

Abstract
Oncogenic K‐RAS has been difficult to target and currently there is no K‐RAS‐based targeted therapy available for patients suffering from K‐RAS‐driven lung adenocarcinoma (AC). Alternatively, targeting K‐RAS‐downstream effectors, K‐RAS‐cooperating signaling pathways or cancer hallmarks, such as tumor‐promoting inflammation, has been shown to be a promising therapeutic strategy. Since the JAK–STAT pathway is considered to be a central player in inflammation‐mediated tumorigenesis, we investigated here the implication of JAK–STAT signaling and the therapeutic potential of JAK1/2 inhibition in K‐RAS‐driven lung AC. Our data showed that JAK1 and JAK2 are activated in human lung AC and that increased activation of JAK–STAT signaling correlated with disease progression and K‐RAS activity in human lung AC. Accordingly, administration of the JAK1/2 selective tyrosine kinase inhibitor ruxolitinib reduced proliferation of tumor cells and effectively reduced tumor progression in immunodeficient and immunocompetent mouse models of K‐RAS‐driven lung AC. Notably, JAK1/2 inhibition led to the establishment of an antitumorigenic tumor microenvironment, characterized by decreased levels of tumor‐promoting chemokines and cytokines and reduced numbers of infiltrating myeloid derived suppressor cells, thereby impairing tumor growth. Taken together, we identified JAK1/2 inhibition as promising therapy for K‐RAS‐driven lung AC.
Keywords: non‐small cell lung cancer, lung adenocarcinoma (AC), Kirsten rat sarcoma viral proto‐oncogene (K‐RAS), Janus kinase (JAK), ruxolitinib, cell‐line derived xenografts, genetically engineered mouse models, tumor microenvironment (TME), tumor promoting inflammation
Short abstract
What's new?
A drug that inhibits the JAK–STAT pathway may score a hit against K‐RAS driven lung cancer. Here, the authors Investigated the JAK STAT pathway as a possible target in lung adenocarcinoma because of its role in inflammation‐mediated tumorigenesis. First, they showed that JAK1 and JAK2 are both activated in lung adenocarcinoma patients with oncogenic mutations in K‐RAS. Next, they treated the tumors with ruxolitinib, which inhibits JAK1/2. The drug successfully slowed tumor proliferation and progression in immunocompetent mouse models. Furthermore, treatment with ruxolitinib reduced the tumor‐promoting factors present in the microenvironment.
Abbreviations
- AC
adenocarcinoma
- ACTB
actin beta
- ANOVA
one‐way analysis of variance
- CXCL1
chemokine (C‐X‐C motif) ligand 1
- EGFR
epidermal growth factor receptor
- GO
gene ontology
- GSEA
gene set enrichment analysis
- IL
interleukin
- JAK
Janus kinase
- K‐RAS
Kirsten rat sarcoma viral proto‐oncogene
- MDSCs
myeloid derived suppressor cells
- mRNA
messenger RNA
- NKs
natural killer cells
- PD‐L1
programmed cell death 1 ligand 1
- RNA‐seq
RNA sequencing
- STAT
signal transducer and activator of transcription
- TBP
TATA‐box binding protein
- TME
tumor microenvironment
- TNF
tumor necrosis factor
- VEGF
vascular endothelial growth factor
- WT
wild‐type
Introduction
Lung cancer is still the leading cause of cancer‐related deaths worldwide in both men and women.1 Histologically, lung cancer can be subdivided into small cell lung cancer and non‐small cell lung cancer, with lung adenocarcinoma (AC) being the most abundant among the non‐small cell lung cancer subtype.2 Roughly 50% of lung ACs harbor oncogenic mutations in the V‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog (K‐RAS) or epidermal growth factor receptor (EGFR).3, 4 While EGFR tyrosine kinase inhibitors, such as erlotinib, gefitinib or afatinib, are part of the clinical routine to treat EGFR‐mutated lung AC patients,5 the development of K‐RAS inhibitors has been less successful so far, with no K‐RAS‐based targeted therapy being clinically approved.6 Hence, targeting alternative tumor drivers fueling K‐RAS‐mediated lung tumorigenesis such as tumor‐promoting inflammation and K‐RAS downstream effector pathways are considered to be a promising treatment strategy.6, 7
Given the pivotal role of Janus kinase (JAK) activation in tumor‐promoting inflammation and its oncogenic functions, JAKs represent attractive therapeutic targets in tumorigenesis.8 The JAK family is composed of four members (JAK1, JAK2, JAK3 and tyrosine kinase 2) and couples extracellular stimuli with gene transcription. Upon cytokine binding to their cognate receptors, JAKs become activated and phosphorylate downstream signal transducer and activator of transcription (STAT) molecules, ultimately leading to nuclear translocation and transcription of various target genes involved in cell cycle regulation, angiogenesis, apoptosis, tumor invasion and metastasis.9 JAK signaling either promotes or suppresses tumorigenesis in a tumor cell intrinsic manner. Indeed, K‐RAS‐mutated lung AC cells secrete pro‐inflammatory cytokines including interleukin (IL)‐6 that activate JAK1 and JAK2 via glycoprotein 130 in an autocrine loop, thereby promoting tumor cell survival.7, 10, 11 On the other hand, genetic deletion of STAT3, a key signaling mediator of JAK1/2, enhances K‐RAS‐driven lung tumorigenesis, and activation of the interferon/JAK/STAT axis induces cell apoptosis and suppresses tumorigenesis in various experimental tumor models.12, 13, 14, 15, 16
In addition to these tumor‐cell intrinsic effects, JAK signaling is also substantially involved in shaping the tumor microenvironment (TME) by modulating T, natural killer (NK) and dendritic cell functions.17 As in tumor cells, JAK activation in the TME and its role in tumorigenesis is controversially discussed. While JAK1/2 inhibition impairs NK‐cell function and NK mediated antitumor immunity and hence indirectly facilitates tumor metastatic spread,18 targeting the JAK–STAT axis has also been proposed as a potential effective strategy to reduce levels of tumor‐promoting immunosuppressive myeloid derived suppressor cells (MDSCs), thereby counteracting malignant progression.19 Furthermore, IL6/JAK/STAT3, as well as interferon‐γ/JAK/STAT1 signaling pathways have also been described to induce programmed cell death 1 and or programmed cell death 1 ligand 1 (PD‐L1) expression and, therefore, to promote tumor immune escape.20, 21 Taken together, these reports indicate that JAKs may have opposite functions in tumorigenesis depending on the cellular context.
Clinical trials investigating JAK inhibition in particular for EGFR‐mutated lung cancers are ongoing. These studies combine the JAK inhibitors ruxolitinib, itacitinib and AZD4205 with the EGFR tyrosine kinase inhibitors erlotinib (NCT02155465), afatinib (NCT02145637) and osimertinib (NCT02917993, NCT03450330). By contrast, JAK targeting in K‐RAS‐mutated lung ACs has not been thoroughly investigated so far. Notably, a recent clinical trial showed positive results when combining ruxolitinib and chemotherapy in non‐small cell lung cancer patients, but the K‐RAS mutation status is not available (NCT02119650). Thus, to complement these clinical studies, we addressed the role of JAK1/2 in K‐RAS‐driven lung AC pathogenesis. By using human tumor samples and experimental mouse models, we show that JAK1/2 are activated in K‐RAS‐driven lung AC and promote tumor growth and progression by establishing a protumorigenic TME. Importantly, pharmacological inhibition of JAK1/2 with the tyrosine kinase inhibitor ruxolitinib reverts the protumorigenic TME and impairs tumorigenesis.
