments. Dashed lines separate different levels of dispersion, and colors denote subpopulations that are underdispersed (gray), dispersed (plum), or highly dispersed (magenta).
-
BRecA‐mCherrywt fluorescence of subpopulations segregated by fluorescence VMR (58 < n < 159). The data shown are from three independent experiments. Black lines indicate means ± SD. Asterisks denote significant difference by Kruskal–Wallis and Dunn's multiple comparison test: ****P < 0.0001.
-
CSingle‐cell time traces of RecA‐mCherrywt fluorescence expressed as percent of generation time of subpopulations segregated by fluorescence VMR. The average fluorescence (solid black lines) and half of the generation time (dashed lines) are indicated. Population statistics (n = 360): 40% (0 < VMR ≤ 1); 44% (1 < VMR ≤ 10); 16% (VMR > 10). The data shown are from three independent experiments.
-
DHistogram showing the distribution of the fluorescence peak time expressed as a percent of generation time. Bars shaded in plum indicate moderately pulsing cells (n = 185), and magenta bars indicate highly pulsing cells (n = 57). Significance by Mann–Whitney U‐test. The data shown are from three independent experiments. Peaks mainly occur between 80% and 100% of the generation time.
-
E, FSingle‐cell size (E) and growth rate (F) averaged over the lifetime of the cell (58 < n < 159). The data shown are from three independent experiments. Black lines indicate means ± SD. Asterisks denote significance by Kruskal–Wallis and Dunn's multiple comparison test: ns, not significant; **P = 0.002; ****P < 0.0001. Cells experiencing more DNA damage (VMR > 10) are slightly larger and grow at slower rates. See also Movie EV5.
RecA pulses and growth rate impact single‐cell fate of M. tuberculosis during ciprofloxacin exposure
Rapid phenotypic switches, independent of genetic changes, have been implicated in bacterial drug tolerance, which is at the root of refractory infections and can eventually lead to drug resistance (Levin–Reisman et al, 2017; Van den Bergh et al, 2017). Here, we probed whether there was an association between preexisting DNA damage events in single M. tuberculosis cells and CIP lethality. We used the same experimental strategy adopted with M. smegmatis (Fig 4), but scaling the temporal kinetics to match the generation time of the pathogen (<Td> = 24 h) (Fig 6A and Movie EV6). We scored four main categories of cellular fates: dead cells with impaired membrane (lysed or SX+, 28.7%); cells with intact membrane (SX−, 69.1%) that never resumed growth; cells that survived only the first treatment (1.9%), referred to as survivors; and cells that survived both treatments (0.3%), referred to as resistant (Fig 6B). Before treatment, survivors exhibited significantly lower growth rates and smaller cell sizes than cells that died during drug exposure, and 22% of survivors stemmed from stalled cells, originated in the pre‐CIP phase (Fig 6B–D). In contrast, cell fate was independent of the microcolony density (Fig 6E).
Figure 6. RecA pulses and growth rate influence M. tuberculosis fate during ciprofloxacin exposure.
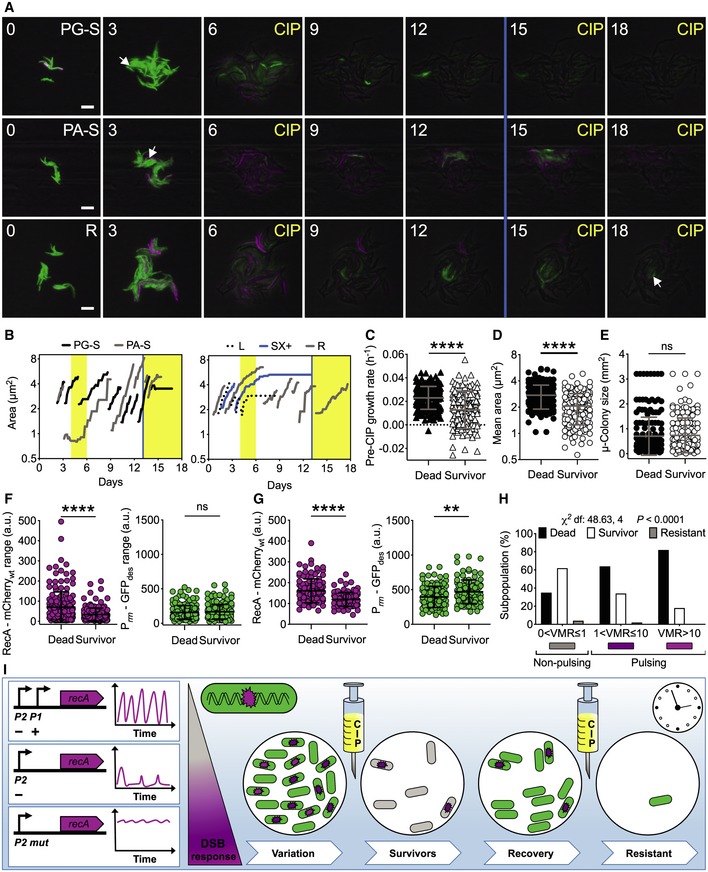
-
ARepresentative time‐lapse image series of the dual reporter of RecA and rRNA expression in exponential phase. Pre‐CIP growing (PG‐S) and arrested (PA‐S) survivors and resistant (R) cells are indicated by arrows. CIP exposure (1 μg/ml, 4X‐MIC) and SX (0.5 μM) death assay (blue line) are indicated. Phase‐contrast (gray), Prrn‐GFPdes (green), and RecA‐mCherrywt (magenta) fluorescence are merged. Fluorescence images of each channel are scaled to the brightest frame. Cells were imaged at 3‐h intervals, and numbers indicate days. Scale bar, 5 μm. See also Movie EV6.
-
BRepresentative time traces of cell lineages with different fates. Vertical bars represent fresh 7H9 medium (white), CIP (yellow), and SX (blue).
-
C–GComparison of growth rate (C), cell size averaged over the lifetime of the cell (D), microcolony size (E) before CIP exposure, fluorescence range over the lifetime of the cell before CIP exposure (F), and mean fluorescence during CIP exposure (G) of cells that died (n = 174) and cells that survived (n = 164). Gray and black lines indicate mean ± SD. The data shown are from two independent experiments. Significance by unpaired Mann–Whitney U‐test: ns, not significant; **P = 0.001; ****P < 0.0001. Most survivors had a moderate growth defect before exposure to ciprofloxacin and lower expression of RecA‐mCherrywt both before and after drug exposure.
-
HSingle‐cell fate as a function of VMR of RecA‐mCherrywt fluorescence prior to CIP exposure. Significance by Chi‐square test of independence: cells that died (n = 174), cells that survived (n = 155), and resistant cells (n = 9). The data shown are from two independent experiments. More survivors arise from the subpopulation that does not experience DNA damage before drug exposure.
-
IThe intact architecture of the recA locus is required for pulsatile expression, which decreases in the absence of positive regulation and is abrogated in the absence of negative regulation. The presence of DNA damage is the main factor triggering RecA pulses. Schematic of M. tuberculosis reporters of DNA damage (magenta) and rRNA expression (green) during CIP treatment. In this study, we classified three subpopulations of bacilli under optimal growth conditions, with no, moderate, or high DNA damage response (magenta gradient scale). The rRNA reporter helped us to visualize the metabolic state of the bacilli and was crucial for early detection of survivors. Following the first drug exposure, 62% of DNA‐healthy cells survived, whereas 64% of cells with moderate and 82% of cells with high DNA damage died, suggesting that preexisting DNA damage in cells treated with ciprofloxacin is detrimental. During the recovery phase, all survivors resumed metabolic activity first and then underwent multiple divisions until they formed a new microcolony. At the time of second drug exposure, most survivors died, indicating that survival of the first drug exposure was not due to drug resistance. However, 0.3% of cells that survived both the first and second drug exposure exhibited drug resistance. In sum, the random state of DNA damage experienced by M. tuberculosis before exposure to a drug targeting DNA replication has consequences for the onset of drug tolerance and the ultimate efficacy of treatment.
