

Abstract
Mammalian homologs of yeast Atg8 protein (mAtg8s) are important in autophagy, but their exact mode of action remains ill‐defined. Syntaxin 17 (Stx17), a SNARE with major roles in autophagy, was recently shown to bind mAtg8s. Here, we identified LC3‐interacting regions (LIRs) in several SNAREs that broaden the landscape of the mAtg8‐SNARE interactions. We found that Syntaxin 16 (Stx16) and its cognate SNARE partners all have LIR motifs and bind mAtg8s. Knockout of Stx16 caused defects in lysosome biogenesis, whereas a Stx16 and Stx17 double knockout completely blocked autophagic flux and decreased mitophagy, pexophagy, xenophagy, and ribophagy. Mechanistic analyses revealed that mAtg8s and Stx16 control several properties of lysosomal compartments including their function as platforms for active mTOR. These findings reveal a broad direct interaction of mAtg8s with SNAREs with impact on membrane remodeling in eukaryotic cells and expand the roles of mAtg8s to lysosome biogenesis.
Keywords: autophagy, GABARAP, lysosome, SNARE, TOR
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport
Introduction
The autophagy pathway, controlled by a conserved set of ATG genes (Mizushima et al, 2011; Levine & Kroemer, 2019), is a cytoplasmic homeostatic process at an interface between quality control and metabolism. This pathway can be activated by metabolic and stress inputs (Noda & Ohsumi, 1998; Scott et al, 2004; Marino et al, 2014; Garcia & Shaw, 2017; Saxton & Sabatini, 2017) or triggered by cargo recognition leading to in situ assembly of ATG factors via receptor regulators (Mandell et al, 2014; Lazarou et al, 2015; Kimura et al, 2016). Autophagy can be bulk or selective (Birgisdottir et al, 2013; Randow & Youle, 2014; Stolz et al, 2014), whereas the multiple biological outputs of autophagy depend on its completion and the type of termination of the autophagic pathway, e.g., degradation or secretion (Ponpuak et al, 2015; Levine & Kroemer, 2019). Bulk autophagy, whereby portions of the cytoplasm of heterogeneous composition are sequestered, occurs during starvation and can be quantified by imaging (An & Harper, 2018) or biochemically (Seglen et al, 2015; Engedal & Seglen, 2016) by following sequestration of the cytosolic enzyme LDH, or ultrastructurally by enumerating partially degraded electron‐dense ribosomes (Tanaka et al, 2000; Eskelinen, 2008). Autophagy can be selectively guided to specific cargo via a number of sequestosome 1/p62‐like receptors (SLRs; Bjorkoy et al, 2005; Birgisdottir et al, 2013; Stolz et al, 2014) or other classes of receptors besides SLRs (Kimura et al, 2016; Levine & Kroemer, 2019). Autophagy machinery is often recruited to damaged or dysfunctional targets after they are marked for autophagic degradation by ubiquitin and galectin tags that in turn are recognized by cytosolic autophagic receptors such as SLRs (Randow & Youle, 2014; Stolz et al, 2014). Autophagy apparatus can also be directly recruited via receptors residing on or within organelles if they become exposed following organellar membrane damage or depolarization or are modified downstream of physiological or developmental signals (Sandoval et al, 2008; Wild et al, 2011; Liu et al, 2012; Bhujabal et al, 2017; Wei et al, 2017).
Degradative autophagy terminates in fusion of autophagosomes with lysosomes whereby the sequestered cargo is degraded. Examples of selective degradative autophagy include autophagy of mitochondria (mitophagy), peroxisomes (pexophagy), intracellular microbes (xenophagy), ribosomes (ribophagy), protein aggregates (aggrephagy), and specific intracellular multiprotein complexes (precision autophagy) (Birgisdottir et al, 2013; Randow & Youle, 2014; Kimura et al, 2016; An & Harper, 2018). Given the diversity of cargo, complexity of the protein components and membrane compartments engaged, as well as the exquisite responsiveness to a variety of cargo triggers (e.g., damaged organelles, aggregates) and stress conditions (e.g., starvation, hypoxia), autophagy is controlled by a collection of subsystems that have to come together in a modular fashion and cooperate in the initiation and execution of autophagy (Mizushima et al, 2011). These include (i) formation of a complex between the ULK1 kinase, FIP200, ATG13, and ATG101, transducing mTOR inhibition (Ganley et al, 2009; Hosokawa et al, 2009; Jung et al, 2009), and AMPK activation (Kim et al, 2011) to induce autophagy; (ii) generation of PI3P by the ATG14‐endowed class III PI3‐kinase complex I (PI3KC3‐CI) containing VPS34 (PI3KC3‐C1) (Petiot et al, 2000; Baskaran et al, 2014; Chang et al, 2019) and Beclin 1 (He & Levine, 2010), which can also be modified by AMPK to specifically activate PI3KC3‐C1 and not the other PI3KC3 forms (Kim et al, 2013) 1; (iii) the ubiquitin‐like conjugation system with ATG5‐ATG12/ATG16L1 (Mizushima et al, 1998a,1998b) acting as an E3 ligase to lipidate mammalian homologs of yeast Atg8 (mAtg8s), some of which like LC3B have become key markers for autophagosomal membranes (Kabeya et al, 2000); and (iv) the only integral membrane ATG protein, ATG9, and the ATG2‐WIPI protein complexes, of still unknown but essential functions (Young et al, 2006; Velikkakath et al, 2012; Bakula et al, 2017). These modules are for the most part interconnected, with FIP200 physically bridging via ATG16L1 the ULK1/2 complex with the mAtg8 conjugation system (Fujita et al, 2013; Gammoh et al, 2013; Nishimura et al, 2013), ATG16L1 and WIPI directly interacting (Dooley et al, 2014), ATG13 connecting the ULK1/2 complex with PI3C3‐C1 via ATG13's HORMA domain binding to ATG14 of PI3KC3‐C1 (Jao et al, 2013; Park et al, 2016), and PI3P, the product of VPS34 (Petiot et al, 2000), being detected on membranes by WIPIs (Bakula et al, 2017).
In yeast, a component of the above systems morphologically equated with autophagy is Atg8, with its appearance as a single punctum defining the pre‐autophagosomal structure (Kirisako et al, 1999). In mammals, this function is spread over a set of six mAtg8s (Weidberg et al, 2010; Mizushima et al, 2011), LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2. Although it is generally accepted that mAtg8s are important for autophagy, and that mAtg8 lipidation and puncta formation herald autophagy induction and isolation membrane formation, the core function of mAtg8s and possibly a direct role in autophagosomal membrane remodeling remains to be defined. Several models, including a direct role of Atg8/mAtg8s in catalyzing membrane fusion (Nakatogawa et al, 2007; Weidberg et al, 2010, 2011), have been considered but later challenged (Nair et al, 2011). Nevertheless, many of the cargos, selective autophagy receptors, and regulatory components (ULK, ATG13, FIP200, ATG14, Beclin 1, VPS34, ATG4) contain LC3‐interacting regions (LIRs) and can either recruit cargo (Bjorkoy et al, 2005; Birgisdottir et al, 2013) or organize core autophagy machinery (Alemu et al, 2012; Skytte Rasmussen et al, 2017; Birgisdottir et al, 2019). Furthermore, recent studies show that even in the complete absence of mAtg8s, autophagosome formation proceeds, albeit with a lower sequestration volume, and that mAtg8s are important for autolysosomal formation (Nguyen et al, 2016a). In keeping with the latter findings, depletion of components participating in mAtg8 conjugation delays autophagic flux progression (Tsuboyama et al, 2016). It remains to be understood precisely how mAtg8s control membrane trafficking and remodeling during phagophore formation and elongation (Xie et al, 2008) and during autophagosome–lysosome fusion (Weidberg et al, 2010; Nguyen et al, 2016a).
Degradative aspects of autophagy depend on autophagosome fusion with organelles of the endolysosomal system, often referred to as autophagic flux or autophagosome–lysosome fusion (Tanaka et al, 2000; Klionsky et al, 2016). Much of the intracellular membrane trafficking and fusion processes are catalyzed by SNARE proteins, which ensure compartment/organelle specificity through pairing of cognate Qa‐, Qb‐, Qc‐, and R‐SNARE partners (Jahn & Scheller, 2006). SNAREs are often found in compartments where they function but also transit through other membranes, and thus, their fusion activities are tightly regulated by tethers, SM (Sec1/Munc18) proteins, Rabs, and additional factors that modulate their recruitment, activation, cycles of reuse, etc. (Hong & Lev, 2014). In the context of autophagy, SNAREs have been studied at different stages along the autophagy pathway (Nair et al, 2011; Itakura et al, 2012; Moreau et al, 2013; Kimura et al, 2017). At the autophagosome–lysosome fusion stage, an initially identified SNARE was the Qa‐SNARE Stx17, forming complexes with Qbc‐SNARE SNAP29 and R‐SNARE VAMP8 (Itakura et al, 2012; Takats et al, 2013; Guo et al, 2014; Diao et al, 2015; Wang et al, 2016). Recent studies have indicated that additional SNAREs may be required or even be dominant in this process (Matsui et al, 2018; Takats et al, 2018). Thus, redundant, complementary or synergistic SNARE‐driven fusion and regulatory events may be at work during autolysosomal fusion. We have recently shown that Stx17 interacts via its LIR motif with mAtg8s, which functions in Stx17's recruitment to autophagosomes (Kumar et al, 2018), and that TBK1‐phosphorylated Stx17 plays a role in autophagy initiation (Kumar et al, 2019), in keeping with studies by others (Hamasaki et al, 2013; Arasaki et al, 2015, 2018). Thus, Stx17 contributes to the autophagy pathway at several stages from initiation to maturation.
A report of direct interaction between mAtg8s and Stx17 (Kumar et al, 2018) has set an unanticipated precedent, and here, we explored whether there is a broader range of interactions between mAtg8s and mammalian SNAREs. Using bioinformatics and biochemical approaches, we found additional candidate SNAREs with LIR motifs that bind mAtg8s. Among the candidates, we focused on a cognate set of SNAREs, Stx16, Vti1a, and Stx6, previously implicated in retrograde transport between endosomes and the trans‐Golgi network (TGN). Through mutational and functional analyses, we found that Stx16 acts synergistically with Stx17 and is important for diverse types of autophagy, including mitophagy, pexophagy, ribophagy, and elimination of intracellular Mycobacterium tuberculosis. We have further found that Stx16 is important for cellular lysosomal content and function, and that mAtg8s modulate Stx16 localization on endolysosomal organelles. This uncovers a mechanism different from the previous views of how mAtg8s help grow autophagosomal and complete autolysosomal membranes. We conclude that mAtg8s control autophagy at least in part by directly binding to SNARE proteins engaged in the biogenesis of the endolysosomal and autolysosomal organelles.
Results
A subset of SNAREs including syntaxin 16 contain LC3‐interacting regions
Previous studies have indicated that at least one SNARE involved in autophagy, Stx17, harbors a LIR motif that affects its distribution (Kumar et al, 2018). Here, we carried out a broader analysis of all SNAREs for the presence of putative LIR motifs using bioinformatics and conventional LIR motif consensus (Birgisdottir et al, 2013) followed up by biochemical assays (Fig 1A and Appendix Table S1). We tested a subset of SNAREs containing putative LIRs in a screen with peptide arrays for binding to GST‐GABARAP as a representative mAtg8 (Fig 1A). Several SNAREs showed positive signals with GST‐GABARAP in peptide arrays. These included only one R‐SNARE, VAMP7, and mostly Qa‐, Qb‐, and Qc‐SNAREs acting in different cellular compartments (Jahn & Scheller, 2006): Stx17, included as a positive control (Kumar et al, 2018) and already implicated in autophagy (Itakura et al, 2012), Stx18 acting in the ER (Hatsuzawa et al, 2000), GOSR1 functioning in the Golgi (Subramaniam et al, 1996; Mallard et al, 2002), Stx3 and Stx4 on plasma membrane (Low et al, 1996), Stx19 with no transmembrane domain and partitioning between cytosol and membranes (Wang et al, 2006), and Stx16, Stx6, and Vti1a, all acting in trafficking between endosomal compartments and TGN (Jahn & Scheller, 2006; Malsam & Söllner, 2011) (Figs 1B and EV1A). Thus, a number of SNAREs may be binding partners for mAtg8s indicating a previously unappreciated potential for broader intersections between mAtg8 and SNARE systems.
Figure 1. Many SNAREs bind mammalian Atg8 proteins.
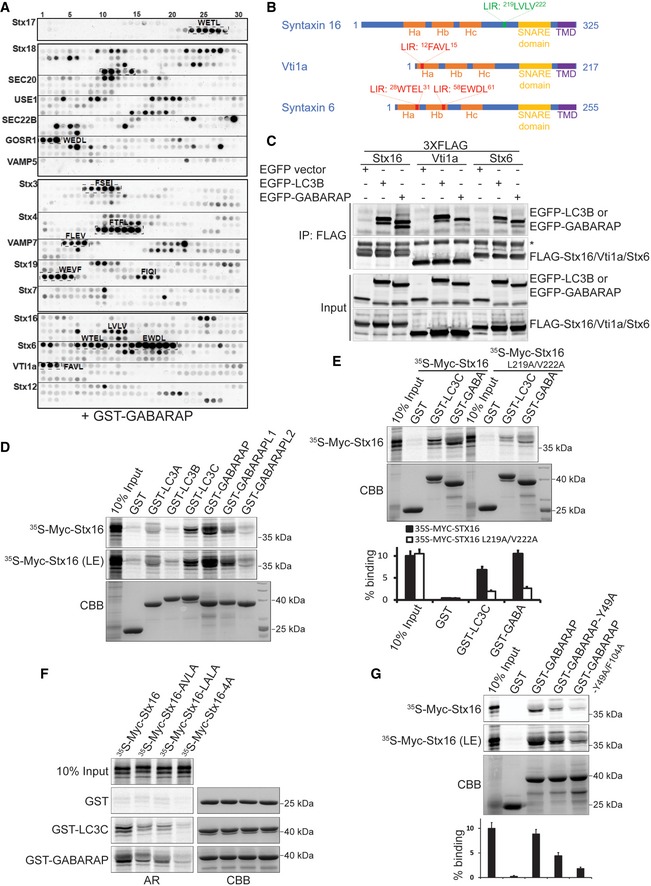
- Peptide array dot blot analysis to identify LIR motifs in the indicated SNARE proteins. Amino acids for the identified LIRs from positive signals are marked on each SNARE. Amino acids are denoted as amino acid single letter codes in the blot.
- Schematics of the functional domains of Stx16/Vti1a/Stx6 cognate SNAREs with the positions of LIR motifs. The LIR motif marked in green in syntaxin 16 indicates established LIR based on following analyses.
- Co‐immunoprecipitation (Co‐IP) analysis of interactions between FLAG‐tagged Stx16/Vti1a/Stx6 and EGFP‐tagged LC3B or GABARAP overexpressed in HEK293T cells. * indicates mouse IgG heavy chain of precipitated mouse anti‐FLAG antibody.