Materials and Methods
Human data
For gene expression analysis, we took advantage of the publicly available Illumina microarray data set, which contains biopsies of K‐RAS‐mutated lung AC tissue and adjacent healthy tissue (GSE75037).22 To determine JAK1 and JAK2 mRNA expression levels, we used probes as depicted in Table S1. As a second cohort, we used gene expression data from K‐RAS‐mutated lung AC patients included in the Cancer Genome Atlas (n = 154).23
Gene set enrichment analysis (GSEA) was performed according to the provider's protocol and permutations were set to 1,000.24 Gene sets were downloaded from the Molecular Signature Database (http://software.broadinstitute.org/gsea/msigdb/index.jsp). To determine the prognostic value of chemo/cytokine related genes upon ruxolitinib treatment for human lung ACs pathogenesis, we used the KM‐Plotter online tool.25 In addition, gene expression data of K‐RAS‐mutated lung AC patients with available survival data included in the Cancer Genome Atlas were analyzed ( n = 150).23 We have run the analysis by evaluating all possible cutoff values and the best performing value was used as the cutoff in the final analysis. False discovery rate was computed to correct for multiple testing, and only results having a false discovery rate below 1% was accepted as statistically significant. Analysis was performed using the mean expression of the genes included in the multigene signature, and all utilized Probe‐IDs are shown in Table S2.
For immunohistochemical analysis, we generated tissue microarrays from two independent lung AC patient cohorts. Cohort A includes biopsies from lung AC patients where information about the mutational K‐RAS status was not available (pJAK1 n = 303, pJAK2 n = 318). Cohort B consists of biopsies of lung AC patients positive for K‐RAS mutation (pJAK1 n = 26, pJAK2 n = 24). Analysis was done with prior approval by the institutional ethical committee. For detection of JAK1 or JAK2 tyrosine phosphorylation the tissue microarrays were stained with a pJAK1 (1:200, Y1022, #PA537617, Thermo Fisher Scientific, Waltham, MA) and a pJAK2 (1:200, Y1007 and Y1008, #32101, Abcam, Cambridge, UK) antibody. Histoscores were determined according to staining intensities and percentage of positive tumor cells by a board‐certified pathologist (H.P.).26
Cell lines
The human K‐RAS‐mutated lung AC cell lines A549 and A427 were grown in RPMI medium (Gibco, Grand Island, NY) supplemented with 10% FBS, 2 mM glutamine (Gibco), penicillin (50 U/ml, Gibco) and streptomycin (50 μg/ml, Gibco). A549 and A427 cell lines were obtained from American Type Culture Collection, and cells were profiled for authentication (Eurofins Genomics, Luxembourg). Cells were tested to be free of mycoplasma contaminations twice a year. For experiments, cells were harvested at ~70% confluency, and the number of viable cells was determined using a CASY counter (Roche, Basel, Switzerland).
Animal husbandry and experiments
For subcutaneous xenograft experiments using the K‐RAS‐mutated human lung AC cell lines A549 and A427, 1 × 106 cells were mixed 1:2 with Matrigel (Corning, Corning, NY) and injected into the right and left flank of immunodeficient NOD scid gamma mice older than 6 weeks of age. Tumor volume was measured using a caliper and tumor volume was calculated using the formula [(length × width2) × 0.52] as previously established in our laboratory.27
K‐ras LSL‐G12D (K) mice as well as K‐ras LSL‐G12D:p53 fl/fl (KP) mice, both bred on a C57Bl/6 background, served as genetically engineered mouse models for autochthonous lung AC.28 Genotyping of the transgenes was performed using primers depicted in Table S3. Induction of lung tumors in these genetically engineered mouse models was achieved through intranasal inhalation of a Cre‐recombinase expressing adenovirus (Ad.Cre) in 8–12 week old mice using 2.5 × 107 plaque‐forming units. Inhalation of K and KP mice with Ad.Cre triggers K‐ras G12D activation in lung epithelial cells resulting in formation of p53 expressing (K) or deficient (KP) lung ACs. Ad.Cre was purchased from the Viral Vector Core (University of Iowa). For treatment of mice with the JAK1/2 inhibitor ruxolitinib (LC Labs, Woburn, MA), the inhibitor was suspended in 0.5% (w/v) methylcellulose solution. Mice were treated via oral gavage with vehicle (ctrl, 0.5% Methylcellulose) or ruxolitinib (Ruxo) at 90 mg/kg twice a day (BID), 7 days per week for the indicated experimental time period. Treatment of mice was paused if any signs of distress occurred.18 All experimental protocols as well as animal maintenance and breeding described above, followed ethical guidelines and were approved by the Austrian Federal Ministry of Science, Research and Economy.
Histology
At the experimental endpoint, subcutaneously engrafted tumor tissue or lung tissue was retrieved and fixed in 2% buffered formaldehyde at 4°C for 24 hr and embedded in paraffin after dehydration. Five micron sections were used for analysis. To determine tumor area, lung sections were stained with hematoxylin and eosin and subsequently scanned using TissueFaxs (TissueGnostics, Vienna, Austria) software. Tumor area and number were quantified using HistoQuest (TissueGnostics) software. Tumor grades were determined by a board‐certified pathologist Katalin Dezso (K.D.) according to DuPage et al.28 For immunohistochemistry, standard protocols were applied, using antibodies against pSTAT3 (1:200,Y705, #9145, Cell Signaling, Danvers, MA), KI67 (1:400, Cell Signaling #12202 or 1:200, #14‐5698‐82, eBioscience, San Diego, CA), cleaved caspase‐3 (1:200, Cell Signaling #9661), cluster of differentiation 31 (1:400, Cell Signaling #77699), PD‐L1 (1:200, Cell signaling #64988). For analysis of stained immunohistochemistry slides HistoQuest (TissueGnostics) software was used.
RNA‐sequencing (RNA‐seq) analysis of total lungs and Gene Expression Omnibus data set analysis
For RNA‐seq analysis, K mice were treated 8 weeks post inhalation with Ad.Cre for four consecutive days with vehicle (0.5% Methylcellulose) or ruxolitinib (90 mg/kg BID). Additionally tumor harboring lungs from KP mice and lungs from wild‐type (WT) mice were included in the analysis. At the experimental endpoint, lungs were harvested and RNA was isolated using the RNeasy Kit (Qiagen, Venlo, Netherlands) following on column DNA digestion using RNase‐Free DNase (Qiagen). Library preparation and sequencing was carried out by the Core facility Genomics, Medical University of Vienna, Vienna, Austria. Briefly, sequencing libraries were prepared using the NEBNext® Ultra™ II RNA Library Prep Kit (NEB, Ipswich, MA) according to manufacturer's instructions and sequenced on a Illumina NextSeq500 platform in a 75 bp single‐read mode. RNA‐Seq data were mapped to the Mus musculus/mmc10 assembly of the murine genome using the STAR Aligner.29 Differential gene expression was analyzed using DESeq2.30
For GSEA, gene sets were downloaded from the Molecular Signature Database (http://software.broadinstitute.org/gsea/msigdb/index.jsp). For gene ontology (GO)‐molecular function analysis, the Enrichr web tool was used (http://amp.pharm.mssm.edu/Enrichr/enrich). The top 350 downregulated genes (based on log2 fold changes) in the ruxolitinib treated group were subjected to pathway analysis with Enrichr.31 To investigate the implications of ruxolitinib downregulated chemo/cytokine related genes for K‐RAS‐driven lung tumorigenesis, we retrieved all genes within the chemo/cytokine related GO gene sets GO0042379, GO0008009, GO0045236, GO0048020 and GO0005125. Data were analyzed using the web‐based online tool InteractiVenn,32 and for hierarchical clustering and heatmap illustration, we used the heatmapper.ca web tool.33 In order to check for the activation status of the JAK–STAT pathway and the expression of chemo/cytokine related genes in primary K‐ras G12D‐mutated lung alveolar type II (K) cells, we analyzed the publically available GSE113146 data set.27
RNA extraction and real‐time quantitative polymerase chain reaction
RNA isolation from the lungs was performed using TRIzol™ Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Complementary DNA was transcribed using the RevertAid H Minus Reverse Transcriptase (Fermentas, Waltham, MA) kit. For detection of the transcript, the iTaq Universal SYBR Green reagent (Biorad, Hercules, CA) and a Biorad CFX Connect Real Time polymerase chain reaction system were utilized, using specific primers depicted in Table S4. For determination of relative gene expression ratios, the Pfaffl method was applied. Calculation of bestkeeper values was performed using actin beta (ACTB), 28S and TATA‐box binding protein (TBP) as housekeeping genes.34
Flow cytometric analysis of total lungs
At the experimental endpoint, K‐RAS‐mutated tumor harboring lungs were harvested, and the lung tissue was mechanically and enzymatically disrupted for cell isolation. Briefly, lungs were put into isolation medium (RPMI with 5% FBS) and minced with scissors. Minced pieces were then transferred into a falcon tube containing isolation medium supplemented with DNase I (50 U/ml, Sigma, St.Louis, MO) and Collagenase I (Gibco, 150 U/ml), and incubated for 1 hr at 37°C. Subsequently, the cell suspension was filtered through 70 and 40 μm cell strainers. For subsequent flow cytometric analysis, cells were stained with the antibodies indicated in Table S5 using a standard protocol. For identification of Th17 and regulatory T cells, the mouse Th17/Treg phenotyping kit (#560767, BD Pharmingen, Franklin Lakes, NJ) was used. All cells are CD45+. Stained samples were measured using the BD FACSCanto™ II system. For data analysis, the FlowJo software (TreeStar, Inc., Ashland, OR) was used.