Next, we quantified the single‐cell fluorescence before, during, and after exposure to a pulse of CIP, focusing on cells that died and cells that survived (Appendix Fig S6A–D). Before treatment, both categories had comparable levels of Prrn‐GFPdes, whereas the range of RecA‐mCherrywt was higher and more heterogeneous in cells that died, indicating preexisting DNA damage (Fig 6F). During treatment, cells that died continued to exhibit higher levels of RecA‐mCherrywt compared to cells that survived, which maintained higher metabolic activity (Fig 6G). This trend was significant at the population level; however, the single‐cell fluorescence values of the two subpopulations partially overlapped. This implies that even though cells with higher levels of RecA‐mCherry fluorescence are more likely to die than cells with lower levels, there is a minority of cells with high RecA‐mCherry fluorescence that may survive and a minority of cells with low RecA‐mCherry fluorescence that may die. Lastly, we tested whether the pulsing state of RecA‐mCherrywt prior to CIP exposure was associated with cell fate. Consistent with M. smegmatis (Fig 4I and J), M. tuberculosis RecA‐pulsing cells were more likely to die and non‐pulsing cells were more likely to survive (Fig 6H). As a control, we found no relationship between Prrn‐GFPdes and cell fate, or between cell fate and cell age (Appendix Fig S6E and F). In sum, we confirmed that random DNA damage events are detrimental when cells are treated with a drug that targets replication, and that a healthier subset exists that will cope with the drug, possibly giving rise to drug resistance (Fig 6I).
Discussion
The fitness of bacterial populations hinges on their propensity to diversify through both transient epigenetic and heritable genetic changes (Ackermann, 2015). M. tuberculosis exhibits a largely clonal population structure mainly due to lack of lateral gene transfer and limited genetic mutability (Gagneux, 2018). As a result, epigenetic variation per se primarily accounts for the rapid diversification into a broad adaptive spectrum (Dhar et al, 2016; Cadena et al, 2017). Nonetheless, due to its lifestyle and intracellular niche its genome integrity is unceasingly jeopardized by endogenous and environmental insults, which are counteracted by detoxification and DNA repair systems (Gengenbacher & Kaufmann, 2012; Singh, 2017). Here we use RecA as a subcellular gauge for DNA damage (Pennington & Rosenberg, 2007), probe its spatiotemporal dynamics at the single‐cell level and investigate its implications for the fitness of drug‐stressed mycobacteria.
Stress‐response regulons as opposed to housekeeping genes are subject to greater variation even at steady state (Ackermann, 2015). Nonlinear behaviors of transcription factors in conjunction with feedback loops bring about multistable phenotypes within clonal populations, which help responding to complex settings and are associated with transient fitness gain (Smits et al, 2006; Veening et al, 2008). For instance, in Bacillus subtilis alternative sigma factors are expressed in pulses (Park et al, 2018), likewise the tumor suppressor p53 in eukaryotic cells (Lahav et al, 2004). In E. coli, the DNA repair methyltransferase Ada is heterogeneously expressed in stress‐free populations, also conferring discrete responsiveness to alkylating agents (Uphoff et al, 2016). In contrast, the expression of recA in unstressed E. coli cells was found to be either negligible (Friedman et al, 2005) or heterogeneous based on snapshots of bacteria (Kamensek et al, 2010).
Here, by monitoring the behavior of fluorescent reporters over long periods of time, we find that different mycobacterial species exhibit intermittent pulses of recA expression at steady state, resulting in marked cell‐to‐cell variation. We demonstrate that the coexistence of non‐pulsing, moderately, and highly pulsing cells is multifactorial. While in model organisms the regulation of the SOS response is based on a single‐input inhibitory module that is derepressed by ssDNA (Alon, 2007), the recA regulatory network in mycobacteria is more complex and yet enigmatic (Gopaul et al, 2003; Gamulin et al, 2004). RecA binding to ssDNA elicits LexA self‐cleavage and activates the distal promoter (Smollett et al, 2012), whereas transcriptional activators allegedly act on the proximal promoter (Wang et al, 2011; Müller et al, 2018). Lastly, RecX inhibits the nucleoprotein filament formation to shutdown the process (Le et al, 2014). This intricate network resembles a multi‐input module, which typically amplifies subcellular noise leading to cell‐to‐cell variation (Alon, 2007). By deconstructing the recA regulatory region, we show that functional LexA binding is required for pulses to occur and for induction by alkylating agents. Moreover, we observe that ClgR exerts a positive regulation of recA basal expression, consistent with bulk‐cell studies (Wang et al, 2011), and with the induction of Clp proteases (Estorninho et al, 2010; McGillivray et al, 2015), which could assist LexA degradation (Cohn et al, 2011). Moreover, by deleting the proximal regulatory region we severely impair the frequency and magnitude of RecA pulses, in agreement with the presence of a second possible positive regulator, recently identified in PafBC (Müller et al, 2018). PafBC binding to the recA P1 promoter region was found to be inconclusive and less specific than ClgR binding (Wang et al, 2011; Fudrini Olivencia et al, 2017). However, using genome‐wide approaches in M. smegmatis exposed to MMC in conjunction with bioinformatics prediction of PafBC binding motifs, Müller and colleagues reported that the PafBC regulon includes several DNA repair pathways (Müller et al, 2018). From our observations, the presence of both recA regulators is plausible, as we observe an incremental change from ΔClgR strain to the strain devoid of the whole P1 region. In summary, we infer that an intricate network of regulators contributes to recA multistability in mycobacteria.
The question arises whether recA variation is exclusively due to intrinsic fluctuations, as a result of its regulatory structure, or whether it is also deterministically triggered. The positive correlation between the native P2P1recA‐GFPdes and the merodiploid RecA‐mCherrywt reporters implies that pulses arise from the activity of the transcriptional regulators on both loci, and are ultimately mirrored at the protein level. RecA was found to form fluorescent foci and bundles in model organisms (Renzette et al, 2005; Simmons et al, 2007; Lesterlin et al, 2014). In contrast, we observe a patchy localization throughout the cell that becomes more intense and uniform following exposure to DNA‐damaging agents, and disappears before resumption of cell division. The phenomenon is less pronounced in M. tuberculosis, whose overall RecA levels are lower than in M. smegmatis (O'Sullivan et al, 2008). We seldom detect dynamic bundles and intense fluorescence, and only in cells that will die, implying that undue amounts of RecA are harmful, and that RecA degradation (Müller et al, 2018) must be fine‐tuned in line with transcription toward DNA damage resolution. Remarkably, we show that different actors of the DNA damage response, sharing only partial regulation with recA, i.e., ruvC via LexA, radA via ClgR, and ssbA via a putative ClgR operator and interacting with RecA at the protein level (Reddy et al, 2001; Davis et al, 2002; Gamulin et al, 2004) are positively correlated to recA expression in single cells, when tested either at transcriptional or translational level. These results imply that cell‐to‐cell fluorescence variation during normal growth is largely driven by a mutual extrinsic stress signal, resulting from spontaneous DNA damage.
DNA replication and cell division are tightly coordinated processes that ensure transmission of intact genetic material to healthy daughter cells, but can also be a source of genetic variation (Santi et al, 2013; Kieser & Rubin, 2014; Reyes‐Lamothe & Sherratt, 2019). RecA pulses reflect the extent of spontaneous DNA damage taking place inside the cell, mainly during chromosomal replication. In this critical phase of the cell cycle, replication forks are formed and ssDNA is more abundant and conceivably more vulnerable to the onset of intracellular alkylating and oxidative stress (Boshoff & Barry, 2005). Having found that the temporal localization of RecA peaks relative to the cell division time is fairly specific, we speculate that pulses could be associated with accumulation of ssDNA at stalled replication forks, which trigger the SOS response and homologous recombination events (Reyes‐Lamothe & Sherratt, 2019). We also quantify a moderate growth defect in highly pulsing subpopulations. A possible interpretation is that undue response to DNA damage causes the induction of growth inhibiting factors, such as toxic peptides (Vogel et al, 2004; Gupta et al, 2015; Torrey et al, 2016), secondary metabolites that cause quiescence through the stringent response (Amato & Brynildsen, 2015) or proteins that irreversibly interfere with cell division (Fonville et al, 2010; Modell et al, 2014; Crew et al, 2015; Shan et al, 2017).