- GST pull‐down analysis of interactions between radiolabeled Myc‐Stx16 and GST‐tagged mAtg8 proteins.
- GST pull‐down analysis of interactions between wild‐type (WT) or LIR‐mutant (L219A/V222A) Stx16 and GST‐tagged LC3C or GABARAP. Lower panel shows percentages of WT or LIR‐mutant Stx16 bound to GST‐LC3C or GST‐GABARAP. Data shown as means ± SEM of precipitated Stx16, n = 3.
- GST pull‐down analysis of interactions between WT or different types of LIR‐mutant Stx16 and GST‐tagged LC3C or GABARAP. Lower panel shows percentages of WT or LIR‐mutant Stx16 bound to GST‐LC3C or GST‐GABARAP.
- GST pull‐down analysis of interactions between radiolabeled Myc‐Stx16 and WT or LDS‐mutant GABARAP (GABARAP‐Y49A and ‐Y49A/F104A). Lower panel shows percentages of Stx16 bound to WT or LDS‐mutant GABARAP. Data shown as means ± SEM of precipitated Stx16, n = 3.
Source data are available online for this figure.
Figure EV1. Peptide arrays identify a set of SNAREs bearing LIR motifs.
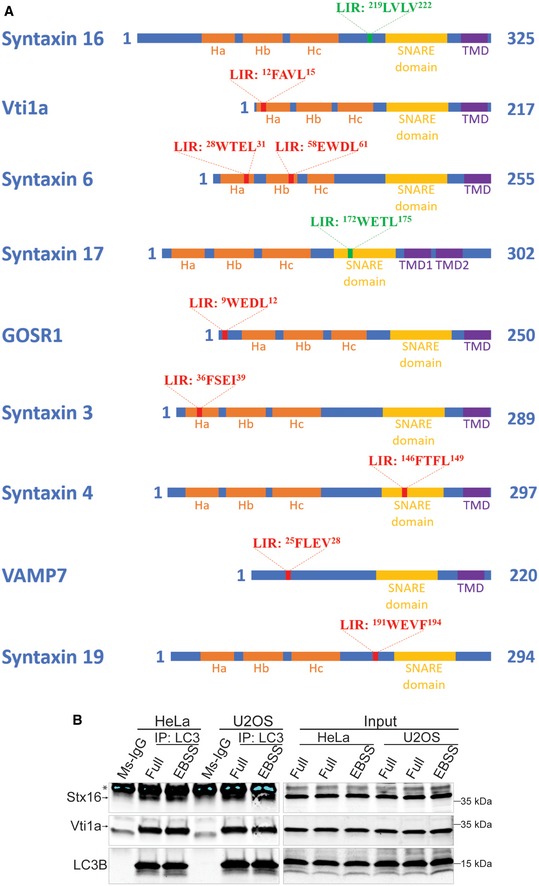
- Schematics of the functional domains of SNAREs bearing LIR motifs identified through bioinformatic and peptide array analyses. LIR motifs with the amino acid sequences are marked as red bars in approximate positions on each SNARE protein. The LIR motifs in Stx16 and Stx17 are marked green as they are validated LIRs through biochemical and functional analyses.
- Endogenous Co‐IP analysis of the interactions between LC3 and Stx16, Vti1a in HeLa and U2OS cells. * indicates IgG heavy chain of precipitated anti‐LC3 antibody.
Source data are available online for this figure.
Syntaxin 16 directly binds mAtg8s through its LIR motif
From the panel of SNAREs showing positive signals with GST‐GABARAP in peptide arrays, we focused on a set of three cognate SNAREs, Stx16, Vti1a, and Stx6, known to form a complex (Malsam & Söllner, 2011). Co‐immunoprecipitation (Co‐IP) analyses of overexpressed fusion proteins confirmed that Stx16, Vti1a, and Stx6 interact with LC3B and GABARAP (Fig 1C). Co‐IP analyses of endogenous proteins in HeLa and U2OS cells confirmed interactions between LC3 and Stx16 and Vti1a (Fig EV1B). In GST pull‐down assays with all 6 mAtg8 proteins, Stx16 showed a positive binding signal with LC3C, GABARAP, and GABARAPL1 (Fig 1D). The peptide array binding results pointed to the 219LVLV222 residues in Stx16 (Fig 1B), which resembled both the LC3C‐preferring LIR of NDP52 (ILVV; von Muhlinen et al, 2012) and the GABARAP interaction motifs with important Val residues in the second and fourth positions of the core LIR motif (Rogov et al, 2017). This motif was found in the linker region between the Habc and SNARE domains of Stx16 matching the requirement to be outside of known structured regions (Popelka & Klionsky, 2015). When we mutated L‐219 and V‐222 in the predicted (albeit atypical) LIR motif (219LVLV222 mutated to 219AVLA222) of Stx16, this reduced Stx16 binding to LC3C and GABARAP (Fig 1E). Additionally, we changed both Val residues within the 219LVLV222 motif to Ala, and the resulting Stx16 variant showed reduction in binding to LC3C and GABARAP comparable to the 219AVLA222 mutant in GST pull‐down experiments (Fig 1F). Finally, when we mutated all 4 residues within the 219LVLV222 motif, the association between Stx16 and LC3C or GABARAP was completely abolished (Fig 1F).
Complementary to the above experiments with LIR mutations, we tested whether the previously defined LIR‐docking site (LDS) in GABARAP (Behrends et al, 2010; Birgisdottir et al, 2013) was responsible for binding to Stx16. Two conserved hydrophobic pockets (HP1 and HP2) are known to define the LIR‐docking site of GABARAP (Behrends et al, 2010). The Y49A mutation in the HP1 pocket of GABARAP reduced its interaction with Stx16, whereas the complete LDS mutant (double HP1 and HP2 Y49A/F104A mutation) abrogated GABARAP's binding to Stx16 in GST pull‐down assays (Fig 1G). Thus, GABARAP and Stx16 interaction depends on the canonical LDS motif in GABARAP.
Syntaxin 16 is required for efficient autophagy flux in response to starvation
Stx17 has been implicated in mammalian autophagosome–lysosome fusion (Itakura et al, 2012). However, additional SNAREs have now been shown to contribute to this process (Matsui et al, 2018), but relative contributions remain to be fully defined. We generated CRISPR/Cas9‐mediated STX17 knockout (KO) in different cell lines (HeLa and Huh7; Fig EV2A and B) and analyzed autophagy flux by monitoring LC3‐II levels (Figs 2A and B, and EV2C and D). In STX17 KO HeLa cells, the autophagic flux continued at almost the same rates as in wild‐type (WT) control cells, as shown by slight increase in LC3‐II levels upon starvation (Fig 2A). A similar effect was observed in hepatocellular carcinoma Huh7 STX17 KO cells, which in general displayed faster LC3‐II flux (Fig EV2C and D). Knocking out Stx17 did not cause any significant change in the levels of p62, an autophagic receptor that is degraded upon starvation (Bjorkoy et al, 2005), and is often used as readout of autophagic flux (Klionsky et al, 2016; Figs 2A and EV2C).
Figure EV2. Stx16 and Stx17 are required for efficient autophagic flux induced by starvation.
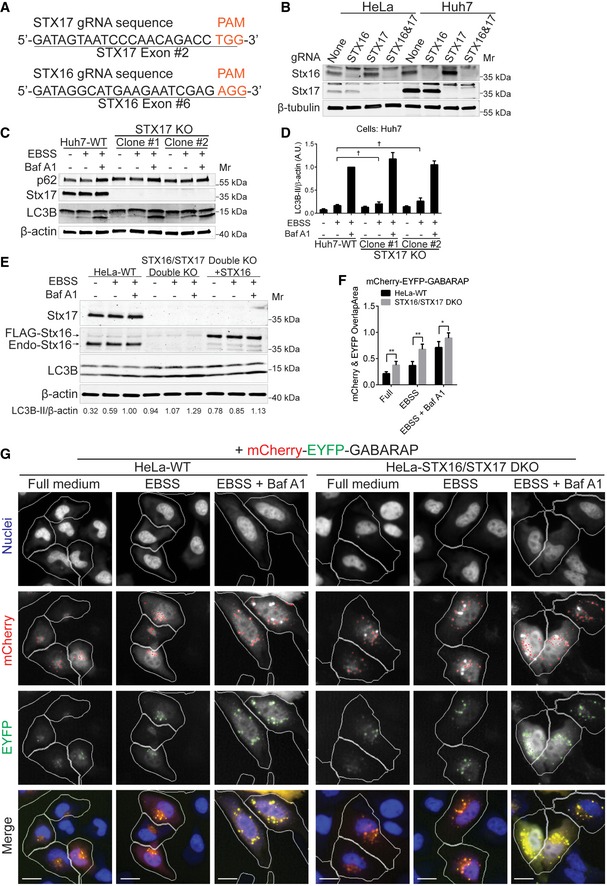
- Sequences of guide RNAs (gRNA) targeting STX17 and STX16. Protospacer adjacent motif (PAM) sequences are shown in orange on the 3′ side of each gRNA. The exon numbers targeted by the gRNAs are shown below each gRNA.
- Validation of STX16 KO, STX17 KO, and STX16/STX17 DKO by Western blot analysis in HeLa and Huh7 cell lines.
- WT or STX17 KO Huh7 cells were starved with or without the presence of bafilomycin A1 (Baf A1, 100 nM) for 2 h, and cell lysates were subjected to Western blot analysis of LC3B and p62.
- Quantifications of LC3B‐II levels normalized to β‐actin from cells treated as in (C). Data shown as means ± SEM of LC3B‐II and β‐actin ratios, n = 3; †, not significant (one‐way ANOVA).
- STX16/STX17 DKO HeLa cells were transfected with 3XFLAG‐STX16, starved with or without the presence of Baf A1 for 2 h, and cell lysates were subjected to Western blot analysis of LC3B. Numbers below the panels are the average of LC3B‐II/β‐actin ratios normalized to the third lane (HeLa WT treated with EBSS plus Baf A1) from two independent experiments.
- WT or STX16/STX17 DKO HeLa cells were transfected with mCherry‐EYFP‐GABARAP (tandem GABARAP), starved in EBSS or EBSS plus Baf A1 for 2 h, and subjected to high‐content microscopic (HCM) analysis of autophagic flux for tandem GABARAP. Data shown as means ± SEM of the overlap area per cell between mCherry and EYFP, minimum 200 transfected cells were counted each well from at least 12 wells, three independent experiments; *P < 0.05; **P < 0.01 (two‐way ANOVA).
- Representative images of tandem GABARAP transfected into WT or STX16/STX17 DKO HeLa cells treated as in (F). Masks: white, cells successfully transfected with tandem GABARAP identified based on average intensity of mCherry; red, mCherry puncta in transfected cells; green, EYFP puncta in transfected cells; yellow, overlap area between mCherry and EYFP. Scale bar: 20 μm.
Source data are available online for this figure.
Figure 2. The mAtg8‐interacting SNAREs Stx16 and Stx17 are required for efficient bulk autophagic flux.
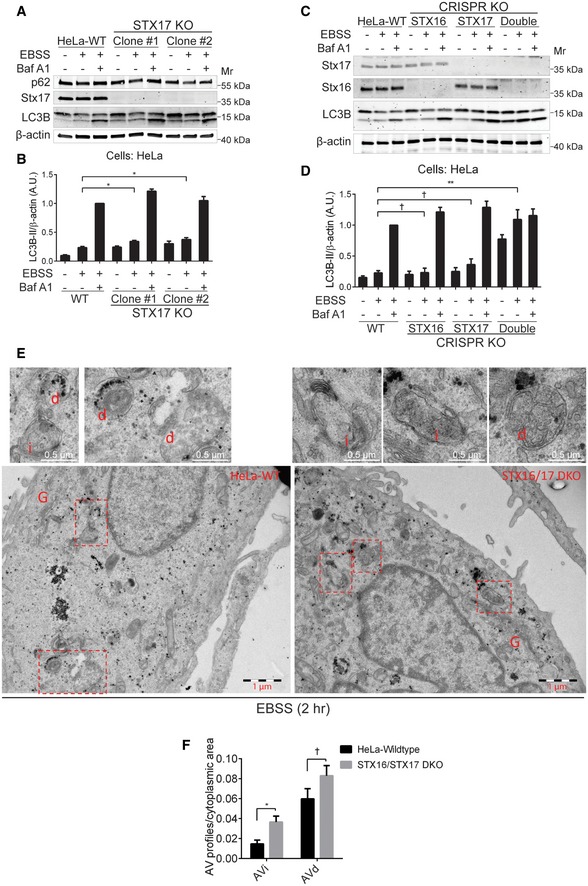
- WT or STX17 knockout (STX17 KO) HeLa cells were starved with or without the presence of bafilomycin A1 (Baf A1, 100 nM) for 2 h, and cell lysates were subjected to Western blot analysis of LC3B and p62.
- Quantifications of LC3B‐II levels normalized to β‐actin from cells treated as in (A); data shown as means ± SEM of LC3B‐II and β‐actin ratios, n = 3; *P < 0.05 (one‐way ANOVA).
- WT, STX16 KO, STX17 KO, or STX16/STX17 double KO (STX16/STX17 DKO) HeLa cells were starved with or without the presence of Baf A1 (100 nM) for 2 h, and cell lysates were subjected to Western blot analysis of LC3B.
- Quantifications of LC3B‐II levels normalized to β‐actin from (C); data shown as means ± SEM of LC3B‐II and β‐actin ratios, n = 3; †, not significant; **P < 0.01 (one‐way ANOVA).
- WT or STX16/STX17 DKO HeLa cells were starved in EBSS for 2 h and subjected to ultrastructural analysis of the autophagic vesicles (AV) with electron microscopy. AVi: initial autophagic vacuoles; AVd: degradative autophagic vacuoles; G: Golgi apparatus. Image acquisition and counting as in Methods. Scale bars, 1 μm and 0.5 μm (top sections).
- Quantifications of autophagic vesicles in WT and STX16/STX17 DKO HeLa cells treated as (E); data shown as means ± SEM of AV profiles relative to cytoplasmic area; †, not significant; *P < 0.05 (two‐way ANOVA). AV profiles from 47 images of each sample were counted.
Source data are available online for this figure.