Statistical analysis
All values are presented as mean ± standard error of mean. For single outlier detection the Grubbs‐test was used, multiple outliers were defined when values were lower than Q1–1.5 × interquartile range or greater than Q3 + 1.5 × interquartile range (difference between the third quartile [Q3] and first quartile [Q1]). Subsequent statistical analyses were either performed utilizing Student's t‐test or Mann–Whitney U‐test calculated by GraphPad Prism 5.0 software. For multiple group comparison, one‐way analysis of variance (ANOVA) in combination with Tukey's multiple comparison test was used. For GO‐molecular function analysis, the results were ranked according to the adjusted p‐value. For Kaplan–Meier analysis, the Log‐rank test was used and false discovery rate was computed to combat for multiple hypothesis testing. For all graphs: *p < 0.05; **p < 0.01; ***p < 0.001.
Results
JAK signaling is activated in human K‐RAS‐mutated lung AC during tumor progression
To investigate the implication of JAK1 and JAK2 for the pathogenesis of human K‐RAS‐mutated lung AC, we took advantage of the publicly available mRNA expression data set GSE75037.22 Comparison of JAK1 and JAK2 mRNA expression levels of stage I classified K‐RAS‐mutated lung AC patients versus those with more advanced disease revealed increased JAK1/2 expression levels in the latter group, which reached significance for JAK1 (Fig. 1 a). In line with these data, we found activation of the JAK–STAT pathway in patients with more advanced lung AC (i.e., in stage II disease), as determined by GSEA (Fig. 1 b).24 Next, we confirmed these data in K‐RAS mutated lung AC patients from a different cohort published by the Cancer Genome Atlas.23 Indeed, we found a significant correlation of JAK1 and JAK2 mRNA expression levels with enrichment of K‐RAS signaling in lung AC patients (Figs. 1 c and 1 d). Moreover, we noticed a significant increase of JAK1 and JAK2 mRNA expression in tumors of late stage patients with oncogenic K‐RAS mutations compared to patients harboring K‐RAS WT tumors, which was not observed in early stage patients (Figs. S1 a and S1 b).
Figure 1.
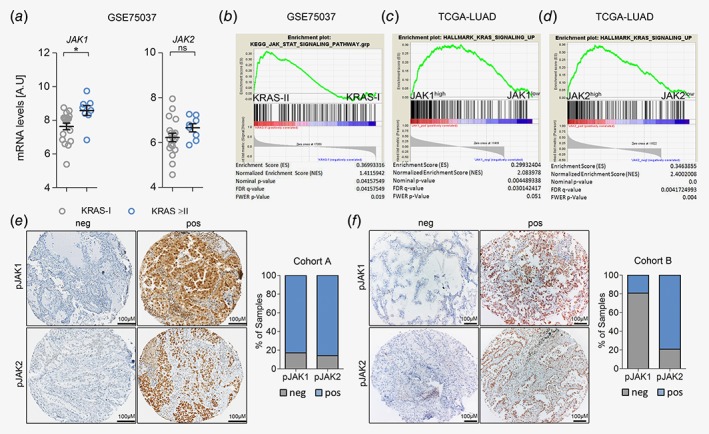
JAK‐mediated signaling is activated in progressing K‐RAS‐mutated human lung AC. (a) Graph showing relative JAK1 and JAK2 mRNA expression levels in human K‐RAS‐mutated lung AC tissue of stage I (n = 19) versus stage II or higher (≥II, n = 8). Data represent mean ± SEM, A.U (arbitrary units), Student's t‐test, *p < 0.05. (b) GSEA for Kyoto encyclopedia of genes and genomes‐JAK–STAT pathway signature geneset comparing human K‐RAS‐mutated tumors of stage I versus stage II, using tumor versus healthy parenchyma mRNA expression ratios. Data in (a) and (b) were retrieved from the Gene Expression Omnibus (GSE75037). (c) GSEA using gene expression data of the Cancer Genome Atlas‐LUAD cohort and HALLMARK_KRAS_SIGNALING_UP geneset, stratifying patients according to JAK1 and (d) JAK2 expression levels (n = 154). (e) Representative pictures of lung AC biopsies of patients included in cohort A (unknown K‐RAS mutation status; pJAK1 n = 303, pJAK2 n = 318) and (f) cohort B (K‐RAS mutated; pJAK1 n = 26, pJAK2 n = 24) with negative and positive staining reactions for pJAK1 and pJAK2 in tumor cells. Graphs represent percentage of positive and negative cases within the respective cohort. Staining intensities and percentage of positive tumor cells were determined by a board‐certified pathologist (H.P.). [Color figure can be viewed at http://wileyonlinelibrary.com]
However, using mRNA expression data of bulk tumor tissue does not allow distinguishing between tumor and stromal cells. Hence, we performed immunohistochemistry for activated JAK1 and JAK2 in biopsies of lung AC patients from two independent cohorts (cohorts A&B, consisting of unselected and K‐RAS mutated samples, respectively). Indeed, JAK1 and JAK2 were activated/phosphorylated in tumor cells of lung AC patients as determined by a pathologist (H.P.). In the stroma, a positive reaction for JAK1 and JAK2 was mainly found in lymphocytes (Figs. 1 e and 1 f). Taken together, these data indicated that JAK signaling is active in human K‐RAS‐driven lung AC.
