

Summary
In developing neurons, phosphoinositide 3-kinases (PI3Ks) control axon growth and branching by positively regulating PI3K/PI(3,4,5)P3, but how neurons are able to generate sufficient PI(3,4,5)P3 in the presence of high levels of the antagonizing phosphatase PTEN is difficult to reconcile. We find that normal axon morphogenesis involves homeostasis of elongation and branch growth controlled by accumulation of PI(3,4,5)P3 through PTEN inhibition. We identify a plasma membrane-localized protein-protein interaction of PTEN with plasticity-related gene 2 (PRG2). PRG2 stabilizes membrane PI(3,4,5)P3 by inhibiting PTEN and localizes in nanoclusters along axon membranes when neurons initiate their complex branching behavior. We demonstrate that PRG2 is both sufficient and necessary to account for the ability of neurons to generate axon filopodia and branches in dependence on PI3K/PI(3,4,5)P3 and PTEN. Our data indicate that PRG2 is part of a neuronal growth program that induces collateral branch growth in axons by conferring local inhibition of PTEN.
Keywords: PTEN, PI3K signaling, neuronal growth homeostasis, axon morphogenesis, phosphoinositide signaling, plasticity-related gene family, branching
Graphical Abstract
Highlights
-
•
Neuronal axon growth and branching is globally regulated by PI3K/PTEN signaling
-
•
PRG2 inhibits PTEN and stabilizes PIP3 and F-actin
-
•
PRG2 localizes to nanoclusters on the axonal membrane and coincides with branching
-
•
PRG2 promotes axonal filopodia and branching dependent on PI3K/PTEN
PTEN globally suppresses growth in multiple cell types and inhibits neuronal axon growth and branching. Brosig et al. describe a mechanism for developing neurons to inhibit PTEN function by upregulating PRG2 in the axon. Although the overall growth capacity of neurons remains constant, PRG2 redirects growth to axon branches.
Introduction
Growth is a key cellular process orchestrated by intrinsic and extrinsic factors that engage signaling pathways and transcriptional programs for the upregulation of macromolecular synthesis and for increases in both cell size and mass (Wullschleger et al., 2006). During nervous system development, cell growth is subject to spatial and temporal constraints because neurons grow in a highly polarized and localized fashion. An example of how spatially segregated neuronal growth can be organized is exemplified during axon morphogenesis. At that time, growth is manifested as axon elongation and axon branching, both of which are developmentally and operationally separated (Kalil and Dent, 2014, Lewis et al., 2013) but, nevertheless, are expected to be coordinated with an overall neuronal growth capacity.
An established growth-promoting signaling pathway in neurons is controlled by the phosphoinositide 3-kinases (PI3Ks) and phosphatase and tensin homolog deleted on chromosome 10 (PTEN). PI3K synthesizes the generation of phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) at the plasma membrane, whereas PTEN hydrolyzes PI(3,4,5)P3 at the D3 position of the inositol ring, thereby directly antagonizing PI3K and downstream AKT/GSK3/mTORC1 growth-associated signaling pathways (Lee et al., 2018). PI3K activity is indispensable for controlling multiple growth processes, and inactivating PTEN mutations are associated with sporadic cancers; overgrowth syndromes, such as PTEN hamartoma tumor syndrome; and autism spectrum disorders (Huang et al., 2016, Kwon et al., 2006).
Precise spatiotemporal regulation of PI3K/PTEN-generated PI(3,4,5)P3 is essential for the proper reorganization of the plasma membrane actin cytoskeleton to support cell morphology and migration in different cell types (Haugh et al., 2000, Iijima et al., 2002, Martin-Belmonte et al., 2007). In neurons, in particular, localized production of PI(3,4,5)P3 is associated with hallmarks of neuronal morphology such as induction and elongation of neurites, dendritic spine morphogenesis and function, and axon branch morphogenesis (Gallo, 2013, Horiguchi et al., 2006, Kreis et al., 2014, Ménager et al., 2004, Shi et al., 2003). In axons, localized production of PI(3,4,5)P3 supports the initiation of F-actin patches, which give rise to filopodia protrusions along the axon shaft (Gallo, 2013, Ketschek and Gallo, 2010, Spillane et al., 2012). These filopodial protrusions are considered precursors for axon branches, which mature by subsequent invasion and stabilization of microtubules and ultimately form the basis of neuronal connectivity in the adult brain (Kalil and Dent, 2014).
Although generation of PI(3,4,5)P3 during axon growth and branching has been well documented (Kalil and Dent, 2014, Ketschek and Gallo, 2010), the mechanisms involving PTEN regulation deserve further attention, given that they can either expand or blunt the signaling output of the PI3K pathway by altering PI(3,4,5)P3 plasma membrane concentration and localization (Kreis et al., 2014). This is particularly relevant because PTEN is highly abundant in neurons and extremely efficient in confining and limiting PI3K-dependent growth in the axon, especially during early development when axons elongate to reach their targets (Chadborn et al., 2006, Christie et al., 2010, Drinjakovic et al., 2010, Zhang et al., 2013). How neurons overcome that growth barrier to allow collateral branching is not fully understood.
Here, we identify and characterize a PTEN membrane protein association, which controls PTEN activity. The protein complex incorporates plasticity-related gene 2 (PRG2), a transmembrane protein belonging to the family of lipid phosphate phosphatase-related (also known as LPPR) proteins. Our data indicate that PRG2 associates with PTEN and organizes PI(3,4,5)P3-mediated cellular responses in neurons during axon filopodia initiation and branch formation.
Results
PTEN Interacts with the Neuronal Membrane Protein PRG2
We set out to identify proteins involved in regulating the sub-cellular localization and/or function of PTEN in neurons using mass spectrometry (van Diepen et al., 2009). Among the protein interactions, we identified PRG2 as a PTEN binding partner. We first confirmed the PRG2-PTEN association by performing endogenous co-immunoprecipitation in embryonic day 18 (E18) rat brain lysates as well as in days in vitro (DIV) 9 cortical neuron cultures (Figure 1A). PRG2 (or LPPR3) is a member of the LPPR protein family whose members show high homology with bioactive lipid-inactivating phosphatases but lack catalytic activity (McDermott et al., 2004, Sigal et al., 2007, Strauss and Bräuer, 2013). PRG2 is closely related to its better functionally characterized homolog PRG1 (Bräuer et al., 2003, Liu et al., 2016, Trimbuch et al., 2009); both proteins share the common domain structure with six transmembrane regions forming an extracellular oriented pseudo-LPP catalytic motif and an additional large C-terminal cytosolic domain (Figure 1B). Unique within this family, PRG2 bears a highly acidic stretch consisting of 20 glutamic acid residues in its C-domain (“poly-E-box”). To characterize whether the PRG2-specific poly-E-box region mediates the PTEN-PRG2 interaction, we generated a PRG2 deletion mutant lacking the poly-E-box as well as the distal C terminus (PRG2ΔC amino acids [aa] 1–407) (Figure 1B). Co-immunoprecipitation experiments demonstrate that the PRG2-PTEN interaction does not require the PRG2 distal C terminus (Figure 1C). However, given the likely formation of complex PRG2 multimers with different members of the PRG family at the cell surface (Figure S1 complementing Yu et al., 2015), one cannot exclude the possibility of indirect interactions with PTEN. Therefore, the PRG2-PTEN interaction was further studied using quantitative microscale thermophoresis (MST). After optimizing the expression and purification of native 1D4-tagged PRG2 from HEK cells, the purity of the tagged PRG2 was characterized (Figures 1D and 1E). Serial dilutions of PTEN were mixed with labeled, purified PRG2-1D4. MST traces identified a dissociation constant for PRG2 binding to PTEN in the low micromolar range, demonstrating that the physical interaction between PRG2 and PTEN is of a direct nature (Figure 1F).
Figure 1.

Interaction of PTEN with PRG2
(A) Immunoprecipitation (IP) with anti-PTEN or anti-PRG2 antibodies was performed on E18 rat brain lysate and DIV 9 cortical cultures, and immunoblotted (western blot [WB]) with the indicated antibodies.
(B) PRG2 belongs to the LPPR family of membrane proteins with six transmembrane (TM) regions. PRG2 shows close homology to PRG1 with 49% amino acid identity and 63% similarity. PRG2 contains a long hydrophilic C-terminal cytoplasmic extension with 20 glutamic acid residues (“poly-E-box”). Prg2 full-length (aa 1–716) and Prg2 deletion construct (PRG2ΔC, aa 1–407) were generated.
(C) IP and WB studies show the independence of the PTEN-PRG2 interaction on the distal C terminus. FLAG-IPs were analyzed with the indicated antibodies.
(D) Purification scheme used to isolate tagged PRG2.
(E) Coomassie-stained gel and western blot showing purity and integrity of PRG2. TL, total cell lysate; SN, unbound fraction; EL, final PRG2 eluate in detergent Fos-Choline-14; EL A8-35, PRG2 in amphipol A8-35.
(F) PRG2-PTEN binding measured by microscale themophoresis (MST); n = 7 replicates of two independent purifications. Plotting the ratio of pre-thermophoresis and during thermophoresis fluorescence (Fnorm) ± SD with the concomitant fitting of the data yielded a dissociation constant KD of 0.73 ± 0.22 μM.
See also Figure S1.
PRG2 Alters PTEN Localization and Activity
Association of PTEN with protein complexes may control cellular responses by confining signaling events to subcellular compartments and/or by altering the PTEN phosphatase enzymatic activity. We deployed two experimental strategies to investigate the molecular framework of PRG2 action in the control of PTEN function. In the first set of experiments, we examined the possibility that PRG2 sequesters PTEN to cellular compartments, using N1E-115 neuroblastoma cells expressing Prg2-Flag. Overexpressed PRG2-FLAG localized to the plasma membrane and was able to redirect endogenous PTEN to the cellular periphery (Figures 2A, 2B, and S7). In the second set of experiments, we tested whether PTEN activity was altered in the presence of PRG2 by establishing a phosphatase assay. After co-transfection of COS-7 cells with GFP-PTEN and control vector or Prg2-Flag, we measured the activity of immunoprecipitated PTEN against PI(3,4,5)P3 in a standard Malachite-Green-based colorimetric assay (Das et al., 2003). Exogenous Prg2 expression resulted in a dose-dependent decrease in PTEN phosphatase activity (Figure 2C), whereas the expression of the Prg2ΔC deletion did not change PTEN activity (Figure 2D). To directly correlate PTEN activity with PTEN protein content in each treatment, we normalized the phosphate produced to relative concentrations of immunoprecipitated PTEN. This demonstrated that PRG2 can reduce PTEN activity to approximately 50% at the highest PRG2 concentration tested (Figure 2E). The specificity of this result was further verified by assaying the activity of the 5′ phosphatase SHIP2, which does not interact with PRG2 (data not shown). Prg2 expression did not affect the amount of SHIP2-produced phosphate (Figure 2F). In conclusion, these experiments established that PRG2 is able to both direct PTEN between cell compartments and to inhibit PTEN phosphatase activity.
Figure 2.

PRG2 Redirects PTEN to the Plasma Membrane and Inhibits PTEN’s Phosphatase Activity
(A) PRG2 redirects PTEN to the plasma membrane in neuroblastoma cells. Panels show Prg2-Flag expressing N1E-115 cells and GFP expressing cells immunolabeled with anti-FLAG (green) and anti-PTEN (white). Scale bar: 5 μm.
(B) Quantification of membrane PTEN relative to cytosolic PTEN. Bars indicate the means ± SEM from n = 16 (GFP) and n = 13 (PRG2) cells from two independent experiments.
(C and D) PRG2 decreased PTEN-activity. COS-7 cells were co-transfected with GFP-PTEN and control vector, or with GFP-PTEN and increasing amounts (μg) of Prg2-Flag (C) or Prg2ΔC-Flag (D). PTEN activity after IP was measured in a colorimetric assay.
(E) Normalized net phosphate release. Phosphate produced (at 10 min) was normalized to relative PTEN concentration in IP. Bars indicate the mean of three experiments ± SD. ∗∗∗p < 0.001 (t test)
(F) PRG2 does not alter phosphate product released by the 5′ phosphatase SHIP2. Bars indicate the mean of three experiments ± SD, n.s. = not significant.
PRG2 Affects Cellular PI(3,4,5)P3 and F-Actin-Rich Protrusions in a PTEN-Dependent Manner
Unlike most protein phosphatases, PTEN preferentially dephosphorylates phosphoinositide substrates. PTEN activity thus decreases levels of plasma membrane PI(3,4,5)P3 and functions as a growth suppressor by negatively regulating the PI3K/AKT signaling pathway. We, therefore, addressed whether the PRG2-PTEN interaction regulates PI3K/AKT signaling strength in cells. HEK293 cells were co-transfected with GFP-PTEN and Prg2-Flag/Prg2-ΔC-Flag. To monitor subtle changes in PI3K/AKT activity in transfected cells only, the co-transfection of AKT-GFP was exploited as an effective and sensitive experimental protocol (An et al., 2015). In these experiments, intact PRG2, but not the C-terminal PRG2-truncated protein missing the poly-E-box (PRG2ΔC), antagonized PTEN’s ability to reduce pAKT (Figures 3A and 3B).