When we knocked out STX16 in HeLa cells, this too had no major effects on LC3‐II flux (Figs 2C and D, and EV2A and B). However, when a double STX16/STX17 knockout was generated (Fig EV2B), the LC3 flux was completely abrogated, with LC3‐II levels in starvation‐induced cells accumulating at similar levels as in cells treated with bafilomycin A1 (Fig 2C and D), usually considered as a complete blocker of autophagic flux (Klionsky et al, 2016). The LC3‐II accumulation could be partially relieved by complementation of STX16/STX17 DKO cells transfected with FLAG‐tagged WT Stx16 (Fig EV2E). Given that this indicated a block in autolysosome production, we examined STX16/STX17 DKO cells using automated quantitative high‐content microscopy (HCM) and tandem mCherry‐EYFP‐GABARAP, by monitoring and quantifying overlap between red (mCherry) and green (EYFP) fluorescence as another measure of autophagic flux. The overlap between mCherry and EYFP was significantly increased in STX16/STX17 DKO cells compared to that in WT cells upon starvation in EBSS (Fig EV2F and G). At the ultrastructural level, electron microscopy quantification of initial (AVi) vs. degradative (AVd) autophagic vacuoles (Tanaka et al, 2000; Eskelinen, 2008) also indicated accumulation of AVi vesicles (Fig 2E and F). In conclusion, the absence of either Stx16 or Stx17 alone cannot block completion of the autophagy pathway, but when both are ablated, this prevents progression to autolysosomes.
Syntaxin 16 and syntaxin 17 are required for completion of diverse selective autophagy processes
Mitophagy is a very well‐studied process of selective autophagy and is often used as an example to study elimination of defunct organelles (Youle & Narendra, 2011). To further characterize effects of STX16/STX17 double knockout, we employed HCM quantification of mitochondrial DNA (mtDNA) as a measure of cellular mitochondrial content often used in mitophagy studies (Lazarou et al, 2015; Nguyen et al, 2016b). We generated STX16/STX17 DKO in HeLa cells stably expressing YFP‐Parkin (Lazarou et al, 2015; Fig EV3A). We then stressed mitochondria using two different depolarizing agents: CCCP or oligomycin A + antimycin A (OA), and quantified Parkin‐dependent mitophagy in STX16/STX17 DKO HeLa/YFP‐Parkin cells. In wild‐type cells, CCCP or OA caused a major reduction in mtDNA (Fig 3A and B). Loss of Stx16 and Stx17 caused significant reduction in mitophagy (Fig 3A and B). Clearly, additional processes were also involved as STX16/STX17 DKO HeLa/YFP‐Parkin cells still eliminated mitochondria probably through additional mechanisms (Nguyen et al, 2016a). Previous studies indicated that Stx17 is not important for mitophagy (Nguyen et al, 2016b). Nevertheless, a loss of Stx16 in the STX17 KO background affected mitophagy significantly.
Figure EV3. Stx16 is required for lysosomal biogenesis.
- Validation of STX16 and STX17 double knockout in HeLa‐YFP‐Parkin cells by Western blot analysis. The expression of YFP‐Parkin was detected by rabbit anti‐GFP antibody (ab290).
- Western blot analysis of LKB1 expression and AMPKα phosphorylation (Thr172) in response to H2O2 treatment in Huh7 cells.
- Western blot analysis of Stx16 and Stx17 protein levels in THP1 cells infected with lentiviruses containing STX16, STX17, or STX16 and STX17 CRISPR gRNAs.
- WT or STX16 KO HeLa cells were starved in EBSS for 2 h and subjected to HCM analysis of the overlap between TGN46 and LAMP2. Masks: white, cells identified based on nuclei; green, TGN46 indicating trans‐Golgi network (TGN); red, LAMP2 puncta; yellow, overlap between TGN46 and LAMP2. Scale bar: 20 μm.
- Quantifications of overlaps between TGN46 and LAMP2 in WT and STX16 KO HeLa cells treated as in (A). Data shown as means ± SEM of percentages of LAMP2 overlapping with TGN46 to compensate reduced LAMP2 puncta in STX16 KO cells (see Fig 5D), minimum 500 cells were counted each well from at least 12 wells, three independent experiments; **P < 0.01 (two‐way ANOVA).
- Co‐IP analysis of the interactions between overexpressed FLAG‐tagged Stx6/Stx16 and endogenous VAMP3/VAMP4/VAMP8/VPS41 in HEK293T cells under full or starved (EBSS for 1 h) conditions.
Source data are available online for this figure.
Figure 3. Stx16 and Stx17 are required for selective autophagy of mitochondria, peroxisomes, and M. tuberculosis .
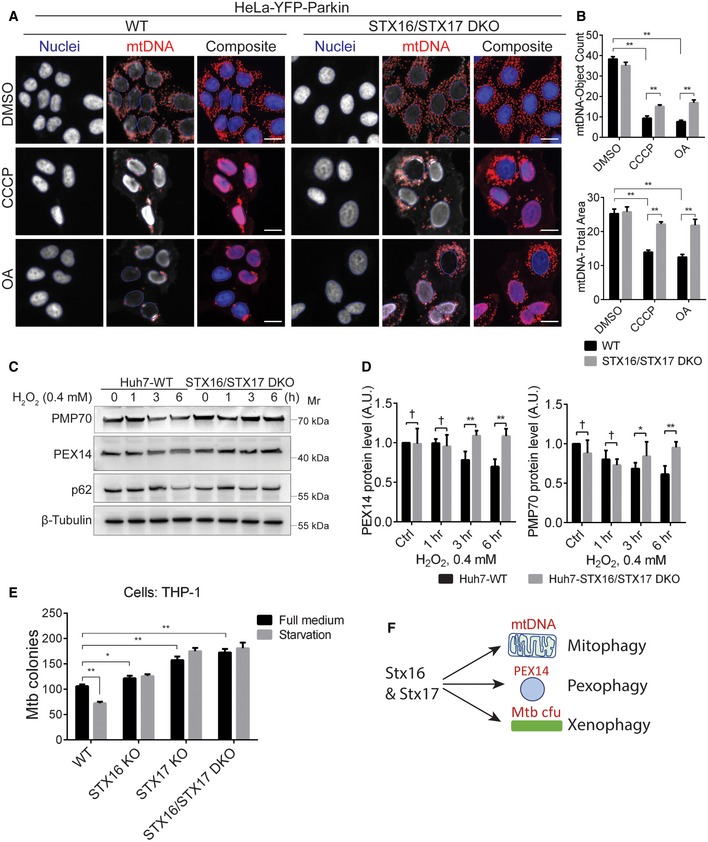
- WT or STX16/STX17 DKO HeLa‐YFP‐Parkin cells were treated with CCCP (10 μM) or oligomycin A (5 μM) and antimycin A (10 μM) (OA) for 16 h, and subjected to high‐content microscopy (HCM) analysis of mitochondria clearance. Masks: blue, nuclei; red, mitochondria stained with mitochondrial DNA (mtDNA) antibody. Scale bar: 20 μm.
- Quantifications of mtDNA by object count or total area per cell in WT or STX16/STX17 DKO HeLa‐YFP‐Parkin cells treated as in (A); data shown as means ± SEM of mtDNA object count (upper panel) or object total area (lower panel) per cell; minimum 500 cells were counted each well from at least 12 wells, three independent experiments; **P < 0.01 (two‐way ANOVA).
- WT or STX16/STX17 DKO Huh7 cells were treated with H2O2 (0.4 mM) for indicated time points, and autophagic clearance of peroxisomes was measured by the protein levels of PMP70, PEX14, and p62.
- Quantifications of peroxisomal proteins PMP70 and PEX14 for WT or STX16/STX17 DKO Huh7 cells treated as in (C); data shown as means ± SEM of PEX14 or PMP70 and β‐actin ratios, n = 3; †, not significant; *P < 0.05; **P < 0.01 (two‐way ANOVA).
- WT, STX16 KO, STX17 KO, or STX16/STX17 DKO THP‐1 cells were infected with Mycobacterium tuberculosis (Mtb), followed by starvation to induce autophagy. Autophagic clearance of infected Mtb was measured by Mtb colonies grown on Middlebrook 7H11 agar plates; data shown as means ± SEM of Mtb colonies, n = 4; *P < 0.05; **P < 0.01 (two‐way ANOVA).
- Schematics for the requirement of Stx16 and Stx17 in autophagic clearance of damaged mitochondria, surplus peroxisomes, and invaded bacterial pathogens.
Pexophagy is another classical example of selective autophagy (Manjithaya et al, 2010; Gatica et al, 2018). We monitored pexophagy in cells treated with H2O2 (Zhang et al, 2015) using liver cell line Huh7, which expresses wild‐type LKB1 that can activate AMPK (Tan et al, 2018; Fig EV3B). We monitored pexophagy by quantifying levels of PEX14 and PMP70 (Zhang et al, 2015) proteins, and detected decrease in these peroxisomal constituents over a period of 6 hours of exposure to H2O2 (Fig 3C). In cells lacking both Stx16 and Stx17, this reduction in peroxisomal protein levels was abrogate (Fig 3C and D). Thus, STX16/STX17 DKO significantly inhibited pexophagy.
Autophagic defense against intracellular microbes, such as Mycobacterium tuberculosis (Mtb) (Gutierrez et al, 2004), sometimes referred to as xenophagy, is considered to be another form of selective autophagy. We tested the survival of M. tuberculosis in macrophage‐like cell line THP1 and compared the effects of Mtb killing in WT, single STX16 KO or STX17 KO and STX16/STX17 DKO cells (Fig EV3C). As previously reported (Gutierrez et al, 2004), induction of autophagy by starvation reduced survival of Mtb in WT THP1 cells (Fig 3E). However, this effect was lost in STX16 KO THP1 cells (Figs 3E and EV3C). Further analyses of a CRISPR KO mutant of STX17 alone, previously reported in THP1 cells to play a role in control of intracellular Mtb (Kumar et al, 2018), or a combination CRISPR KO mutant of both STX16 and STX17, showed no further additive effects between Stx16 and Stx17 (Fig 3E).
Taken together, these data indicate that STX16/STX17 DKO significantly reduces mitochondrial (Lazarou et al, 2015), peroxisomal (Manjithaya et al, 2010), and bacterial (Mtb) (Gutierrez et al, 2004) clearance under conditions known to induce autophagy (Fig 3F).
Keima‐based ribophagy probes indicate a role of Stx16 in autophagic maturation
Keima fluorescent protein has become an important tool for detection of the arrival of autophagy substrates to autolysosomes (Katayama et al, 2011). In functional autolysosomes, despite being in a degradative compartment, Keima protein is stable but changes its spectral properties (illustrated in Fig 4A) from maximum excitation wavelength at 440 nm in neutral pH to 586 nm in acidic pH, with emission remaining constant at 620 nm (Violot et al, 2009). These shifts can be monitored using 440‐nm and 560‐nm excitation filters and emission at 620 nm in an HCM Cellomics system (see Materials and Methods). We applied Keima to monitor ribophagy (An & Harper, 2018) and generated STX16/STX17 DKO using CRISPR/Cas9 (Fig 4B) in HEK293 cells stably expressing RPS3‐Keima (fusion between 40S ribosomal protein S3 and mKeima) protein (An & Harper, 2018). RPS3‐Keima was diffuse cytoplasmic in untreated cells. Under starvation conditions, we examined ribophagy as previously reported (An & Harper, 2018), by monitoring progression of RPS3‐Keima into acidic autolysosomal compartments via quantification of cytoplasmic puncta at 560 nm excitation and 620 emission wavelengths (Fig 4C and D). The HEK293 STX16/STX17 DKO cells showed significantly fewer autolysosomal puncta originating from RPS3‐Keima than in parental WT cells (Fig 4C and D). An independent set of experiments was carried out with HCT116 cells stably expressing a fusion between 60S ribosomal protein L28 and mKeima (RPL28‐Keima). We generated STX16/STX17 DKO in these cells as well using CRISPR/Cas9 (Fig 4B). Progression of ribophagy in HCT116 RPL28‐Keima STX16/STX17 DKO cells was diminished relative to parental WT cells (Fig 4E and F). Thus, the use of previously characterized Keima ribophagy probes (An & Harper, 2018) further confirmed the importance of having both Stx16 and Stx17 for autophagic cargos to arrive into acidified compartments, including during ribophagy.
Figure 4. Stx16 and Stx17 cooperate in ribophagy.
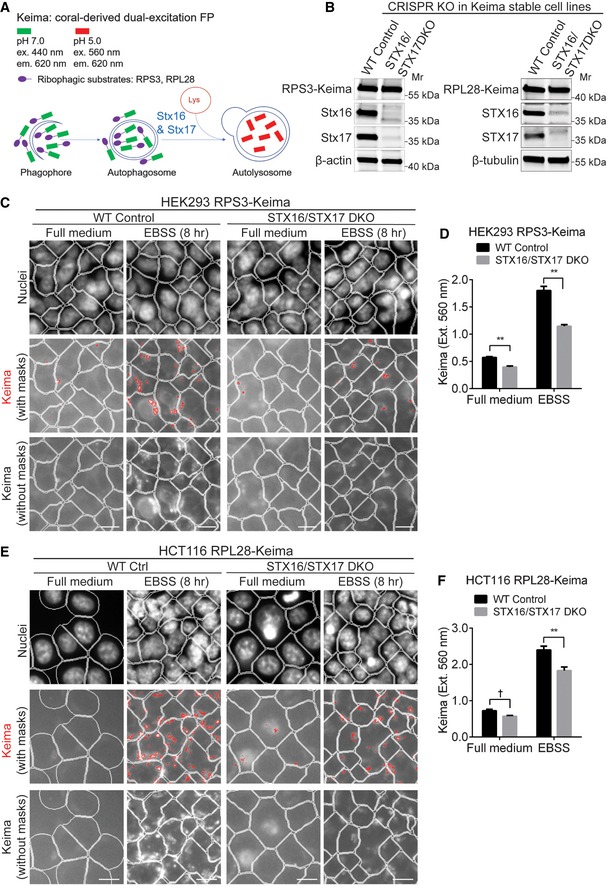
- Schematic of ribophagy detection with Keima‐based reporter system; FP: fluorescent protein.
- Validation of STX16/STX17 DKO by Western blot analysis in HEK293 cells stably expressing RPS3‐Keima (left panel) and HCT116 cells stably expressing RPL28‐Keima (right panel).
- WT or STX16/STX17 DKO HEK293 RPS3‐Keima cells were cultured in full medium or starved for 8 h, and subjected to HCM analysis of Keima puncta accumulated in autolysosomes with the excitation wavelength at 560 nm. Masks: white, cells identified based on nuclei; red, Keima puncta detected with the excitation and emission wavelengths of 560 nm and 620 nm, respectively; lower panel shows Keima puncta without masks. Scale bar: 20 μm.
- Quantifications of autolysosomal Keima puncta in WT or STX16/STX17 DKO HEK293 RPS3‐Keima cells treated as in (C). Data shown as means ± SEM of Keima puncta per cell, minimum 1,000 cells were counted each well from at least 12 wells, three independent experiments; **P < 0.01 (two‐way ANOVA).
- WT or STX16/STX17 DKO HCT116 RPL28‐Keima cells were cultured in full medium or starved for 8 h, and subjected to HCM analysis of Keima puncta accumulated in autolysosomes with the excitation wavelength at 560 nm. Masks: white, cells identified based on nuclei; red, Keima puncta detected with the excitation and emission wavelengths of 560 nm and 620 nm, respectively; lower panel shows Keima puncta without masks. Scale bar: 20 μm.