JAK inhibition impairs human K‐RAS‐mutated lung AC cell growth in vivo
Next, we assessed the potential benefits of pharmacological JAK1/2 inhibition in K‐RAS‐driven lung AC using the JAK1/2 selective tyrosine kinase inhibitor ruxolitinib. We performed subcutaneous engraftments of human lung AC cell lines harboring oncogenic K‐RAS mutations into immunodeficient NOD scid gamma mice. Intriguingly, ruxolitinib treatment significantly attenuated growth of the human K‐RAS G12S‐mutated lung AC cell line A549 (Figs. 2 a–2 c), and impaired growth of A427 (K‐RAS G12D) transplanted cells (Figs. S2 a and S2 b). Immunohistochemistry analysis of proliferation and apoptosis markers within tumors revealed that JAK inhibition significantly reduced proliferation and slightly increased apoptosis of transplanted lung AC cells (Figs. 2 d and 2 e). JAK1/2 inhibition in tumor cells of ruxolitinib treated mice was efficient, since activation of the downstream effector STAT3 was almost absent after tyrosine kinase inhibitor treatment (Figs. 2 d and 2 e). Accordingly, ruxolitinib treatment impaired a well‐described autocrine oncogenic activation loop in this model.10 Indeed, tumor cell‐derived expression of pro‐inflammatory cytokines IL‐6 and IL‐1β was significantly decreased in the ruxolitinib treated group, as verified by quantitative real‐time PCR using primers specific for the human transcripts of these cytokines (Fig. 2 f). Notably, the ruxolitinib dose used in our experiments was generally well tolerated by the experimental mice, and we did not observe any significant body weight loss in the treated groups (Fig. S2 c). Since abrogation of JAK signaling may also impair angiogenesis,35 we checked expression of the angiogenic markers Vegfα and Hif1α and evaluated numbers of cluster of differentiation 31+ vessels, but we did not observe changes upon ruxolitinib treatment (Figs. S2 d and S2 e).
Figure 2.
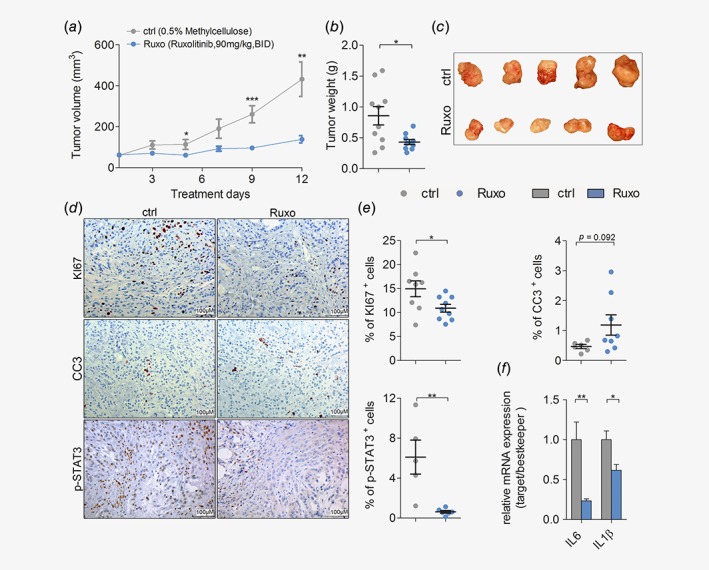
JAK inhibition impairs growth of human K‐RAS‐mutated lung AC cells in vivo. (a) Mean volumes of xenografted A549 derived tumors in mice treated with vehicle control (ctrl) or ruxolitinib (Ruxo) at 90 mg/kg body weight, seven times per week, BID, and (b) the endpoint tumor weight. (c) Representative pictures of A549 tumor‐derived xenografts after ctrl or Ruxo treatment. (d) Representative pictures of immunohistochemical stainings for KI67, cleaved caspase 3 (CC3) and p‐STAT3 staining of A549 cell line derived xenograft tumors upon ctrl and Ruxo treatment (scale bar: 100 μm), and (e) quantitation of positive cells for respective stainings. (f) Relative mRNA expression of human transcript variants of indicated genes normalized to human housekeeping genes (28S, TBP, ACTB) in A549 derived xenografted tumors. (d–f) n = ≥ 5 tumors per group. For all graphs Student's t‐test: *p < 0.05, **p < 0.01, ***p < 0.001. For all graphs data presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
Ruxolitinib attenuates tumorigenesis of autochthonous K‐RAS‐driven lung AC
Although cell‐line‐derived xenograft models have the advantage to study tumors of human origin, a major drawback of these models is the absence of a functional immune system.36 However, this is a critical factor in our study, since ruxolitinib was reported to promote immunosuppression and could thereby eventually enhance tumor progression.18 Thus, we addressed the effects of JAK1/2 inhibition in fully immunocompetent mouse models of autochthonous K‐RAS‐driven lung AC. Therefore, we took advantage of the K‐ras LSL‐G12D (K) and K‐ras LSL‐G12D:p53 fl/fl (KP) mice. Inhalation of K and KP mice with a Cre‐recombinase expressing adenovirus (Ad.Cre) triggers K‐ras G12D activation in lung epithelial cells, resulting in formation of p53 expressing (K) or deficient (KP) lung ACs. Of note, KP mice develop lung ACs showing features of advanced grade tumors.28, 37 First, we validated the mouse models by isolating primary lung alveolar type II cells derived from K mice and inducing the expression of oncogenic KRAS in vitro. Intriguingly, global gene expression analysis revealed that K‐ras G12D expression resulted in activation of the JAK–STAT pathway (Figs. S3 a and S3 b).27 Encouraged by these data, we treated K and KP mice 1 week after tumor induction with ruxolitinib or vehicle control for a period of 10 weeks, euthanized them and analyzed their lungs. We noticed a significant decrease in tumor burden in ruxolitinib treated mice compared to vehicle‐treated control mice, as indicated by reduced lung weight to body weight and tumor to lung area ratios, concomitant with reduced tumor numbers (Fig. 3 a). Of note, sex differences in treatment response were not observed in our mouse models (Fig. S4 a). Furthermore, grading of tumors by a pathologist (K.D.) revealed that ruxolitinib blocked the progression of K‐RAS‐driven lung tumors. Indeed, ruxolitinib treatment resulted in a significant reduction of high grade tumors compared to vehicle control, which reached significance for grade III tumors (Fig. 3 b and Fig. S4 b). No differences in body weights were observed between control and treatment groups (Figs. S4 c and S4 d). Similar to the engrafted human lung AC cells, ruxolitinib significantly attenuated tumor cell proliferation in these mice (Fig. 3 c). Intriguingly, we observed a higher rate of tumor cell apoptosis in the vehicle treated compared to ruxolitinib group in both K and KP mouse models (Fig. 3 d). Importantly, there were no differences in the expression levels of the angiogenic factors Hif1α and Vegfα between control and ruxolitinib treatment groups (Figs. S4 e and S4 f), suggesting that impairment of tumor cell proliferation is responsible for the reduced tumor burden upon ruxolitinib treatment in these models.
Figure 3.