Figure 3.

PRG2 Regulates pAKT and PI(3,4,5)P3 in Dependence of PTEN
(A) PRG2 rescues decreased pAKT-levels induced by PTEN overexpression. HEK293 cells were triple transfected with AKT-GFP, GFP-PTEN (or control vector), and Prg2-Flag/Prg2ΔC-Flag (or control vector). Cell lysates were analyzed with indicated antibodies.
(B) Bars represent normalized pAKT(S473) over total AKT ratios across treatments ± SEM of n = 5 independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA with Bonferroni post hoc test).
(C) PRG2 rescues the loss of PI(3,4,5)P3-accumulations induced by PTEN overexpression. U87MG cells transfected with PH-AKT-mRFP (red) and GFP, GFP-PTENC124S, GFP-PTEN (green, insets), and Prg2-Flag (white, insets). Panels show single confocal sections. Arrowheads indicate PH-AKT foci at plasma membranes (PM). Scale bar: 10 μm.
(D) Cells were scored for the presence of peripheral PH-AKT foci. Bars show the means ± SD of two independent experiments performed in duplicate, n ≥ 50. ∗∗p < 0.01 (t test).
(E) PRG2 protects from the loss of peripheral F-actin accumulations induced by PTEN overexpression. Cells were stained with Phalloidin (F-actin, red top panels), anti-GFP (for detection of GFP or GFP-PTEN, green bottom panels) and anti-FLAG antibodies (for detection of PRG2-FLAG or PRG2ΔC-FLAG, white insets in lower magnification). Panels show maximum intensity confocal projections. Arrowheads indicate peripheral F-actin foci. Scale bar: 10 μm.
(F) Cells were scored for presence of peripheral F-actin foci. Bars show means ± SD of three independent experiments performed in duplicates, n ≥ 60. ∗∗p < 0.01 (t test).
In a direct approach to characterize possible regulation of plasma membrane PI(3,4,5)P3, by PRG2, we performed a pleckstrin homology (PH)-domain translocation assay in PTEN-deficient U87MG glioblastoma cells (Pavlovic et al., 2016). Under basal conditions, the PI(3,4,5)P3-binding AKT PH-domain was found to sporadically enrich at the plasma membrane in U87MG cells expressing control GFP or catalytically inactive GFP-PTENC124S. PH-domain membrane recruitment was significantly reduced after expression of catalytically active GFP-PTEN (Figure 3C). In contrast, co-expression of Prg2 with PTEN antagonized the PTEN effect on PH-AKT accumulation (Figures 3C and 3D). These results indicate that PRG2 is able to antagonize the PI(3,4,5)P3-hydrolyzing activity of PTEN in cells.
U87MG cells are highly migratory cells that extend dynamic F-actin-rich protrusions (Figure 3E). Given the established function of PI(3,4,5)P3 to drive spontaneous cytoskeletal and membrane dynamics, we examined the effect of the PRG2-PTEN interaction on the actin cytoskeletal network in U87MG cells. Control transfected cells maintain robust F-actin protrusions, which remain unaffected by expression of Prg2 (Figures 3E and 3F). As expected, PTEN expression resulted in a decreased percentage of cells exhibiting F-actin-enriched membrane protrusions, confirming the validity of this assay. Co-expression of full-length Prg2 with PTEN antagonized the PTEN-induced decrease in membrane protrusions, whereas the Prg2-deletion mutant missing the poly-E-box (PRG2ΔC) failed to rescue the PTEN effect (Figure 3F). Collectively, these results establish a causal link for PRG2 as a positive regulator of both membrane PI(3,4,5)P3 and F-actin, in dependency on PTEN. They further demonstrate the ability of PRG2 to protect PI(3,4,5)P3 from PTEN hydrolysis.
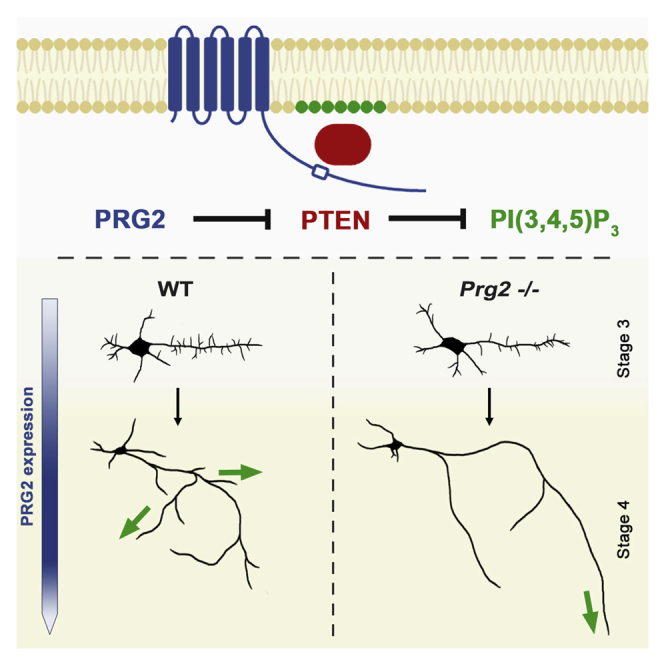
PRG2 Expression Correlates with Growth and Branching of Axons and Dendrites
Next, we determined the pattern of Prg2 expression at different stages in the developing rodent brain using a custom-made anti-PRG2 antibody. This antibody is specific for detecting PRG2 by western blotting and immunocytochemistry because reductions in Prg2 expression accurately reproduced the decrease in protein abundance in both experimental paradigms (Figures S2A and S2B). Analyses of the expression of PRG2 protein in rat brain lysates obtained at different developmental (E13–E19) and postnatal/adult stages (P1–W40) revealed high protein expression during stages corresponding broadly to cortical migration and branch formation of axon projections (E17–P1; Figure 4A). In contrast, the related PRG1 was first detected, albeit weakly, at P1, and its expression peaks at P15. Western blotting of different brain regions (cortex, hippocampus, cerebellum) with the anti-PRG2 antibody confirmed dynamic expression with high PRG2 levels found at young postnatal stages, whereas embryonic or late postnatal/adult stages show less PRG2 abundance (Figure S3A). Immunohistochemistry using the anti-PRG2 antibody on mouse brain sections identified expression of the protein in the cortical plate and in axonal tracts at E16.5 (Figure 4B). Cortical neurons cultured in vitro demonstrated a steep increase in the expression of PRG2 protein between DIV 3 and 5 in culture, which coincide with a general phase of complex neuronal branch behavior of axonal and dendritic processes (Figure 4C). Indeed, our direct analyses of axon morphogenesis in time identified that the steepest increase in axon branch growth occurs after DIV3 (Figure S3B). The control of the dynamic expression of Prg2 most likely occurs at the level of the protein because analyses of mRNA expression at different stages revealed high, relatively constant mRNA levels throughout DIV1–DIV6; in sharp contrast, Prg1 mRNA levels were initially low but increased significantly at later stages (Figure S3C).
Figure 4.

Profile of Developmental Expression in the Forebrain and Subcellular Distribution of PRG2 in Neurons
(A) Prg2 expression in vivo peaks at birth. Rat brain lysates isolated at different developmental stages were analyzed by WB using the indicated antibodies. E, embryonic day; P, postnatal day; W, weeks. Prg2 and Prg1 expression was quantified at three time points from five independent animals. Bars represent protein levels relative to tubulin ± SEM. ∗∗∗p < 0.001, n.s. = not significant (one-way ANOVA with Bonferroni post hoc test).
(B) Coronal sections through the mouse forebrain at E16.5 labeled with anti-L1 and anti-PRG2 antibodies show axonal localization of PRG2. Nuclei were visualized by Hoechst. Scale bar: 200 μm.
(C) Prg2 expression in primary neurons. PRG2 and PTEN levels in cortical neurons at different days in vitro (DIV) analyzed by WB.
(D) Subcellular distribution of PRG2 in hippocampal neurons cultured for 9 DIV and stained with anti-MAP2, anti-PRG2, and Phalloidin (F-actin). A single, confocal section at high magnification reveals punctate labeling of PRG2 at the membrane of axons. Scale bar: 2 μm. PRG2 membrane enrichment was measured as a percentage of processes with plasma-membrane-enriched PRG2 and the ratio of cell edge to middle intensity. Results are means ± SEM of 21 (axonal) and 27 (dendritic) measurements from 12 axonal and dendritic processes. ∗∗∗p < 0.001 (t test).
(E) Structured illumination microscopy (SIM) of PRG2 and F-actin in 9 DIV hippocampal neurons. Cells were stained with anti-PRG2 antibody and Phalloidin. Single sections show regular PRG2 puncta at the plasma membrane. Scale bar: 0.5 μm.
(F) SIM of 9 DIV hippocampal neurons treated with 5 μm latrunculin B for 1 h or 3 h and labeled with anti-Tau and anti-PRG2 antibodies. Scale bar: 1 μm. Bar graph demonstrates the increase in mean distance between two consecutive PRG2 spots following latrunculin B treatment. Results are means ± SEM from n = 9 neurons per condition, with ≥10 μm axon lengths per neuron analyzed. ∗∗∗p < 0.001 (one-way ANOVA with Bonferroni post hoc test).
See also Figures S2 and S3.
We then investigated the endogenous distribution of PRG2 in neurons and identified membrane localization of PRG2 in hippocampal neurons cultured for 9 days in vitro. The presence of prominent PRG2 puncta at the plasma membrane, predominantly in axons, was striking, whereas most dendrites showed intracellular PRG2 localization (Figures 4D and S7B). To further characterize the axonal PRG2 distribution, we imaged neurons using three-dimensional structured illumination microscopy (3D-SIM), a super-resolution imaging method that reaches a 2-fold increase in axial and lateral resolution compared with conventional confocal microscopy (Gustafsson, 2000, Schermelleh et al., 2010). Membrane-localized PRG2 adopted a regular pattern along the axon with a periodicity of approximately 300 nm (Figures 4E, 4F, and S7D–S7F for quantification). The periodic distribution of PRG2 does not correlate with F-actin ring-like structures that wrap around the circumference of axons (Xu et al., 2013), which are evenly spaced along the axon shaft approximately every 190 nm (Figure 4E). However, depolymerizing actin filaments altered the periodicity of PRG2 clusters, indicating that PRG2 nanoscale localization on the axonal plasma membrane is regulated by F-actin (Figures 4F and S2C–S2E). Interestingly, although both short (1 h) and long (3 h) treatment with latrunculin B resulted in broader distance distributions of axonal PRG2 puncta, only long-term treatment caused a net decrease in plasma membrane PRG2 (Figure S2E). Control experiments supported both treatments as preserving overall normal morphologies (Figure S2C). This, together with the results shown in Figure 3, suggests a reciprocal relationship for PRG2 and the F-actin cytoskeleton. In summary, these experiments establish PRG2 as a neuronal plasma membrane protein, which—through the interaction and inhibition of PTEN—may facilitate signaling processes that are confined to sub-micrometer-sized clusters at the axon cell membrane.
PRG2 Induces Filopodia and Branches in ESC-Derived Neurons
Several PRG membrane proteins have previously been shown to control neuronal growth responses, such as filopodia formation, as well as axon growth and guidance after CNS lesions (Bräuer et al., 2003, Fink et al., 2017, Savaskan et al., 2004, Velmans et al., 2013). When overexpressed, PRG family members are distributed to the plasma membrane and induce the formation of filopodia (Broggini et al., 2010, Velmans et al., 2013, Yu et al., 2015). Here, we confirmed this to be the case: overexpression of Prg2 in N1E-115 cells results in a strong induction of filopodia with a characteristic punctate distribution of PRG2 revealed by SIM (Figures S4A–S4C). Given the prominent localization of PRG2 in axons, we tested the possibility that PRG2 may cooperate with PTEN during the generation of axon filopodia in neurons. For this, we used embryonic stem cell (ESC)-derived motor neurons (ESCMNs), which can rapidly enter a robust axonal growth phase. Importantly, young ESCMNs do not express detectable Prg2 at these early phases of rapid growth (Figure 5A). We generated doxycycline-inducible cell lines expressing Prg2 after neural differentiation into embryoid bodies. We then re-plated doxycycline-induced (or non-induced) ESCMNs at low density and assessed neuronal morphologies after 2 h (or after 24 h for subsequent experiments when branching was analyzed) (Figure 5B). ESCMN-Prg2-Flag clones responded to induction of Prg2 expression with a significant increase in the number of filopodia (Figures 5C and 5D). Interestingly, filopodia induction was confined to the axon compartment and not seen around neuronal cell soma or growth cones (data not shown).
Figure 5.

Prg2 Overexpression in ES-Cell-Derived Neurons Induces Axon Filopodia Involving PI3K/PI(3,4,5)P3
(A) Induction of Prg2-Flag expression after doxycycline (DOX) application. Lysates of ES-cell-derived motor neurons (ESCMNs) treated with 2 μg/mL doxycycline for 24 h were analyzed using the indicated antibodies.
