

Abstract
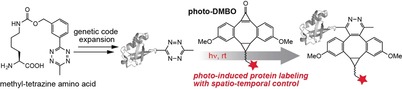
Inverse electron‐demand Diels–Alder cycloadditions (iEDDAC) between tetrazines and strained alkenes/alkynes have emerged as essential tools for studying and manipulating biomolecules. A light‐triggered version of iEDDAC (photo‐iEDDAC) is presented that confers spatio‐temporal control to bioorthogonal labeling in vitro and in cellulo. A cyclopropenone‐caged dibenzoannulated bicyclo[6.1.0]nonyne probe (photo‐DMBO) was designed that is unreactive towards tetrazines before light‐activation, but engages in iEDDAC after irradiation at 365 nm. Aminoacyl tRNA synthetase/tRNA pairs were discovered for efficient site‐specific incorporation of tetrazine‐containing amino acids into proteins in living cells. In situ light activation of photo‐DMBO conjugates allows labeling of tetrazine‐modified proteins in living E. coli. This allows proteins in living cells to be modified in a spatio‐temporally controlled manner and may be extended to photo‐induced and site‐specific protein labeling in animals.
Keywords: bioorthogonal reactions, photo-induced labeling, protein labeling, tetrazine, unnatural amino acids
Light‐induced protein labeling: Cyclopropenone‐caged dibenzoannulated bicyclononynes (photo‐DMBO) are photo‐activatable dienophiles that engage in rapid inverse electron‐demand Diels–Alder cycloadditions with tetrazines upon light‐induced decarbonylation. Site‐specific incorporation of methyl‐tetrazine amino acids allows photo‐induced protein labeling in living cells with spatio‐temporal control using photo‐DMBO fluorophore conjugates.
Introduction
Bioorthogonal chemistries offer a versatile platform to probe and control biomolecules.1 Progress over the last two decades in the formulation of more selective and faster bioorthogonal reagents has led to far‐reaching applications in biology, medicine, and materials.2 An especially exciting class of bioorthogonal reactions is represented by inverse electron‐demand Diels–Alder cycloadditions (iEDDAC) between s‐tetrazines and alkene or alkyne dienophiles.3 In combination with site‐specific incorporation of unnatural amino acids (UAAs) into proteins via genetic code expansion, iEDDAC reactions have emerged as indispensable tools for labeling and manipulating proteins in living systems.1a, 3a Highly specific and orthogonal Pyrrolysyl‐tRNA synthetase (PylRS)/tRNACUA pairs have allowed the site‐specific incorporation of UAAs bearing strained alkene/alkyne moieties, such as norbornene (Nor),4 1,3‐disubstituted cyclopropene (Cp),5 bicyclo[6.1.0]nonyne (BCN),6 and trans‐cyclooctene (TCO)6a, 7 into proteins in E. coli and mammalian cells and their subsequent chemoselective labeling with tetrazine conjugates.8 iEDDAC reactions have found application for imaging of cell‐surface and intracellular proteins, for labeling and identifying proteomes in E. coli, mammalian cells, and multicellular organisms5 as well as for selectively inhibiting a specific target protein within living cells.9 Furthermore, TCO‐bearing UAAs have enabled tetrazine‐triggered protein decaging and activation in living cells.10 Likewise, tetrazine‐bearing amino acids have been incorporated into proteins in E. coli using engineered variants of the Methanocaldococcus jannaschii (Mj) Tyrosyl‐tRNA synthetase (TyrRS)/tRNACUA pair, and tetrazine‐modified proteins have been labeled with strained TCO fluorophores in ultra‐rapid iEDDAC (second order rate constants up to 105 m −1 s−1).11
Recent advancements in bioorthogonal chemistries have shown a growing interest in using external stimuli to induce bioorthogonal reactivity. In particular, photo‐inducible reactions have emerged as a method to exert control over when and where bioorthogonal partners react with each other.12 Key advances include light‐triggered tetrazole‐alkene photoclick chemistry,13 visible‐light induced [4+2] cycloaddition between 9,10‐phenanthrenequinone and vinyl ethers14 as well as photo‐induced activation of cyclopropenone‐caged cyclooctynes for strain‐promoted azide–alkyne cycloadditions (photo‐SPAAC).15 Importantly, also versions of photo‐induced iEDDAC have been reported, such as light/enzyme‐triggered redox‐activation of dihydrotetrazines to tetrazines16 and the modular caging of cyclopropenes.17 As the former approach is so far limited to dihydrotetrazines that are stable to spontaneous air oxidation, and photo‐induced iEDDAC with described caged cyclopropenes show very slow reaction rates (k 2≈10−2–10−4 m −1 s−1), there is however a pressing need for novel, photo‐inducible iEDDAC reactions with fast kinetics.
Herein we report a rapid light‐triggered tetrazine ligation by developing a photo‐activatable BCN‐based probe that confers spatial and temporal control to labeling of tetrazine‐bearing proteins in living cells. Inspired by innovative work from Popik and co‐workers on photo‐activated cyclooctynes,18 we designed and synthesized a cyclopropenone‐caged dibenzoannulated BCN derivative (photo‐DMBO) that is quantitatively decarbonylated to the corresponding alkyne (DMBO) by short UV irradiation at 365 nm. While photo‐DMBO is inert towards tetrazines, the decaged version reacts rapidly and selectively with tetrazine‐bearing proteins via iEDDAC (Scheme 1).
