

Abstract
N‐ethylmaleimide‐sensitive factor (NSF) plays a critical role in intracellular vesicle transport, which is essential for neurotransmitter release. Herein, we, for the first time, document human monogenic disease phenotype of de novo pathogenic variants in NSF, that is, epileptic encephalopathy of early infantile onset. When expressed in the developing eye of Drosophila, the mutant NSF severely affected eye development, while the wild‐type allele had no detectable effect under the same conditions. Our findings suggest that the two pathogenic variants exert a dominant negative effect. De novo heterozygous mutations in the NSF gene cause early infantile epileptic encephalopathy.
Introduction
Intracellular vesicle transport and endo/exocytosis are fundamental processes underlying a wide range of biological activities, including neurotransmitter release and hormone secretion. The delivery and release of cargo proteins are precisely regulated by a number of proteins.1 N‐ethylmaleimide‐sensitive factor (NSF) is a homo‐hexameric AAA ATPase that is involved in membrane fusion.2 The membrane receptors for NSF were identified and named as the soluble NSF attachment protein receptor (SNARE) complex.3 Assembly and disassembly of NSF and the SNARE complex, along with calcium triggering at appropriate location and time, are critical steps in vesicular transport and membrane fusion. The discovery and understanding of this trafficking machinery were awarded the Nobel Prize in Physiology or Medicine in 2013.4
Dysregulation of this intracellular vesicular transport/membrane fusion process has been increasingly recognized as the mechanistic basis for the development of diabetes and neurological disorders. Indeed, pathogenic variants of the SNARE complex are known to be associated with the development of Alzheimer's disease, Parkinsonism, autism, and psychiatric disorders.5 However, monogenic disease phenotype of NSF mutations has not been described previously. In invertebrates, the Drosophila homolog of NSF, that is, comatose (comt), was first mapped and cloned in association with temperature‐sensitive paralysis four decades ago.6, 7 The comt mutant flies show a burst of electrical discharges of the thoracic flight muscles at high temperatures,8 akin to febrile seizures in humans. Here, we describe two patients with de novo pathogenic variants in NSF with early infantile epileptic encephalopathy, to demonstrate that pathogenic variants of NSF cause the monogenic epileptic phenotype in humans.
Materials and Methods
Subjects and exome sequencing
The present research protocol was approved by the local ethics committees. Written informed consent was obtained from the parents for the analyses. As part of nationwide clinical projects, that is, Initiative on Rare and Undiagnosed Diseases (IRUD) and Rapid Genetic Diagnosis toward Neonatal Precision Medicine, conducted by Japan Agency for Medical Research and Development, exome analyses in trios were conducted in subjects with suspected genetic diseases who fulfilled the enrollment criteria.9 The enrollment criteria included the presence of multiple organ symptoms and history of familial inheritance. Since July 2015, more than 1300 undiagnosed families were enrolled in these projects. Genomic DNA was extracted from the peripheral blood leukocytes of patients 1 and 2 and their parents. Whole‐exome sequencing in patients and their parents was performed, as described previously.10
Functional assay using the Drosophila model
To assess the functional relevance of the amino acid changes identified in the two patients, we introduced exogenous human NSF alleles carrying p.Ala459Thr and p.Pro563Leu to Drosophila. We ectopically expressed the wild‐type and two mutant NSF genes in the nervous system of the Drosophila using the GAL4/UAS system. A full‐length cDNA clone of the human NSF gene (clone 4812117) was obtained from DNAFORM (Yokohama, Japan). The entire coding sequence was amplified with and without a stop codon by PCR and subcloned into the pENTR221 vector using the In‐Fusion® HD Cloning Kit (Takara Bio USA, Inc, CA). The subclones were sequenced and confirmed to have the correct sequence. The two proband mutations (c.1375G>A, p.Ala459Thr and c.1688C>T, p.Pro563Leu) were introduced into the NSF coding sequence by site‐directed mutagenesis. Based on the Gateway technology, the cloned genes were transferred into the destination vectors, pUASg‐attB and pUASg‐HA_attB.11 The plasmid DNAs were injected into embryos carrying the attP40 landing site for phiC31 integrase‐mediated transformation (y 1 v 1 P{y[+t7.7]=nos‐phiC31\int.NLS}iedX; P{y[+t7.7]=CaryP}attP40). Five virgin females of GAL4 driver strains were crossed with five males of the UAS‐NSF strains and transferred every 3 days to a new vial. In order to detect dying cells in the developing eye discs, a TUNEL (terminal deoxynucleotidyl transferase‐mediated dUTP nick end‐labeling) assay was performed.12 The host strain for transformation and the nSyb‐GAL4 strains (y1 w1118; P{y[+t7.7] w[+mC]=nSyb‐GAL4.P}attP2 and y1 w; P{w[+m*]=nSyb‐GAL4.S}3) were obtained from Bloomington Drosophila Stock Center and the P{GAL4‐ninaE.GMR} driver strain (w P{w[+mC]=GAL4‐ninaE.GMR}12) was procured from the KYOTO Stock Center. These flies carried intact intrinsic comt/NSF1 and NSF2.