Stochastic gene expression favors the emergence of phenotypic variants that withstand drugs in the absence of genetic mutations and are implicated in drug persistence, which is also being increasingly associated to drug resistance (Ackermann, 2015; Levin–Reisman et al, 2017; Sebastian et al, 2017). The consensus view argues that cellular quiescence is more likely to promote drug persistence, which was however reported in replicating cells too (Dhar et al, 2016; Van den Bergh et al, 2017). Fluoroquinolones are a case in point of how complex drug persistence can be. The efficacy of fluoroquinolones in bulk assays is inversely proportional to the bacterial metabolic state and cell density, not merely due to target unavailability and decreased permeability (Sarathy et al, 2013; Gutierrez et al, 2017), but also for the ability of both replicating and non‐replicating cells to repair DNA (Volzing and Brynildsen, 2015). In line with this, we observe that single cells cultured in spent medium grow slowly but dynamically respond to CIP by inducing RecA and produce almost 60 times more survivors than cells growing rapidly in fresh medium, an advantage that is lost upon restoration of fresh medium. In model microorganisms, the formation of cells persistent to fluoroquinolones and increased mutation rate were associated with constitutive induction of the SOS response (Dörr et al, 2009). Conversely, M. tuberculosis expresses relatively low levels of recA, also following exposure to fluoroquinolones, and its mutation rate is generally low (O'Sullivan et al, 2008; Zignol et al, 2016; Gagneux, 2018). This could result in a different persistence strategy.
Prompted by the finding that mycobacteria exhibit marked RecA variation during optimal growth, we probed whether this phenomenon was detrimental or beneficial. By monitoring the behavior of individual bacilli before, during, and after CIP treatment, we discovered a mechanism of persistence, which is shared by non‐pathogenic and pathogenic mycobacteria (Fig 6I). Fluoroquinolones act similarly to the most common anticancer strategies that inflict DNA damage to bring cancer cells to death, although a fraction evades the treatment (Fonville et al, 2010; Torgovnick & Schumacher, 2015). This strategy is influenced by stochastic events in mycobacteria. Surprisingly, if a cell is dealing with DNA damage at the time of drug exposure, it mounts a disproportionate response that is more conducive to death, instead of leveraging the active DNA repair arsenal. Conversely, genetically healthy cells mount a moderate response compatible with survival that favors the onset of resistance (Zignol et al, 2016) in a small fraction of M. tuberculosis survivors. Importantly, the more fit M. tuberculosis cells exhibit a moderate growth defect and one‐fifth of them becomes quiescent prior to treatment, implying that different metabolic states are compatible with the persistent phenotype (Manina et al, 2015; Stapels et al, 2018), and that variation in several cellular factors influences the ability to tolerate drug pressure (Wakamoto et al, 2013). In the future, we will delve into the molecular alterations between non‐pulsing and pulsing subsets and how they deal with other drug classes and the host microniche.
Mycobacterial heterogeneity is a major challenge in tuberculosis eradication and intermittent exposure to drugs in vivo fosters persistence (Cadena et al, 2017). Demystifying the probabilistic and deterministic roots of variation will aid not only to clarify the dynamics of drug lethality but also to design strategies that elude persistence and prevent resistance in hard‐to‐treat infections. The search for anti‐persistence strategies is an expanding field, which requires knowledge of the underlying causes (Defraine et al, 2018; Meylan et al, 2018). Targeting variation can help potentiating existing drugs and represents a viable scenario to shorten the antitubercular therapy and to fight other recurrent diseases, such as cancer.
Materials and Methods
Bacterial strains and growth conditions
All cloning procedures and DNA sequencing were carried out using TOP10 chemically competent E. coli grown in LB medium containing the appropriate antibiotic. Transformants with constructs derived from pCR2.1‐TOPO plasmid, and from pND200KmR, pTTP1A, pND200HygR, and pJG1100 vectors were selected, respectively, on 100 μg/ml ampicillin; 50 μg/ml kanamycin for E. coli; 20 μg/ml kanamycin for mycobacteria; 150 μg/ml hygromycin for E. coli; 50 μg/ml hygromycin for mycobacteria; and 50 μg/ml hygromycin plus 20 μg/ml kanamycin. LexA‐His protein induction was achieved in E. coli BL21 (DE3) grown in LB medium containing 50 μg/ml kanamycin and 1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG).
All mycobacterial strains were cultured at 37°C. Middlebrook 7H9 broth was supplemented with 0.5% BSA, 0.2% glucose, 0.085% NaCl, 0.5% glycerol, and 0.05% Tween‐80. Middlebrook 7H10 agar was supplemented with 10% OADC enrichment and 0.5% glycerol. Exponentially growing cultures were obtained from single colonies and aliquots were supplemented with 15% glycerol, stored at −80°C, and used once to start primary cultures.
M. smegmatis and M. tuberculosis were grown in Middlebrook 7H9 broth at 37°C in shaking conditions to mid‐log phase (OD600 0.5–0.8) prior to bulk‐ and single‐cell assays, unless specified otherwise. Primary cultures of the strains containing chromosomal integrative vectors were grown in the presence of the selective antibiotic, which was removed in the secondary cultures used for final experiments.
Strains construction
Oligonucleotides, recombinant plasmids, and strains used in this study are listed in Appendix Table S1, also indicating their respective application.
Transcriptional reporter strains were constructed using either stable or destabilized variants of gfp and mCherry (Hailey et al, 2002; Manina et al, 2015), fused upstream to a mycobacterial Shine‐Dalgarno sequence. Knock‐in strains (KI) were obtained by inserting the fluorescent marker into the chromosomal native locus, downstream the transcriptional start site. KI and knock‐out (KO) strains were generated by two events of homologous recombination using the suicide vector pJG1100. Regions immediately upstream (UPR) and downstream (DNR) the insertion site or the site to be deleted were PCR‐amplified producing amplicons of 600 bp on average, which were flanked by appropriate restriction sites. Genes encoding the fluorescent protein variants were also PCR‐amplified and flanked by restriction sites compatible with the UPR and DNR regions. Amplicons were digested and ligated in pJG1100, and final clones were confirmed by restriction enzyme profiling. M. smegmatis was transformed by electroporation (2,500 V; 25 μF; 1,000 Ω; 2 mm path), and transformants were selected on Middlebrook 7H10 agar containing 50 μg/ml hygromycin and 20 μg/ml kanamycin. The first homologous recombination event was confirmed by PCR analysis of genomic DNA extracted from single colonies. Confirmed strains were grown in 7H9 medium devoid of antibiotics, and serial dilutions were plated on Middlebrook 7H10 agar containing 5% sucrose, to select for the second event of homologous recombination. Successful recombination events, resulting either in the insertion of the gene coding for a fluorescent protein or in the deletion of a targeted chromosomal region, were confirmed by PCR analysis of genomic DNA extracted from single colonies.
Final KI and KO strains were named: GMS2 (P2P1recA‐GFPdes); GMS3 (P2P1recA‐GFPwt); GMS9 (P2wt‐mCherry des‐P1wt‐gfp des); GMS13 (P2P1recA‐GFPdes _PradA‐mCherrydes); GMS16 (P2P1recA‐GFPdes _PruvC‐mCherrydes); GMS10 (P2wt‐mCherry desΔP1); GMS11 (P2P1recA‐GFPdes_ΔClgR) and GMS15 (P2mut‐mCherry desΔP1).
Translational reporter strains were constructed using stable variants of mCherry wt and mCitrine wt (Hailey et al, 2002; Reddy et al, 2001) fused upstream to an oligopeptide linker, so as not to affect the folding of the protein to be tagged. To construct the recA translational reporters both in M. smegmatis and in M. tuberculosis, the respective recA upstream regulatory region and the recA open reading frame were fused in‐frame at the C‐terminus to the mCherry wt open reading frame, and cloned into the chromosomal integrative vector pND200. The final vectors were confirmed by restriction enzyme profiling and inserted by electroporation into the chromosome of M. smegmatis P2P1recA‐GFPdes strain or M. tuberculosis Prrn‐GFPdes strain (Manina et al, 2015), via the L5 phage attachment site attB (Pena et al, 1996). The recA‐merodiploid P2P1recA‐GFPdes_attB::P2P1recA‐linker‐mCherrywt M. smegmatis strain was named GMS17, and the Prrn‐GFPdes_attB::P2P1recA‐linker‐mCherrywt M. tuberculosis strain was named LKT2.