(B) ESCMN culture scheme.
(C) Images show representative examples of neurons stained with Phalloidin (F-actin) after Prg2 induction with or without the PI3K inhibitor LY294002 (15 μM, 2 h). Scale bar: 10 μm. Doxycycline-induced Prg2 expression results in an increase in the number of axon filopodia. The PRG2-induced effect is reversed by PI3K inhibition using LY294002.
(D) Quantification of filopodia. Bars show the mean number of filopodia per neuron, and the average length of filopodia/neuron ± SEM of 2–3 independent experiments performed in triplicates, n ≥ 100. **p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA with Bonferroni post hoc test).
(E) Prg2-induced ESCMNs cultured for 24 h in the presence or absence of LY294002 (15 μM). PRG2 increases the number of axons and axon branches in dependence of PI3K/PI(3,4,5)P3. Cells were stained with anti-tubulin antibody (green) and Phalloidin (red). Scale bar: 30 μm.
(F) Quantification of axons per cell and axonal branching. Bars show the mean number of branches ± SEM of two to three independent experiments performed in triplicates, n ≥ 120. ∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA with Bonferroni post hoc test).
See also Figure S4.
Filopodia are dynamic membrane extensions generated by bundles of F-actin that are essential for numerous cellular processes involved in the establishment of neuronal morphology (Heckman and Plummer, 2013). Axon filopodia are thought to initiate the formation of collateral branches and synaptic structures and arise from specialized cytoskeletal structures termed F-actin patches (Kalil and Dent, 2014, Lewis et al., 2013). Because PI(3,4,5)P3 has been shown to drive the formation of axonal F-actin patches, filopodia, and axon branches (Ketschek and Gallo, 2010, Spillane et al., 2012), we asked whether PRG2-induced axon filopodia are sensitive to PI3K/PI(3,4,5)P3 inhibition. To test that idea, we applied the PI3K inhibitor LY294002 immediately after re-plating the ESCMNs following Prg2 induction with doxycycline. Quantification of filopodia revealed a PI3K signaling dependency for the PRG2-induced number of filopodia (Figure 5D). Interestingly, Prg2 induction did not affect the average length of filopodia per neuron, suggesting that PRG2 is involved in initiating filopodia, rather than influencing the filopodia growth phase (Figures 5C and 5D).
We also tested the effect of induction of Prg2 expression in ESCMN-Prg2-Flag clones on axon branching (Figure 5E). After incubation of cells for 24 h, we found a significant increase in both the number of axons extending from a single neuronal soma as well as in the number of axon branches in cultures treated with doxycycline (Figures 5E and 5F). In the presence of the PI3K inhibitor, the axon phenotype was again reversed to that in untreated controls. Together, these results provide evidence for a requirement of PI(3,4,5)P3 in PRG2-induced axon filopodia formation and indicate PI(3,4,5)P3 as an essential signaling component of PRG2 function. Given the identified association of PTEN and PRG2 causing inhibition of the phosphatase, the protein interaction is likely involved in stabilizing phosphoinositides within axon membrane compartments required for filopodia and branch formation.
PRG2 Redistributes Growth by Controlling Axon Branching Morphogenesis
To directly examine the role of PRG2 in axon morphogenesis and branching, we performed in vitro experiments using neurons from wild-type (WT) and Prg2 knockout (Prg2−/−) mice. Prg2−/− were established through CRISPR/Cas9-mediated excision of the first exon in the Prg2 gene (Figures 6A and S5). Prg2−/− mice were backcrossed to wild-type Bl6/N for several generations. As a result of homozygosity for the null allele, the PRG2 protein was not detected by western blotting (Figure 6A). Hippocampal neurons of WT and Prg2−/− mice were dissociated, and axon morphology was analyzed at different developmental stages. In the first set of experiments, after in vitro culture for 1 day, neurons had clearly adopted early polarized (stage 3) morphologies. Quantification revealed that PRG2 loss did not affect early neuronal growth and polarization (Figures S6A–S6C) but led to significant decreases in the density of axon filopodia compared with controls, again without affecting filopodia length (Figures 6B and 6C).
Figure 6.

PRG2 Is Required for Filopodia and Branch Formation in Axons
(A) Validation of Prg2 knockout (Prg2−/−) mouse by WB using P8 brain lysates.
(B) Hippocampal neurons from WT or Prg2−/− mice imaged at 1.5 DIV. Scale bar: 20 μm.
(C) Bars show the mean number of filopodia per axon length, the mean length of filopodia, and the mean length of the longest filopodium/neuron ± SEM of four independent experiments, n ≥ 100. ∗∗∗p < 0.001 (t test).
(D) Hippocampal neurons from WT or Prg2−/− mice imaged at 5 DIV. Scale bar: 50 μm.
(E) Bar graphs show mean ± SEM of 3 independent experiments, n ≥ 100. ∗p < 0.05, ∗∗∗p < 0.001 (t test).
(F) Schematic representation of Prg2−/− neuronal growth at two developmental stages.
(G) Correlation of branches and total axon length. Transparent dots represent individual neurons of either WT (blue) or Prg2−/− (red); darker dots represent means for equal clusters of neurons to reduce the complexity of the dataset.
(H) Representative filopodium emerging from a local PRG2 spot. Panels show F-actin probe GFP-Utrophin and Halo-tagged PRG2 over 40 s in 16-color Look-Up Table (LUT; FIJI), scale bar: 2 μm.
(I) Boxplots of nearest-neighbor analyses show smaller distances of emerging filopodia to PRG2 clusters when compared with randomly distributed clusters. Whiskers show the 5%–95% interval, ∗∗∗p < 0.001 (Wilcoxon signed rank test), n = 73 from four independent experiments.
See also Figures S5 and S6.
In a second set of experiments, we cultured neurons for 5 days to developmental stages when they adopt more complex, branched morphologies. Those experiments showed that loss of PRG2 significantly reduced overall axon branching compared to controls (Figures 6D and 6E), whereas dendrite morphology was unaffected (Figure S6D). To exclude the possibility that this phenotype was a consequence of a general impairment in neuronal growth, we quantified axon length, as well as the length of the primary (longest) axonal process in each genotype. This analysis found that PRG2-deficient neurons showed no significant change in overall length of axonal processes in individual cells but exhibited significant increases in the length of primary axons (Figure 6E). This suggests that, through the ability to control axon branching, PRG2 may direct cell growth toward primary or collateral axons, respectively. This indeed seems to be the case: although both WT and Prg2−/− neurons demonstrate a high degree of correlation between overall axon length and number of branches, Prg2−/− neurons show a decreased probability of branching with increased axon length (Figures 6F and 6G).
To address the specific regulatory role of PRG2 during formation of filopodia in neuronal axons, a core process of axon branch formation, we exploited live cell imaging of tagged PRG2 in Prg2−/− neurons. We expressed Halo-Prg2 in combination with the F-actin probe GFP-Utrophin and noticed a consistent co-localization (73%) of newly extending filopodia with their described precursors, F-actin patches. Interestingly, Halo-PRG2 showed a punctate pattern along the axon (Figure 6H). Although PRG2 enrichment did not predict a site of de novo filopodia formation, per se, 55% of filopodia emerged directly from PRG2 clusters and 33% within the vicinity of PRG2 clusters. To exclude the possibility that filopodia emerging from PRG2 clusters as random events, we developed a semi-automatic nearest-neighbor analysis of emerging filopodia. Filopodia were found to form preferentially in proximity (<1 μm) to intensity maxima of PRG2 when compared with the same number of randomly distributed spots (Figures 6I and S6E). Our combined results identify that PRG2 clusters are able to locally induce filopodia to facilitate axon branch formation.
The Effect of PRG2 on Axon Branching Morphogenesis Involves PTEN
In the final set of experiments, we tested whether the PRG2 effect on branching involves the function of PTEN. We used Pten small hairpin (shRNA) virus particles to test epistasis between PRG2 and PTEN in Prg2−/− neurons cultured for 5 days (Figures 7A and 7B). PTEN inhibition did not significantly alter overall axon length in either WT or Prg2−/− neurons (Figure 7C). However, knockdown of Pten reversed the increase in primary axon length and also corrected the deficit in axon branch number induced by Prg2−/− (Figure 7C), suggesting that PTEN indeed cooperates with PRG2 during axon branching. To test that idea further, we compared the ability of full-length PRG2 to restore branching in Prg2−/− neurons, with that of the PRG2 tail deletion (PRG2ΔC) that is unable to inhibit PTEN (Figure 7D). Although full-length PRG2 rescued branching in Prg2−/− neurons, the PRG2ΔC mutant failed to do so (Figures 7E and 7F), which indicates that PRG2’s involvement during axon branching depends on its ability to antagonize PTEN activity. Given its specific upregulation at developmental stages coinciding with branching morphogenesis, we propose that PRG2 functions as an efficient internal “off” switch for the PTEN-mediated hydrolysis of PI(3,4,5)P3 essential for establishing and segregating membrane phosphoinositides required for axon branching in hippocampal neurons.
Figure 7.

PTEN Cooperates with PRG2 during Branch Formation
(A) Scheme of experiments.
(B) Validation of shRNA specificity to target Pten.
(C) Knockdown of Pten rescues branching deficit of Prg2−/− neurons at 5 DIV. Bar graphs show mean total axon length, mean primary axon length, and mean branches per axon length ± SEM of five independent experiments, n ≥ 130. ∗p < 0.05, ∗∗∗p < 0.001 (one-way ANOVA with Bonferroni post hoc test).
(D) Validation of viral expression of Prg2-Flag or Prg2ΔC-Flag by WB.
(E) Hippocampal Prg2−/− neurons expressing Prg2-Flag or Prg2ΔC-Flag imaged at 5 DIV. Scale bar: 50 μm.
(F) PRG2-FLAG (but not PRG2ΔC-FLAG) increases branch density in Prg2−/− neurons. Bars show mean total axon length and mean branches per axon length ± SEM of 88–97 cells per condition from four independent cultures. ∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA with Bonferroni post hoc test).
Discussion
Here, we uncover a role for the membrane protein PRG2 in axon branch growth, via the regulation of PTEN localization and activity. We identify PI(3,4,5)P3 as a critical signaling molecule for PRG2 function and establish its importance in mediating PRG2 effects on actin cytoskeleton organization and filopodia generation. Our results indicate that PRG2 is temporally and locally poised to inhibit global growth suppression mediated by PTEN. In principle, PRG2 control of PTEN allows the introduction of a PI(3,4,5)P3 amplification system that may instruct cytoskeletal organization in controlling neuronal growth behavior.
Spatiotemporal Control of PI(3,4,5)P3 by PRG2
Recent studies suggest a common mechanistic step in initiating nanoscale membrane protrusions in close apposition with F-actin accumulations as a filopodia initiation mechanism in both axons and dendrites (Hou et al., 2015, Ketschek and Gallo, 2010, Saarikangas et al., 2015). These filopodia-initiating steps have been shown to require the presence of local PI(4,5)P2 and/or PI3K products PI(3,4,5)P3 and PI(3,4)P2 at plasma membranes, to recruit and regulate proteins involved in sensing membrane curvature and/or orchestrating F-actin nucleation and assembly (Ketschek and Gallo, 2010, Saarikangas et al., 2015, Zhang et al., 2017). It is still unclear whether a mechanism exists to mark “competent” plasma-membrane micro-domains for initiation of these early events. Moreover, whether membrane proteins per se are involved in this activity has yet to be elucidated.
To date, only a few membrane proteins, including members of the PRG family, have been linked to filopodia and neurite or branch formation in neurons and other cells (Broggini et al., 2010, Coiro et al., 2014, Liu et al., 2016, Sigal et al., 2007, Velmans et al., 2013, Yu et al., 2015). At least one member of the family, PRG3 (LPPR1), has been shown to promote axonal growth after injury (Broggini et al., 2016, Fink et al., 2017), suggesting that PRGs—in general—may boost intrinsic growth capacity in neurons. In this study, we present evidence for additional functions of PRGs in orchestrating axon branching during development. We demonstrate that an axonal membrane complex consisting of the transmembrane protein PRG2 and PTEN organizes the complex branching behavior of neurons. In this way, gain-of-function and loss-of-function of PRG2 results in increases and decreases, respectively, in axon filopodia and branch numbers in neurons. This mechanism likely involves local accumulations of PRG2 that instruct emerging filopodia to form branches and the ability to antagonize PTEN. In molecular terms, PRG2 recruits PTEN and inhibits its PI(3,4,5)P3 phosphatase activity, thus resulting in local stabilization of PI(3,4,5)P3 at the plasma membrane. Unlike upstream activation of PI3K by growth factors, disinhibition of PTEN would suggest milder phenotypes in downstream signaling. This model implies the presence of altered dynamics and/or lower abundance of microscale PI(3,4,5)P3 domains along the axonal membrane as a result of the Prg2 deletion. We have extensively analyzed PI(3,4,5)P3 domains along the axonal membrane with PH-domain probes and anti-PI(3,4,5)P3 antibodies, combined with pharmacological or genetic approaches (J.F., G.L., and B.J.E., unpublished data). However, the low sensitivity and low dynamic range afforded by these approaches, together with the inherent difficulties associated with small-diameter axons in hippocampal neurons and the stabilization of PI(3,4,5)P3 by high-affinity PH domain probes (Yip et al., 2008) have precluded us from quantifying reliable and consistent changes in PI(3,4,5)P3-rich domains in PRG2-deficient axons. Nevertheless, the fact that PRG2-dependent alterations in the filopodia and branching capacity of neurons are reversed by correcting PI(3,4,5)P3 levels via PI3K or PTEN activity provides a strong argument for PI(3,4,5)P3 as a critical signaling component of PRG2 function along the neuronal axon.