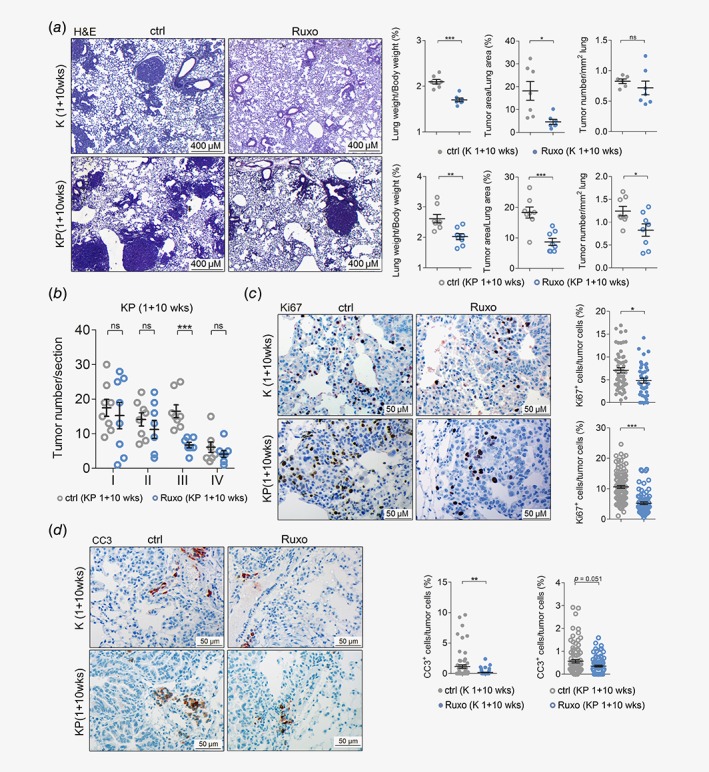
Ruxolitinib attenuates tumorigenesis of autochthonous K‐RAS‐driven lung AC. (a) Left panel: representative pictures of hematoxylin and eosin stained lung sections of vehicle control (ctrl) or ruxolitinib (Ruxo) treated K‐ras G12D (K) mice (n = 7 per group) and K‐ras G12D:p53 fl/fl (KP) mice (n = 8 per group). Treatment was started 1 week post tumor initiation, and continued for 10 weeks, with ruxolitinib being administered at 90 mg/kg body weight, seven times per week, BID (scale bars: 400 μm). Right panel: quantitation of hematoxylin and eosin stained lung sections from K mice (upper) and KP‐mice (lower) treated with ctrl or Ruxo started 1 week post tumor initiation and continued for 10 weeks. (b) Graph depicts tumor numbers per section stratified by tumor grades. Per mouse, one section was analyzed (n = 8 mice per group). (c) Left panel: representative pictures of immunohistochemical stainings for KI67 positive cells of lungs stemming from ctrl and Ruxo treated K and KP mice. Right panel: quantitation of indicated stainings. (d) Left panel: cleaved caspase‐3 positive cells of lungs of ctrl and Ruxo treated K and KP mice. Right panel: quantitation of indicated stainings (scale bars: 50 μm). (c, d) Mann–Whitney U‐test, *p < 0.05, **p < 0.01, ***p < 0.001. Others: Student's t‐test. *p < 0.05, **p < 0.01, ***p < 0.001. For all graphs data presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
JAK inhibition impairs progression of established K‐RAS‐mutated lung AC and modulates the TME
Having demonstrated that ruxolitinib effectively reduces tumorigenesis when administered at early stages of tumor formation, we questioned whether ruxolitinib prevents progression of already established K‐RAS‐driven lung AC in a therapeutic setting. Therefore, we treated K mice with ruxolitinib starting 8 weeks post Ad.Cre inhalation for five consecutive weeks. In this model, we observed a reduction in lung to body weight ratios and total tumor area per lung section after ruxolitinib treatment as compared to vehicle control treatment, while tumor numbers remained unaltered (Fig. 4 a). Notably, analysis of tumor to lung area ratios did not show differences between the treatment groups, most likely because total lung areas were smaller upon ruxolitinib treatment (Figs. S5 a–S5 c). Strikingly, grading of tumors revealed a reduction of high‐grade tumors, namely tumors of grades II and III (Fig. 4 b). In addition, ruxolitinib treatment of mice with established tumors also triggered significant reduction in tumor cell proliferation and increased apoptosis (Fig. 4 c). Similar to the previous models, differences in mRNA expression levels of angiogenic markers Hif1α and Vegfα were not observed and body weights showed no significant differences between control and ruxolitinib treatment groups (Figs. S5 d and S5 e). These data suggests that ruxolitinib impairs the progression of established K‐RAS‐driven lung AC. Since JAK signaling plays a crucial role in regulating the immune system, we analyzed if JAK1/2 inhibition affected the composition of the TME by performing flow cytometric analysis of lung tumors.17 Indeed, the abundance of tumor promoting granulocytic CD45+CD11b+LY6G+ and monocytic CD45+CD11b+LY6C+ MDSC38 was significantly reduced in the lungs of tumor‐bearing, ruxolitinib‐treated mice. Furthermore, the number of tumor promoting CD45+CD11b+CD206+ M2 macrophages was lowered upon JAK1/2 inhibition (Fig. 4 d). With respect to the lymphoid lineage, our analysis revealed significantly diminished NK cell numbers upon ruxolitinib administration in tumor bearing lungs of these mice (Fig. 4 d). These data are in line with a previous report using a preclinical breast cancer model.18 In addition, we observed a slight reduction in infiltrating CD8+ and CD4+ T cells upon ruxolitinib treatment, but an increase in CD4+IL17A+ Th17 cells as well as CD4+Foxp3+ regulatory T cells (Fig. 4 d). Moreover, JAK inhibition lead to a significant impairment of NK and CD8+ T cell activation, but on the other hand also abrogated interferon‐γ‐mediated STAT1 signaling and hence reduced PD‐L1 expression (Figs. 4 e–4 g). In summary, these data indicated that ruxolitinib altered the myeloid compartment of the TME toward an antitumorigenic state, which out‐competed the suppressing effects of ruxolitinib on infiltrating cytotoxic NK and CD8+T cells.
Figure 4.
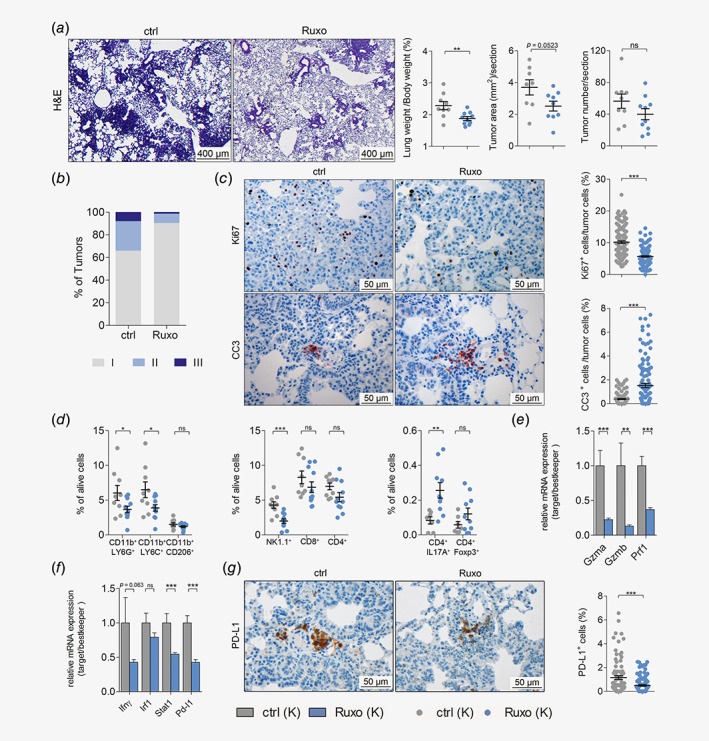
JAK inhibition impairs progression of established K‐RAS‐mutated lung AC and alters TME. (a) Left panel: representative pictures of hematoxylin and eosin stained lung sections of vehicle control (ctrl) or ruxolitinib (Ruxo) treated K‐ras G12D (K) mice. Right panel: quantitation of hematoxylin and eosin stained lung sections of ctrl or Ruxo treated K‐ras G12D (K) mice. Treatment was started 8 week post tumor initiation, and continued for 5 weeks (8 + 5wks), with ruxolitinib being administered at 90 mg/kg body weight, seven times per week, BID (scale bars: 400 μm) (n ≥ 9 per group). (b) Graph represents the percentage of tumors with indicated grades in vehicle control and ruxolitinib treated K mice. (c) Left panel: representative pictures of immunohistochemical stainings for KI67 and cleaved caspase‐3 positive cells, comparing lungs of ctrl or Ruxo treated K mice (8 + 5 weeks). Right panel: quantitation of indicated stainings (scale bars: 50 μm). (d) Results of flow cytometric analysis depicting the percentage of myeloid cells (left), lymphoid cells (middle) and