Results
Clinical description
Patient 1: The first proband was a Japanese girl. She was born at 37 weeks of gestation by uneventful delivery after an uneventful pregnancy. There was no significant family medical history. Her birth weight was 2,930 g (+0.42 SD) and her head circumference was 33.5 cm (+0.38 SD). Immediately after birth, she developed continuous vomiting and tonic seizures. Electroencephalography on the ninth day after birth showed a continuous burst‐suppression pattern, regardless of the sleep–wake stage. The burst phase lasted for a duration of 2–5 sec and consisted of diffuse irregular spikes and sharp waves measuring 100–250 µV in amplitude. The suppression phase lasted for 5–20 sec and was nearly isoelectric (Fig. 1). Based on the findings, this female infant was diagnosed as having early infantile epileptic encephalopathy. Despite maximum medical support in the pediatric intensive care unit, the infant died on day 36 after birth of respiratory failure.
Figure 1.
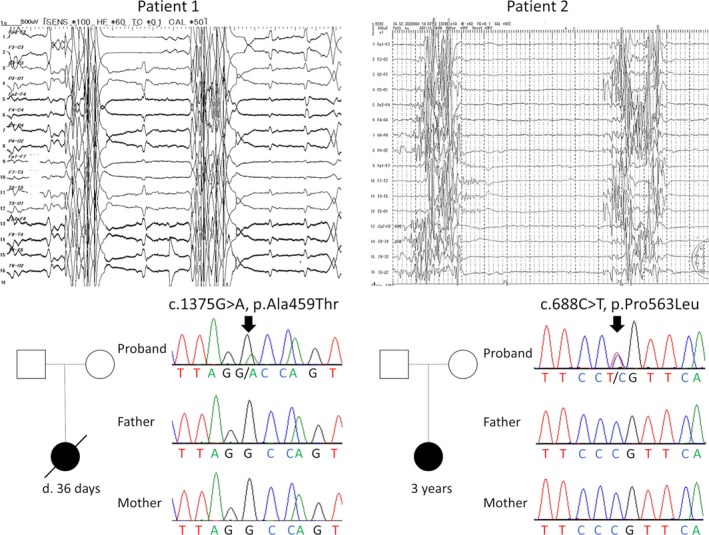
Clinical characteristics of the two patients. Top: The electroencephalographs of both patients 1 and 2 showed a continuous burst‐suppression pattern, regardless of sleep–wake stage. The burst phase lasted for a duration of 2–5 sec and consisted of diffuse irregular spikes and sharp waves measuring 100–250 µV in amplitude. The suppression phase lasted for 5–20 sec and was nearly isoelectric. Bottom: Family pedigree analysis of patients 1 and 2 showed that no other family members were affected. The chromatograms of patient 1 showed c.1375G>A p.Ala459Thr in NSF in a heterozygous state (an arrow). Neither of the parents carried this pathogenic variant. The chromatograms of patient 2 showed c.1688C>T p.Pro563Leu in NSF in a heterozygous state (an arrow). Neither of the parents carried this pathogenic variant.