PRG2 Regulates Axonal F-Actin Cytoskeleton and Induces Filopodia
Integration of neurons into functional circuits requires both robust formation as well as flexible remodeling of their morphology. This complex regulation involves multiple pathways, including phosphoinositides, calcium and Rho-family GTPases (reviewed in Gallo, 2011, Kalil and Dent, 2014) to instruct the formation of dynamic F-actin-rich structures that are commonly stabilized by microtubule to mature into specialized compartments, such as branches (Dogterom and Koenderink, 2019). Our studies show that, in addition to regulating PTEN localization and activity, PRG2 is highly likely to impose additional and complementary mechanisms for controlling local F-actin remodeling along the axon to regulate branching. First, PRG2 exhibits a striking localization pattern of sub-micrometer size clusters along the axonal membrane. Its periodic localization, revealed by super-resolution is controlled by axonal F-actin, as suggested by latrunculin experiments. Second, PRG2 has recently been found to interact with, and to activate, radixin (Cheng et al., 2016). Radixin, an ERM family protein, crosslinks F-actin to the plasma membrane and has been shown to stabilize membrane protrusions in neuronal growth cones (Menon and Gupton, 2016). Although the PRG2-radixin interaction was studied in the context of lysophosphatidic acid (LPA)-induced growth cone guidance in thalamocortical neurons (Cheng et al., 2016), it is possible that a similar mechanism may operate along the axon shaft. Indeed, our live cell imaging identified F-actin rearrangements—specifically during the formation of filopodia—to occur preferentially in the vicinity of PRG2 clusters along the axon.
PRGs Partake in Dynamic Axonal Scaffolds
An additional mode of functioning for PRGs is suggested by their ability to form multimeric complexes (Yu et al., 2015). We have verified the existence of heteromeric PRG2 complexes at the plasma membrane. It is highly likely that PRG2-PTEN complexes that include different PRGs exert non-redundant functions in neuronal axon growth at distinct developmental stages. Several PRGs exhibit partially overlapping expression patterns in the developing brain (Wu et al., 2009). Notably, PRG3, but not PRG5 or PRG1, is enriched in developing axons (Velmans et al., 2013) and has been proposed to promote axonal growth and sprouting after injury (Broggini et al., 2016, Fink et al., 2017). Thus, although we have extensively characterized the effects of PRG2 loss in developing neurons in vitro, it is likely that the robust regulation of neuronal branching, as well as redundancy within the PRG family, will dampen gross differences in neuronal morphology of Prg2−/− mice in vivo.
Local Inhibition of Global PTEN
In neurons, PTEN appears to be active and dynamically interacting with the plasma membrane in a constitutive manner (Vazquez et al., 2006), contributing as gatekeeper to limiting basal levels of PI(3,4,5)P3 and thus also preventing aberrant formation of branches during early development (Kreis et al., 2014, Song et al., 2012). In addition to constitutive dynamic interactions with the plasma membrane, PTEN can be actively recruited by membrane PDZ-domain proteins (Valiente et al., 2005) or other transmembrane proteins and receptors (Kreis et al., 2014, Lee et al., 2018). In most cases, these enhanced membrane interactions of PTEN appear to prime PTEN activity toward PI(3,4,5)P3 and, thus, to antagonize PI3K signaling in a spatial manner (Lee et al., 2018). Nevertheless, mechanisms that inhibit PTEN are thought necessary to control against silencing of PI(3,4,5)P3 by abundant unrestricted access of PTEN to the plasma membrane (Kreis et al., 2014).
This would be particularly relevant when steep gradients of PI(3,4,5)P3/PI(3,4)P2 are needed along the plasma membrane for initiation of neurites or filopodia/branch outgrowth. In axons specifically, PTEN is abundant and active during both embryonic development and in the adult (Chadborn et al., 2006, Christie et al., 2010), and local inhibition counteracts its growth-restricting function (Drinjakovic et al., 2010, Zhang et al., 2013). In this way, a developmentally regulated, membrane-associated PRG2-PTEN complex at the axonal membrane provides an efficient intrinsic factor that is interposed to restrict PTEN activity in axons. Consistent with this, the primary phenotype of our PRG2 gain-of-function and loss-of-function experiments was restricted to axon filopodia and branches with virtually absent effects in dendrites.
This control mechanism likely operates in concert with extrinsic factors, including growth factors, such as brain-derived neurotrophic factors (BDNFs) and nerve growth factors (NGFs) that activate upstream PI3K signaling (Ketschek and Gallo, 2010), thus creating PI(3,4,5)P3 “hotspots” along the axonal membrane that are, in turn, stabilized or even enhanced by local PRG2-mediated PTEN inhibition. These PI(3,4,5)P3 hotspots are instrumental for controlling local F-actin cytoskeleton reorganization (Kakumoto and Nakata, 2013), via recruitment of small GTPases of the Rho family and F-actin regulators (Ketschek and Gallo, 2010, Spillane and Gallo, 2014), toward the formation of filopodia and branches (Armijo-Weingart and Gallo, 2017). This way, the mechanism we describe adds an additional layer of precise spatial and temporal control to axon morphogenesis.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti PRG2, custom made, anti: CAESYYRRMQARRYQD | Eurogentec | N/A |
| rabbit anti-PRG1 | R&D Systems | Cat#MAB2874; RRID:AB_2138597 |
| Rabbit anti-PTEN | Cell signaling | Cat#9559; RRID:AB_390810 |
| rabbit anti-AKT | Cell signaling | Cat#9272; RRID:AB_329827 |
| rabbit anti-pSer473AKT | Cell signaling | Cat#4060; RRID:AB_2315049 |
| Goat anti-PTEN (N19) | Santa Cruz Biotech | Cat#sc-6818; RRID:AB_654895 |
| mouse anti human CD14 (clone 26ic) | ATCC | Cat#HB-246; RRID:AB_2782995 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| di-C8-PI(3,4,5)P3 | Echelon Biosciences | Cat#P-3908 |
| di-C8-PI(4,5)P2 | Echelon Biosciences | Cat#P-4508 |
| PTEN (GeneBank: NM_000314) Human Recombinant Protein | OriGene | Cat#TP302627 |
| Latrunculin B | Millipore | Cat#42820; CAS: 76343-94-7 |
| LY294002 | Millipore | Cat#440202; CAS: 154447-36-6 |
| Janelia Fluor 646 HaloTag ligand | Promega | Cat#GA1120 |
| Human recombinant BDNF | R&D systems | Cat#248-BD |
| Retinoic acid | Sigma | Cat#R2625; CAS 302-79-4 |
| Smoothened agonist (SAG) | Merck | Cat#566660; CAS: 364590-63-6 |
| Doxycycline (DOX) | Sigma | Cat#D9891; CAS: 564-25-0 |
| G418 | Invitrogen | Cat#10131027; CAS: 49863-47-0 |
| Amphipol (A8-35) | Anatrace | Cat#A835 |
| Fos-Choline-14 | Anatrace | Cat#F312 |
| Critical Commercial Assays | ||
| MEGAshortscript T7-Kit | Life Tech. | Cat#AM1354 |
| mMessage mMachine Kit | Life Tech. | Cat#AM1344 |
| MEGAclear Kit | Life Tech. | Cat#AM1908 |
| Monolith Protein Labeling Kit RED-NHS | NanoTemper Technologies | Cat#MO-L001 |
| Malachite green reagent | Echelon Biosciences | Cat#K-1500 |
| Experimental Models: Cell Lines | ||
| HEK293TN | BioCAT/SBI | Cat#LV900A-1; RRID:CVCL_UL49 |
| HEK293S GnTI- | ATCC | Cat#CRL-3022; RRID:CVCL_A785 |
| COS-7 | ATCC | Cat#CRL-1651; RRID:CVCL_0224 |
| U87MG | ATCC | Cat#HTB-14; RRID:CVCL_0022 |
| N1E-115 | ATCC | Cat#CRL-2263; RRID:CVCL_0451 |
| Mouse embryonic fibroblasts, Neomycin-Resistant, Strain FVB, passage 3 | Millipore | Cat#PMEF-NL |
| A2lox mouse ESC clone H14IG#E3 | Derivative of E14Tg2a (Machado et al., 2014) | N/A |
| A2lox mouse ESC H14IG#E3_PRG2-Flag_clone #B3 | This paper | N/A |
| A2lox mouse ESC H14IG#E3_PRG2-Flag_clone #B4 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Prg2−/−: C57 Bl/6NCrl Prg2−/− | This paper | N/A |
| Oligonucleotides | ||
| sgRNA1: GTGGCCTGGTTCCGCGGTGC | This paper | N/A |
| sgRNA2: GCCGCGGGCACGGAAGGCGT | This paper | N/A |
| shRNA PTEN: CGACTTAGACTTGACCTATAT | Shi et al., 2014 | N/A |
| shRNA scrambled: CAACAAGATGAAGAGCACCAA | Shi et al., 2014 | N/A |
| Recombinant DNA | ||
| pCAGGS-PRG2-Flag | This paper | N/A |
| pCAGGS-PRG2ΔC-Flag | This paper | N/A |
| pCAX-GFP-PTEN | An et al., 2015, Kreis et al., 2013 | N/A |
| pCAX-GFP-PTENC124S | An et al., 2015, Kreis et al., 2013 | N/A |
| mRFP-PH-Akt | Idevall-Hagren et al., 2012 | N/A |
| pCMV-GFP-Akt | An et al., 2015, Kreis et al., 2013 | N/A |
| p2Lox-PRG2-Flag | This paper | N/A |
| pX330 | Cong et al., 2013 | Addgene plasmid #42230 |
| pCA-Halo-PRG2 | This paper | N/A |
| GFP-Utrophin(CH) | Burkel et al., 2007 | Addgene plasmid #26737 |
| f(syn)-NLS-RFP_(U6)shPTEN | This paper | N/A |
| f(syn)-NLS-RFP_(U6)shCTRL | This paper | N/A |
| f(CAG)-PRG2-Flag-w | This paper | N/A |
| f(CAG)-PRG2ΔC-Flag-w | This paper | N/A |
| Software and Algorithms | ||
| FIJI (ImageJ 1.51n) | Schindelin et al., 2012 | https://imagej.net/Fiji |
| GraphPad Prism 5 | GraphPad Software, La Jolla, USA | https://www.graphpad.com |
| NeuronJ (version 1.4.3) | Meijering et al., 2004 | https://imagescience.org/meijering/software/neuronj/ |
| Imaris x64 (version 8.1.2) | Bitplane AG | https://imaris.oxinst.com |
| MO.Affinity Analysis v2.3 | NanoTemper Technologies | https://nanotempertech.com/ |
| Timelapse nearest neighbor analysis | This paper | https://github.com/ngimber/NearestNeighborWorkflow_Filopodia |
| Other | ||
| Monolith NT. 115 Series Standard Treated Capillaries | NanoTemper Technologies | Cat#MO-K002 |
| High-precision #1.5 coverslips | Carl Roth | Cat#LH24.1 |
| Vectashield | Vector Laboratories | Cat#H1000 |
| 100 nm fluorescent beads (Tetraspeck) | Thermo Fischer Scientific | Cat#T7284 |
Lead Contact and Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Britta Eickholt (britta.eickholt@charite.de).
Experimental Model and Subject Details
Animal procedures
All animal procedures were conducted in accordance with local ethical guidelines and approved animal care protocols. Experiments were carried out according to the institutional guidelines of the animal welfare of the Charité Universitätsmedizin Berlin, under the licenses T0347/11 of the Landesamt for Gesundheit and Soziales Berlin (LaGeSo). All mice were maintained in a C57 Bl6/NCrl background and kept under standard conditions in a 12 hours day-night-cycle with water and food available ad libitum. Experiments were performed with animals of both sexes.
Cell lines
HEK293TN (female), HEK293S GnTI- (female), COS-7 (male), U87MG (male) and N1E-115 cells (male) were purchased from ATCC and cultured in DMEM-high glucose with GlutaMAX, 10% FCS, and 1% penicillin/streptomycin (Invitrogen) at 5% CO2 and 37°C. A2lox mouse ESC clone H14IG#E3 (Machado et al., 2014) was cultured on mitomycin-C-treated mouse embryonic fibroblasts (PMEFNL, Millipore) in knockout DMEM medium (Invitrogen) supplemented with 15% FBS (Hyclone), 1% non-essential amino acids (NEAA; Invitrogen), 2 mM L-glutamine (Invitrogen), 10 nM PD173074 (FGF/VEGF receptor tyrosine kinase inhibitor; Tocris Scientific), 0.1 mM 2-mercaptoethanol (Sigma), 1:500 dilution of LIF supernatant, 1% EmbryoMax ESCell Qualified Nucleosides (Millipore), 5 μg/ml plasmocin (Invivogen) and 1% penicillin/streptomycin (Invitrogen).