CD4+ subsets (right) in tumor‐harboring lung lysates of ctrl and Ruxo treated K mice (8 + 5 weeks). (e) Graph displaying relative mRNA expression of the indicated genes normalized to housekeeping genes (28s, Tbp, Actb) in tumor harboring lungs of ctrl versus Ruxo treated K mice (8 + 5 weeks). (f) Graph indicating relative mRNA expression of the indicated genes normalized to housekeeping genes (28s, Tbp, Actb) in tumor harboring lungs of ctrl versus Ruxo treated K mice (8 + 5 weeks). (g) Left panel: representative pictures of immunohistochemical staining for PD‐L1 positive cells of lungs stemming from ctrl and Ruxo treated K mice (8 + 5 weeks). Right panel: quantitation of indicated staining. (c, g) Mann–Whitney U‐test. Others: Student's t‐test. For all graphs data presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. [Color figure can be viewed at http://wileyonlinelibrary.com]
JAK inhibition abrogates expression of oncogenic chemokines and cytokines
Next, we thought to unravel the transcriptional changes, leading to the establishment of an antitumorigenic TME upon ruxolitinib treatment. Therefore, we performed RNA‐seq analysis of short‐term (4 days) treated tumor‐bearing lungs from K mice with already established lung ACs. As expected, short‐term administration of ruxolitinib significantly diminished activation of JAK–STAT signaling in the lung lysates of these mice (Fig. 5 a and Fig. S6 a). Intriguingly, GSEA analysis also revealed that ruxolitinib administration significantly reduced K‐RAS activity in these lungs (Fig. 5 b). Notably, within the top 350 genes negatively regulated by ruxolitinib (Table S6), we noticed an over‐representation of factors found within GO clusters for chemokine and cytokine activity (Fig. 5 c and Fig. S6 b). Accordingly, most of the genes annotated in these gene sets were downregulated upon ruxolitinib treatment compared to vehicle control (Fig. 5 d and Table S6). Importantly, 84 of these 109 genes downregulated by ruxolitinib were either upregulated in K‐ras G12D‐mutated lung alveolar type II (K) cells compared to WT cells (67 genes) or in tumor harboring lungs of KP mice compared to healthy WT lungs (52 genes). Notably, 35 hits (Tables S2 and S6) were common in all three groups (Fig. 5 e and Figs. S6 c and S6 d). Among these 35 genes, key pro‐inflammatory and pro‐tumorigenic factors including ll6, Tnfα and chemokine (C‐X‐C motif) ligand 1 (Cxcl1) were found. The anti‐inflammatory properties of ruxolitinib were also observed in the long‐term treatment models, as evidenced by decreased expression of these genes in treated K and KP mice (Fig. 5 f).
Figure 5.
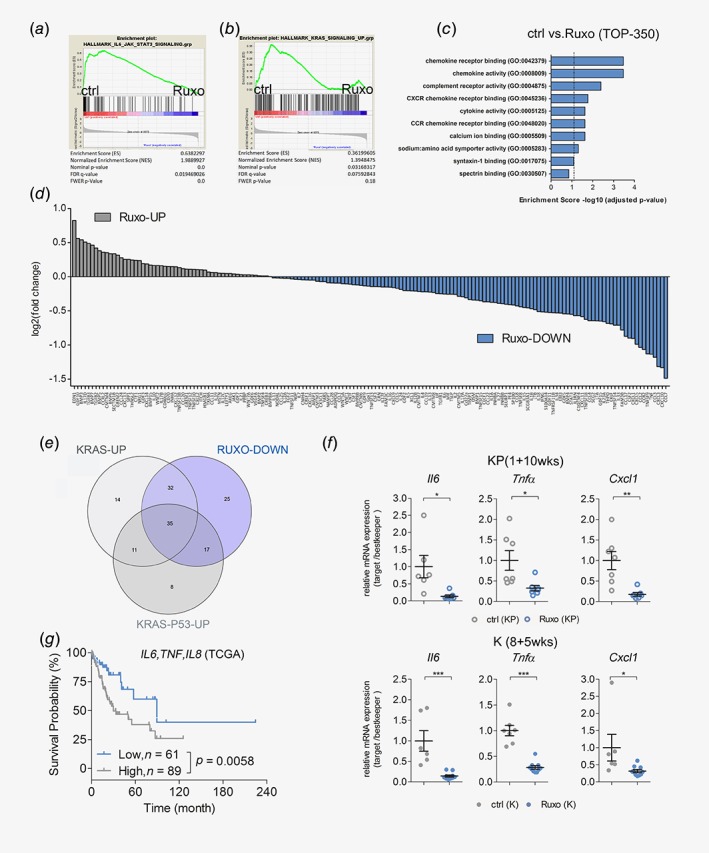
JAK inhibition abrogates expression of oncogenic chemokines and cytokines. (a) GSEA of RNA‐seq data of lungs of K‐ras G12D (K) mice 8 weeks post tumor induction, treated with vehicle control (ctrl) or ruxolitinib (Ruxo) for four consecutive days before harvesting (n = 4 per group). The used genesets are HALLMARK_IL6_STAT3_SIGNALING and (b) HALLMARK_KRAS_SIGNALING_UP, comparing ctrl and Ruxo treatment. (c) Graph depicting the adjusted p‐values of enrichment scores of indicated GO‐molecular function pathways in ctrl versus Ruxo treated K mice. The top 350 genes downregulated in lungs of Ruxo treated mice compared to ctrl treated mice were included in the analysis. (d) Graph shows DESeq calculated log2 fold changes in mRNA expression of indicated up‐ or downregulated chemokine/cytokine related genes in tumor bearing lungs of Ruxo treated compared to ctrl treated K mice. (e) Venn diagram depicting amount of overlapping chemokine and cytokine related genes in all three groups. The defined 35 common genes were found by intersecting the after gene data sets: genes downregulated in tumor bearing lungs of Ruxo treated mice compared to ctrl treated mice (RUXO‐DOWN), genes upregulated in K‐ras G12D mutated alveolar type‐II cells compared to WT alveolar type‐II cells (KRAS‐UP, GSE113146) and genes upregulated in K‐ras‐p53‐mutated tumor‐harboring lungs compared to WT lungs from KP mice (KRAS‐P53‐UP). (f) Relative mRNA expression of the indicated genes normalized to housekeeping genes (28s, Tbp, Actb) in tumor harboring lungs of vehicle control versus ruxolitinib treated KP mice (upper) and K mice (lower). (g) Graph depicts prognostic value of indicated genes in human K‐RAS‐mutated lung AC samples. Patients were stratified according to auto best cut‐off selection. Data were retrieved from the Cancer Genome Atlas (n = 150). Log‐rank test was used for statistical analysis. For (f) Student's t‐test. *p < 0.05, **p < 0.01, ***p < 0.001. Data presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
Next, we evaluated the implication of these chemo/cytokines for human lung AC pathogenesis. Therefore, we took advantage of a meta‐analysis based tool for the assessment of potential biomarkers in lung AC.25 Multigene classifier analysis revealed that lower mRNA expression of the signature comprising of 35 genes was associated with a better survival prognosis in these patients at a false discover rate < 1% (Fig. S6 e and Table S2). Moreover, lower mRNA levels of lL6, TNFα and IL8, the human orthologue to mouse Cxcl1 (Fig. 5 g), could be associated with a better prognosis in lung AC patients harboring K‐RAS mutations.39 Taken together, we provide here evidence that ruxolitinib treatment succesfully impairs K‐RAS activity in lung tumors and leads to the establishment of an anti‐inflammatory and antitumorgenic tumor microenviroment.