- Quantifications of autolysosomal Keima puncta in WT or STX16/STX17 DKO HCT116 RPL28‐Keima cells treated as in (E). Data shown as means ± SEM of Keima puncta per cell, minimum 1,000 cells were counted each well from at least 12 wells, three independent experiments; †, not significant; **P < 0.01 (two‐way ANOVA).
Source data are available online for this figure.
Syntaxin 16 plays a role in lysosomal biogenesis
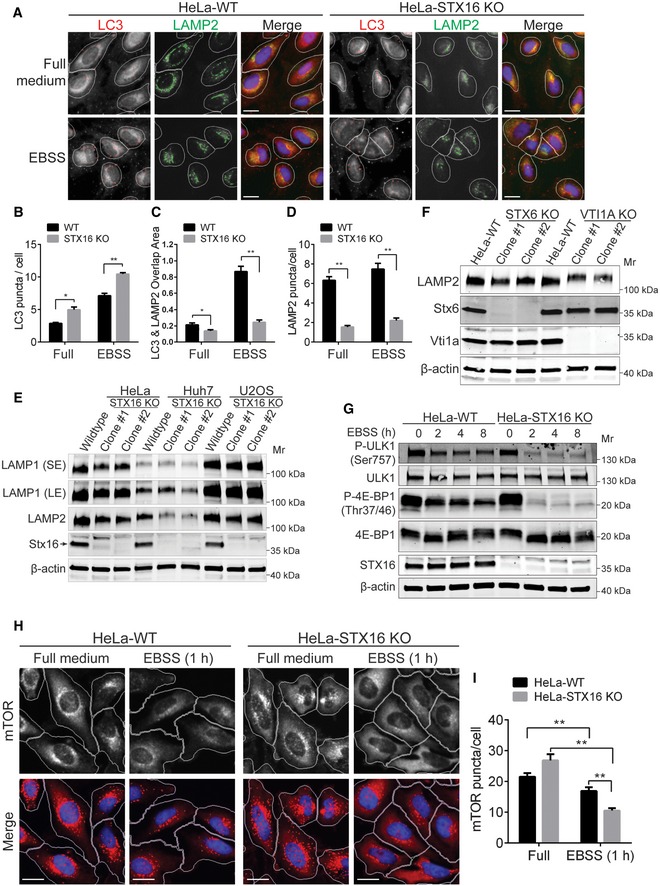
When we tested autophagy response by monitoring endogenous LC3 puncta, we found an increase in LC3 dots in STX16 knockout cells (HeLa STX16 KO) relative to WT cells, under basal conditions and in EBSS‐starved cells (Fig 5A and B). Next, we determined by HCM the overlap area between LC3 and LAMP2 profiles, and found that the overlap was strongly reduced in HeLa STX16 KO vs WT cells (Fig 5A and C). When we quantified LAMP2 puncta/cell, we found that LAMP2+ puncta were diminished in HeLa STX16 KO cells (Fig 5A and D). Thus, the appearance of reduced colocalization (overlap area by HCM) between LC3 and LAMP2 was not a result of fewer autolysosomes but was primarily due to the overall reduction of LAMP2+ organelles in STX16 KO cells (Fig 5A and D), a phenotype that could be complemented by reintroducing WT Stx16 but less so with a LIR mutant of Stx16 (Appendix Fig S1A and B). The reduction in cytoplasmic content of LAMP2+ organelles in HeLa STX16 KO cells was also seen by immunoblots, showing reduced total LAMP1 and LAMP2 protein levels in cells knocked out for Stx16 (Fig 5E). This was further tested by generating STX16 knockout in different cell lines (Fig EV2A and B). As with different clones of HeLa STX16 KO cells, different single clones of Huh7 STX16 KO and osteosarcoma U2OS STX16 KO cells had reduced LAMP1 and LAMP2 protein levels (Fig 5E). Thus, Stx16 is important for the maintenance of homeostatic LAMP levels. We also generated CRISPR knockouts for Stx6 and Vti1a, and found reduced levels of LAMP2 in STX6 KO and VTI1A KO HeLa cells (Fig 5F). Thus, Stx16 and its cognate Qb‐ and Qc‐SNAREs affect cellular LAMP levels.
Figure 5. Stx16 is required for the maintenance of lysosomal homeostasis and mTOR activity.
-
AWT or STX16 KO HeLa cells were starved in EBSS for 2 h and subjected to HCM analysis of LAMP2 and LC3 puncta. Masks: white, cells identified based on nuclei; red, LC3 puncta; green, LAMP2 puncta; yellow, overlap of LC3 and LAMP2. Scale bar: 20 μm.
-
B–DQuantifications of LC3 puncta per cell (B), LC3, and LAMP2 overlap area per cell (C), and LAMP2 puncta per cell (D) in WT or STX16 KO cells treated as in (A). Data shown as means ± SEM of puncta or overlap area per cell, minimum 500 cells were counted each well from at least 12 wells, three independent experiments; *P < 0.05; **P < 0.01 (two‐way ANOVA).
-
EWestern blot analysis of LAMP1 and LAMP2 protein levels in WT and separate clones of STX16 KO HeLa, Huh7, and U2OS cells.
-
FWestern blot analysis of LAMP2 protein levels in WT and separate clones of STX6 KO or VTI1A KO HeLa cells.
-
GTime course of mTOR inactivation during EBSS starvation in WT vs. STX16 KO HeLa cells analyzed by Western blotting for mTOR substrates ULK1 and 4E‐BP1.
-
HWT or STX16 KO HeLa cells were starved in EBSS for 1 h and subjected to HCM analysis of mTOR puncta. Masks: white, cells identified based on nuclei; red, mTOR puncta. Scale bar: 20 μm.
-
IQuantifications of mTOR puncta in WT or STX16 KO cells treated as in (H). Data shown as means ± SEM of mTOR puncta per cell, minimum 500 cells were counted each well from at least 12 wells, three independent experiments; **P < 0.01 (two‐way ANOVA).
Source data are available online for this figure.
Prior ultrastructure studies have implicated HOPS component VPS41 in LAMP1 and LAMP2 transport from the TGN to late endosomal compartments (Pols et al, 2013). In STX16 KO HeLa cells, there was increased colocalization of detectable LAMP2 with the TGN marker TGN46, normalized for LAMP puncta in HCM quantifications (Fig EV3D and E). We thus tested whether Stx16 and its cognate Qb‐ and Qc‐SNAREs, Vti1a, and Stx6 (both showing positivity in mAtg8 peptide binding arrays; Fig 1A) interact with VPS41. Using FLAG‐tagged Stx6 and Stx16, we detected VPS41 in FLAG immunoprecipitates (Fig EV3F). We found VPS41 but not VPS33A (a component of the HOPS complex) in endogenous protein complexes with Stx6, and Vti1a (Appendix Fig S1C). These observations suggest a connection of Stx16 (plus Vti1a and Stx6) to VPS41 and the transport of lysosomal proteins such as LAMP1 and LAMP2, previously proposed specifically for VPS41 and not other components of HOPS (Pols et al, 2013).
Starvation increases interactions of Stx16 with the lysosomal R‐SNARE VAMP8
We found that Stx16 interacts with VAMP3 and VAMP4 (Fig EV3F), consistent with the known literature regarding VAMP3 and VAMP4 as cognate R‐SNAREs during retrograde transport from endosomes to TGN (Mallard et al, 2002; Ganley et al, 2008). However, upon starvation another R‐SNARE, VAMP8, increased its presence in Stx16 complexes (Fig EV3F). The lysosomal SNARE VAMP8 (Jahn & Scheller, 2006) has not been previously implicated in Stx16‐dependent trafficking, and our findings suggest a new relationship detected only under starvation conditions. Similarly, Stx6, a Qc‐SNARE working together with Stx16, showed increased interactions with VAMP8 in starved cells (Fig EV3F). Thus, similar relationship to VAMP8 was observed with both Stx16 and Stx6. We also noticed increased Stx6‐VAMP3 interactions in starved cells (Fig EV3F) and interpret this as an overall increase in trafficking between TGN and the lysosomal/endosomal pathway.
Stx16 affects mTOR regulation
Lysosomes are not only digestive organelles but also serve as a principal cytoplasmic location for active mTOR (Betz & Hall, 2013; Saxton & Sabatini, 2017; Lawrence & Zoncu, 2019). Given that we detected defects in LAMP1/2 levels and profiles (Fig 5D and E) in STX16 KO cells, we wondered whether mTOR would be affected and serve as another reporter of the lysosomal status in this context. We tested mTOR activity by monitoring phosphorylation of its downstream targets, such as 4E‐BP1 (Gingras et al, 1999), and ULK1S757, with latter being a principal phospho‐site for ULK1 and repression of autophagy by mTOR (Kim et al, 2011). Under resting conditions, no differences in mTOR activation status were observed in STX16 KO vs WT cells (Fig 5G). Under starvation conditions, as expected, mTOR activity was reduced, but this reduction was much more pronounced in STX16 KO cells (Fig 5G).
In resting cells, active mTOR forms cytoplasmic puncta and localizes to lysosomes (Betz & Hall, 2013; Saxton & Sabatini, 2017), but in starved cells mTOR is less punctate and translocates from lysosomes into the cytoplasm (Sancak et al, 2010). Under resting conditions (full medium), mTOR puncta (quantified by HCM) were not reduced in STX16 KO vs. WT cells (Fig 5H and I). One hour of starvation reduced mTOR puncta, as quantified by HCM (Fig 5H and I), reflecting mTOR inactivation. This effect was more pronounced in STX16 KO cells relative to WT cells (Fig 5H and I). When the cells were starved for 6 h, a time point that coincided with mTOR persistent inactivation assessed by phosphorylation of its targets (Fig 5G), we found an even stronger reduction in total mTOR puncta (Appendix Fig S2A and B). Localization of mTOR to LAMP2+ profiles was reduced by starvation (examined at 6 h; Fig EV4A and B), an effect that was more pronounced in STX16 KO relative to WT cells. Thus, Stx16 affects mTOR activation state, correlating with the role of Stx16 in cellular lysosomal status.
Figure EV4. Stx16 affects lysosomal homeostasis and mTOR activity.
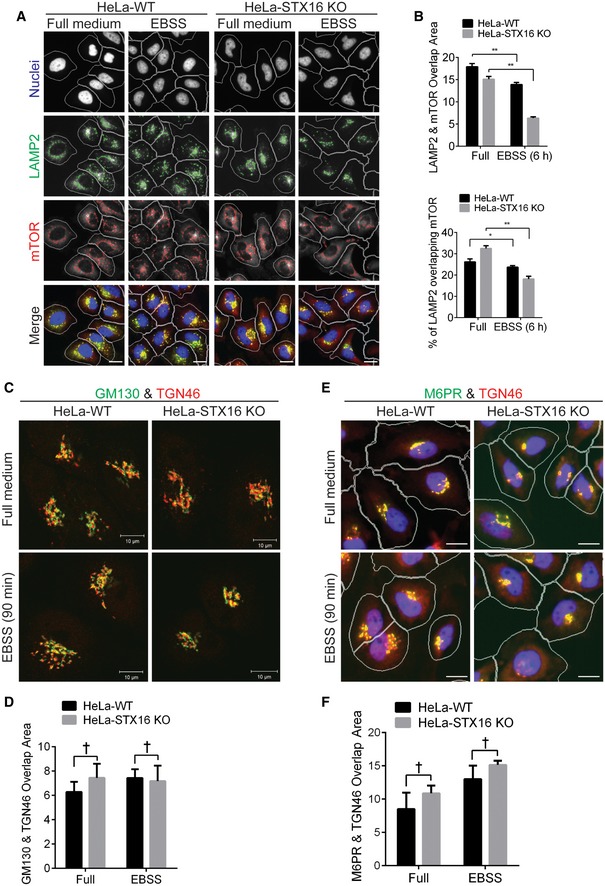
- WT or STX16 KO HeLa cells were starved in EBSS for 6 h and subjected to HCM analysis of the overlap between LAMP2 and mTOR. Masks: white, cells identified based on nuclei; green, LAMP2 puncta; red, mTOR puncta; yellow, overlap between LAMP2 and mTOR. Scale bar: 20 μm.
- Quantifications of overlaps between LAMP2 and mTOR in WT and STX16 KO HeLa cells treated as in (D). Data shown as means ± SEM of overlap area per cell between LAMP2 and mTOR (upper panel), and percentages of LAMP2 overlapping with mTOR to compensate reduced LAMP2 puncta in STX16 KO cells (see Fig 5D), minimum 500 cells were counted each well from at least 12 wells, three independent experiments; *P < 0.05; **P < 0.01 (two‐way ANOVA).
- WT or STX16 KO HeLa cells were starved in EBSS for 90 min, fixed, and stained with GM130 and TGN46 antibodies, followed by confocal microscopy analysis of the GM130 (Golgi marker, green) and TGN46 (TGN marker, red) profiles. Scale bar, 10 μm.
- HCM quantifications of overlaps between GM130 and TGN46 in WT and STX16 KO HeLa cells treated as in (A). Data shown as means ± SEM of overlap area per cell between GM130 and TGN46, minimum 500 cells were counted each well from at least 12 wells, three independent experiments; †, not significant (two‐way ANOVA).
- WT or STX16 KO HeLa cells were starved in EBSS for 90 min, fixed, and stained with M6PR and TGN46 antibodies, followed by HCM analysis of the M6PR and TGN46 profiles. Masks: white, cells identified based on nuclei; green, M6PR puncta; red, TGN46; yellow, overlap between M6PR and TGN46. Scale bar: 20 μm.
- HCM quantifications of overlaps between M6PR and TGN46 in WT and STX16 KO HeLa cells treated as in (C). Data shown as means ± SEM of overlap area per cell between M6PR and TGN46, minimum 500 cells were counted each well from at least 12 wells, three independent experiments; †, not significant (two‐way ANOVA).