Patient 2: The second proband was a 1‐year‐old Japanese girl with no significant family medical history. In her prenatal period, she exhibited failure to thrive, hydrops, and anemia, for which blood transfusion via the placenta was performed. She was born at 33 weeks of gestation via emergent cesarean section due to fetal distress. Her birth weight was 1,334 g (−2.1 SD) and her head circumference was 23.5 cm (−3.4 SD). After birth, she had no spontaneous respiration, which needed mechanical ventilation, and had frequent myoclonic seizures. Her electroencephalography showed continuous burst‐suppression patterns highly similar to that in patient 1 (Fig. 1). At the age of 3 years, she had profound intellectual disability, severe motor developmental delay, and no spontaneous respiration. Her electroencephalogram persistently showed burst‐suppression patterns. She continued to have frequent myoclonic seizures and epileptic spasms.
Molecular analysis
Whole‐exome sequencing in the patients and their parents revealed a de novo heterozygous variants in the NSF gene (NM_006178.3) in both probands: c.1375G>A chr17:44782125G>A (GRCh37) p.Ala459Thr in patient 1 and c.1688C>T chr17:44791279C>T (GRCh37) p.Pro563Leu in patient 2. The results were confirmed by Sanger sequencing. Neither of the two variants were present in the exome datasets of more than 60,000 individuals without severe pediatric diseases (the ExAC database (http://exac.broadinstitute.org/)) or in a cohort of 2049 Japanese normal individuals (Integrative Japanese Genome Variation Database (https://ijgvd.megabank.tohoku.ac.jp/)). The codons Ala459 and Pro563 in the NSF gene are highly conserved among many species (Fig. 2). No candidate genes were identified in the autosomal recessive model.
Figure 2.
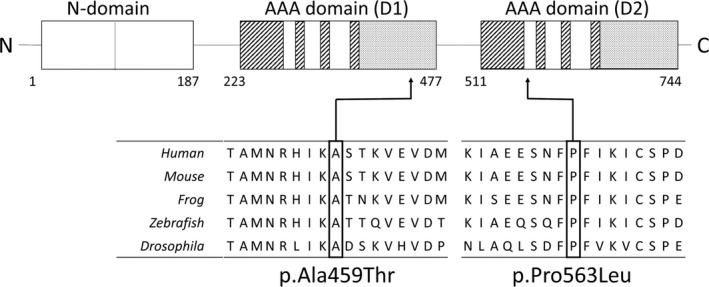
Top: A schematic representation of the NSF molecule. Ala459 is located in the AAA domain (D1). Pro563 is located in the AAA domain (D2). Bottom: Amino acid alignments at and around Ala459 and Pro563. Note that Ala459 and Pro563 are highly conserved among species.
In silico analyses showed that the combined annotation‐dependent depletion score, which reflects the relative pathogenicity of human variants,13 was 30 for p.Ala459Thr, and 34 for p.Pro563Leu. According to the American College of Medical Genetics guideline for the pathogenicity of variants,14 the two variants were classified as “pathogenic.”
Functional analysis of the pathogenic variants using Drosophila.
Although the wild‐type UAS‐NSF gene combined with the GAL4 driver gene did not result in any detectable phenotype, both flies carrying the mutant UAS‐NSF and P{GAL4‐ninaE.GMR} genes exhibited defective eyes. Especially, the P{GAL4‐ninaE.GMR}/UAS‐NSFP563L flies exhibited complete obliteration of the eyes, while massive cell death was observed in the developing eye discs (Fig. 3). Ectopic expression of the mutated NSF genes under the control of the pan‐neuronal nSyb‐GAL4 driver genes resulted in embryonic or the first instar larval lethality (data not shown).
Figure 3.