To construct the ssbA translational reporter in M. smegmatis, the ssbA locus fused at its C‐terminus to the yellow fluorescent marker mCitrine wt was cloned into a plasmid carrying the Tweety phage attachment site attT (Pham et al, 2007), and co‐transformed in M. smegmatis already carrying the recA translational reporter at the attB site, generating a dual‐translational reporter of RecA‐mCherrywt (at the attB site) and SSBa‐mCitrinewt (at the attT site), which was named GMS25.
M. smegmatis expressing stable and unstable GFP and mCherry fluorescent protein variants, under the control of the UV15 strong promoter, from the attB site were named GMS4, GMS6, GMS7, GMS8, and GMS18.
Measurement of proteins half‐life
GMS4, GMS6, GMS7, and GMS8 strains were cultured in Middlebrook 7H9 broth to exponential phase (OD600 0.5) and labeled with 1.5 mCi of [35S] EasyTag Express Protein Labeling Mix (PerkinElmer) for 1 h. Cells were collected at 4,200 g for 10 min at 4°C, washed once, and resuspended in an equal volume of 7H9 medium supplemented with 100 μM of cold L‐methionine and L‐cysteine. Culture volumes corresponding to OD600 10.0 were withdrawn at each time point and cells collected by centrifugation at 4,200 g for 10 min at 4°C. Pellets were washed with 0.9 ml of protein buffer containing 10 mM Tris–Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% glycerol, and 1× Protease Inhibitor Cocktail (Roche), centrifuged at 13,500 g for 5 min at 4°C, resuspended in 350 μl of protein buffer, and transferred to a vial prefilled with 0.5‐mm glass beads (Precellys). Cells suspensions were lysed by bead‐beating three times at 4,234 g for 60 s, with 30‐s intervals on ice. Cell lysates were centrifuged at 13,500 g for 10 min to remove cell debris, and total protein concentration was quantified using Bradford reagent (Sigma). Fluorescent proteins were immunoprecipitated using either GFP‐Trap_A or RFP‐Trap_A beads (Chromotek). Beads were washed three times with ice‐cooled protein buffer and incubated with 250 μg of total protein extract for 4 h at 4°C. Beads were washed once with ice‐cooled protein buffer and a second time with protein buffer containing 500 mM NaCl. GFP and mCherry immunocomplexes were dissociated from the beads using 1/10 (vol/vol) of 0.2 M glycine buffer pH 2.5 and neutralized using 0.1 M Tris‐base pH 10.8. Equal amounts of immunoprecipitated proteins were run on a NuPAGE 4–12% Bis‐Tris Gel (Life Technologies), Coomassie stained, exposed to an Amersham Hyperfilm MP (GE Healthcare). Band intensities were measured from the autoradiography film using ImageJ software (Schneider et al, 2012). The proteins half‐life times were measured by subtracting the dilution rate from the decay rate of intensity of the bands.
Growth‐curve assay
Exponentially growing primary cultures were diluted to OD600 0.05 in 25 ml of pre‐warmed Middlebrook 7H9 broth and incubated at 37°C for 24 h in the case of M. smegmatis and for 30 days in the case of M. tuberculosis, during which OD600 was measured at regular intervals.
MIC evaluation by resazurin assay
Exponentially growing cultures were diluted to OD600 0.005 using pre‐warmed Middlebrook 7H9 broth and used to fill a 96‐well plate with 100 μl of cell suspension per well. In the first well of each row, where a different drug was tested, 200 μl of cell suspension was dispensed and the highest concentration of drug was added and then twofold serially diluted, except for the last well that was used as the positive control. M. smegmatis plates were incubated at 37°C for 24 h, and M. tuberculosis plates were incubated at 37°C for 1 week. On completion of the incubation period, 5 μl of 0.01% resazurin was added to each well and reincubated at 37°C for 24 h before readout. Wells where the blue color turned to pink indicated bacterial viability, whereas wells with unchanged blue color indicated cidality. The MIC was scored as the lowest drug concentration causing cidality.
Real‐time quantitative PCR
M. smegmatis WT and fluorescent reporter strains were cultured in Middlebrook 7H9 broth until OD600 0.2 and split in two samples, which were reincubated at 37°C for 2 h either without stress or in the presence of 0.5 μg/ml MMC. At the end of the incubation period, culture volumes corresponding to OD600 5.0 were withdrawn and cells collected by centrifugation at 4,200 g for 10 min at 4°C. Cell pellets were resuspended in 500 μl TRIzol (Ambion) and 0.1% polyacryl carrier (MRC) and transferred to vials prefilled with glass beads (Precellys). Cell suspensions were lysed by bead‐beating three times at 4,234 g for 60 s, with 30‐s intervals on ice and centrifuged at 9,300 g for 2 min to recover the supernatant. RNA was precipitated with 1/10 (vol/vol) of 3 M NaOAc pH 5.2 and 0.7 volumes of isopropanol, washed in 75% ethanol, and resuspended in 50 μl of DEPC‐treated water. DNA was depleted from the RNA samples by dual treatment with Turbo DNase (Ambion). cDNA was generated starting from 200 ng of RNA using the SuperScript First‐Strand Synthesis System and random hexamers (Invitrogen), according to manufacturer's instructions. Primers specific for sigA, recA, radA, ruvC, gfp, and mCherry are listed in Appendix Table S1. qRT–PCR products were carried out using the SYBR Green PCR Master Mix (Applied Biosystems), 0.5 μM primers, and 2.2 μl cDNA. Absolute quantifications were run on ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). The melting curves were checked to confirm amplicon specificity. Transcripts copy numbers were calculated from the standard curves generated with each primer pair using serial dilutions of M. smegmatis genomic DNA. Measurements were performed at least in duplicate for each biological sample.
LexA overexpression and purification
Primary cultures were obtained from single colonies of GME53 and E. coli transformed with pET28a+ inoculated into LB selective broth and diluted to OD600 0.1 until they reached OD600 0.6. Secondary cultures were induced with 1 mM IPTG at 37°C for 2 h, and 200 ml were collected at 4,200 g for 15 min at 4°C obtaining about 0.5 g of wet cell pellet. Pellets were processed under native conditions in 2.5 ml of lysis buffer containing 50 mM NaH2PO4, 300 mM NaCl, pH 8.0, 1 mg/ml lysozyme, 5 μg/ml DNase I, and 1× Protease Inhibitor Cocktail (Roche). Cell suspensions were incubated on ice for 30 min before 10 sonication cycles of 15 s at 40% amplitude, with 15‐s intervals on ice. Cell lysates were centrifuged at 10,000 g for 30 min at 4°C, the supernatant was transferred to a clean tube, adding 0.5 g of PrepEase His‐Tagged High Specificity Purification Resin (Affymetrix, UBS Products), and incubated in an orbital shaker for 20 min at 4°C. The resin was sedimented by centrifugation at 500 g for 2 min, and the supernatant discarded. The resin was washed three times for 5 min in washing buffer containing 50 mM NaH2PO4, 300 mM NaCl, 5% glycerol, pH 8.0, and 1× Protease Inhibitor Cocktail (Roche) and eluted three times for 5 min with 0.5 ml of elution buffer containing 50 mM NaH2PO4, 300 mM NaCl, 5% glycerol, 250 mM imidazole, pH 8.0, and 1× Protease Inhibitor Cocktail (Roche). Elutions were dialyzed overnight against 1 l dialysis buffer containing 50 mM Tris–Cl pH 7.5, 50 mM NaCl and 5% glycerol, using a dialysis membrane of 8K MWCO. Samples were checked by SDS–PAGE, quantified using Bradford reagent (Sigma), and stored at −80°C.