Method Details
CRISPR/Cas9-mediated generation of Prg2 −/− mice
Prg2 −/− were generated using CRISPR/Cas9 mediated non-homologous end-joining (NHEJ). Six sgRNAs targeting non-coding regions 5′ and 3′ of exon 1 were designed and cloned into the plasmid pX330 (a gift from Feng Zhang (Cong et al., 2013), Addgene plasmid #42230). Efficiency of the sgRNAs was tested in vitro using a plasmid containing overlapping parts of eGFP cDNA (Mashiko et al., 2013) spaced by the genomic target region of Prg2. Efficient sgRNA targeting induces homologous recombination of GFP cDNA and was monitored by fluorescence microscopy and western blot. The most efficient sgRNAs with the least predicted off-targets (determined by Tools from Sanger (https://www.sanger.ac.uk/htgt/wge/) and Feng Zhang’s lab (http://zlab.bio/guide-design-resources) were used in further experiments. In vitro transcribed sgRNAs (MEGAshortscript T7-Kit, AM1354, Life Tech.) and Cas9 mRNA (mMessage mMachine Kit, AM1344, Life Tech.) were purified (MEGAclear Kit, AM1908, Life Tech.) and subjected to CRISPR/Cas9 gene editing in single-cell zygotes by the Transgenic Technology Core Unit of the Charité – Universitätsmedizin Berlin, Germany. Offspring were genotyped by PCR and sequenced using primers 5′ and 3′ of exon 1 outside the sgRNA PAM sequence (Figure S5). NHEJ resulted in a deletion of exon 1 that led to loss of Prg2 expression. Prg2 −/− mice were backcrossed to WT Bl6/N for several generations.
Generation of PRG2 ESCMNs
Doxycycline-inducible Prg2-Flag expressing-motor neurons were established from a transgenic A2lox mouse ESC line (clone H14IG#E3), which allows insertion of doxycycline-inducible genes by Cre/loxP-mediated cassette exchange. This parental ESC line also contains a motor neuron-specific reporter transgene (Hb9::CD14-IRES-GFP transgene) (Machado et al., 2014). Prg2-Flag was subcloned into a p2Lox plasmid and after electroporation of parental ESC cells, positive clones were grown on G418-resistant mitomycin C-treated mouse embryonic fibroblasts and selected by 350 μg/ml G418 (Invitrogen). Prg2-Flag ESCs were differentiated in embryoid body cultures for 5 days, and motor neuron identity was induced by exposure to retinoic acid (1 μM) and smoothened agonist (SAG, 0.5 μM) between day 3 and day 5, as described previously (Machado et al., 2014).
Cells were dissociated after induction of Prg2-Flag expression by 2 μg/mL doxycycline during the last day of differentiation. Motor neurons were isolated from mixed cultures by magnetically activated cell sorting (MACS) using a mouse antibody (clone 26ic, ATCC) raised against human CD14 (Adolf et al., 2016, Machado et al., 2014). Prg2-Flag motor neurons were plated on poly-lysine/laminin-coated coverslips at a density of 12,000 cells/cm2 for morphometric analyses and 100,000 cells/cm2 for biochemical experiments and incubated for indicated periods in the presence of 2 μg/mL doxycycline. As a control, motor neurons were isolated and cultured in parallel, but in the absence of doxycycline. Two representative Prg2-Flag ESC clones (H14IG#E3B3 and H14IG#E3B4) were used for axon filopodia/branch measurements and results were pooled.
Brain tissue and neuronal culture
Whole rat brain tissue as well as tissue of individual brain regions were isolated at different ages from male and female Wistar rats and lysed in RIPA buffer (Sigma-Aldrich) with protease and phosphatase inhibitors (Schrötter et al., 2016). Primary cortical or hippocampal neurons were dissected from male and female embryonic day 16.5 WT C57 BL/6NCrl or Prg2 −/− mice as described previously (van Diepen et al., 2009, Kreis et al., 2013). Briefly, cortices and hippocampi were isolated, extracellular matrix was degraded for 15 min with 10% Trypsin in HBSS (Life Technologies), washed with HBSS and neurons were triturated to single cells with glass pipets. Neurons were plated on poly-ornithine(15 μg/ml)-coated or poly-ornithine/laminin(20 μg/ml)-coated coverslips and cultured in Neurobasal A medium (Life Technologies) containing 2% B27 (Life Technologies), 1% penicillin/streptomycin (Life Technologies), 100 μM β-mercaptoethanol (Applichem) and 1% GlutaMAX (Life Technologies). For morphometric analyses, neurons were plated at a density of 15,000/cm2. For biochemical experiments, neurons were plated at a density of 80,000 cells/cm2. Neurons were transfected using a modified Lipofectamine or a modified calcium phosphate protocol (Vadhvani et al., 2013) as described previously. Briefly, for lipofection 1-1.5 μg of DNA with 1μl Lipofectamine 2000 per well (12-well-plate or 4-well Ibidi dish) were diluted in OptiMEM and incubated for 10min at 37°C. The cell culture medium was collected, replaced by transfection mix, and the cells were incubated at 37°C 5% CO2 for 20 min. After removal of the transfection mix the cells were grown in their conditioned medium for 2-3 days. For calcium phosphate transfection, 4 μg plasmid DNA per well (12-well-plate) were mixed with 250 mM CaCl2, then gently taken up in an equal volume of 2xBBS buffer (50 mM BES, 280 mM NaCl, 1.5 mM Na2HPO4, pH 7.26) and finally incubated in Neurobasal A medium with B27 and Glutamax for 15 min at room temperature. This transfection mix replaced the medium of primary neurons for 45 min and was washed off with washing buffer (135 mM NaCl, 4 mM KCl, 1 mM Na2HPO4, 2 mM CaCl2, 1 mM MgCl2, 20 mM HEPES, 20 mM d-Glucose, pH 7.3) before the neurons were put into their previous culture medium until further processing.
For viral transductions of cultured neurons, lentiviral vectors expressing nuclear localized RFP controlled by the human synapsin-1 promotor, and the appropriate shRNA controlled by the U6 promoter, were used. Lentiviral particles were produced by the viral core facility of the Charité – Universitätsmedizin Berlin, Germany (https://vcf.charite.de/en/).
Transfection of cell lines
Cell lines were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions for plasmid DNA or siRNA. For mRFP-PH-AKT translocation and F-actin foci assays, U87MG cells were plated on coverslips in 24-well plates (10,000/cm2) and co-transfected with the appropriate plasmids. For antibody test, increasing amounts of siPrg2 (Thermo Fisher Scientific, ON TARGET plus SMARTpool Prg2: GAAGCGAGCCAGCGUGGAU, CCAGGCAGCUUAUCGGUGA, UGUCUACGUGUCGAUGUA, CCUCAAUCAUGGUCGGCGA) or ON-Target plus Non-Targeting pool (D-001810-10) was cotransfected with GFP or Prg2-Flag in HEK293 cells. For colocalization studies of PRGs or SIM of PRG2, N1E cells were plated on 12-well coverslips or high-precision #1.5 coverslips (10,000 cells/cm2), for co-immunoprecipitations, cells were plated in 6-well plates (300,000 cells/cm2). Transfections were performed using Lipofectamine 2000 according to the manufacturer’s protocol. In all experiments, the total amount of plasmid DNA was normalized with empty pcDNA or pCAGGS plasmids. Cells were cultured for 2 days after transfection and fixed with 4% PFA/ 4% sucrose in PBS.
Antibodies
A rabbit polyclonal antiserum was raised against a cytosolic C-terminal PRG2 peptide (CAESYYRRMQARRYQD; Eurogentec). Antibodies were affinity-purified by Sulfolink column chromatography (Thermo Scientific). A rabbit antibody against PRG2 (AP5732c) from Abgent was also used in some experiments. The rabbit anti-PRG1 antibody was from R&D Systems (#MAB2874). Rabbit anti-PTEN (#9559), rabbit anti-AKT (#9272) and rabbit anti-pSer473AKT (#4060) were from Cell Signaling. Goat anti-PTEN (N19) was from Santa Cruz Biotechnology. Mouse anti-Flag antibody (F1804), mouse anti-MAP2 (M9942), mouse anti-α-tubulin (T6199) and anti-Flag Affinity Gel (A2220) was from Sigma. Chicken anti-GFP antibody (ab13970) was from Abcam, the mouse anti-Tau1 (MAB3420) and mouse anti-GAPDH (CB1001) from Millipore. Rabbit anti-Tubulin β-3 (Tuj1) (PRB-435P) was from Covance. HRP-conjugated anti-rabbit and anti-mouse secondary antibodies (#PI1000, #PI2000) were from Vector Labs and HRP-conjugated anti-chicken antibodies (G135A) from Promega. All fluorophore conjugated secondary antibodies were purchased from Jackson lab.
DNA constructs and plasmids
Prg2 cDNA (obtained from ABgene Surrey, UK; clone ID 6808849, GenBank: BC066006) was amplified by PCR using the primers P1: 5′- AAGCTTgtcaccATGCTTGCTATGAAGGAGAA-3′ and P2: 5′-tggttaactcgagGTCGACGTCCTGGTACCTC-3′, introducing a HindIII site and a Kozak sequence at the 5′ and a SalI site at the 3′ of Prg2 cDNA, eliminating the stop codon. 3xFlag sequence was amplified by PCR from a PTEN-3xFlag plasmid (Kreis et al., 2013) using the primers P3: 5′-GAGGTACCAGGACGTCGACctcgagttaacca-3′ and P4: 5′-ACCGGTTTAggtacccttgtcatcgtca-3′. The P4 primer introduced an in-frame stop codon followed by an AgeI site. The amplified Prg2-Flag sequence was cloned into a pGEM vector and subcloned into pCAGGS (provided by I. Lieberam, King’s College London) using an SphI/NotI fragment; subcloning to HindIII/XmaI-digested p2Lox plasmid for generation of ESC clones (Machado et al., 2014) used a HindIII/AgeI fragment. The Prg2ΔC- sequence (1-1221 bp corresponding to the first 407 amino acids of PRG2) was amplified similarly using P1 and P2′ primers: 5′-tggttaactcgagGTCGACACCGATAAGCT-3′ and subcloned as a HindIII/SalI fragment in-frame into the pGEM- Prg2-Flag plasmid generating pGEM- Prg2ΔC-Flag and further subcloned int pCAGGS-plasmid generating pCAGGS-Prg2ΔC-Flag. Prg2-YFP plasmid was constructed using P1 and P2yfp primers (P2yfp: 5′-tggttaactcgagGTCGACTTGTCCTGGTACCTC-3′) for amplification of Prg2 cDNA and cloning of a HindIII/SalI fragment in frame to pEYFP-N1 vector. For viral infection under CAG promoter, Prg2-Flag and Prg2ΔC-Flag fragments were PCR-amplified (P5: 5′-GCTAGCgtcaccATGCTTGCTATG-3′ and P6: 5′-AAGGCGCGCCTTACTTGTCATCGTCATCC-3′) introducing NheI and AscI restriction sites into the modified FUGW lentiviral shuttle vector comprising a CAG promoter. For live-cell imaging, Prg2 was tagged N-terminally with Halo amplified from pFC14A HaloTag® CMV Flexi® Vector (Promega, #G9651) using P7 (5′-gagCTAGCgccaccatggggtccgaaatcg-3′) and P8: (5′-CATggatccgccgccaccagacccacctccgccggagccaccgccggaaccggaaatctc-3′) primers and subcloned using NheI/BamHI into a pCAGGS plasmid. Prg2 harboring a C-terminal Stop codon was amplified with primers P9: 5′-GAGCTGTACAAGtccggcggtggctccggcggaggtgggtctggtggcggcggatccATGCTTGCTATG-3′ and P10: 5′-gtcgcggccgctTTAGTCCTGGTACCTC-3′ and subcloned as a BamHI/NotI-fragment into pCAGGS-Halo. GFP-UtrCH was a gift from William Bement (Addgene plasmid # 26737). Prg1-GFP, Prg3-GFP, Prg4-GFP and Prg5-GFP were kindly provided by Antje Bräuer (Carl von Ossietzky University Oldenburg).
pCAX plasmids expressing GFP-PTEN wild-type, the phosphatase-inactive PTENC124S (CS) mutant and pCMV-GFP-AKT plasmids were described previously (An et al., 2015, Kreis et al., 2013). mRFP-PHAKT (Idevall-Hagren et al., 2012) was a gift from P. De Camilli (Yale School of Medicine, New Haven, CT, USA), F-GFP plasmid expressing a farnesylated version of GFP was provided by Kai Murk (Charité-Universitätsmedizin Berlin, Germany) and GFP-SHIP2 was provided by Nick Leslie (Heriot-Watt University, Edinburgh, Scotland). GFP-UtrCH was a gift from William Bement (Addgene plasmid # 26737, Burkel et al., 2007). The Pten RNAi (5′ CGACTTAGACTTGACCTATAT 3′) targeting mouse Pten, and scrambled RNAi (5′ CAACAAGATGAAGAGCACCAA 3′) sequences were used as previously described (Shi et al., 2014). The constructs were generated by sub-cloning an inverted repeat, separated by 9-nucleotide loop, into the modified FUGW lentiviral shuttle vector using BamHI/PacI restriction sites under U6 promoter. As a transduction marker, the shuttle vector contained a synapsin promoter-controlled RFP cassette with nuclear localization signal (NLS).