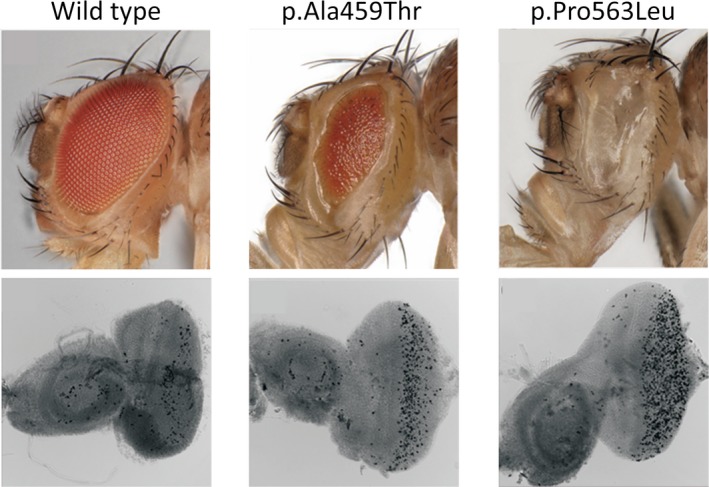
Functional effects of the two NSF pathogenic variants on the eye discs of Drosophila. Top: Photographs of the eyes of Drosophila females expressing the NSF gene under the control of the P{GAL4‐ninaE.GMR} driver. Note that the mutants, but not the wild‐type NSF genes, led to defective eye phenotypes when expressed in the developing eyes. Left: P{GAL4‐ninaE.GMR}/UAS‐NSF (the wild‐type allele); Middle: P{GAL4‐ninaE.GMR}/UAS‐NSFA459T; Right: P{GAL4‐ninaE.GMR}/UAS‐NSFP563L. Bottom: TUNEL images of the eye imaginal discs of larvae expressing the NSF gene under the control of the P{GAL4‐ninaE.GMR} driver. Left: P{GAL4‐ninaE.GMR}/UAS‐NSF (wild‐type allele); Middle: P{GAL4‐ninaE.GMR}/UAS‐NSFA459T; Right: P{GAL4‐ninaE.GMR}/UAS‐NSFP563L. TUNEL‐positive cells are seen in black and also in white (out‐of‐focus). Note that TUNEL‐positive dying cells are more abundant in the developing eye discs expressing the mutant NSF genes than in those expressing the wild‐type NSF gene.
Discussion
In the present report, we, for the first time, document epileptic encephalopathy of infantile onset caused by de novo heterozygous pathogenic variants in the gene encoding NSF, a molecule that is critical for intracellular vesicle transport and membrane fusion. To analyze the functional relevance of the human NSF pathogenic variants observed in the patients, the human mutant alleles were overexpressed in the Drosophila eye discs using the GAL4‐UAS system. The flies expressing the human mutant NSF alleles in the developing eye discs exhibited defective eye development, suggesting that the pathogenic variants exerted a dominant negative effect. Collectively, we herein establish that pathogenic variants in the NSF cause early infantile epileptic encephalopathy.
Functional analysis of mutant Drosophila is a well‐established method in the evaluation of pathogenic variants.15 In the Drosophila model, overexpression of a loss‐of‐function allele results in no overt phenotype, whereas overexpression of gain‐of‐function alleles results in overt phenotypes.16 In the present study, while overexpression of the two mutant alleles resulted in defective eye development, overexpression of the wild‐type allele had no such effect. These observations suggest that the amino acid substitution changes (p.Ala459Thr and p.Pro563Leu) in the two patients exerted a dominant negative effect. Yet, it still remains to be explored how mutant proteins exert a dominant negative effect. NSF forms a homomeric hexamer.17 Given the similarity of the amino acid alignment between exogenous NSF alleles and intrinsic comt/NSF alleles, the proteins arising from exogenous mutant alleles likely interfere with the intrinsic NSF product during the formation of multimers.