Electrophoretic mobility shift assay
Oligonucleotides containing either wild‐type or mutated M. smegmatis recA Cheo box variants are listed in Appendix Table S1 and were either unlabeled or biotinylated. DNA duplexes were produced in 50 μl of annealing reaction containing 10 mM Tris pH 8.0, 50 mM NaCl, 1 mM EDTA, and 25 pmol of each primer pair, incubating at 95°C for 5 min then cooling down at room temperature, and stored on ice. Binding reactions were prepared, using the LightShift Chemiluminescent EMSA Kit (Thermo Scientific), in 20 μl containing 1× binding buffer, 2.5% glycerol, 5 mM MgCl2, 50 ng/μl Poly (dI•dC), 0.05% NP‐40, 20 fmol of either wild‐type or mutated biotinylated DNA duplex, LexA protein extract ranging from 0 to 2.5 pmol per reaction, and 4 pmol of the corresponding cold DNA duplex in a control reaction. Binding reactions were gently mixed and incubated at room temperature for 30 min, and then, 5 μl of 5× loading buffer was added. Protein–DNA complexes were resolved from free DNA duplexes on a 6% native polyacrylamide gel by electrophoresis in 0.5× TBE buffer at 100 V for 45 min (until the bromophenol blue dye had migrated to three quarters the length of the gel), and transferred to positively charged nylon membrane using the iBlot Transfer Apparatus (Invitrogen). The biotinylated DNA duplexes and DNA–protein complexes were detected using the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Scientific) and an Amersham Hyperfilm MP (GE Healthcare).
Western blot assay
Western blot assays were carried out on whole‐cell extracts from exponentially growing M. smegmatis WT, GMS2, GMS17, M. tuberculosis WT and LKT2, and from the same strains exposed to 0.5 μg/ml MMC for 2 h in the case of M. smegmatis, or exposed to 0.25 μg/ml MMC for 24 h in the case of M. tuberculosis. Culture volumes corresponding to OD600 10.0 were withdrawn and cells collected by centrifugation at 4,200 g for 10 min at 4°C. Pellets were washed with 1 ml of protein buffer containing 50 mM Tris–Cl pH 7.5, 50 mM NaCl, 0.5 mM EDTA, 5% glycerol, and 1× Protease Inhibitor Cocktail (Roche), centrifuged at 13,500 g for 5 min at 4°C, resuspended in 300 μl of protein buffer, and transferred to vials prefilled with 0.5‐mm glass beads (Precellys). Cell suspensions were lysed by bead‐beating three times at 4,234 g for 60 s, with 30‐s intervals on ice. Cell lysates were centrifuged at 13,500 g for 10 min to remove cell debris. Additionally, M. tuberculosis lysates were filtered through a 0.2‐μm centrifugal device (Pall Life Sciences). Cell‐free extracts were quantified using Bradford reagent (Sigma), and 25 μg were used in Western blot assays. Following electrophoresis at 100 V for 2 h, proteins were transferred onto a PVDF membrane using the iBlot Transfer Apparatus (Invitrogen) and equal loading was confirmed by Ponceau S staining (Sigma). Membranes were blocked with 5% skim milk in 1× TBS and 0.1% Tween‐20 (TBS‐T) at 4°C overnight and washed five times with TBS‐T for 5 min. Primary antibodies were diluted in TBS‐T and 1% skim milk as follows: anti‐GFP (1:5,000); anti‐tRFP (1:5,000); anti‐Ag85 (1:1,000); and anti‐HSP 65 (1:5,000), and incubated at room temperature for 2 h. Membranes were washed five times with TBS‐T for 5 min and incubated with secondary antibodies diluted in TBS‐T and 1% skim milk as follows: HRP‐conjugated anti‐rabbit (1:2,000) and HRP‐conjugated anti‐mouse (1:10,000). Membranes were incubated at room temperature for 2 h and washed five times with TBS‐T for 5 min. Proteins were detected using the Pierce ECL Western Blotting Substrate (Thermo Scientific) and an Amersham Hyperfilm MP (GE Healthcare).
Snapshot microscopy
Phase contrast and fluorescence snapshot imaging were carried out using an inverted DeltaVision Elite Microscope (GE Healthcare) equipped with an UPLFLN100XO2/PH3/1.30 objective (Olympus). Exponential‐ and stationary‐phase GMS2 batch cultures were stained with 20 μM DRAQ5 for 1 h, and 0.5 μl of cells were dispensed between two #1.5 coverslips. Exposure conditions: phase 50% T, 100 ms; FITC (Ex 475/28, Em 525/48) 50% T, 150 ms; and Cy5 (Ex 632/22, Em 679/34) 100% T, 1 s. Exponential‐ and 56‐days stationary‐phase LKT2 batch cultures and exponential‐phase LKT2 batch cultures exposed for 24 h to 0.25 μg/ml MMC; 5 mM H2O2; nutrient starvation in Hartmans‐de Bont minimal medium (HdB) (Smeulders et al, 1999); HdB using 2 g/l sodium propionate as a sole carbon source; and 10% human serum were dispensed between two #1.5 coverslips and sealed with glue. Exposure conditions: phase 50% T, 150 ms; FITC (Ex 475/28, Em 525/48) 50% T, 100 ms; and TRITC (Ex 542/27, Em 597/45), 50% T, 250 s.
Microfabrication of the hexa‐device
The hexa‐device was designed using Layout Editor software, and hexa‐device.gds file was sent to SELBA S.A. for photomask manufacturing. A 100‐mm‐diameter silicon wafer was placed on a hot plate at 200°C for 30 min. SU8 2150 photoresist was spin‐coated on the wafer at 350 g for 30 s with an acceleration of 8 g/s, soft‐baked at 65°C for 10 min followed by incubation at 95°C for 60 min. Lithography was achieved using a MA/BA6 mask aligner (SUSS MicroTec). The coated wafer was exposed to a 12 mW/cm2 UV light for 37 s and baked at 65°C for 5 min followed by incubation at 95°C for 30 min. The wafer was developed by immersion in PGMEA twice for 10 min, rinsed with PGMEA, washed with isopropanol, and air‐dried with nitrogen. The microstructures were hard‐baked at 180°C for 2 h and silanized in a vacuum chamber overnight.
For soft‐lithography, the wafer was secured to the bottom of a square petri dish, covered with a degassed mixture of 120 g of Sylgard 184 pre‐polymer and 12 g of cross‐linker, and placed inside a vacuum chamber until degassing was completed (about 30 min). The petri dish was incubated at 80°C overnight and then transferred to room temperature. The polymerized PDMS layer was gently separated from the silicon mold, and the device was cut out in the same size as a 25 × 50 mm coverslip. The device was punched laterally to insert the input and output tubing, which were connected by means of metal connectors, fixed with a drop of PDMS, and incubated again at 80°C for 2 h. Female luer connectors were inserted into the inlets to be able to screw on 50‐ml syringes.
Membrane preparation
The Spectra/Por 6 Dialysis Tubing, 25 kD MWCO, was cut into a few 10‐cm strips and washed twice in ddH2O for 2 h with gentle stirring. With the help of a scalpel blade and a glass support, strips of 11 × 50 mm in size were cut and transferred to a clean beaker containing ddH2O for 1 h with gentle stirring. The membrane strips were fished and dried from the excess water and transferred to a clean beaker, which contained methanol under a chemical hood, and kept in gentle stirring overnight. The membrane strips were fished and stacked between sheets of filter paper, clamped between two glass slides, and dried in a vacuum chamber for 3 days.