Expression analysis and immunoprecipitation
Total RNA from primary mouse cortical neurons of different days in vitro (DIV) was purified using the BioReagents SurePrep RNA/DNA/Protein Purification Kit (Thermo Fisher Scientific; #BP2802) according to the manufacturer’s protocol. Contaminating genomic DNA was degraded with DNase I (Roche; #04716728001) prior to cDNA synthesis from 2 μg total RNA using random hexamer primers from the Transcriptor First Strand cDNA Synthesis Kit (Roche; #04379012001). Intron spanning primers for Prg2 (fw: 5′ gaataaaacgccgaaggaca 3′, rev: 5′ aagccacgatgggtagctc 3′), Prg1 (fw: 5′ accatccagtcgatgtctattgt 3′, rev: 5′ agcatactgtcttcactaggcaaa 3′) and Tubulin (fw: 5′ gcgcatcagcgtatactacaa 3′, rev: 5′ catggttccaggttccaagt 3′), and corresponding recommended probes (Prg1: UPL #63, Prg2: UPL #51, Tubulin: UPL #81) were selected using the Roche Universal ProbeLibrary assay design center. Each primer set was tested for efficiency and real-time PCRs were performed with primers with efficiency close to 100% using the LightCycler® 480 Probes Master kit (Roche; #04707494001) on LightCycler® 480 (Roche). Abundance of Prg2 and Prg1 transcripts in neurons was quantified relative to Tubulin at different ages using the dCt-Method.
Protein lysates from cell lines, neurons and brain tissue were prepared in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulfate), supplemented with EDTA 2 mM and protease and phosphatase inhibitor cocktails from Calbiochem (539134) and Sigma (P5726). For co-immunoprecipitations and PTEN phosphatase assay, a PTEN lysis buffer consisting of 50 mM Tris pH 7.8, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, supplemented with 0.1% β-mercaptoethanol and protease inhibitor cocktail was additionally used. Protein samples for western blotting were prepared with 4x Roti-Load 1 (Roth), loaded on 8% SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were blocked with 5% skim milk in TBST for 30 min. Primary antibodies were prepared at a dilution of 1:1000 in 5% skim milk/TBST and incubated overnight at 4°C. After washing and incubation with secondary HRP-conjugated antibodies, the ECL signal (Promega) was detected and imaged using the Fusion SL system from Vilber Lourmat.
Flag-tagged proteins were immunoprecipitated by incubating pre-cleared protein lysates with anti-Flag M2 affinity gel for 2 h at 4°C and after three to four washes with RIPA buffer, precipitated protein complexes were eluted by boiling in 2x Roti-Load buffer. For PTEN and SHIP2 immunoprecipitation, protein lysates were first pre-cleared with protein G agarose for 1 h at 4°C and then incubated overnight with 2 μg anti-PTEN goat antibody (N19, Santa Cruz) or 0.2 μg anti-GFP rabbit antibody (Roche). Protein immunocomplexes were captured by incubation with protein-G-agarose beads for 2 h, washed and eluted as above.
PTEN and SHIP2 phosphatase assays
GFP-PTEN immunoprecipitates from transfected COS-7 cells were washed four times with PTEN lysis buffer and three times with PTEN assay buffer (100 mM Tris pH 8.0, 2 mM DTT). After the final wash, beads were resuspended in assay buffer, distributed in 25 μL aliquots and placed on ice. In preliminary experiments, we established the specificity of the assay using wild-type and phosphatase-inactive PTEN with di-C8-PI(3,4,5)P3 (P-3908, Echelon Biosciences) as substrate. We also established that assaying 5% of total immunoprecipitated PTEN from a 6-well plate results in linear kinetics of PI(3,4,5)P3 hydrolysis for up to 30 min. We followed this protocol in all subsequent experiments to ensure assay linearity. One-tenth of the immunoprecipitation reactions was kept for analysis of PTEN levels by western blotting. Reactions were started by the addition of 3 μL of 1 mM di-C8-PI(3,4,5)P3 to a 25 μL-aliquot of immunoprecipitated PTEN and samples were incubated at 37°C under constant rotation for up to 60 min. Reactions were terminated by addition of 100 μL malachite green reagent, and after a further incubation for 15 min at room temperature, absorbance at 620 nm was measured. Released inorganic phosphate was calculated using a phosphate (KH2PO4) standard curve. For SHIP2 phosphatase assays, we used an assay buffer consisting of 50 mM Tris, pH 7.2, 5 mM MgCl2. The specificity of this assay was also determined using wild-type and phosphatase-inactive SHIP2 (lacking the catalytic domain) with both di-C8-PI(3,4,5)P3 and di-C8-PI(4,5)P2 as substrates (data not shown).
Purification of PRG2 and MST binding assays
Prg2 was tagged with a C-terminal Rhodopsin 1D4 epitope (Rho1D4) in a pMT4 vector and expressed in HEK293S GnTI- cells using polyethylenimine (PEI; Sigma) with 3:1 (v/v) ratio of PEI:DNA for transient transfection. Cells were centrifuged at 4,000 g and the pellet was washed twice with PBS, and lysed in 20 mM HEPES pH 7.4, 150 mM NaCl, 1% (w/v) Fos-Choline-14 (Anatrace) supplemented with protease inhibitors, using gentle agitation for 1 h at 4°C. Solubilized proteins were incubated overnight with Rho1D4 Agarose (Cube Biotech) and after washing with 20 mM HEPES pH 7.4, 150 mM NaCl, 0.01% (w/v) Fos-Choline-14, Rho1D4-tagged PRG2 was eluted by addition of 200 μM Rho1D4 peptide (Cube Biotech, 16201) overnight. Following collection of the Rho1D4-tagged PRG2 eluate, the detergent was exchanged with amphipol (A8-35; Anatrace). For the substitution process, 1% (w/v) A8-35 was added and incubated for 1 h. Active Bio-Beads were then added and detergent was removed overnight. The purity of PRG2 was characterized with SDS-PAGE followed by Coomassie staining and western blotting. The tagged protein was concentrated to 20 μM using a 100 kDa cut-off Amicon Ultra-0.5 mL centrifugal filter (Merck), and fluorescently labeled using the Monolith Protein Labeling Kit RED-NHS (NanoTemper Technologies) according to the manufacturer’s protocol. Excess dye was eliminated and protein purified by size-exclusion chromatography on a Superose 12 10/300 GL column (GE Healthcare), pre-equilibrated with 20 mM HEPES pH 7.4, 150 mM NaCl. The protein peak was collected for MST measurements.
MST was measured on a Monolith NT.115 device (NanoTemper Technologies) using 32.5 nM fluorescently labeled PRG2 in 20 mM HEPES pH 7.4, 150 mM NaCl, 0.05% (v/v) Tween 20. Samples were added to standard capillaries (NanoTemper Technologies), and MST was measured at 9-10°C with 20% IR laser power using medium MST power settings. Sixteen-fold serial dilutions of 2.5 μM PTEN (OriGene) in 25 mM Tris-HCl pH 7.3, 100 mM glycine, 10% (v/v) glycerol were prepared, mixed with a constant concentration of labeled PRG2. Measurements were repeated seven times in two independent PRG2 sample preparations. Data were analyzed using the MO.Affinity analysis software (NanoTemper Technologies). The dissociation constant KD between PRG2 and PTEN was calculated using the software’s standard fitting mode derived from the law of mass action with a fixed lower border at the lowest measured Fnorm.
Immunochemistry and image acquisition
Paraformaldehyde-fixed cells (N1E-115 cells, U87MG cells, primary neurons and ESCMNs) were washed three times with PBS, permeabilized in 0.2% Triton X-100 for 5 min and then blocked in blocking buffer (5% bovine serum albumin in PBS or 4% normal goat serum in PHEM) for 1 h. Cells were then incubated with primary antibodies in blocking buffer and after washes with PBS, were incubated with appropriate Alexa dye-conjugated secondary antibodies (Jackson labs and Invitrogen). Actin filaments were labeled using Alexa-conjugated Acti-stain (Cytoskeleton) or Phalloidin-Alexa647 (Invitrogen). Proximity ligation assays (PLA) were performed using Duolink reagents (Sigma). Briefly, GFP-coupled proteins were detected using rabbit anti-GFP, Flag-coupled PRG2 was detected using mouse anti-Flag with corresponding secondary anti-rabbit PLA-Plus and anti-mouse PLA-Minus antibodies. Duolink PLA reagents were used according to the manufacturer’s protocol for ligation, amplification and mounting. Images were acquired with a Leica TCS SP5 confocal microscope or a Nikon Eclipse Ti epifluorescence microscope. For 3D structured illumination microscopy (SIM), N1E cells as well as hippocampal or cortical neurons were plated on acid-washed, poly-ornithine/laminin-coated high-precision #1.5 coverslips (Carl Roth LH24.1). N1E cells were transfected with Prg2-Flag as described above and fixed two days later with 0.2% GA, 4% PFA, 4% sucrose in PHEM buffer and stained for Flag, Tubulin and F-actin. Neurons were fixed at DIV7-9 with 4% PFA/15% sucrose in PBS and stained for PRG2, Tau and F-actin. Neurons were mounted with Vectashield (H1000, Vector Laboratories), N1E cells with ProLong Gold antifade reagent (Invitrogen). Image acquisition was undertaken on an OMX V4 Blaze system (GE Healthcare) using the 488 nm, 568 nm and 647 nm laser lines, standard filter sets and 125 nm z sectioning. 100 nm fluorescent beads (Tetraspeck, T7284, Thermo Fischer Scientific) were used for registration of the detection channels, achieving less than 40 nm registration error for all three channels.
For immunohistology, brains of E16.5 mice were dissected on ice and fixed overnight in 4% PFA in PBS at 4°C. Brains were then washed with PBS and incubated with 15% sucrose in PBS for 8 h at 4°C before exchanging the 15% sucrose with 30% sucrose in PBS for overnight incubation. Brains were then embedded in OCT and 20 μm coronal sections were cut on a cryostat. Sections were permeabilized with 0.2% Triton X-100 in blocking buffer at room temperature for 1 h, and stained using anti-PRG2 (dilution 1:200), anti-L1 (Merck, MAB5272, dilution 1:500). Sections were mounted in 80% glycerol with 2.5% DABCO.
All live-imaging of primary hippocampal neurons was performed at 3-4 DIV. Neurons were transfected at DIV1 as described above with 0.3 μg of UTR-GFP and 0.5 μg of Halo-Prg2 per well. Prior to imaging, cells were labeled with Janelia Fluor 646 HaloTag ligand (Promega) at a final concentration of 100 nM. Briefly, one-tenth of the culture medium was replaced with the HaloTag ligand and incubated at 37°C 5% CO2 for 15 min. The medium was then replaced with Neurobasal A medium minus phenol red (GIBCO) (supplemented as described above). Additionally, 50 ng/ml of human recombinant BDNF (R&D systems) was added to the imaging medium to promote filopodia formation. Healthy-looking cells with medium-level expression of the constructs were selected and only the most distal part of the main axon was imaged. Imaging was performed on Nikon Spinning Disk Confocal CSU-X under controlled temperature (37°C), CO2 (5%) and humidity conditions, using an oil-immersed 100x 1.45 NA objective and ND Acquisition software. 488nm and 647 nm laser lines with 300 ms exposure time were used for triggered acquisition of z stacks. Each axon was imaged for 10 minutes with no delay between frames resulting in a focal plane interval of 8 s.
Quantification and Statistical Analysis
Morphometry analyses
For morphometric analyses of U87MG cells expressing Prg2 and PTEN variants, maximum intensity projection images were acquired of random fields on a Leica TCS SP5 confocal microscope. U87MG cells were scored for the presence or absence of peripheral F-actin foci, most frequently appearing as peripheral lamellipodia at the cell edges, or for the presence or absence of PH-AKT plasma membrane accumulations/foci, depending on the experiment. Results from 2-4 transfections of 2-3 different cell batches were pooled. For PH-AKT plasma membrane accumulation (Figure 3C), data is from n = 60 (GFP), 52 (PTENC124S), 50 (PTENWT) and 64 (PTENWT+PRG2) cells. For peripheral F-actin foci (Figure 3E), data is from n = 100 (GFP), 50 (PRG2), 91 (PTEN), 64 (PTEN+PRG2) and 66 (PTEN+PRG2ΔC) cells.