Stx16 is required for appropriate distribution of acidified compartments in the cell
Since STX16 knockout resulted in reduced cellular content of LAMP1/2, which is in lysosomal and additional acidified endosomal compartments (Lippincott‐Schwartz & Fambrough, 1987) (Cheng et al, 2018), we anticipated that the cells might have diminished content of acidified organelles. We used LysoTracker Red DND‐99 (LTR) as a standard cell biological probe for acidification of intracellular compartments (Wubbolts et al, 1996). Surprisingly, we found no change in total LTR fluorescence by number of profiles/cell or total area/cell (Fig 6A and B) utilizing HCM (Cellomics) and comparing WT and STX16 KO HeLa cells under autophagy‐inducing conditions (starvation). This was confirmed by total LysoTracker Green DND‐26 (LTG) fluorescence and LTG puncta/cell using ImageStream flow cytometry (Amnis) in WT vs STX16 KO cells induced for autophagy (Appendix Fig S3A–C). However, when we examined localization and morphology of LTR+ profiles in STX16 KO cells, under autophagy‐inducing conditions when LTR+ organelles were strongly enhanced (relative to the resting conditions), we found that the LTR labeling redistributed from its usual presentation as cytoplasmic puncta into a compartment clustering in a perinuclear region colocalizing with the TGN marker TGN46 (Fig 6C and D). To ensure that the Golgi apparatus was not perturbed, we re‐examined the ultrastructural images in Fig 2E and found that the overall morphology of the Golgi stacks was similar in WT and mutant cells, but that the volume fraction of the Golgi compartment was increased (Fig 6E), consistent with an increased retention of acidified compartments in the Golgi. Furthermore, TGN46 did not show major changes in distribution relative to the Golgi marker GM130 (Fig EV4C and D) and relative to another factor in TGN‐endolysosomal trafficking, mannose 6‐phosphate receptor/M6PR (Fig EV4E and F). Thus, the effects of Stx16's absence entail redistribution of acidified compartments, in keeping with a reduction in levels of the integral lysosomal membrane proteins LAMP1 and LAMP2.
Figure 6. Stx16 regulates proper distribution of acidified compartments in the cell.
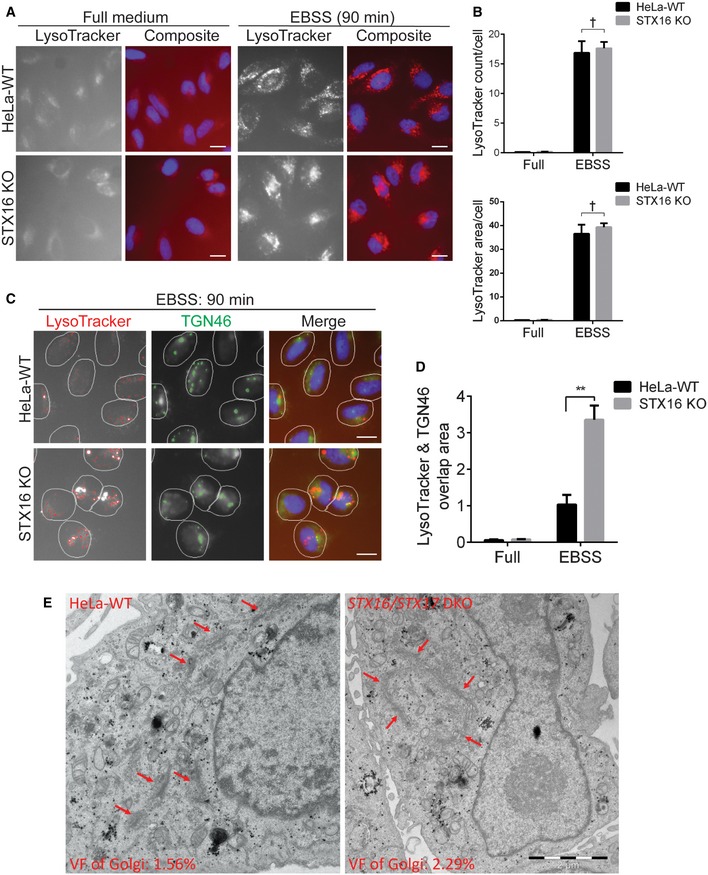
- WT or STX16 KO HeLa cells were starved in EBSS for 1 h, followed by starvation with the presence of LysoTracker Red DND‐99 (LTR) (100 nM) for additional 30 min, and subjected to HCM analysis of LTR. Scale bar: 20 μm.
- Quantifications of LTR count per cell or total area per cell in WT and STX16 KO HeLa cells treated as in (A). Data shown as means ± SEM of LTR puncta per cell or object total area per cell, minimum 500 cells were counted each well from at least 12 wells, three independent experiments; †, not significant (two‐way ANOVA).
- WT or STX16 KO HeLa cells were starved in EBSS for 1 h, followed by starvation with the presence of LTR (100 nM) for additional 30 min, and subjected to HCM analysis of the colocalization between LTR and TGN46. Masks: white, cells identified based on nuclei; red, LTR puncta; green, TGN46 puncta; yellow, overlap between LTR and TGN46. Scale bar: 20 μm.
- Quantifications of overlaps between LysoTracker Red and TGN46 in WT and STX16 KO cells treated as in (C). Data shown as means ± SEM of LTR and TGN46 overlap area per cell, minimum 500 cells were counted each well from at least 12 wells, three independent experiments; **P < 0.01 (two‐way ANOVA).
- WT or STX16/STX17 DKO HeLa cells were starved in EBSS for 2 h and subjected to ultrastructural analysis of the Golgi apparatus with electron microscopy. VF of Golgi: volume fraction of Golgi relative to total cytoplasmic area. Scale bar, 2 μm. Red arrows indicate Golgi apparatus.
Mammalian Atg8s regulate Stx16 localization to endosomal and lysosomal compartments
Since Stx16 is an mAtg8‐binding SNARE (Fig 1), we tested whether mAtg8s influenced Stx16 localization in the cell. We used the previously characterized triple LC3A, LC3B, LC3C knockout (Tri‐LC3KO), triple GABARAP, GABARAPL1, GABARAPL2 knockout (Tri‐GBRPKO), and all six mATG8 knockout (HexaKO) HeLa cells (Nguyen et al, 2016a) (Fig EV5A). First, we tested LAMP1 and LAMP2 protein levels in Tri‐LC3KO, Tri‐GBRPKO, and HexaKO cells. LAMP levels were not reduced under resting conditions in mAtg8‐mutant cells by quantifying LAMP2+ organelles (HCM, number of puncta/cell and total area/cell) (Appendix Fig S4A and B) as well as by Western blot analysis of LAMP1/2 proteins (Appendix Fig S4C). We nevertheless observed a minor LAMP reduction, possibly reflecting “exhaustion” during long‐term (8 h) starvation, in Tri‐GBRPKO and HexaKO cells (Appendix Fig S4D). When we assessed localization of Stx16 to LAMP2 (lysosome) and LBPA (late endosome), we observed significantly reduced colocalizations between Stx16 and LAMP2 as well as Stx16 and LBPA by several HCM measures: (i) LAMP2‐Stx16 overlap (Fig 7A and B); and (ii) LBPA‐Stx16 overlap (Fig 7C; Appendix Fig S5A). Comparing STX16 WT and its 219LVLV222, 219AVLA222, or LIR‐4A mutants, the LIR mutants showed reduced localization to LAMP2 profiles when compared to WT (Fig EV5B and C). Thus, mAtg8s and their binding site (LIR) on Stx16 play a role in placement of Stx16 on lysosomes.
Figure EV5. mAtg8s affect acidification of the endolysosomal system and mTOR activity.
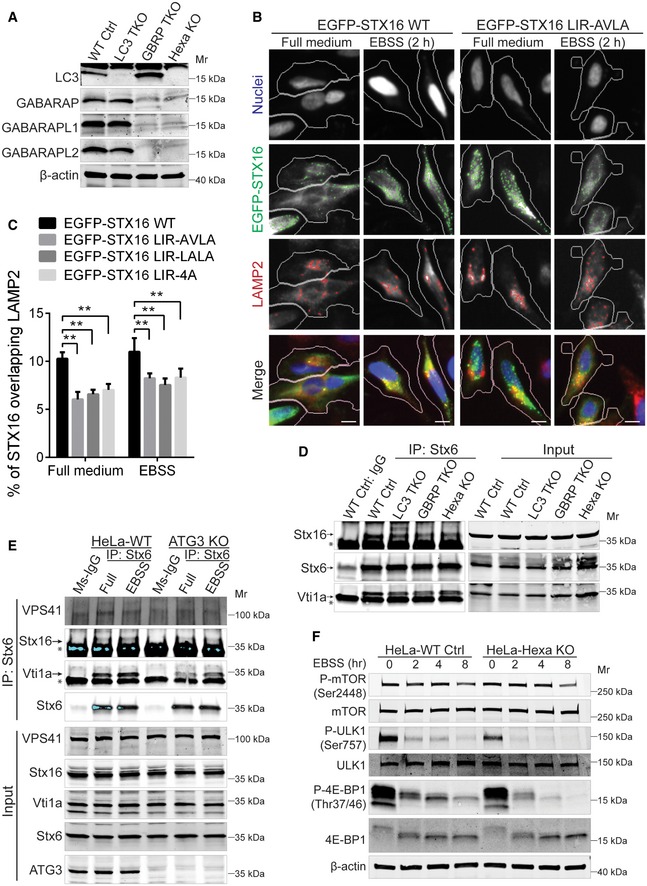
- Western blot analysis of LC3 (LC3A, LC3B, LC3C), GABARAP, GABARAPL1, and GABARAPL2 in WT HeLa or cells knocked out for LC3A, LC3B, LC3C (LC3 TKO), GABARAP, GABARAPL1, and GABARAPL2 (GBRP TKO) or all 6 mATG8s (Hexa KO).
- HeLa cells were transfected with WT or LIR‐mutant EGFP‐tagged Stx16, and subjected to HCM analysis of overlaps between Stx16 and LAMP2. Masks: white, cells transfected with Stx16 identified based on the average intensity of EGFP‐Stx16; green, EGFP‐Stx16 puncta; red, LAMP2 puncta; yellow, overlap between EGFP‐Stx16 and LAMP2. Scale bar: 20 μm.
- HCM quantifications of overlaps between LAMP2 and WT or different types of LIR‐mutant EGFP‐Stx16 transfected into HeLa cells and treated as in (B). Data shown as means ± SEM of percentages of EGFP‐Stx16 overlapping with LAMP2, minimum 200 transfected cells were counted each well from at least 12 wells, 3 independent experiments; **P < 0.01 (one‐way ANOVA).
- Endogenous Co‐IP analysis of the interactions between Stx16, Vti1a, and Stx6 in WT, LC3 TKO, GABARAP TKO, or Hexa KO HeLa cells. * indicates mouse IgG of precipitated mouse anti‐Stx6 antibody.
- Endogenous Co‐IP analysis of the interactions between VPS41, Stx16, Vti1a, and Stx6 in WT, or ATG3 KO cells. * indicates mouse IgG of precipitated mouse anti‐Stx6 antibody.
- WT or Hexa KO HeLa cells were starved in EBSS for indicated time points, and cell lysates were subjected to Western blot analysis of mTOR activity by mTOR substrate phosphorylation.
Source data are available online for this figure.
Figure 7. Mammalian Atg8s regulate Stx16 localization and acidification of endolysosomal organelles.
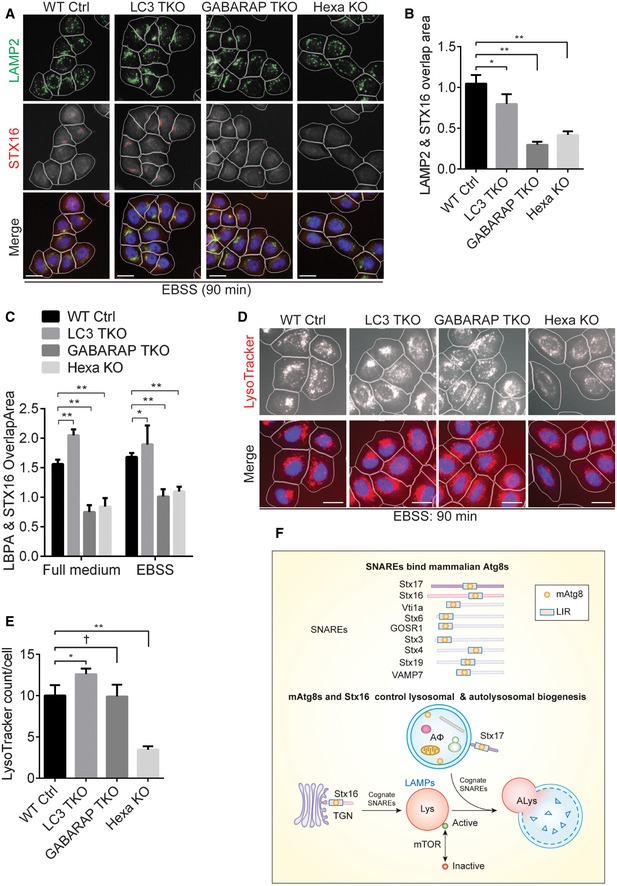
- WT, LC3TKO, GABARAPTKO, or HexaKO HeLa cells were starved in EBSS for 90 min and subjected to HCM analysis of overlaps between LAMP2 and Stx16. Masks: white, cells identified based on nuclei; green, LAMP2 puncta; red, Stx16 puncta; yellow, overlap of LAMP2 and Stx16. Scale bar: 20 μm.
- Quantifications of overlaps between LAMP2 and Stx16 in WT, LC3TKO, GABARAPTKO, or HexaKO HeLa cells treated as in (C). Data shown as means ± SEM of LAMP2 and Stx16 overlap area per cell, minimum 500 cells were counted each well from at least 12 wells, three independent experiments; *P < 0.05; **P < 0.01 (one‐way ANOVA).
- Quantifications of overlaps between LBPA and Stx16 in WT, LC3TKO, GABARAPTKO, or HexaKO HeLa cells grown in full medium or treated as in (A). Data shown as means ± SEM of LBPA and Stx16 overlap area per cell, minimum 500 cells were counted each well from at least 12 wells, three independent experiments; *P < 0.05; **P < 0.01 (one‐way ANOVA). Representative images are shown in Appendix Fig S5A.
- WT, LC3TKO, GABARAPTKO, or HexaKO HeLa cells were starved in EBSS for 1 h, followed by starvation with the presence of LTR (100 nM) for additional 30 min, and subjected to HCM analysis of LTR. Scale bar: 20 μm.
- Quantifications of LTR puncta in WT, LC3TKO, GABARAPTKO, or HexaKO HeLa cells treated as in (D). Data shown as means ± SEM of LTR puncta per cell, minimum 500 cells were counted each well from at least 12 wells, three independent experiments; †, not significant; *P < 0.05; **P < 0.01 (one‐way ANOVA).
- Schematic model of this study. Mammalian Atg8 proteins directly bind Stx16 through its LIR motif, which controls the proper localization of the Stx16 SNARE complex at the endolysosomal compartments. The Stx16 SNARE complex functions in the maintenance of lysosomal homeostasis upon autophagy induction, thus controlling mTOR activity.