Time‐lapse microscopy
Exponential‐phase batch cultures were passed through a 5‐μm filter, pre‐equilibrated with 1 ml of fresh Middlebrook 7H9 Broth, to remove clumps. Stationary‐phase batch cultures were diluted 1:10,000 using spent medium. Spent medium was recovered from stationary‐phase cultures by centrifugation at 4,200 g for 10 min at room temperature and cleared from the macroscopic cellular debris by filtration using a 0.22‐μm filter. A 25 × 50 mm #1 coverslip was placed on the metal frame and centered (Fig 1B). The hexa‐device was positioned and centered on the acrylic layer with the channels facing upwards. Two membranes were wetted with culture medium and positioned in correspondence of the two serpentine channels. 2 μl of bacterial suspensions was dispensed at each of the six observation areas of the device and let settle for 5 min. The device was turned upside down, superimposed on the glass coverslip, and the stack was secured with eight screws. Two syringes containing culture medium were connected to the inlets and driven by a syringe pump set at 15 μl/min. The assembled device was mounted on the microscope stage of a DeltaVision Elite Microscope (GE Healthcare). Images were recorded at 15‐ or 30‐min intervals for M. smegmatis and at 3‐h intervals for M. tuberculosis, using an UPLFLN100XO2/PH3/1.30 objective (Olympus) and a high‐speed sCMOS camera—2,560 × 2,160 pixels, pixel size 6.5 × 6.5 μm, 15 bit, spectral range of 370–1,100 nm. Exposure conditions for M. smegmatis: phase 50% T, 150 ms; FITC (Ex 475/28, Em 525/48) 50% T, 150 ms; YFP (Ex 513/17, Em 548/22) 50% T, 150 ms; and mCherry (Ex 575/25, Em 625/45) 50% T, 150 ms. Exposure conditions for M. tuberculosis: phase 50% T, 150 ms; FITC (Ex 475/28, Em 525/48) 50% T, 100 ms; and TRITC (Ex 542/27, Em 597/45), 50% T, 250 s. Drugs or SX were added to the medium flow when required. SX was recorded on the same green fluorescence channel due to a spectral overlap. From 180 to 300 XY, fields were imaged per experiment and at least two microscopy experiments per sample type and condition were conducted and analyzed. In particular, different areas of the microfluidic system were used to seed either different reporter strains or independent cultures of the same reporter strain. All experimental configurations were repeated independently at least twice. For each reporter strain, we analyzed individual XY fields from the same experiment (technical repeats) as well as from different experiments (biological and technical repeats). Single‐cell analysis data from independent experiments were compared to verify reproducibility and pooled to generate plots.
Single‐cell analysis
Single‐cell segmentation of snapshot and time‐lapse images was performed using the ROI Manager Macro of ImageJ 1.51s software (Schneider et al, 2012). The Selection Brush Tool was used to drag polygons over the profile of individual cells, adjusting the thickness between 7 and 9 for M. smegmatis, and between 3 and 5 for M. tuberculosis. Polygons were used to extract the size of individual cells and the total fluorescence, expressed as the sum of the pixels intensity normalized to the area of the polygon. The background signal was also subtracted at each measurement. The cell annotation with respect to the position in the lineage and pole age was manually attributed through a binary code, and a customized R script was used for data post‐processing from the.csv files extracted from ROI Manager. The standard deviation derived from DRAQ5‐fluorescence values of each pixel within the area of a cell was used to distinguish evenly spread fluorescence (low standard deviation) from localized fluorescence (high standard deviation), and the latter was used as a proxy for DNA condensation.
Statistics
Prism 7.0 (GraphPad Software) was used to generate plots and to perform statistical analyses. D'Agostino‐Pearson normality test was used to test whether values derived from Gaussian distributions. Lognormal and sum of three Gaussians were used to evaluate the distributions of subpopulations (Fig 1C and D). Spearman r correlation coefficient was computed on (x, y) data sets, unless otherwise specified. Mann–Whitney U‐test was used to compare two datasets, and nonparametric paired Wilcoxon test to compare siblings, unless otherwise specified. Kruskal–Wallis followed by Dunn's multiple comparison test was computed to compare more than two datasets, unless otherwise specified. Chi‐square test of independence was used to analyze contingency tables, unless otherwise specified. P values < 0.05 were considered significant.
Calculation of parameters and subpopulations
Growth rate was calculated by fitting an exponential curve to single‐cell size measurements over the generation time (Td) of the cell from birth to division. The fluorescence range was expressed as the difference between maximum and minimum fluorescence values from birth to division. Subpopulations were identified by computing the single‐cell index of dispersion of fluorescence or noise strength (Raser & O'Shea, 2004), equal to ratio of the variance (squared standard deviation) to the mean fluorescence (VMR) calculated from birth to division. VMR is a measure of the variability of a distribution normalized to the mean of the distribution and helps quantifying the level of dispersion of a given dataset. A distribution is underdispersed for VMR values between 0 and 1 and overdispersed for VMR values greater than 1. We used this parameter to uniquely define the extent of fluorescence variation or dispersion during the entire generation time of a cell, and to understand whether a cell had homogeneous fluorescence during its lifetime, which is suggestive of the absence of pulsing, or if it had fluorescence fluctuations during its lifetime, which is suggestive of pulsing. In particular, we classified the mycobacterial populations in three subsets based on the following cutoffs: underdispersed or non‐pulsing (0 < VMR ≤ 1); dispersed or moderately pulsing (1 < VMR ≤ 10); and highly dispersed or highly pulsing (VMR > 10). Pulses within lineages were identified by computing the area under the curve integrated measurement of the normalized fluorescence over 24 h, using Prism 7.0 and setting the following parameters: baseline (y = 1); minimum peak height (y ≥ 2); minimum peak width defined by at least 2 adjacent points; peaks above the baseline. Cellular age was defined by the age of the cell's poles. To define the age of a given cell, at least two prior generations must be known. From the division of any cell (generation 0) of unknown age (n), two cells are generated (generation 1), endowed with a preexisting older pole of unknown age (n + 1), and a newer pole of known age (0) produced at division. In turn, the daughter cells (generation 2) derived from a mother cell (generation 1) are datable as follows: the so‐called “old” daughter cell has an older pole of unknown age (n + 2) and a younger pole of known age (0), and the so‐called “new” daughter cell has an older pole of known age (1) and a younger pole of known age (0).
Author contributions
GM conceived the study and designed the methodology. GM conducted the experiments and performed data analysis and interpretation. AG contributed to data analysis and protein experiments. LKS constructed the M. tuberculosis reporter strain. ND provided some reagents. GM, ND, and JDM discussed the data. GM wrote the manuscript. GM and ND edited the manuscript. GM, ND, and JDM revised the manuscript. All authors commented on the manuscript. GM and JDM supervised the study and secured funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File
Acknowledgements
We would like to thank the Center of Bioinformatics, Biostatistics and Integrative Biology of the Institut Pasteur for the support in writing the R script for data processing. We are grateful to Pietro Slavich (LPTHE) for comments and discussion. We thank Olivia Mariani (EPFL) for her initial advice on single‐cell segmentation. This work was initially funded by a grant to J.D.M. from the Innovative Medicines Initiative (115337), a joint undertaking of the EU 7th Framework Programme and EFPIA. This work was subsequently funded by grants to G.M. from the French National Research Agency (ANR‐16‐TERC‐0011‐01 and ANR‐17‐CE11‐0007‐01); the French Government's Investments for the future, Laboratoire d'Excellence “Integrative Biology of Emerging Infectious Diseases” (ANR‐10‐LABX‐62‐IBEID); and from the Fondation pour la Recherche Médicale (FRM ING20160435202). Work in G.M.'s laboratory also received financial support from the Institut Pasteur. The content is the sole responsibility of the authors and does not necessarily represent the views of the funders.