Analysis of PRG2 localization on axonal and dendritic processes was undertaken by acquiring high magnification images of PRG2-stained hippocampal neurons on a Leica TCS SP5 confocal microscope. Selected individual processes identified as dendritic (MAP2-positive) or axonal (MAP2-negative or Tau-positive) of similar widths were analyzed by drawing vertical lines crossing the process, running line intensity profiles and normalizing intensity values using the quantify mode of LAS AF software (as shown in Figure S2). We calculated the ratio of maximum peak to maximum valley intensities along these vertical lines; depending on whether these occur toward the edge or the middle of the process the ratio provides a measure of membrane localization, with high values (> 1) indicating membrane enrichment.
Analysis of super-resolution images of PRG2 and F-actin localization was undertaken using FIJI (Schindelin et al., 2012). Tau-positive axons were manually traced sequentially using the measurement point function. For analysis of the frequency distribution of PRG2 spots (Figure S7D-F) and the average spacing of consecutive PRG2 spots (Figure 4F), regions of an axonal process longer than 1.5 μm were included. For analysis of the effect of Latrunculin treatment on PRG2 localization and membrane abundance, control and treated DIV9 hippocampal neurons were stained and tracked as above. Results were pooled from a total axonal length of 174.36 μm / 553 spots (control), 171.95 μm / 377 spots (Latrunculin, 1h) and 183.00 μm / 364 spots (Latrunculin, 3h) from 9 neurons per condition. In all cases, example images were adjusted for brightness/contrast within linear ranges using Fiji/ImageJ when necessary. Control and experiment conditions were adjusted with the same parameters.
Analysis of axonal filopodia in ESCMNs was undertaken by acquiring images of GFP- or Tuj1-stained and F-actin-stained neurons. Axons were traced in the GFP or Tuj1 images and numbers and lengths of filopodia were traced in the F-actin images using the NeuronJ plug-in of FIJI (Meijering et al., 2004, https://imagescience.org/meijering/software/neuronj/). Only filopodia longer than 2.5 μm were included in the analysis. The number, total filopodia length, length of the longest filopodium, as well as the average length per traced axon were calculated. In ESCMNs grown for 20 h and stained for Tuj1, we traced axons (defined as the longest process) and number of branches/axon. Results from 2-3 separate ESCMN differentiation/isolation experiments with neurons plated in triplicate coverslips were pooled. For axonal filopodia measurements (Figure 5D) data is from 102-167 cells. For branch measurements (Figure 5F), we included data from 155-227 cells.
Morphometric analysis of hippocampal neurons was performed on F-actin and Tau-stained stage 3 neurons grown on poly-ornithine/laminin-coated coverslips imaged on a Nikon Eclipse Ti epifluorescence microscope with a 40x lens. All processes of the neurons were traced and classified as axons (defined as the Tau-positive and longest neurite), dendrites (defined as ‘other’ neurites). Axonal branches and axonal filopodia were measured using the NeuronJ plug-in (FIJI), processes longer than 10 μm were considered branches. Mean and maximal lengths of axons, dendrites and filopodia as well as filopodia density on the axon shaft and number of dendrites per cell were calculated using a custom script in the statistics software R. In each genotype, 61-69 cells were quantified in four independent cultures derived from Prg2 x Het crossings.
Morphometric analysis of branching in stage 4 hippocampal neurons was performed in GFP-transfected neurons (transfection at DIV3) fixed at DIV5 and stained for GFP and Tau. Prg2-Flag or Prg2ΔC-Flag were reconstituted in separate but similarly treated Prg2 −/− cultures at DIV1 using viral infection. Effects of Pten-knockdown were analyzed in separate cultures additionally infected with shRNA against Pten (shPten) or a scrambled control (Scr) at DIV1, shPten virus was used in identical (3rd) or double volume (4th lane) compared to Scr virus (2nd lane in Figure 7 B). All neurites were traced in the GFP-images and classified as axons (defined as the Tau-positive and longest neurite), dendrites (defined as other neurites), as well as primary, secondary and tertiary axonal branches using the NeuronJ plug-in. All data were calculated using a custom script in the statistics software R. To reduce the complexity of the dataset in Figure 6G, neurons of equal axon lengths were grouped into clusters of equal size and means of branches and axon length for these clusters were used to determine the correlation of total branches and total axon length using Excel. The branching analysis of WT against Prg2 −/− was performed on 99-119 cells per genotype from three independent cultures of Prg2-Het x Het crossings. The effect of Pten shRNA was assessed in 131-148 cells per genotype from five independent cultures. PRG2 and PRG2-ΔC reconstitution experiments were performed with 88-97 cells per condition from four independent cultures.
Classification of PRG2- and F-actin clusters
To assess local effects of PRG2 on Filopodia formation, we first quantified co-localization of newly forming filopodia with both PRG2- and F-actin clusters (“patches”). Initially, confocal z stacks were reduced by max projection, fluorophore beaching was corrected by histogram matching (https://imagej.net/Bleach_Correction) and drift was corrected by the “MultiStackReg” plugin (https://github.com/miura/MultiStackRegistration) of FIJI. Movies were cropped to 20 μm axon stretches with (if possible) emerging filopodia in the center.
Signals of both Halo-PRG2 and GFP-Utrophin were classified as clusters if their mean intensity was at least 20% brighter than the intensity of an adjacent area in the axon. Filopodia were classified to (a) emerge directly from a cluster, (b) in vicinity to a cluster (< 1.5 μm distance to the border) or (c) without a nearby cluster.
Nearest neighbor analysis of timelapse data
To test for specificity of co-localization, we performed a nearest neighbor analysis on time-lapse data. Coordinates from filopodia and PRG2 spots were determined from max-projected movies in FIJI and the nearest neighbor (NN) analysis was conducted in a custom written python script (https://github.com/ngimber/NearestNeighborWorkflow_Filopodia), which was run in a Jupyter Notebook.
Newly emerging filopodia were identified from both the actin and PRG2 channel. In the frame before protrusion of the filopodium, the brightest points of all PRG2 clusters were detected with the local maxima function of FIJI and refined manually to exclude noise. The coordinates of emerging filopodia were determined manually as the center of the filopodium base on the plasma membrane in the PRG2 channel.
The distance to the nearest PRG2 maximum in the axon was calculated for each newly formed filopodium. To test, whether the resulting nearest neighbor distribution reports a spatial relation between PRG2 and filopodia, or spatial randomness, all PRG2 spots from the frame of filopodium appearance were randomly placed inside a binary mask of the corresponding axon shaft, created from the merge of actin and PRG2 channels by excluding non-neuronal background with a manual intensity threshold. Nearest neighbor distances to the random spots were calculated for each filopodium (with 1000 iterations). Attraction and repulsion at a certain nearest neighbor distance were plotted as the ratio (measured data – random) / random. Confidence intervals were calculated from all randomized nearest neighbor distributions and Bonferroni corrected. The difference between the medians of the measured and the simulated data were calculated for each single filopodium and this distribution was tested against zero using a Wilcoxon Signed Rank Test. Analyses included 73 newly formed filopodia from 4 independent experiments.
Statistics
Sample sizes were not chosen based on pre-specified effect size. Instead, multiple independent experiments were carried out using several biological replicates as detailed in the figure legends and Method Details sections. The imaging and analysis of filopodia and branching in hippocampal axons was undertaken blinded to both genotypes and shRNA-treatment.
Statistical analysis was performed using GraphPad Prism 5 (Graph Pad Software, La Jolla, USA), and the significance level was set to p < 0.05. Data are expressed as mean ± SD or means ± SEM; N describes the number of mice or independent cultures or transfections; n number of cells. Data were tested for normality and equality of variances. If not stated explicitly, Student’s t tests were used to analyze differences between two conditions. One-way ANOVA with post hoc Bonferroni tests were used to analyze differences between multiple conditions.
Data and Code Availability
Critical analysis tools used in the study are specified in the key resource table as well as the corresponding method sections, tools generated for this study were uploaded to Github. Specifically, the script for nearest neighbor analysis performed in Figure 7 was deposited to https://github.com/ngimber/NearestNeighborWorkflow_Filopodia.
Acknowledgments
We thank Kerstin Schlawe, Kristin Lehmann, Beate Diemar, Anja Koch, and Dr. Foteini Delis for excellent technical assistance. We would like to thank the Viral Core Facility for the production of the viruses and the Advanced Medical BioImaging Core Facility (AMBIO) and the NeuroCure Multi-user Microscopy Core Facility for usage of confocal microscopes. We thank the Transgenic Technology Core Unit of the Charité–Universitätsmedizin Berlin and Tilman Breiderhoff for support during the generation of the Prg2−/− mouse line. We would also like to thank Andrew Newman for excellent technical support and James Fawcett for fruitful discussions. Funding was provided by the DFG SFB 958, A16 (awarded to B.J.E.), A05 (to C.R.), Z02 (to J.S.), and Z05 (to C.S.); SFB TRR 186, A10 (to B.J.E.) and A04 (to C.R.); SFB 1078, B6 (to P.S.), SFB 740, B6 (to P.S.), and SFB 1365, A03 (to P.S.); EXC311 2008/1 (UniSysCat)-390540038, Research Unit E (to P.S.); NeuroCure EXC257 (to B.J.E.); and the BBSRC BB/I022392/1 (to B.J.E. and I.L.).
Author Contributions
A.B., J.F., C.K., F.I., S.S., M.V., A.P., J.L., H.G.H., N.G., and G.L. performed experiments and analysis. J.F. and T.T. established the mouse line, N.G. and J.S. performed super-resolution imaging, and N.G. developed the nearest-neighbor analysis. M.v.D. isolated and initially verified the protein-protein interaction. I.L. supported ESC work. B.J.E., G.L., P.S., M.S., C.R., and C.S. designed experiments and supervised the study. B.J.E., G.L., J.F., and A.B. wrote the manuscript. All authors reviewed the final version of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: November 12, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.039.
Contributor Information
George Leondaritis, Email: gleondar@uoi.gr.
Britta J. Eickholt, Email: britta.eickholt@charite.de.