Our animal model had intact intrinsic comt/NSF1 and NSF2. We introduced human mutant alleles using the GAL4‐UAS driver to express human mutant alleles exclusively in the Drosophila eye discs. Generation and functional analysis of a transgenic model may provide further insight into the pathogenetic mechanism arising from the two NSF mutations that were identified in the patients. Nonetheless, the observation that both flies with abnormal function in comt and infants with abnormal function in NSF exhibited epileptic phenotype illustrates that two orthologs (comt and NSF) in the two different species are related to the abnormal electrical discharges and epilepsy phenotype.
Pathogenic variants in proteins involved in the regulation of intracellular vesicular transport and membrane fusion are known to cause epileptic encephalopathies. STXBP1 (MUNC18‐1) and NECAP1 are regulatory proteins that are involved in synaptic neurotransmitters release. Pathogenic variants in these genes are known to cause epileptic encephalopathy of early infantile onset.18, 19 Considering that NSF is involved in the intracellular vesicle transport and recycling of vesicles20 and the development of central nervous system malformation in the mutant Drosophila, the epileptic phenotype observed in the two reported patients is likely caused by dysregulation of synaptic neurotransmission.
Besides playing roles in intracellular vesicle transport and synaptic transmission of neurotransmitters, neurogenesis, and neuroprotection, NSF may also play a role in the epileptic phenotype. Drosophila carrying mutant NSF/comt exhibited a reduced life‐span and progressive neurodegeneration of the dopaminergic neurons, suggesting involvement of NSF in autophagy and neuroprotection.21 Moreover, the lethal mutation in comt resulted in a reduction in the size and branching of synapses, suggesting that NSF plays a critical role in neurogenesis.22 In our Drosophila model, ectopic expression of mutant NSF (i.e., p.Ala459Thr and p.Pro563Leu) under the control of the pan‐neuronal nSyb‐GAL4 driver showed embryonic or first instar larval lethality, and the finding of TUNEL‐positive dying cells observed in the developing eye discs appeared to be consistent with the critical roles of NSF in neurogenesis and perhaps in neuroprotection. Although the paralytic phenotype in flies does not prove but suggests that NSF dysfunction causes the epilepsy phenotype, we could not determine which mechanisms, that is, neurogenesis and/or neuroprotection, of the NSF are relevant to the patients' epileptic phenotype. Further studies employing anatomical and physiological analyses are needed.
From a clinical standpoint, defining the exact phenotypic spectrum of this presumably new early infantile epileptic encephalopathy would have implications in the clinical management. Both patients exhibited frequent myoclonic seizures accompanied by burst‐suppression patterns on electroencephalogram within 1 week after birth. These clinical features during the neonatal period were essentially nondistinguishable from those in early infantile epileptic encephalopathy due to KCNQ2 and SCN2A mutations. However, it was notable that the electroencephalogram in patient 2 showed persistent burst‐suppression patterns even beyond 3 years of age, which has not so far been described in cases of early infantile epileptic encephalopathy caused by KCNQ2 or SCN2A mutations.23 Further investigations in a larger cohort of patients are warranted to explore characteristic electroencephalographic progression and optimal antiepileptic strategies for patients with NSF‐related early infantile epileptic encephalopathy.
In summary, two unrelated infants carrying de novo heterozygous pathogenic variants in the NSF, that is, p.Ala459Thr and p.Pro563Leu, presented with epileptic encephalopathy of early infantile onset. The functional analysis using the Drosophila eye disc model suggested the pathogenicity of the variants, that is, that they exerted dominant negative effects. We conclude that de novo heterozygous mutations in the NSF cause early infantile epileptic encephalopathy.
Conflict of Interest
None of the authors have any conflict of interest to disclose.
Acknowledgments
This work was supported by the Japan Agency for Medical Research and Development (Grant number JP18ek0109301, JP19gk0110038, and JP18ek0109288h0002).
Funding information
This work was supported by the Japan Agency for Medical Research and Development (Grant number JP18ek0109301, JP19gk0110038, and JP18ek0109288h0002).
Funding Statement
This work was funded by Japan Agency for Medical Research and Development grants JP18ek0109288h0002, JP18ek0109301, and JP19gk0110038.