The EMBO Journal (2019) 38: e101876
References
- Ackermann M (2015) A functional perspective on phenotypic heterogeneity in microorganisms. Nat Rev Microbiol 13: 497–508 [DOI] [PubMed] [Google Scholar]
- Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L (2011) Drug tolerance in replicating mycobacteria mediated by a macrophage−induced efflux mechanism. Cell 145: 39–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U (2007) Network motifs: theory and experimental approaches. Nat Rev Genet 8: 450–461 [DOI] [PubMed] [Google Scholar]
- Amato SM, Brynildsen MP (2015) Persister heterogeneity arising from a single metabolic stress. Curr Biol 25: 2090–2098 [DOI] [PubMed] [Google Scholar]
- Andersen JB, Sternberg C, Poulsen LK, Bjorn SP, Givskov M, Molin S (1998) New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl Environ Microbiol 64: 2240–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharoglu Z, Mazel D (2014) SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev 38: 1126–1145 [DOI] [PubMed] [Google Scholar]
- Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S (2004) Bacterial persistence as a phenotypic switch. Science 305: 1622–1625 [DOI] [PubMed] [Google Scholar]
- Bergmiller T, Andersson AMC, Tomasek K, Balleza E, Kiviet DJ, Hauschild R, Tkacik G, Guet CC (2017) Biased partitioning of the multidrug efflux pump AcrAB−TolC underlies long−lived phenotypic heterogeneity. Science 356: 311–315 [DOI] [PubMed] [Google Scholar]
- Boshoff HI, Barry CE III (2005) Tuberculosis − metabolism and respiration in the absence of growth. Nat Rev Microbiol 3: 70–80 [DOI] [PubMed] [Google Scholar]
- Cadena AM, Fortune SM, Flynn JL (2017) Heterogeneity in tuberculosis. Nat Rev Immunol 17: 691–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DE, Collins JJ (2014) Tunable protein degradation in bacteria. Nat Biotechnol 32: 1276–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn MT, Kjelgaard P, Frees D, Penades JR, Ingmer H (2011) Clp−dependent proteolysis of the LexA N−terminal domain in Staphylococcus aureus . Microbiology 157: 677–684 [DOI] [PubMed] [Google Scholar]
- Crew R, Ramirez MV, England K, Slayden RA (2015) MadR1, a Mycobacterium tuberculosis cell cycle stress response protein that is a member of a widely conserved protein class of prokaryotic, eukaryotic and archeal origin. Tuberculosis 95: 251–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EO, Dullaghan EM, Rand L (2002) Definition of the Mycobacterial SOS Box and Use To Identify LexA−Regulated Genes in Mycobacterium tuberculosis . J Bacteriol 184: 3287–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defraine V, Fauvart M, Michiels J (2018) Fighting bacterial persistence: current and emerging anti−persister strategies and therapeutics. Drug Resist Updat 38: 12–26 [DOI] [PubMed] [Google Scholar]
- Dhar N, Manina G (2015) Single−cell analysis of mycobacteria using microfluidics and time−lapse microscopy. Methods Mol Biol 1285: 241–256 [DOI] [PubMed] [Google Scholar]
- Dhar N, McKinney J, Manina G (2016) Phenotypic heterogeneity in Mycobacterium tuberculosis . Microbiol Spectr 4: 671–697 [DOI] [PubMed] [Google Scholar]
- Dörr T, Lewis K, Vulic M (2009) SOS response induces persistence to fluoroquinolones in Escherichia coli . PLoS Genet 5: e1000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estorninho M, Smith H, Thole J, Harders−Westerveen J, Kierzek A, Butler RE, Neyrolles O, Stewart GR (2010) ClgR regulation of chaperone and protease systems is essential for Mycobacterium tuberculosis parasitism of the macrophage. Microbiology 156: 3445–3455 [DOI] [PubMed] [Google Scholar]
- Flentie K, Garner AL, Stallings CL (2016) Mycobacterium tuberculosis transcription machinery: ready to respond to host attacks. J Bacteriol 198: 1360–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonville NC, Bates D, Hastings PJ, Hanawalt PC, Rosenberg SM (2010) Role of RecA and the SOS response in thymineless death in Escherichia coli . PLoS Genet 6: e1000865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N, Vardi S, Ronen M, Alon U, Stavans J (2005) Precise temporal modulation in the response of the SOS DNA repair network in individual bacteria. PLoS Biol 3: e238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudrini Olivencia B, Müller AU, Roschitzki B, Burger S, Weber‐Ban E, Imkamp F (2017) Mycobacterium smegmatis PafBC is involved in regulation of DNA damage response. Sci Rep 7: 13987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S (2018) Ecology and evolution of Mycobacterium tuberculosis . Nat Rev Microbiol 16: 202–213 [DOI] [PubMed] [Google Scholar]
- Gamulin V, Cetkovic H, Ahel I (2004) Identification of a promoter motif regulating the major DNA damage response mechanism of Mycobacterium tuberculosis . FEMS Microbiol Lett 238: 57–63 [DOI] [PubMed] [Google Scholar]
- Gengenbacher M, Kaufmann SH (2012) Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev 36: 514–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopaul KK, Brooks PC, Prost JF, Davis EO (2003) Characterization of the two Mycobacterium tuberculosis recA promoters. J Bacteriol 185: 6005–6015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta KR, Kasetty S, Chatterji D (2015) Novel functions of (p)ppGpp and Cyclic di−GMP in mycobacterial physiology revealed by phenotype microarray analysis of wild−type and isogenic strains of Mycobacterium smegmatis . Appl Environ Microbiol 81: 2571–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A, Jain S, Bhargava P, Hamblin M, Lobritz MA, Collins JJ (2017) Understanding and sensitizing density−dependent persistence to quinolone antibiotics. Mol Cell 68: 1147–1154.e3 [DOI] [PubMed] [Google Scholar]
- Hailey DW, Davis TN, Muller EG (2002) Fluorescence resonance energy transfer using color variants of green fluorescent protein. Meth Enzymol 351: 34–49 [DOI] [PubMed] [Google Scholar]
- Hasty J, McMillen D, Collins JJ (2002) Engineered gene circuits. Nature 420: 224–230 [DOI] [PubMed] [Google Scholar]
- Hung‐Yi W, Chih‐Hao L, Hung‐Wen L (2017) RecA‐SSB interaction modulates RecA nucleoprotein filament formation on SSB‐wrapped DNA. Sci Rep 7: 11876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Fukui K, Fujii Y, Nakagawa N, Yano T, Kuramitsu S, Masui R (2017) The Lon protease‐like domain in the bacterial RecA paralog RadA is required for DNA binding and repair. J Biol Chem 292: 9801–9814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kærn M, Elston TC, Blake WJ, Collins JJ (2005) Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet 6: 451–464 [DOI] [PubMed] [Google Scholar]
- Kamensek S, Podlesek Z, Gillor O, Zgur−Bertok D (2010) Genes regulated by the Escherichia coli SOS repressor LexA exhibit heterogeneous expression. BMC Microbiol 10: 283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser KJ, Rubin EJ (2014) How sisters grow apart: mycobacterial growth and division. Nat Rev Microbiol 12: 550–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, Sigal A, Geva–Zatorsky N, Levine AJ, Elowitz MB, Alon U (2004) Dynamics of the p53–Mdm2 feedback loop in individual cells. Nat Genet 36: 147–150. [DOI] [PubMed] [Google Scholar]
- Le S, Chen H, Zhang X, Chen J, Patil KN, Muniyappa K, Yan J (2014) Mechanical force antagonizes the inhibitory effects of RecX on RecA filament formation in Mycobacterium tuberculosis . Nucleic Acids Res 42: 11992–11999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesterlin C, Ball G, Schermelleh L, Sherratt DJ (2014) RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature 506: 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin–Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ (2017) Antibiotic tolerance facilitates the evolution of resistance. Science 355: 826–830 [DOI] [PubMed] [Google Scholar]
- Manina G, Dhar N, McKinney JD (2015) Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17: 32–46 [DOI] [PubMed] [Google Scholar]
- McGillivray A, Golden NA, Kaushal D (2015) The Mycobacterium tuberculosis Clp gene regulator is required for in vitro reactivation from hypoxia–induced dormancy. J Biol Chem 290: 2351–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan S, Andrews IW, Collins JJ (2018) Targeting antibiotic tolerance, pathogen by pathogen. Cell 172: 1228–1238 [DOI] [PubMed] [Google Scholar]