Supplemental Information
References
- Adolf A., Leondaritis G., Rohrbeck A., Eickholt B.J., Just I., Ahnert-Hilger G., Höltje M. The intermediate filament protein vimentin is essential for axonotrophic effects of Clostridium botulinum C3 exoenzyme. J. Neurochem. 2016;139:234–244. doi: 10.1111/jnc.13739. [DOI] [PubMed] [Google Scholar]
- An W., Jackson R.E., Hunter P., Gögel S., van Diepen M., Liu K., Meyer M.P., Eickholt B.J. Engineering FKBP-based destabilizing domains to build sophisticated protein regulation systems. PLoS ONE. 2015;10:e0145783. doi: 10.1371/journal.pone.0145783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armijo-Weingart L., Gallo G. It takes a village to raise a branch: Cellular mechanisms of the initiation of axon collateral branches. Mol. Cell. Neurosci. 2017;84:36–47. doi: 10.1016/j.mcn.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräuer A.U., Savaskan N.E., Kühn H., Prehn S., Ninnemann O., Nitsch R. A new phospholipid phosphatase, PRG-1, is involved in axon growth and regenerative sprouting. Nat. Neurosci. 2003;6:572–578. doi: 10.1038/nn1052. [DOI] [PubMed] [Google Scholar]
- Broggini T., Nitsch R., Savaskan N.E. Plasticity-related gene 5 (PRG5) induces filopodia and neurite growth and impedes lysophosphatidic acid- and nogo-A-mediated axonal retraction. Mol. Biol. Cell. 2010;21:521–537. doi: 10.1091/mbc.E09-06-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broggini T., Schnell L., Ghoochani A., Mateos J.M., Buchfelder M., Wiendieck K., Schäfer M.K., Eyupoglu I.Y., Savaskan N.E. Plasticity Related Gene 3 (PRG3) overcomes myelin-associated growth inhibition and promotes functional recovery after spinal cord injury. Aging (Albany N.Y.) 2016;8:2463–2487. doi: 10.18632/aging.101066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkel B.M., von Dassow G., Bement W.M. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton. 2007;64:822–832. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadborn N.H., Ahmed A.I., Holt M.R., Prinjha R., Dunn G.A., Jones G.E., Eickholt B.J. PTEN couples Sema3A signalling to growth cone collapse. J. Cell Sci. 2006;119:951–957. doi: 10.1242/jcs.02801. [DOI] [PubMed] [Google Scholar]
- Cheng J., Sahani S., Hausrat T.J., Yang J.W., Ji H., Schmarowski N., Endle H., Liu X., Li Y., Böttche R. Precise Somatotopic Thalamocortical Axon Guidance Depends on LPA-Mediated PRG-2/Radixin Signaling. Neuron. 2016;92:126–142. doi: 10.1016/j.neuron.2016.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie K.J., Webber C.A., Martinez J.A., Singh B., Zochodne D.W. PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J. Neurosci. 2010;30:9306–9315. doi: 10.1523/JNEUROSCI.6271-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coiro P., Stoenica L., Strauss U., Bräuer A.U. Plasticity-related gene 5 promotes spine formation in murine hippocampal neurons. J. Biol. Chem. 2014;289:24956–24970. doi: 10.1074/jbc.M114.597880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S., Dixon J.E., Cho W. Membrane-binding and activation mechanism of PTEN. Proc. Natl. Acad. Sci. USA. 2003;100:7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogterom M., Koenderink G.H. Actin-microtubule crosstalk in cell biology. Nat. Rev. Mol. Cell Biol. 2019;20:38–54. doi: 10.1038/s41580-018-0067-1. [DOI] [PubMed] [Google Scholar]
- Drinjakovic J., Jung H., Campbell D.S., Strochlic L., Dwivedy A., Holt C.E. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron. 2010;65:341–357. doi: 10.1016/j.neuron.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink K.L., López-Giráldez F., Kim I.-J., Strittmatter S.M., Cafferty W.B.J. Identification of Intrinsic Axon Growth Modulators for Intact CNS Neurons after Injury. Cell Rep. 2017;18:2687–2701. doi: 10.1016/j.celrep.2017.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G. The cytoskeletal and signaling mechanisms of axon collateral branching. Dev. Neurobiol. 2011;71:201–220. doi: 10.1002/dneu.20852. [DOI] [PubMed] [Google Scholar]
- Gallo G. Mechanisms underlying the initiation and dynamics of neuronal filopodia. from neurite formation to synaptogenesis. Int. Rev. Cell Mol. Biol. 2013;301:95–156. doi: 10.1016/B978-0-12-407704-1.00003-8. [DOI] [PubMed] [Google Scholar]
- Gustafsson M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Haugh J.M., Codazzi F., Teruel M., Meyer T. Spatial sensing in fibroblasts mediated by 3′ phosphoinositides. J. Cell Biol. 2000;151:1269–1280. doi: 10.1083/jcb.151.6.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman C.A., Plummer H.K., 3rd Filopodia as sensors. Cell. Signal. 2013;25:2298–2311. doi: 10.1016/j.cellsig.2013.07.006. [DOI] [PubMed] [Google Scholar]
- Horiguchi K., Hanada T., Fukui Y., Chishti A.H. Transport of PIP3 by GAKIN, a kinesin-3 family protein, regulates neuronal cell polarity. J. Cell Biol. 2006;174:425–436. doi: 10.1083/jcb.200604031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W., Izadi M., Nemitz S., Haag N., Kessels M.M., Qualmann B. The actin nucleator cobl is controlled by calcium and calmodulin. PLoS Biol. 2015;13:e1002233. doi: 10.1371/journal.pbio.1002233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.-C., Chen Y., Page D.T. Hyperconnectivity of prefrontal cortex to amygdala projections in a mouse model of macrocephaly/autism syndrome. Nat. Commun. 2016;7:13421. doi: 10.1038/ncomms13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idevall-Hagren O., Dickson E.J., Hille B., Toomre D.K., De Camilli P. Optogenetic control of phosphoinositide metabolism. Proc. Natl. Acad. Sci. USA. 2012;109:E2316–E2323. doi: 10.1073/pnas.1211305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima M., Huang Y.E., Devreotes P. Temporal and spatial regulation of chemotaxis. Dev. Cell. 2002;3:469–478. doi: 10.1016/s1534-5807(02)00292-7. [DOI] [PubMed] [Google Scholar]
- Kakumoto T., Nakata T. Optogenetic control of PIP3: PIP3 is sufficient to induce the actin-based active part of growth cones and is regulated via endocytosis. PLoS ONE. 2013;8:e70861. doi: 10.1371/journal.pone.0070861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalil K., Dent E.W. Branch management: mechanisms of axon branching in the developing vertebrate CNS. Nat. Rev. Neurosci. 2014;15:7–18. doi: 10.1038/nrn3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketschek A., Gallo G. Nerve growth factor induces axonal filopodia through localized microdomains of phosphoinositide 3-kinase activity that drive the formation of cytoskeletal precursors to filopodia. J. Neurosci. 2010;30:12185–12197. doi: 10.1523/JNEUROSCI.1740-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis P., Hendricusdottir R., Kay L., Papageorgiou I.E., van Diepen M., Mack T., Ryves J., Harwood A., Leslie N.R., Kann O. Phosphorylation of the actin binding protein Drebrin at S647 is regulated by neuronal activity and PTEN. PLoS ONE. 2013;8:e71957. doi: 10.1371/journal.pone.0071957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis P., Leondaritis G., Lieberam I., Eickholt B.J. Subcellular targeting and dynamic regulation of PTEN: implications for neuronal cells and neurological disorders. Front. Mol. Neurosci. 2014;7:23. doi: 10.3389/fnmol.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon C.H., Luikart B.W., Powell C.M., Zhou J., Matheny S.A., Zhang W., Li Y., Baker S.J., Parada L.F. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.R., Chen M., Pandolfi P.P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol. 2018;19:547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- Lewis T.L., Jr., Courchet J., Polleux F. Cell biology in neuroscience: Cellular and molecular mechanisms underlying axon formation, growth, and branching. J. Cell Biol. 2013;202:837–848. doi: 10.1083/jcb.201305098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Huai J., Endle H., Schlüter L., Fan W., Li Y., Richers S., Yurugi H., Rajalingam K., Ji H. PRG-1 regulates synaptic plasticity via intracellular PP2A/β1-integrin signaling. Dev. Cell. 2016;38:275–290. doi: 10.1016/j.devcel.2016.06.019. [DOI] [PubMed] [Google Scholar]
- Machado C.B., Kanning K.C., Kreis P., Stevenson D., Crossley M., Nowak M., Iacovino M., Kyba M., Chambers D., Blanc E., Lieberam I. Reconstruction of phrenic neuron identity in embryonic stem cell-derived motor neurons. Development. 2014;141:784–794. doi: 10.1242/dev.097188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Belmonte F., Gassama A., Datta A., Yu W., Rescher U., Gerke V., Mostov K. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–397. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiko D., Fujihara Y., Satouh Y., Miyata H., Isotani A., Ikawa M. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep. 2013;3:3355. doi: 10.1038/srep03355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott M.I., Sigal Y.J., Sciorra V.A., Morris A.J. Is PRG-1 a new lipid phosphatase? Nat. Neurosci. 2004;7 doi: 10.1038/nn0804-789a. 789, author reply 789–790. [DOI] [PubMed] [Google Scholar]
- Meijering E., Jacob M., Sarria J.-C.F., Steiner P., Hirling H., Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Ménager C., Arimura N., Fukata Y., Kaibuchi K. PIP3 is involved in neuronal polarization and axon formation. J. Neurochem. 2004;89:109–118. doi: 10.1046/j.1471-4159.2004.02302.x. [DOI] [PubMed] [Google Scholar]
- Menon S., Gupton S.L. Building blocks of functioning brain: cytoskeletal dynamics in neuronal development. Int. Rev. Cell Mol. Biol. 2016;322:183–245. doi: 10.1016/bs.ircmb.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic I., Thakor D.T., Vargas J.R., McKinlay C.J., Hauke S., Anstaett P., Camuña R.C., Bigler L., Gasser G., Schultz C. Cellular delivery and photochemical release of a caged inositol-pyrophosphate induces PH-domain translocation in cellulo. Nat. Commun. 2016;7:10622. doi: 10.1038/ncomms10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarikangas J., Kourdougli N., Senju Y., Chazal G., Segerstråle M., Minkeviciene R., Kuurne J., Mattila P.K., Garrett L., Hölter S.M. MIM-Induced Membrane Bending Promotes Dendritic Spine Initiation. Dev. Cell. 2015;33:644–659. doi: 10.1016/j.devcel.2015.04.014. [DOI] [PubMed] [Google Scholar]
- Savaskan N.E., Bräuer A.U., Nitsch R. Molecular cloning and expression regulation of PRG-3, a new member of the plasticity-related gene family. Eur. J. Neurosci. 2004;19:212–220. doi: 10.1046/j.1460-9568.2003.03078.x. [DOI] [PubMed] [Google Scholar]
- Schermelleh L., Heintzmann R., Leonhardt H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010;190:165–175. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrötter S., Leondaritis G., Eickholt B.J. Capillary isoelectric focusing of akt isoforms identifies highly dynamic phosphorylation in neuronal cells and brain tissue. J. Biol. Chem. 2016;291:10239–10251. doi: 10.1074/jbc.M115.700138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.H., Jan L.Y., Jan Y.N. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/s0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- Shi Y., Wang J., Chandarlapaty S., Cross J., Thompson C., Rosen N., Jiang X. PTEN is a protein tyrosine phosphatase for IRS1. Nat. Struct. Mol. Biol. 2014;21:522–527. doi: 10.1038/nsmb.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal Y.J., Quintero O.A., Cheney R.E., Morris A.J. Cdc42 and ARP2/3-independent regulation of filopodia by an integral membrane lipid-phosphatase-related protein. J. Cell Sci. 2007;120:340–352. doi: 10.1242/jcs.03335. [DOI] [PubMed] [Google Scholar]
- Song M.S., Salmena L., Pandolfi P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- Spillane M., Gallo G. Involvement of Rho-family GTPases in axon branching. Small GTPases. 2014;5:e27974. doi: 10.4161/sgtp.27974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillane M., Ketschek A., Donnelly C.J., Pacheco A., Twiss J.L., Gallo G. Nerve growth factor-induced formation of axonal filopodia and collateral branches involves the intra-axonal synthesis of regulators of the actin-nucleating Arp2/3 complex. J. Neurosci. 2012;32:17671–17689. doi: 10.1523/JNEUROSCI.1079-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss U., Bräuer A.U. Current views on regulation and function of plasticity-related genes (PRGs/LPPRs) in the brain. Biochim. Biophys. Acta. 2013;1831:133–138. doi: 10.1016/j.bbalip.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Trimbuch T., Beed P., Vogt J., Schuchmann S., Maier N., Kintscher M., Breustedt J., Schuelke M., Streu N., Kieselmann O. Synaptic PRG-1 modulates excitatory transmission via lipid phosphate-mediated signaling. Cell. 2009;138:1222–1235. doi: 10.1016/j.cell.2009.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadhvani M., Schwedhelm-Domeyer N., Mukherjee C., Stegmüller J. The centrosomal E3 ubiquitin ligase FBXO31-SCF regulates neuronal morphogenesis and migration. PLoS ONE. 2013;8:e57530. doi: 10.1371/journal.pone.0057530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M., Andrés-Pons A., Gomar B., Torres J., Gil A., Tapparel C., Antonarakis S.E., Pulido R. Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J. Biol. Chem. 2005;280:28936–28943. doi: 10.1074/jbc.M504761200. [DOI] [PubMed] [Google Scholar]
- van Diepen M.T., Parsons M., Downes C.P., Leslie N.R., Hindges R., Eickholt B.J. MyosinV controls PTEN function and neuronal cell size. Nat. Cell Biol. 2009;11:1191–1196. doi: 10.1038/ncb1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F., Matsuoka S., Sellers W.R., Yanagida T., Ueda M., Devreotes P.N. Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc. Natl. Acad. Sci. USA. 2006;103:3633–3638. doi: 10.1073/pnas.0510570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmans T., Battefeld A., Geist B., Farrés A.S., Strauss U., Bräuer A.U. Plasticity-related gene 3 promotes neurite shaft protrusion. BMC Neurosci. 2013;14:36. doi: 10.1186/1471-2202-14-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C.L., Haase J., Janes J., Huss J.W., 3rd, Su A.I. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S., Loewith R., Hall M.N. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Xu K., Zhong G., Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339:452–456. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip S.-C., Eddy R.J., Branch A.M., Pang H., Wu H., Yan Y., Drees B.E., Neilsen P.O., Condeelis J., Backer J.M. Quantification of PtdIns(3,4,5)P(3) dynamics in EGF-stimulated carcinoma cells: a comparison of PH-domain-mediated methods with immunological methods. Biochem. J. 2008;411:441–448. doi: 10.1042/BJ20071179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P., Agbaegbu C., Malide D.A., Wu X., Katagiri Y., Hammer J.A., Geller H.M. Cooperative interactions of LPPR family members in membrane localization and alteration of cellular morphology. J. Cell Sci. 2015;128:3210–3222. doi: 10.1242/jcs.169789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.X., Duan L.H., He S.J., Zhuang G.F., Yu X. Phosphatidylinositol 3,4-bisphosphate regulates neurite initiation and dendrite morphogenesis via actin aggregation. Cell Res. 2017;27:253–273. doi: 10.1038/cr.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Ueno Y., Liu X.S., Buller B., Wang X., Chopp M., Zhang Z.G. The microRNA-17-92 cluster enhances axonal outgrowth in embryonic cortical neurons. J. Neurosci. 2013;33:6885–6894. doi: 10.1523/JNEUROSCI.5180-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Critical analysis tools used in the study are specified in the key resource table as well as the corresponding method sections, tools generated for this study were uploaded to Github. Specifically, the script for nearest neighbor analysis performed in Figure 7 was deposited to https://github.com/ngimber/NearestNeighborWorkflow_Filopodia.