References
- 1. Rothman JE. Mechanisms of intracellular protein transport. Nature 1994;372:55–63. [DOI] [PubMed] [Google Scholar]
- 2. Glick BS, Rothman JE. Possible role for fatty acyl‐coenzyme A in intracellular protein transport. Nature 1987;326:309–312. [DOI] [PubMed] [Google Scholar]
- 3. Sollner T, Bennett MK, Whiteheart SW, et al. A protein assembly‐disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993;75:409–418. [DOI] [PubMed] [Google Scholar]
- 4. Ferro‐Novick S, Brose N. Nobel 2013 physiology or medicine: traffic control system within cells. Nature 2013;504:98. [DOI] [PubMed] [Google Scholar]
- 5. Ramakrishnan NA, Drescher MJ, Drescher DG. The SNARE complex in neuronal and sensory cells. Mol Cell Neurosci 2012;50:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pallanck L, Ordway RW, Ganetzky B. A Drosophila NSF mutant. Nature 1995;376:25. [DOI] [PubMed] [Google Scholar]
- 7. Siddiqi O, Benzer S. Neurophysiological defects in temperature‐sensitive paralytic mutants of Drosophila melanogaster. Proc Natl Acad Sci USA 1976;73:3253–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanyal S, Basole A, Krishnan KS. Phenotypic interaction between temperature‐sensitive paralytic mutants comatose and paralytic suggests a role for N‐ethylmaleimide‐sensitive fusion factor in synaptic vesicle cycling in Drosophila. J Neurosci 1999;19:RC47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adachi T, Kawamura K, Furusawa Y, et al. Japan's initiative on rare and undiagnosed diseases (IRUD): towards an end to the diagnostic odyssey. Eur J Hum Genet 2017;25:1025–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takenouchi T, Yamaguchi Y, Tanikawa A, et al. Novel overgrowth syndrome phenotype due to recurrent de novo PDGFRB mutation. J Pediatr 2015;166:483–486. [DOI] [PubMed] [Google Scholar]
- 11. Bischof J, Bjorklund M, Furger E, et al. A versatile platform for creating a comprehensive UAS‐ORFeome library in Drosophila. Development 2013;140:2434–2442. [DOI] [PubMed] [Google Scholar]
- 12. Togane Y, Ayukawa R, Hara Y, et al. Spatio‐temporal pattern of programmed cell death in the developing Drosophila optic lobe. Dev Growth Differ 2012;54:503–518. [DOI] [PubMed] [Google Scholar]
- 13. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jeibmann A, Paulus W. Drosophila melanogaster as a model organism of brain diseases. Int J Mol Sci 2009;10:407–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herskowitz I. Functional inactivation of genes by dominant negative mutations. Nature 1987;329:219–222. [DOI] [PubMed] [Google Scholar]
- 17. Zhao M, Wu S, Zhou Q, et al. Mechanistic insights into the recycling machine of the SNARE complex. Nature 2015;518:61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18‐1) cause early infantile epileptic encephalopathy. Nat Genet 2008;40:782–788. [DOI] [PubMed] [Google Scholar]
- 19. Alazami AM, Hijazi H, Kentab AY, Alkuraya FS. NECAP1 loss of function leads to a severe infantile epileptic encephalopathy. J Med Genet 2014;51:224–228. [DOI] [PubMed] [Google Scholar]
- 20. Ryu JK, Min D, Rah SH, et al. Spring‐loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science 2015;347:1485–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Babcock DT, Shen W, Ganetzky B. A neuroprotective function of NSF1 sustains autophagy and lysosomal trafficking in Drosophila. Genetics 2015;199:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanyal S, Krishnan KS. Lethal comatose mutation in Drosophila reveals possible role for NSF in neurogenesis. NeuroReport 2001;12:1363–1366. [DOI] [PubMed] [Google Scholar]
- 23. Olson HE, Kelly M, LaCoursiere CM, et al. Genetics and genotype‐phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann Neurol 2017;81:419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]