- Mills KV, Lew BM, Jiang S, Paulus H (1998) Protein splicing in trans by purified N– and C–terminal fragments of the Mycobacterium tuberculosis RecA intein. Proc Natl Acad Sci USA 95: 3543–3548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Kambara TK, Perchuk BS, Laub MT (2014) A DNA damage–induced, SOS–independent checkpoint regulates cell division in Caulobacter crescentus . PLoS Biol 12: e1001977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller AU, Imkamp F, Weber–Ban E (2018) The mycobacterial LexA/RecA–independent DNA damage response is controlled by PafBC and the Pup–proteasome system. Cell Rep 23: 3551–3564 [DOI] [PubMed] [Google Scholar]
- Nautiyal A, Rani PS, Sharples GJ, Muniyappa K (2016) Mycobacterium tuberculosis RuvX is a Holliday junction resolvase formed by dimerisation of the monomeric YqgF nuclease domain. Mol Microbiol 100: 656–674 [DOI] [PubMed] [Google Scholar]
- O'Sullivan DM, Hinds J, Butcher PD, Gillespie SH, McHugh TD (2008) Mycobacterium tuberculosis DNA repair in response to subinhibitory concentrations of ciprofloxacin. J Antimicrob Chemother 62: 1199–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Dies M, Lin Y, Hormoz S, Smith–Unna SE, Quinodoz S, Hernandez–Jimenez MJ, Garcia–Ojalvo J, Locke JCW, Elowitz MB (2018) Molecular time sharing through dynamic pulsing in single cells. Cell Syst 6: 216–229.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena CE, Stoner JE, Hatfull GF (1996) Positions of strand exchange in mycobacteriophage L5 integration and characterization of the attB site. J Bacteriol 178: 5533–5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington JM, Rosenberg SM (2007) Spontaneous DNA breakage in single living Escherichia coli cells. Nat Genet 39: 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham TT, Jacobs‐Sera D, Pedulla ML, Hendrix RW, Hatfull GF (2007) Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site‐specific integration vectors for mycobacteria. Microbiology 153: 2711–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu Y, Zhao Z, Li Y, Zou J, Ma Q, Zhao Y, Ke Y, Zhu Y, Chen H, Baker MAB et al (2016) Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol Cell 62: 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand L, Hinds J, Springer B, Sander P, Buxton RS, Davis EO (2003) The majority of inducible DNA repair genes in Mycobacterium tuberculosis are induced independently of RecA. Mol Microbiol 50: 1031–1042 [DOI] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK (2004) Control of stochasticity in eukaryotic gene expression. Science 304: 1811–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MS, Guhan N, Muniyappa K (2001) Characterization of single‐stranded DNA‐binding proteins from Mycobacteria. The carboxyl‐terminal of domain of SSB is essential for stable association with its cognate RecA protein. J Biol Chem 276: 45959–45968 [DOI] [PubMed] [Google Scholar]
- Renzette N, Gumlaw N, Nordman JT, Krieger M, Yeh SP, Long E, Centore R, Boonsombat R, Sandler SJ (2005) Localization of RecA in Escherichia coli K−12 using RecA−GFP. Mol Microbiol 57: 1074–1085 [DOI] [PubMed] [Google Scholar]
- Reyes‐Lamothe R, Sherratt DJ (2019) The bacterial cell cycle, chromosome inheritance and cell growth. Nat Rev Microbiol 17: 467–478 [DOI] [PubMed] [Google Scholar]
- Russo S, Schweitzer JE, Polen T, Bott M, Pohl E (2009) Crystal structure of the caseinolytic protease gene regulator, a transcriptional activator in actinomycetes. J Biol Chem 284: 5208–5216 [DOI] [PubMed] [Google Scholar]
- Sala A, Bordes P, Genevaux P (2014) Multiple toxin−antitoxin systems in Mycobacterium tuberculosis . Toxins (Basel) 6: 1002–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi I, Dhar N, Bousbaine D, Wakamoto Y, McKinney JD (2013) Single−cell dynamics of the chromosome replication and cell division cycles in mycobacteria. Nat Commun 4: 2470 [DOI] [PubMed] [Google Scholar]
- Santi I, McKinney JD (2015) Chromosome organization and replisome dynamics in Mycobacterium smegmatis. mBio 6: e01999‐14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarathy J, Dartois V, Dick T, Gengenbacher M (2013) Reduced drug uptake in phenotypically resistant nutrient−starved nonreplicating Mycobacterium tuberculosis . Antimicrob Agents Chemother 57: 1648–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian J, Swaminath S, Nair RR, Jakkala K, Pradhan A, Ajitkumar P (2017) De Novo emergence of genetically resistant mutants of Mycobacterium tuberculosis from the persistence phase cells formed against antituberculosis drugs in vitro . Antimicrob Agents Chemother 61: e01343‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, Brown Gandt A, Rowe SE, Deisinger JP, Conlon BP, Lewis K (2017) ATP−dependent persister formation in Escherichia coli MBio 8: e02267‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons LA, Grossman AD, Walker GC (2007) Replication is required for the RecA localization response to DNA damage in Bacillus subtilis . Proc Natl Acad Sci USA 104: 1360–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A (2017) Guardians of the mycobacterial genome: a review on DNA repair systems in Mycobacterium tuberculosis . Microbiology 163: 1740–1758 [DOI] [PubMed] [Google Scholar]
- Smeulders MJ, Keer J, Speight RA, Williams HD (1999) Adaptation of Mycobacterium smegmatis to stationary phase. J Bacteriol 181: 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits WK, Kuipers OP, Veening JW (2006) Phenotypic variation in bacteria: the role of feedback regulation. Nat Rev Microbiol 4: 259–271 [DOI] [PubMed] [Google Scholar]
- Smollett KL, Smith KM, Kahramanoglou C, Arnvig KB, Buxton RS, Davis EO (2012) Global analysis of the regulon of the transcriptional repressor LexA, a key component of SOS response in Mycobacterium tuberculosis . J Biol Chem 287: 22004–22014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapels DAC, Hill PWS, Westermann AJ, Fisher RA, Thurston TL, Saliba AE, Blommestein I, Vogel J, Helaine S (2018) Salmonella persisters undermine host immune defenses during antibiotic treatment. Science 362: 1156–1160 [DOI] [PubMed] [Google Scholar]
- Torgovnick A, Schumacher B (2015) DNA repair mechanisms in cancer development and therapy. Front Genet 6: 157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrey H, Keren I, Via LE, Lee JS, Lewis K (2016) High persister mutants in Mycobacterium tuberculosis . PLoS ONE 11: e0155127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphoff S, Lord ND, Okumus B, Potvin–Trottier L, Sherratt DJ, Paulsson J (2016) Stochastic activation of a DNA damage response causes cell–to–cell mutation rate variation. Science 351: 1094–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bergh B, Fauvart M, Michiels J (2017) Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol Rev 41: 219–251 [DOI] [PubMed] [Google Scholar]
- Veening JW, Smits WK, Kuipers OP (2008) Bistability, epigenetics, and bet−hedging in bacteria. Annu Rev Microbiol 62: 193–210 [DOI] [PubMed] [Google Scholar]
- Vijayan M (2005) Structural biology of mycobacterial proteins: the Bangalore effort. Tuberculosis 85: 357–366 [DOI] [PubMed] [Google Scholar]
- Vogel J, Argaman L, Wagner EG, Altuvia S (2004) The small RNA IstR inhibits synthesis of an SOS−induced toxic peptide. Curr Biol 14: 2271–2276 [DOI] [PubMed] [Google Scholar]
- Volzing KG, Brynildsen MP (2015) Stationary−phase persisters to ofloxacin sustain DNA damage and require repair systems only during recovery. MBio 6: e00731‐15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamoto Y, Dhar N, Chait R, Schneider K, Signorino–Gelo F, Leibler S, McKinney JD (2013) Dynamic persistence of antibiotic–stressed mycobacteria. Science 339: 91–95 [DOI] [PubMed] [Google Scholar]
- Wang Y, Huang Y, Xue C, He Y, He ZG (2011) ClpR protein−like regulator specifically recognizes RecA protein−independent promoter motif and broadly regulates expression of DNA damage−inducible genes in mycobacteria. J Biol Chem 286: 31159–31167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zignol M, Dean AS, Alikhanova N, Andres S, Cabibbe AM, Cirillo DM, Dadu A, Dreyer A, Driesen M, Gilpin C et al (2016) Population−based resistance of Mycobacterium tuberculosis isolates to pyrazinamide and fluoroquinolones: results from a multicountry surveillance project. Lancet Infect Dis 16: 1185–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla AI, Gillespie SH, Hoelscher M, Philips PPJ, Cole ST, Abubakar I, McHugh TD, Schito M, Maeurer M, Nunn AJ (2014) New antituberculosis drugs, regimens, and adjunct therapies: needs, advances, and future prospects. Lancet Infect Dis 14: 327–340 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File