Discussion
Being a well‐defined culprit in myeloproliferative disorders, the JAK/STAT pathway is a well‐known target for pharmaceutical industry. Ruxolitinib was already Food and Drug Administration approved in 2011 based on the positive results of the COMFORT‐I clinical trial.40 Somatic mutations rendering JAK2 constitutively active like the V617F mutation are often found in hematologic malignancies. However, solid cancers including lung cancer lack such activating somatic mutations.23, 41 Nevertheless, clinical trials for lung AC patients were launched using JAK inhibitors ruxolitinib, in particular for patients with activating EGFR‐mutations upstream of JAK1 and JAK2 (NCT02155465, NCT02145637, NCT02917993, NCT03450330). A recent clinical trial specifically excluding patients with EGFR‐mutations showed a longer overall survival (5.9 vs. 7.5 months for placebo treated vs. ruxolitinib treated, hazard ratio = 0.877) when combining ruxolitinib and chemotherapy in non‐small cell lung cancer patients (NCT02119650). However, the K‐RAS mutation status of the involved patients was not disclosed. Therefore the potential benefit of a JAK1/2 inhibition for K‐RAS‐driven lung AC pathogenesis cannot be extrapolated. Hence, as in other clinical trials when using for example mitogen‐activated protein kinase kinase 1 or rapidly accelerated fibrosarcoma inhibitors, it would be important for JAK1/2 targeting clinical trials to stratify for the K‐RAS mutational status.
Here, we found increased expression of JAKs and activation of downstream STAT signaling in patients harboring advanced K‐RAS lung AC. Increased JAK levels also correlated with enhanced K‐RAS activity in human lung AC patient samples, suggesting that JAK/STAT signaling fuels the proliferation of K‐RAS‐mutated lung AC. Accordingly, ruxolitinib treatment markedly reduced tumor cell proliferation of human K‐RAS‐mutated A549 cells engrafted in immunodeficient NOD scid gamma mice and reduced a K‐RAS activation gene signature. The engrafted tumors showed decreased expression of tumor cell‐derived, pro‐oncogenic cytokines IL‐1β and IL‐6, suggesting that ruxolitinib interferes with feed‐forward cytokine signaling which promotes K‐RAS tumorigenesis.10 Furthermore, ruxolitinib treatment resulted in tumor growth reduction in immunocompetent experimental mouse models. Ruxolitinib impaired K‐RAS‐driven lung AC not only at early tumor stages but also in established tumors, independently of the p53 status. Early treatment of tumors with ruxolitinib resulted in reduced tumor cell proliferation and reduced apoptosis levels. By contrast, we observed reduced cell proliferation and increased apoptosis rates in established tumors upon ruxolitinib treatment. This discrepancy can be interpreted as the result of a higher cell turnover in early tumorigenesis or by a tumor‐compensatory mechanism upon JAK1/2 inhibition present at early tumor stages. In agreement with previous reports, ruxolitinib treatment diminished tumor infiltrating CD8+ and CD4+ T cells and significantly lowered NK cell numbers but did not result in tumor metastasis, even in the absence of p53.17, 18, 42 In agreement with others, JAK inhibition abrogated interferon‐γ‐mediated STAT1 signaling and reduced PD‐L1 expression levels.43 Nevertheless, NK and CD8+ T cell activation markers were reduced in lungs of ruxolitinib treated mice. Furthermore, ruxolitinib elevated numbers of tumor infiltrating CD4+IL17A+ Th17 and CD4+Foxp3+ regulatory T cells. Of note, T cells potentially play a minor role in our experimental mouse models due to a lack of tumor immunogenicity. In this sense, assessment of JAK inhibitors in T‐cell‐dependent lung AC experimental models44 may be a better option and can be more informative to design and interpret future and ongoing clinical trials, especially those combining JAK inhibitors and immune checkpoint blockers (e.g., NCT03425006).
On the other hand, systemic JAK inhibition triggered a slight reduction of tumor promoting M2 macrophages and significantly reduced the abundance of infiltrating granulocytic and monocytic MDSCs, which is in agreement with an induction of an antitumorigenic TME and the antitumor effects of ruxolitinib. Importantly, MDSCs and M2 tumor‐associated macrophages not only impede tumor immune surveillance but also support tumor cell proliferation via cytokine production, and could thereby foster growth of K‐RAS‐driven lung AC.38, 45 In line with these data, ruxolitinib treatment of mouse autochthonous lung tumors mainly affected the expression of pro‐tumorigenic chemokines and cytokines. This chemokine/cytokine milieu is well‐known to be a central modulator of the cancer microenvironment and it has been established as one of the hallmark drivers of cancer.46 Among these prominently downregulated chemokines and cytokines upon ruxolitinib treatment, we found ll6, Tnfα and Cxcl1. All of them are well‐described pro‐inflammatory and pro‐tumorigenic chemo‐ and cytokines. Reduced IL6 levels were shown to trigger a switch in the TME toward an antitumoral phenotype.47 TNFα directly endorses tumor progression and metastasis.48 Furthermore, CXCL1 has been reported to facilitate lung cancer cell growth due to the recruitment of MDSCs and tumor associated neutrophils.13, 49 Importantly, all of these factors were also found to be upregulated in K‐ras G12D‐mutated lung alveolar type II (K) cells compared to WT cells or in tumor harboring lungs from KP mice compared to non‐tumorous lungs from KP mice. In agreement with this data, low mRNA expression levels of lL6, Tnfα and IL8 (the human orthologue to murine Cxcl1) were also associated with a better prognosis in lung AC patients harboring K‐RAS mutations.39 Thus, these data indicate that lL6, Tnfα and Cxcl1 play a central role for K‐RAS driven lung AC both in human and mice and provides a solid rationality for using agents blocking inflammation‐driven tumorigenesis, such as ruxolitinib, to treat lung AC.7
In summary, we demonstrated here that JAK‐mediated signaling is involved in progression of K‐RAS‐driven lung tumorigenesis and that pharmacological inhibition of JAK/STAT signaling blocks the cytokine‐mediated feed‐forward loop responsible for tumor cell proliferation. Additionally, JAK inhibition reprograms the TME toward an anti‐inflammatory/antitumorigenic state. Therefore, our data suggest that ruxolitinib treatment represents an attractive therapeutic opportunity for treatment of K‐RAS‐mutated lung AC.
Supporting information
Fig. S1: JAK mediated signaling is activated in progressing K‐RAS‐mutated human lung AC.
Figure S2: JAK inhibition impairs human K‐RAS‐mutated lung AC cell growth in vivo.
Figure S3: K‐RASG12D transformed primary pneumocytes exhibit JAK–STAT pathway activation in vitro
Figure S4: Ruxolitinib attenuates tumorigenesis of autochthonous K‐RAS‐driven lung AC
Figure S5: JAK inhibition impairs progression of established K‐RAS‐mutated lung AC
Figure S6: JAK inhibition abrogates expression of oncogenic chemokines and cytokines
Table S1. Probeset‐IDs‐GSE75037
Table S2. Probeset‐IDs‐Cytokines‐Chemokines
Table S3. Genotyping primers
Table S4. Quantitative PCR analysis primers
Table S5. Antibodies for Flow Cytometric analysis
Table S6 Chemokine & cytokine gene names
Acknowledgements
This project was supported by the Austrian Science Fund (FWF): P 25599‐B19 to EC. H.P.M. was supported by the Initiative Krebsforschung research grant and the Fellinger Krebsforschungsverein. B.G. was supported by the NVKP_16‐1‐2016‐0037 and KH‐129581 grants of the National Research, Development and Innovation Office, Hungary. R.E. was supported by the Austrian Science Fund Doktoratskolleg grant “Inflammation and Immunity” and P29222‐B28, and K.D. was supported by the Jànos Bolyai Research Scholarship, Hungarian Academy of Sciences. Funders had no role in study design, data collection and analysis, decision to publish or in the preparation of the manuscript. The authors thank Safia Zahma for help with the preparation of tissue sections and Markus Jeitler and Sophia Derdak (Core Facilities, Medical University of Vienna, Vienna, Austria) for help with RNA‐Seq analysis.