Endogenous Co‐IP analyses showed reduced interactions between components of the Stx16/Vti1a/Stx6 SNARE complex in Tri‐GBRPKO and HexaKO cells (Fig EV5D), suggesting a regulatory role for mAtg8s in SNARE complex assembly or stability. A lipidation status of mAtg8s may be of significance as well, since we observed reduced interactions between Stx6 and Stx16, Vti1a or VPS41 in ATG3 KO HeLa cells generated by CRISPR/Cas9 (Fig EV5E); ATG3 is a key factor required for lipidation of mAtg8s (Mizushima et al, 2011). We next tested cellular acidified compartments by HCM and Amnis, and quantified effects of mAtg8 mutants. In HexaKO cells, the overall LTR+ profiles were reduced by HCM (Fig 7D and E) and LTG+ profiles were reduced by ImageStream flow cytometry (Appendix Fig S5B–D). mAtg8s also had an effect on mTOR inactivation in response to starvation, as evidenced by a more pronounced loss of phosphorylation of mTOR targets ULK1 (phospho‐Ser757) and 4E‐BP1 (phospho‐Thr37/46) in HexaKO cells relative to WT HeLa cells (Fig EV5F). In summary, albeit mAtg8s do not completely parallel Stx16 phenotype, they do have an effect on Stx16 distribution and mTOR inactivation. Additionally, mAtg8s show strong effects on acidified, LTR+ compartments in cells under autophagy‐inducing conditions. Thus, mAtg8s and Stx16 have partially overlapping effects on the endolysosomal system.
Discussion
Here, we uncovered a broad potential for interactions between the core autophagy components, mAtg8s, and the core membrane fusion system composed of SNAREs. The bioinformatics analyses and follow‐up biochemical experiments indicate that mAtg8s and several SNAREs work together in membrane trafficking and organelle biogenesis. One subset of SNAREs interacting with mAtg8s, Stx16‐Vti1a‐Stx6, was examined in detail, and Stx16 was fully characterized for mAtg8 binding, as has been done for Stx17 (Kumar et al, 2018). This leads us to propose a model departing from the conventional views of how LC3 or other mAtg8s work in autophagy, suggesting a new mechanism of control of membrane trafficking/fusion through direct action of mAtg8s on SNARE molecules (Fig 7F).
Our findings with Stx16 indicate a function for mAtg8s in the maintenance of lysosomal compartments (Fig 7F), through binding directly and regulating SNAREs of the Stx16 complex, which leads up to autolysosome biogenesis. This amends the conventional view of mAtg8s expanding the autophagosomal membranes (Carlsson & Simonsen, 2015) and complements more recent views of mAtg8s working in the recruitment of various components of the autophagic apparatus. These include (i) mAtg8 binding to autophagic receptors from the SLR (Birgisdottir et al, 2013; Rogov et al, 2014) and TRIM (Mandell et al, 2014; Keown et al, 2018) families of receptors; (ii) the ULK1‐FIP200‐ATG13 autophagy initiation complex (Alemu et al, 2012); (iii) a large number of ancillary components facilitating the autophagosome–lysosome fusion (McEwan et al, 2015; Olsvik et al, 2015; Wang et al, 2016; Kriegenburg et al, 2018); and (iv) non‐autophagic roles of mAtg8s, such as LAP (Sanjuan et al, 2007), LAP‐like processes (Florey et al, 2015), MVB/exosome formation (Guo et al, 2017), and via binding to TECPR2 in the COPII‐dependent ER export (Stadel et al, 2015).
Human cells have 6 mAtg8s, with proposed but not fully delineated specialized functions (Shpilka et al, 2011). Their role has been established in the recruitment of several important ancillary factors facilitating membrane fusion at the autophagosome–lysosome fusion stage of the autophagic pathway (Nakamura & Yoshimori, 2017). An early study has shown GABARAPL2/GATE‐16 interaction with NSF enhancing its ATPase activity, thus implicating it in activation of the Golgi SNARE GOS‐28/GOSR1 (Sagiv et al, 2000), of consequence for post‐mitotic Golgi reformation (Muller et al, 2002) and intra‐Golgi transport (Sagiv et al, 2000). This is in keeping with the results of our screen of mAtg8‐interacting SNAREs and is compatible with the concept of mAtg8s directly acting on SNAREs.
Stx16 and its cognate SNAREs have been shown to play a role in retrograde trafficking from endosomal compartments to TGN with two distinct pairings of Qa‐, Qb‐, and Qc‐SNAREs: (i) Stx16, Vti1a, and Stx10, required specifically for M6PR transport (we did not observe effects on M6PR relative to TGN46 in our studies), which impacts trafficking of lysosomal hydrolases (Ganley et al, 2008); and (ii) Stx16, Vti1a, and Stx6, which is independent of the Stx10/M6PR pathway and controls trafficking to TGN of endocytosed incoming toxins bound to membrane glycoconjugates (Mallard et al, 2002). There is also a Stx16‐independent retrograde pathway that instead depends on GOS‐28/GOSR1 (Tai et al, 2004), another SNARE that incidentally binds mAtg8s (see Fig 1A, 6th peptide array from the top). Of note, GOS‐28/GOSR1 has been first described as a GATE‐16‐binding SNARE (Sagiv et al, 2000) with Golgi functions. Most of the prior studies with Stx16 complexes have been focused on retrograde trafficking to the TGN, and thus, our observations could be due to perturbed retrograde membrane and cargo flow. However, our findings that Stx16 is in complexes with VPS41, previously implicated in anterograde transport of LAMP1/2 (Pols et al, 2013), and reduced abundance of LAMP1/2+ organelles in STX16 KO cells suggest that Stx16 defect blocks the prograde delivery of LAMP1/2 during lysosomal biogenesis.
Trafficking of various components that make up functional lysosomes is only partially understood (Luzio et al, 2000; Saftig & Klumperman, 2009; Bright et al, 2016) especially regarding anterograde pathways to lysosomes of integral membrane proteins such as LAMP1/2 and trafficking to and assembly at the lysosome of the V1V0 holoenzyme vacuolar H+‐ATPase (Saftig & Klumperman, 2009). Our findings suggest that Stx16 is responsible for LAMP1/2 trafficking to lysosomes with secondary consequences on the overall levels of LAMPs when this delivery is disrupted. It is likely that Stx16 does not affect vacuolar H+‐ATPase's ability to function but these acidified compartments may be displaced due to a lack of clearly defined lysosomal destination. Some of STX16 KO phenotypes (perturbation of acidified compartments) are echoed in mAtg8s null HexaKO cells, which show diminished overall content of acidified organelles. These phenomena can in part be explained by mAtg8s affecting Stx16 localization. Since HexaKO effects on acidification are more pronounced than the effects of STX16 KO, this indicates additional functions for mAtg8s, specifically in H+‐ATPase function. For example, it has recently been shown that LC3 affects ATP6V1E1 subunit of the V1 domain of vacuolar H+‐ATPase (Guo et al, 2017). Although mAtg8s do not phenocopy in full the Stx16 phenotype, they do affect Stx16 distribution and mTOR activity. Thus, mAtg8s and Stx16 have only partially overlapping effects on the endolysosomal system most likely due to mAtg8 action exceeding the reach of the Stx16 function.
Since lysosomes are the location of active mTOR (Betz & Hall, 2013; Saxton & Sabatini, 2017), we used mTOR activity as another readout of Stx16 effects on the lysosomal function. Somewhat surprisingly, given the strong reduction of LAMP1/2 levels and diminished number of LAMP+ organelles, STX16 KO did not show effects on basal mTOR activity. However, during starvation, which inactivates mTOR and triggers its translocation from the lysosomes, mTOR was inhibited far more prominently in STX16 KO cells and this was also reflected in fewer mTOR+ cytoplasmic puncta. One interpretation is that the reduced LAMP1/2 content differentially affects subpopulations of lysosomes (Bright et al, 2016) and that the ones that are platforms for active mTOR are preferentially preserved/maintained. Nevertheless, upon stress and perturbance, inactivation of mTOR is more effective and more durable in STX16 KO cells. This could be a reflection of altered trigger thresholds dependent on diminished lysosomal sources of nutrients and/or difficulties in re‐establishing nutrient equilibrium and mTOR activity. Alternatively, Stx16 may be important in transport to lysosomes of additional mTOR regulatory components important for its reactivation.
The Qa‐SNARE Stx17 was first appreciated as a key SNARE in autophagosome–lysosome fusion (Itakura et al, 2012; Takats et al, 2013; Guo et al, 2014; Diao et al, 2015; Wang et al, 2016). Recent studies have uncovered the yeast SNARE, Ykt6, as important in autophagosome–vacuole/lysosome fusion in yeast (Bas et al, 2018; Gao et al, 2018), and that mammalian Ykt6 ortholog (Matsui et al, 2018; Takats et al, 2018) plays a potentially dominant (over Stx17) role in mammalian autophagosome–lysosome fusion. Whereas the exact mechanism of Ykt6 in maturation has not been agreed upon, with differences in proposed models (Matsui et al, 2018; Takats et al, 2018), it is worth noting that Ykt6 is also part of one of the retrograde trafficking routes from endosomes to TGN that includes GOS‐28/GOSR1 (Tai et al, 2004), an mAtg8‐binding SNARE as discussed above. Nevertheless, our data here suggest that Stx17 still plays a very important role in autophagosomal flux, revealed in the requirement for a STX16/STX17 double KO to block autophagic flux using the conventional and well‐accepted LC3 flux assay (Klionsky et al, 2016), and reflected in autophagic degradation of a diverse panel of substrates: mitochondria, peroxisomes, M. tuberculosis, and ribosomes.
Stx17 interacts via its LIR motif with mAtg8s, and this may play a role in its recruitment to autophagosomes as a prelude to fusion with the lysosomes (Kumar et al, 2018). However, Stx17's role in autophagy begins much earlier, during initiation (Hamasaki et al, 2013; Arasaki et al, 2015, 2018; Kumar et al, 2019) ensuring that once autophagosomes are formed they are enabled for fusion with endolysosomal compartments. The present screen for other SNAREs that bind mAtg8s yielded a number of candidates in addition to Stx16, such as Vti1a and Stx6, which partner with Stx16, and Stx3 and Stx4, SNAREs that are engaged in secretory autophagy (Kimura et al, 2017). This prompts us to suggest that mAtg8s’ function in autophagy should be reconsidered and some of its mechanistic principles revisited. We propose that mAtg8s act by reshaping overall membrane trafficking in the cell through direct interactions and effects on specific SNAREs and redirect membrane flow toward autophagy at its initiation and termination stages.
Materials and Methods
Antibodies and reagents
Antibodies: Rabbit anti‐LC3B (L7543, for WB), rabbit anti‐STX17 (HPA001204), and mouse anti‐FLAG (F1804, for WB and IP) were purchased from Sigma‐Aldrich; mouse anti‐LC3 (M152‐3, for WB and IP) and rabbit anti‐LC3 (PM036, for IF) were purchased from MBL International; mouse anti‐p62 (#610833) and mouse anti‐GM130 (#610823) were purchased from BD Biosciences; rabbit anti‐STX16 (NBP1‐92467), rabbit anti‐VAMP3 (NB300‐510), rabbit anti‐VAMP4 (NBP2‐13512), rabbit anti‐VPS33A (NBP2‐20872), and rabbit anti‐TGN46 (NBP1‐49643) were purchased from Novus Biologicals; mouse anti‐STX6 (H00010228‐M01, for WB and IP) was purchased from Abnova; mouse anti‐Vti1a (sc‐136117), mouse anti‐VAMP8 (sc‐166820), mouse anti‐ATG3 (sc‐393660), and mouse anti‐β‐actin (sc‐47778) were purchased from Santa Cruz Biotechnology; rabbit anti‐VPS41 (ab181078), rabbit anti‐PEX14 (ab183885), rabbit anti‐PMP70 (ab85550), rabbit anti‐GFP (ab290), and rabbit anti‐β‐tubulin (ab18251) were purchased from Abcam; rabbit anti‐LAMP1 (D2D11) (#9091), Phospho‐AMPKα (Thr172) (#2531), AMPKα (#2532), Phospho‐ULK1 (Ser757) (#6888), ULK1 (D8H5) (#8054), Phospho‐4EBP1 (Thr37/46) (#2855), 4E‐BP1 (#9452), Phospho‐mTOR (Ser2448) (#2971), rabbit anti‐mTOR (7C10) (#2981), rabbit anti‐LKB1 (27D10) (#3050), and Autophagy Atg8 Family Antibody Sampler Kit (#64459) were purchased from Cell Signaling Technology; mouse anti‐LBPA (C64) (MABT837) was purchased from EMD Millipore; mouse anti‐DNA antibody (#61014) was purchased from Progen; and mouse anti‐LAMP2 (H4B4) and mouse anti‐M6PR (22d4) were purchased from Developmental Studies Hybridoma Bank (DSHB) at the University of Iowa.
Earle's Balanced Salt Solution (EBSS) (E3024) and hydrogen peroxide solution (H1009) were purchased from Sigma‐Aldrich. Bafilomycin A1 was purchased from InvivoGen (Cat. Code: tlrl‐baf1). CCCP (C2759), oligomycin A (#75351), and antimycin A (A8674) were purchased from Sigma‐Aldrich. LysoTracker™Red DND‐99 (L7528) and LysoTracker™ Green DND‐26 (L7526) were purchased from Thermo Fisher Scientific and used at 100 nM final concentration for 30 min.
Cell culture and transfection
HeLa, HEK293T, U2OS, and THP‐1 cells were purchased from the American Type Culture Collection (ATCC). HeLa, HEK293T, and U2OS cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L‐glutamine, 10 mM HEPES, 1.0 mM sodium pyruvate, and 1× penicillin–streptomycin (Thermo Fisher Scientific) at 37°C in a 5% CO2 atmosphere. THP‐1 cells were grown in RPMI 1640 with 2 mM l‐glutamine adjusted to contain 1.5 g/l sodium bicarbonate, 4.5 g/l glucose, 10 mM HEPES, and 1.0 mM sodium pyruvate and supplemented with 0.05 mM β‐mercaptoethanol (M6250, Sigma‐Aldrich) and 10% FBS. The Huh7 cell line was purchased from Rocky Mountain Laboratory. Keima reporter cell lines HEK293 RPS3‐Keima and HCT116 RPL28‐Keima were obtained from J. Wade Harper, Harvard University (An & Harper, 2018).
For Co‐IP analysis of the interactions between overexpressed Stx16/Vti1a/Stx6 and mAtg8s, HEK293T cells in 10‐cm petri dishes were transfected with corresponding plasmids via the ProFection Mammalian Transfection System (E1200, Promega). For other experiments, cells were transfected with Lipofectamine 2000 Transfection Reagent (# 11668‐019, Thermo Fisher Scientific) according to the manufacturer's manual.
Plasmid constructs
STX16 plasmid was obtained from DNASU (#HsCD00396980); STX6 was obtained from Addgene (#31581); VTI1A DNA was PCR amplified from cDNA reverse transcribed from total RNAs isolated from HeLa cells. All constructs were transferred into pDEST‐EGFP or pDEST‐3XFLAG vectors using Gateway cloning system (#12535‐019, Thermo Fisher Scientific) and verified by sequencing at GENEWIZ. EGFP‐tagged LC3B and GABARAP constructed in pDEST vector were described before (Alemu et al, 2012). LIR mutants of STX16 were generated with QuikChange Lightning Site‐Directed Mutagenesis Kit (#210519, Agilent).
GST pull‐down assay and peptide array analysis
Recombinant GST and GST‐fusion proteins were expressed in competent Escherichia coli SoluBL21 (Genlantis, #C700200) by inducing overnight bacterial cultures with 50–75 μg/ml isopropyl β‐D‐1‐thiogalactopyranoside (IPTG). Expressed proteins were purified by immobilization on Glutathione Sepharose 4 Fast Flow beads (GE Healthcare, #17‐5132‐01). For GST pull‐down assays, myc‐tagged proteins were in vitro translated in the presence of radioactive methionine (35S‐methionine) using the TNT T7 Reticulocyte Lysate System (Promega, #l4610). 10 μl of in vitro translated proteins was first precleared to remove unspecific binding with 10 μl of empty Glutathione Sepharose beads in 100 μl of NETN buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5% NP‐40) supplemented with cOmplete™ EDTA‐free Protease Inhibitor Cocktail (Roche, #1183617001) for 30 min at 4°C. This was followed by incubation of the precleared mixture with purified GST or GST‐fusion proteins for 1–2 h at 4°C. The mixture was washed five times with NETN buffer by centrifugation at 2,500 g for 2 min followed by addition of 2XSDS gel‐loading buffer (100 mM Tris pH 7.4, 4% SDS, 20% glycerol, 0.2% bromophenol blue, and 200 mM dithiothreitol (DTT) (Sigma, #D0632) and heating for 10 min. The reaction was then resolved by SDS–PAGE and the gel stained with Coomassie Brilliant Blue R‐250 Dye (Thermo Fisher Scientific, #20278) to visualize the GST and GST‐fusion proteins. The gel was then vacuum‐dried and radioactive signal detected by Bioimaging analyzer BAS‐5000 (Fujifilm).
Peptide arrays of 20‐mer amino acids of each SNARE protein were synthesized and immobilized on a membrane with the MultiPrep peptide synthesizer (Intavis Bioanalytical Instruments AG). The membrane was first blocked with 5% non‐fat dry milk and then incubated with GST‐GABARAP for 24 h. The membrane was washed three times and visualized by immunoblotting with anti‐GST antibody as described (Rasmussen et al, 2019).
Generation of knockout cells with CRISPR/Cas9 gRNA
Sequences of gRNAs targeting STX16 and STX17 are shown in Fig EV2A. ATG3 gRNA target sequence: TAGTCCACCACTGTCCAACA; STX6 gRNA target sequence: ACATGTCCCAGCGCATCGGA; VTI1A gRNA target sequence: AGATACCACCCCAAAGTCGA. All the single KO and STX16/STX17 DKO HeLa, Huh7, U2OS, THP1, and HeLa‐YFP‐Parkin cells were generated by infecting target cells with single‐guide RNA (sgRNA) lentiviral vectors, lentiCRISPRv2, as described in Sanjana et al (2014). Briefly, HEK293T cells were transfected with lentiCRISPRv2 sgRNA vectors together with psPAX2 and pMD2.G at the ratio of 4 μg, 2.5 μg, and 1.5 μg/6‐cm dish. Sixty hours later, the supernatants containing lentiviruses were collected and spun down at 300 g for 5 min to clear cell debris. Lentiviruses were diluted with DMEM full medium at 1:2 ratio and used to infect target cells overnight with the presence of 8 μg/ml of polybrene (Hexadimethrine bromide, H9268, Sigma‐Aldrich) in 12‐well plates. Then, the medium with lentivirus was removed and changed to fresh medium for another 24 h. Cells were selected with puromycin (1 μg/ml) for 5 days before validation of the knockout. Single clones of HeLa, Huh7, and U2OS KO cells were isolated by seeding single cells in 96‐well plates after serial dilutions. For THP1 KO cells, it is impossible to isolate single clones due to their suspension nature, and thus, depletion of Stx16 and Stx17 was not complete. Data are shown in Fig EV3C. Generation of LC3 TKO, GABARAP TKO, and Hexa KO HeLa cells was described as before (Nguyen et al, 2016b).
Western blotting and co‐immunoprecipitation (Co‐IP)
Cell lysates were prepared with a standard procedure using RIPA buffer (Pierce, #89900) supplemented with cOmplete™ EDTA‐free Protease Inhibitor Cocktail (Roche, #1183617001). Protein concentrations were determined using Pierce™ BCA Protein Assay Kit (#23225). Protein electrophoresis was carried out using TGX™ SDS Gels (Bio‐Rad, #4561091), followed by blotting to a nitrocellulose membrane (Bio‐Rad, #1620112). Membranes were incubated with primary antibodies diluted in blocking buffer (3% BSA diluted in 1XTBS buffer plus 0.05% Tween‐20) overnight at 4°C. After incubation with fluorescently labeled secondary antibodies goat‐anti‐mouse IRDye 680LT or goat‐anti‐rabbit IRDye 800CW (LI‐COR Biosciences) diluted 1:10,000 in blocking buffer, membranes were scanned using a fluorescence scanner Odyssey (LI‐COR Biosciences).
For Co‐IP of overexpressed proteins, HEK293T cells cultured in 10‐cm dishes were lysed with 1 ml NP‐40 cell lysis buffer (Invitrogen, FNN0021). Cell lysates were spun down at top speed for 10 min at 4°C to clear cell debris. The supernatants were collected and incubated with 2.5 μl anti‐FLAG antibody (M2) for 4 h, followed by incubation with Dynabeads® Protein G for 1 h, and precipitated proteins were subjected to Western blot analysis with rabbit anti‐GFP antibody. For endogenous Co‐IP analysis, cell lysates were prepared from cells cultured in 15‐cm dishes and incubated with 2.5 μg mouse anti‐Stx6 or mouse anti‐LC3 antibody overnight at 4°C, followed by Dynabeads® Protein G (Thermo Fisher Scientific, #10004D) binding and Western blot analysis of bound proteins.
Immunostaining and staining of acidified compartments for HCM and ImageStream flow cytometry
For HCM, cells were seeded into 96‐well plates. After treatments, cells were washed once with 1× PBS, followed by fixation with 4% paraformaldehyde (PFA) for 5 min at room temperature. Cells were then permeabilized with 0.2% Triton X‐100 for 10 min at room temperature. After incubation with blocking buffer (3% goat serum in 1× PBS), cells were stained with primary antibodies in blocking buffer for 1 h at room temperature or overnight at 4°C, followed by staining with secondary antibodies (Alexa Fluro conjugates, Thermo Fisher Scientific) diluted 1:1,000 in blocking buffer for 1 h at room temperature. For LysoTracker Red DND‐99, cells in 96‐well plates were cultured in full medium or starved for 1 h in EBSS, followed by adding LTR for another 30 min at the final concentration of 100 nM. For ImageStream flow cytometry analysis of LysoTracker Green DND‐26, cells in 6‐cm dishes were starved in EBSS for 1 h, followed by adding LTG for another 30 min at the final concentration of 100 nM. Cells were collected by trypsinization and resuspended in 200 μl 1× PBS for Amnis analysis.
High‐content microscopy (HCM) computer‐based image acquisition and quantification
High‐content microscopy with automated image acquisition and quantification was carried out using a Cellomics ArrayScan VTI platform (Thermo Fisher Scientific). A minimum of 500 primary objects (valid cells) per well were imaged for the quantifications of intracellular targets (regions of interest, ROI). Scanning parameters and object masks were preset with HCS Studio Scan software for algorithm‐defined automated image acquisition and analysis. The images were viewed, and ROI quantified with HCS View software iDEV provided with the Cellomics (Mandell et al, 2014).
ImageStream flow cytometry analysis
ImageStreamX Mark II flow cytometer Amnis® (EMD Millipore) was used to analyze LysoTracker Green DND‐26 (LTG) staining after autophagy induction by starvation. The 488‐nm laser was used for excitation. Debris and doublets were gated out. Bright field (430–480 nm, Channel 01) and LTG (505–560 nm, Channel 02) channels were measured, and at least 8,000 events of single cells per sample were collected. For analysis, the IDEAS version 6.0 software was used. Gating was applied to focused single cells using gradient root mean square of the bright field image then bright field area and aspect ratio, respectively. Fifty cells with low/no fluorescent intensity and 50 cells with high fluorescent intensity and verified punctate LTG staining by image collection from WT cells were selected to create a template. The template was then applied to all other samples in the same experiment to generate quantifications (counts of LTG/cell) for each sample.
Electron microscopy
Wild‐type or STX16/STX17 DKO HeLa cells were grown in 6‐cm dishes until they became semi‐confluent. The cells were starved for 2 h in EBSS, followed by fixation with 2% glutaraldehyde (EM grade) in 0.2 M HEPES, pH 7.4. After 30‐min initial incubation, cells were scraped under a small volume of fixative and transferred to Eppendorf tubes. The tubes were spun at full speed for 10 min at room temperature to get a firm pellet. The pellets were continued being fixed until total fixation time is 2 h. Thin sections were cut using an ultramicrotome, collected onto electron microscopy grids, and stained with uranyl acetate and lead citrate. In order to count autophagic compartments or Golgi apparatus, 47 images for each sample were taken at primary magnification of 5,000×, using the principles of uniform random sampling. The images were zoomed on computer screen. Autophagic compartments and the Golgi apparatus were counted, and the cytoplasmic area was estimated by point counting (Eskelinen, 2008).
Mycobacterium tuberculosis killing assay
Mycobacterium tuberculosis killing assay was carried out according to previously described (Kumar et al, 2018). Briefly, Mycobacterium tuberculosis Erdman (Erdman) culture was prepared by thawing frozen stock aliquot and grown in 7H9 Middlebrook liquid medium supplemented with oleic acid, albumin, dextrose, and catalase (OADC, Becton Dickinson, Inc., Sparks, MD, USA), 0.5% glycerol, and 0.05% Tween 80. Cultures were grown at 37°C. WT, STX16 KO, STX17 KO, and STX16/STX17 DKO THP1 cells (Fig EV3C) were infected with Erdman at MOI 10 and incubated with full medium for 18 h or 16 h followed by starvation in EBSS for 2 h to induce autophagy, lysed, and plated on 7H11 agar plates. CFU was enumerated 3 weeks later.
Quantifications and statistical analysis
Data, means ± SEM (n ≥ 3), were analyzed with paired and unpaired two‐tailed Student's t‐test, or by one‐way or two‐way ANOVA (analysis of variance) followed by post hoc Tukey's test. Statistical significance was defined as: † P ≥ 0.05; *P < 0.05; **P < 0.01.
Author contributions
Conceptualization, YG, TJ, SK, and VD; Investigation and Validation, YG, VD, and TJ; Methodology, YG, YPA, SK, BB, SWC, JJ, and E‐LE; Formal Analysis, YG, SK, VD, and TJ; Resources, ML; Writing—Original Draft, YG and VD; Writing—Review and Editing, YG, VD, ML, and TJ; Supervision, VD and TJ; Funding Acquisition, VD and TJ.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank John Weaver and the AIM center, and J. Wade Harper for Keima cell lines, and Aurore Claude‐Taupin for discussions. This work was supported by NIH grants R37AI042999, R01AI042999 and a center grant P20GM121176 to V.D. The work in the laboratory of T.J. was supported by Grant No. 190214 from the Norwegian Cancer Society and Grant No. 249884 from the Research Council of Norway to T.J. M.L. is supported by an ARC future fellowship (FT1601100063) and by the NHMRC (GNT1106471 and GNT1160315).
The EMBO Journal (2019) 38: e101994
Note
Correction added on 24 October 2019, after first online publication: the sentence has been corrected.