Conflict of interest: The authors declare no potential conflict of interest.
Data availability
All materials will be made available to the scientific community. RNAseq data are available within the sequence read archive (SRA) with the accession number PRJNA518030.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Zappa C, Mousa SA. Non‐small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res 2016;5:288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Swanton C, Govindan R. Clinical implications of genomic discoveries in lung cancer. N Engl J Med 2016;374:1864–73. [DOI] [PubMed] [Google Scholar]
- 5. Yang Z, Hackshaw A, Feng Q, et al. Comparison of gefitinib, erlotinib and afatinib in non‐small cell lung cancer: a meta‐analysis. Int J Cancer 2017;140:2805–19. [DOI] [PubMed] [Google Scholar]
- 6. Roman M, Baraibar I, Lopez I, et al. KRAS oncogene in non‐small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer 2018;17:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Golay HG, Barbie DA. Targeting cytokine networks in KRAS‐driven tumorigenesis. Expert Rev Anticancer Ther 2014;14:869–71. [DOI] [PubMed] [Google Scholar]
- 8. Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 2016;35:939–51. [DOI] [PubMed] [Google Scholar]
- 9. O'Shea JJ, Schwartz DM, Villarino AV, et al. The JAK‐STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med 2015;66:311–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu Z, Aref AR, Cohoon TJ, et al. Inhibition of KRAS‐driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov 2014;4:452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kitajima S, Thummalapalli R, Barbie DA. Inflammation as a driver and vulnerability of KRAS mediated oncogenesis. Semin Cell Dev Biol 2016;58:127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Looyenga BD, Hutchings D, Cherni I, et al. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non‐small cell lung carcinoma. PLoS One 2012;7:e30820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grabner B, Schramek D, Mueller KM, et al. Disruption of STAT3 signalling promotes KRAS‐induced lung tumorigenesis. Nat Commun 2015;6:6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caetano MS, Hassane M, Van HT, et al. Sex specific function of epithelial STAT3 signaling in pathogenesis of K‐ras mutant lung cancer. Nat Commun 2018;9:4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song L, Rawal B, Nemeth JA, et al. JAK1 activates STAT3 activity in non‐small‐cell lung cancer cells and IL‐6 neutralizing antibodies can suppress JAK1‐STAT3 signaling. Mol Cancer Ther 2011;10:481–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meissl K, Macho‐Maschler S, Muller M, et al. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017;89:12–20. [DOI] [PubMed] [Google Scholar]
- 17. McLornan DP, Khan AA, Harrison CN. Immunological consequences of JAK inhibition: friend or foe? Curr Hematol Malig Rep 2015;10:370–9. [DOI] [PubMed] [Google Scholar]
- 18. Bottos A, Gotthardt D, Gill JW, et al. Decreased NK‐cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat Commun 2016;7:12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Haas N, de Koning C, Spilgies L, et al. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology 2016;5:e1196312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL‐6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 2018;15:234–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Girard L, Rodriguez‐Canales J, Behrens C, et al. An expression signature as an aid to the histologic classification of non‐small cell lung cancer. Clin Cancer Res 2016;22:4880–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 2016;48:607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gyorffy B, Surowiak P, Budczies J, et al. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non‐small‐cell lung cancer. PLoS One 2013;8:e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ali A, Brown V, Denley S, et al. Expression of KOC, S100P, mesothelin and MUC1 in pancreatico‐biliary adenocarcinomas: development and utility of a potential diagnostic immunohistochemistry panel. BMC Clin Pathol 2014;14:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moll HP, Pranz K, Musteanu M, et al. Afatinib restrains K‐RAS‐driven lung tumorigenesis. Sci Transl Med 2018;10:eaao2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 2009;4:1064–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heberle H, Meirelles GV, da Silva FR, et al. InteractiVenn: a web‐based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 2015;16:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Babicki S, Arndt D, Marcu A, et al. Heatmapper: web‐enabled heat mapping for all. Nucleic Acids Res 2016;44:W147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pfaffl MW, Tichopad A, Prgomet C, et al. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper‐‐Excel‐based tool using pair‐wise correlations. Biotechnol Lett 2004;26:509–15. [DOI] [PubMed] [Google Scholar]
- 35. Xue C, Xie J, Zhao D, et al. The JAK/STAT3 signalling pathway regulated angiogenesis in an endothelial cell/adipose‐derived stromal cell co‐culture, 3D gel model. Cell Prolif 2017;50:e12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hidalgo M, Amant F, Biankin AV, et al. Patient‐derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 2014;4:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jackson EL, Olive KP, Tuveson DA, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res 2005;65:10280–8. [DOI] [PubMed] [Google Scholar]
- 38. Ostrand‐Rosenberg S, Sinha P. Myeloid‐derived suppressor cells: linking inflammation and cancer. J Immunol 2009;182:4499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol 2006;7:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Verstovsek S, Mesa RA, Gotlib J, et al. A double‐blind, placebo‐controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–97. [DOI] [PubMed] [Google Scholar]
- 42. Heine A, Held SAE, Daecke SN, et al. The JAK‐inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013;122:1192–202. [DOI] [PubMed] [Google Scholar]
- 43. Luo N, Formisano L, Gonzalez‐Ericsson PI, et al. Melanoma response to anti‐PD‐L1 immunotherapy requires JAK1 signaling, but not JAK2. Oncoimmunology 2018;7:e1438106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. DuPage M, Cheung AF, Mazumdar C, et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 2011;19:72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Caux C, Ramos RN, Prendergast GC, et al. A milestone review on how macrophages affect tumor growth. Cancer Res 2016;76:6439–42. [DOI] [PubMed] [Google Scholar]
- 46. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 47. Unver N, Delgado O, Zeleke K, et al. Reduced IL‐6 levels and tumor‐associated phospho‐STAT3 are associated with reduced tumor development in a mouse model of lung cancer chemoprevention with myo‐inositol. Int J Cancer 2018;142:1405–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shang GS, Liu L, Qin YW. IL‐6 and TNF‐alpha promote metastasis of lung cancer by inducing epithelial‐mesenchymal transition. Oncol Lett 2017;13:4657–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan M, Zhu H, Xu J, et al. Tumor‐derived CXCL1 promotes lung cancer growth via recruitment of tumor‐associated neutrophils. J Immunol Res 2016;2016:6530410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: JAK mediated signaling is activated in progressing K‐RAS‐mutated human lung AC.
Figure S2: JAK inhibition impairs human K‐RAS‐mutated lung AC cell growth in vivo.
Figure S3: K‐RASG12D transformed primary pneumocytes exhibit JAK–STAT pathway activation in vitro
Figure S4: Ruxolitinib attenuates tumorigenesis of autochthonous K‐RAS‐driven lung AC
Figure S5: JAK inhibition impairs progression of established K‐RAS‐mutated lung AC
Figure S6: JAK inhibition abrogates expression of oncogenic chemokines and cytokines
Table S1. Probeset‐IDs‐GSE75037
Table S2. Probeset‐IDs‐Cytokines‐Chemokines
Table S3. Genotyping primers
Table S4. Quantitative PCR analysis primers
Table S5. Antibodies for Flow Cytometric analysis
Table S6 Chemokine & cytokine gene names
Data Availability Statement
All materials will be made available to the scientific community. RNAseq data are available within the sequence read archive (SRA) with the accession number PRJNA518030.