References
- Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Overvatn A, Kirkin V, Johansen T (2012) ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3‐interacting region (LIR) motifs. J Biol Chem 287: 39275–39290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Harper JW (2018) Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol 20: 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, Kudo Y, Baba M, Baba N, Cheng J et al (2015) A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell 32: 304–317 [DOI] [PubMed] [Google Scholar]
- Arasaki K, Nagashima H, Kurosawa Y, Kimura H, Nishida N, Dohmae N, Yamamoto A, Yanagi S, Wakana Y, Inoue H et al (2018) MAP1B‐LC1 prevents autophagosome formation by linking syntaxin 17 to microtubules. EMBO Rep 19: e45584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakula D, Muller AJ, Zuleger T, Takacs Z, Franz‐Wachtel M, Thost AK, Brigger D, Tschan MP, Frickey T, Robenek H et al (2017) WIPI3 and WIPI4 beta‐propellers are scaffolds for LKB1‐AMPK‐TSC signalling circuits in the control of autophagy. Nat Commun 8: 15637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas L, Papinski D, Licheva M, Torggler R, Rohringer S, Schuschnig M, Kraft C (2018) Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome‐vacuole fusion. J Cell Biol 217: 3656–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskaran S, Carlson LA, Stjepanovic G, Young LN, Kim do J, Grob P, Stanley RE, Nogales E, Hurley JH (2014) Architecture and dynamics of the autophagic phosphatidylinositol 3‐kinase complex. Elife 3: e05115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW (2010) Network organization of the human autophagy system. Nature 466: 68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz C, Hall MN (2013) Where is mTOR and what is it doing there? J Cell Biol 203: 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhujabal Z, Birgisdottir AB, Sjottem E, Brenne HB, Overvatn A, Habisov S, Kirkin V, Lamark T, Johansen T (2017) FKBP8 recruits LC3A to mediate Parkin‐independent mitophagy. EMBO Rep 18: 947–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgisdottir AB, Lamark T, Johansen T (2013) The LIR motif – crucial for selective autophagy. J Cell Sci 126: 3237–3247 [DOI] [PubMed] [Google Scholar]
- Birgisdottir AB, Bhujabal Z, Wirth M, Sjøttem E, Evjen G, Zhang W, Lee R, O'Reilly N, Tooze SA, Lamark T et al (2019) Members of the autophagy class III phosphatidylinositol 3‐kinase complex I interact with GABARAP and GABARAPL1 via LIR motifs. Autophagy 15: 1333–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171: 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright NA, Davis LJ, Luzio JP (2016) Endolysosomes are the principal intracellular sites of acid hydrolase activity. Curr Biol 26: 2233–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson SR, Simonsen A (2015) Membrane dynamics in autophagosome biogenesis. J Cell Sci 128: 193–205 [DOI] [PubMed] [Google Scholar]
- Chang C, Young LN, Morris KL, von Bulow S, Schoneberg J, Yamamoto‐Imoto H, Oe Y, Yamamoto K, Nakamura S, Stjepanovic G et al (2019) Bidirectional control of autophagy by BECN1 BARA domain dynamics. Mol Cell 73: 339–353.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XT, Xie YX, Zhou B, Huang N, Farfel‐Becker T, Sheng ZH (2018) Characterization of LAMP1‐labeled nondegradative lysosomal and endocytic compartments in neurons. J Cell Biol 217: 3127–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S et al (2015) ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520: 563–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12‐5‐16L1. Mol Cell 55: 238–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engedal N, Seglen PO (2016) Autophagy of cytoplasmic bulk cargo does not require LC3. Autophagy 12: 439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL (2008) Fine structure of the autophagosome. Methods Mol Biol 445: 11–28 [DOI] [PubMed] [Google Scholar]
- Florey O, Gammoh N, Kim SE, Jiang X, Overholtzer M (2015) V‐ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 11: 88–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S et al (2013) Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 203: 115–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammoh N, Florey O, Overholtzer M, Jiang X (2013) Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex‐dependent and ‐independent autophagy. Nat Struct Mol Biol 20: 144–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Espinosa E, Pfeffer SR (2008) A syntaxin 10‐SNARE complex distinguishes two distinct transport routes from endosomes to the trans‐Golgi in human cells. J Cell Biol 180: 159–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, du Lam H, Wang J, Ding X, Chen S, Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284: 12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Reggiori F, Ungermann C (2018) A novel in vitro assay reveals SNARE topology and the role of Ykt6 in autophagosome fusion with vacuoles. J Cell Biol 217: 3670–3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D, Shaw RJ (2017) AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell 66: 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatica D, Lahiri V, Klionsky DJ (2018) Cargo recognition and degradation by selective autophagy. Nat Cell Biol 20: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N (1999) Regulation of 4E‐BP1 phosphorylation: a novel two‐step mechanism. Genes Dev 13: 1422–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovacs AL, Zhang Z et al (2014) O‐GlcNAc‐modification of SNAP‐29 regulates autophagosome maturation. Nat Cell Biol 16: 1215–1226 [DOI] [PubMed] [Google Scholar]
- Guo H, Chitiprolu M, Roncevic L, Javalet C, Hemming FJ, Trung MT, Meng L, Latreille E, Tanese de Souza C, McCulloch D et al (2017) Atg5 disassociates the V1V0‐ATPase to promote exosome production and tumor metastasis independent of canonical macroautophagy. Dev Cell 43: 716–730.e7 [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766 [DOI] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER‐mitochondria contact sites. Nature 495: 389–393 [DOI] [PubMed] [Google Scholar]
- Hatsuzawa K, Hirose H, Tani K, Yamamoto A, Scheller RH, Tagaya M (2000) Syntaxin 18, a SNAP receptor that functions in the endoplasmic reticulum, intermediate compartment, and cis‐golgi vesicle trafficking. J Biol Chem 275: 13713–13720 [DOI] [PubMed] [Google Scholar]
- He C, Levine B (2010) The Beclin 1 interactome. Curr Opin Cell Biol 22: 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W, Lev S (2014) Tethering the assembly of SNARE complexes. Trends Cell Biol 24: 35–43 [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N et al (2009) Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell 20: 1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi‐Itakura C, Mizushima N (2012) The hairpin‐type tail‐anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151: 1256–1269 [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH (2006) SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol 7: 631–643 [DOI] [PubMed] [Google Scholar]
- Jao CC, Ragusa MJ, Stanley RE, Hurley JH (2013) A HORMA domain in Atg13 mediates PI 3‐kinase recruitment in autophagy. Proc Natl Acad Sci USA 110: 5486–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK‐Atg13‐FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20: 1992–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18: 1042–1052 [DOI] [PubMed] [Google Scholar]
- Keown JR, Black MM, Ferron A, Yap M, Barnett MJ, Pearce FG, Stoye JP, Goldstone DC (2018) A helical LC3‐interacting region mediates the interaction between the retroviral restriction factor Trim5alpha and mammalian autophagy‐related ATG8 proteins. J Biol Chem 293: 18378–18386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Mandell M, Deretic V (2016) Precision autophagy directed by receptor regulators – emerging examples within the TRIM family. J Cell Sci 129: 881–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, Dupont N, Jiang S, Peters R, Farzam F et al (2017) Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J 36: 42–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y (1999) Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol 147: 435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 12: 1–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegenburg F, Ungermann C, Reggiori F (2018) Coordination of autophagosome‐lysosome fusion by Atg8 family members. Curr Biol 28: R512–R518 [DOI] [PubMed] [Google Scholar]
- Kumar S, Jain A, Farzam F, Jia J, Gu Y, Choi SW, Mudd MH, Claude‐Taupin A, Wester MJ, Lidke KA et al (2018) Mechanism of Stx17 recruitment to autophagosomes via IRGM and mammalian Atg8 proteins. J Cell Biol 217: 997–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Gu Y, Abudu YP, Bruun JA, Jain A, Farzam F, Mudd M, Anonsen JH, Rusten TE, Kasof G et al (2019) Phosphorylation of Syntaxin 17 by TBK1 controls autophagy initiation. Dev Cell 49: 130–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence RE, Zoncu R (2019) The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 21: 133–142 [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G (2019) Biological functions of autophagy genes: a disease perspective. Cell 176: 11–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott‐Schwartz J, Fambrough DM (1987) Cycling of the integral membrane glycoprotein, LEP100, between plasma membrane and lysosomes: kinetic and morphological analysis. Cell 49: 669–677 [DOI] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185 [DOI] [PubMed] [Google Scholar]
- Low SH, Chapin SJ, Weimbs T, Komuves LG, Bennett MK, Mostov KE (1996) Differential localization of syntaxin isoforms in polarized Madin‐Darby canine kidney cells. Mol Biol Cell 7: 2007–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Rous BA, Bright NA, Pryor PR, Mullock BM, Piper RC (2000) Lysosome‐endosome fusion and lysosome biogenesis. J Cell Sci 113: 1515–1524 [DOI] [PubMed] [Google Scholar]
- Mallard F, Tang BL, Galli T, Tenza D, Saint‐Pol A, Yue X, Antony C, Hong W, Goud B, Johannes L (2002) Early/recycling endosomes‐to‐TGN transport involves two SNARE complexes and a Rab6 isoform. J Cell Biol 156: 653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malsam J, Söllner TH (2011) Organization of SNAREs within the Golgi Stack. Cold Spring Harb Perspect Biol 3: a005249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell MA, Jain A, Arko‐Mensah J, Chauhan S, Kimura T, Dinkins C, Silvestri G, Munch J, Kirchhoff F, Simonsen A et al (2014) TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell 30: 394–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjithaya R, Nazarko TY, Farre JC, Subramani S (2010) Molecular mechanism and physiological role of pexophagy. FEBS Lett 584: 1367–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso‐Santano M et al (2014) Regulation of autophagy by cytosolic acetyl‐coenzyme A. Mol Cell 53: 710–725 [DOI] [PubMed] [Google Scholar]
- Matsui T, Jiang P, Nakano S, Sakamaki Y, Yamamoto H, Mizushima N (2018) Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J Cell Biol 217: 2633–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K et al (2015) PLEKHM1 regulates autophagosome‐lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57: 39–54 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y (1998a) A protein conjugation system essential for autophagy. Nature 395: 395–398 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Sugita H, Yoshimori T, Ohsumi Y (1998b) A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 273: 33889–33892 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- Moreau K, Renna M, Rubinsztein DC (2013) Connections between SNAREs and autophagy. Trends Biochem Sci 38: 57–63 [DOI] [PubMed] [Google Scholar]
- von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, Freund SM, Komander D, Randow F (2012) LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell 48: 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JM, Shorter J, Newman R, Deinhardt K, Sagiv Y, Elazar Z, Warren G, Shima DT (2002) Sequential SNARE disassembly and GATE‐16‐GOS‐28 complex assembly mediated by distinct NSF activities drives Golgi membrane fusion. J Cell Biol 157: 1161–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T et al (2011) SNARE proteins are required for macroautophagy. Cell 146: 290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Yoshimori T (2017) New insights into autophagosome‐lysosome fusion. J Cell Sci 130: 1209–1216 [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Ichimura Y, Ohsumi Y (2007) Atg8, a ubiquitin‐like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130: 165–178 [DOI] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Lazarou M (2016a) Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol 26: 733–744 [DOI] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, Lazarou M (2016b) Atg8 family LC3/GABARAP proteins are crucial for autophagosome‐lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215: 857–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, Virgin HW, Mizushima N (2013) FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep 14: 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y (1998) Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 273: 3963–3966 [DOI] [PubMed] [Google Scholar]
- Olsvik HL, Lamark T, Takagi K, Bowitz Larsen K, Evjen G, Overvatn A, Mizushima T, Johansen T (2015) FYCO1 contains a C‐terminally extended, LC3A/B‐preferring LC3‐interacting region (LIR) motif required for efficient maturation of autophagosomes during basal Autophagy. J Biol Chem 290: 29361–29374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Jung CH, Seo M, Otto NM, Grunwald D, Kim KH, Moriarity B, Kim YM, Starker C, Nho RS et al (2016) The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 12: 547–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiot A, Ogier‐Denis E, Blommaart EF, Meijer AJ, Codogno P (2000) Distinct classes of phosphatidylinositol 3′‐kinases are involved in signaling pathways that control macroautophagy in HT‐29 cells. J Biol Chem 275: 992–998 [DOI] [PubMed] [Google Scholar]
- Pols MS, van Meel E, Oorschot V, ten Brink C, Fukuda M, Swetha MG, Mayor S, Klumperman J (2013) hVps41 and VAMP7 function in direct TGN to late endosome transport of lysosomal membrane proteins. Nat Commun 4: 1361 [DOI] [PubMed] [Google Scholar]
- Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V (2015) Secretory autophagy. Curr Opin Cell Biol 35: 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popelka H, Klionsky DJ (2015) Analysis of the native conformation of the LIR/AIM motif in the Atg8/LC3/GABARAP‐binding proteins. Autophagy 11: 2153–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F, Youle RJ (2014) Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15: 403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen MS, Birgisdottir AB, Johansen T (2019) Use of Peptide Arrays for Identification and Characterization of LIR Motifs. Methods Mol Biol 1880: 149–161 [DOI] [PubMed] [Google Scholar]
- Rogov V, Dotsch V, Johansen T, Kirkin V (2014) Interactions between autophagy receptors and ubiquitin‐like proteins form the molecular basis for selective autophagy. Mol Cell 53: 167–178 [DOI] [PubMed] [Google Scholar]
- Rogov VV, Stolz A, Ravichandran AC, Rios‐Szwed DO, Suzuki H, Kniss A, Lohr F, Wakatsuki S, Dotsch V, Dikic I et al (2017) Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep 18: 1382–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10: 623–635 [DOI] [PubMed] [Google Scholar]
- Sagiv Y, Legesse‐Miller A, Porat A, Elazar Z (2000) GATE‐16, a membrane transport modulator, interacts with NSF and the Golgi v‐SNARE GOS‐28. EMBO J 19: 1494–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar‐Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010) Ragulator‐Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S et al (2007) Toll‐like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450: 1253–1257 [DOI] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168: 960–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RC, Schuldiner O, Neufeld TP (2004) Role and regulation of starvation‐induced autophagy in the Drosophila fat body. Dev Cell 7: 167–178 [DOI] [PubMed] [Google Scholar]
- Seglen PO, Luhr M, Mills IG, Saetre F, Szalai P, Engedal N (2015) Macroautophagic cargo sequestration assays. Methods 75: 25–36 [DOI] [PubMed] [Google Scholar]
- Shpilka T, Weidberg H, Pietrokovski S, Elazar Z (2011) Atg8: an autophagy‐related ubiquitin‐like protein family. Genome Biol 12: 226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skytte Rasmussen M, Mouilleron S, Kumar Shrestha B, Wirth M, Lee R, Bowitz Larsen K, Abudu Princely Y, O'Reilly N, Sjottem E, Tooze SA et al (2017) ATG4B contains a C‐terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy 13: 834–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadel D, Millarte V, Tillmann KD, Huber J, Tamin‐Yecheskel BC, Akutsu M, Demishtein A, Ben‐Zeev B, Anikster Y, Perez F et al (2015) TECPR2 cooperates with LC3C to regulate COPII‐dependent ER export. Mol Cell 60: 89–104 [DOI] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- Subramaniam VN, Peter F, Philp R, Wong SH, Hong W (1996) GS28, a 28‐Kilodalton golgi SNARE that participates in ER‐Golgi transport. Science 272: 1161–1163 [DOI] [PubMed] [Google Scholar]
- Tai G, Lu L, Wang TL, Tang BL, Goud B, Johannes L, Hong W (2004) Participation of the syntaxin 5/Ykt6/GS28/GS15 SNARE complex in transport from the early/recycling endosome to the trans‐Golgi network. Mol Biol Cell 15: 4011–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takats S, Nagy P, Varga A, Pircs K, Karpati M, Varga K, Kovacs AL, Hegedus K, Juhasz G (2013) Autophagosomal Syntaxin17‐dependent lysosomal degradation maintains neuronal function in Drosophila . J Cell Biol 201: 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takats S, Glatz G, Szenci G, Boda A, Horvath GV, Hegedus K, Kovacs AL, Juhasz G (2018) Non‐canonical role of the SNARE protein Ykt6 in autophagosome‐lysosome fusion. PLoS Genet 14: e1007359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Liao Z, Liang H, Chen X, Zhang B, Chu L (2018) Upregulation of liver kinase B1 predicts poor prognosis in hepatocellular carcinoma. Int J Oncol 53: 1913–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann‐Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P (2000) Accumulation of autophagic vacuoles and cardiomyopathy in LAMP‐2‐deficient mice. Nature 406: 902–906 [DOI] [PubMed] [Google Scholar]
- Tsuboyama K, Koyama‐Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N (2016) The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354: 1036–1041 [DOI] [PubMed] [Google Scholar]
- Velikkakath AK, Nishimura T, Oita E, Ishihara N, Mizushima N (2012) Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell 23: 896–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violot S, Carpentier P, Blanchoin L, Bourgeois D (2009) Reverse pH‐dependence of chromophore protonation explains the large Stokes shift of the red fluorescent protein mKeima. J Am Chem Soc 131: 10356–10357 [DOI] [PubMed] [Google Scholar]
- Wang Y, Foo LY, Guo K, Gan BQ, Zeng Q, Hong W, Tang BL (2006) Syntaxin 9 is enriched in skin hair follicle epithelium and interacts with the epidermal growth factor receptor. Traffic 7: 216–226 [DOI] [PubMed] [Google Scholar]
- Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J et al (2016) The vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell 63: 781–795 [DOI] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B (2017) Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168: 224–238.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z (2010) LC3 and GATE‐16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29: 1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z (2011) LC3 and GATE‐16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 20: 444–454 [DOI] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C et al (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wubbolts R, Fernandez‐Borja M, Oomen L, Verwoerd D, Janssen H, Calafat J, Tulp A, Dusseljee S, Neefjes J (1996) Direct vesicular transport of MHC class II molecules from lysosomal structures to the cell surface. J Cell Biol 135: 611–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Nair U, Klionsky DJ (2008) Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 19: 3290–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12: 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott‐Schwartz J, Tooze SA (2006) Starvation and ULK1‐dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 119: 3888–3900 [DOI] [PubMed] [Google Scholar]
- Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait‐Mulder J, Lee JH, Paull TT et al (2015) ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol 17: 1259–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5