Scheme 1.
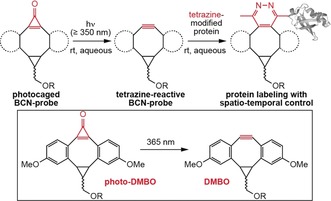
Photochemical decarbonylation of a cyclopropenone‐caged BCN‐probe (photo‐DMBO) activates reactivity towards tetrazine‐bearing proteins, conferring spatial and temporal control to protein labeling.
To use this approach for rapid, photo‐induced protein labeling in living cells, we have synthesized novel tetrazine‐bearing amino acids and found a highly efficient PylRS mutant for their site‐specific incorporation into proteins in E. coli. Tetrazine‐modified proteins react with dibenzoannulated BCN with second‐order rate constants of about 50 m −1 s−1, putting them among the fastest bioorthogonal photo‐induced reactions. Light‐triggered iEDDAC employing photo‐DMBO and our novel tetrazine‐amino acids works selectively and rapidly (completion within a few minutes at low μm concentrations) on purified proteins, in complex mixtures as well as on living E. coli. The strong dependence on light allows for both sequential dual modification of proteins as well as for protein labeling with spatio‐temporal control in living systems.
Results and Discussion
Genetic Encoding and Rapid Labeling of Methyl‐tetrazine Amino Acids
To guarantee efficient and quantitative protein labeling in living cells we aimed at genetically encoding flexible UAAs bearing small and physiologically stable tetrazine moieties. As methyl‐substituted tetrazines have been identified as the most stable derivatives with an optimal stability to reactivity balance,11b, 19 we designed lysine‐based methyl‐substituted tetrazine amino acids TetK(1) and mTetK(2) (Figure 1 a). The amino acids were synthesized by reacting the corresponding nitriles (3‐hydroxyproprionitrile for 1 and 3‐(hydroxymethyl)benzonitrile for 2) with acetonitrile in the presence of hydrazine and zinc‐ or nickel‐triflate to form the respective dihydrotetrazine alcohols, which were oxidized and coupled to the ϵ‐amino group of lysine to yield 1 and 2 (Supporting Information).20 With efficient access to flexible tetrazine‐modified amino acids at hand, we set out to site‐specifically incorporate 1 and 2 into proteins in cellulo. A large variety of structurally diverse UAAs have been genetically encoded in response to an amber codon introduced into a gene of interest by employing PylRS/tRNACUA pairs from Methanosarcina species.1a, 21 None of the so far developed PylRS variants accepts however lysine derivatives with bulky polar side chains such as 1 and 2. Guided by structural analyses of the C‐terminal catalytic center of wt PylRS and its mutants,22 we screened a panel of >50 different M. barkeri PylRS mutants for their ability to direct the selective and site‐specific incorporation of 1 and 2. Gratifyingly, Mb PylRS mutant (Y271G and C313V, Mb numbering, dubbed TetRS) that has not been described so far led to the efficient incorporation of both 1 and 2 into C‐terminally His‐tagged superfolder green fluorescent protein (sfGFP) containing a premature amber codon. SDS‐PAGE and α‐His6 western blot (WB) analysis confirmed very good incorporation efficiencies for both 1 and 2 and full‐length sfGFP‐1‐His6 and sfGFP‐2‐His6 were isolated in good yields (Figure 1 b, ca. 80 mg L−1 of culture). Similarly, myoglobin (Myo) and ubiquitin (Ub) bearing site‐specifically introduced amber codons produced good yields of protein in presence, but not absence of 1 and 2. The selective and site‐specific incorporation of tetrazine‐bearing amino acids was further confirmed by electrospray ionization mass spectrometry (ESI‐MS) of purified proteins (Figure 1 c; Supporting Information, Figure S1).
Figure 1.
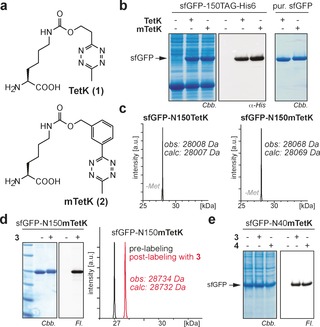
Site‐specific incorporation of tetrazine‐bearing amino acids into proteins in bacteria and selective protein labeling via iEDDAC with BCN‐ (3) and TCO‐ (4) fluorophore conjugates. a) Structures of TetK (1) and mTetK (2). b) Incorporation of 1 and 2 into sfGFP‐150TAG‐His6. left: Coomassie stained SDS‐PAGE; middle: α‐His6 WB, right: Coomassie stained SDS‐PAGE of purified sfGFP mutants. c) ESI‐MS‐characterization of purified sfGFP‐N150TetK‐His6 and sfGFP‐N150mTetK‐His6. d) SDS‐PAGE fluorescence imaging and ESI‐MS confirm selective and quantitative labeling of sfGFP‐mTetK with BCN‐TAMRA e) Selective labeling of mTetK bearing sfGFP in E. coli cell lysate with fluorophore conjugates 3 and 4. Structures of 3 and 4 are shown in the Supporting Information, Figure S2. Cbb: Coomassie brilliant blue, Fl: Fluorescence.
As expected, purified proteins that bear site‐specifically incorporated methyl‐tetrazines reacted readily and quantitatively with BCN‐ (3) or TCO‐modified (4) fluorophores as shown by SDS‐PAGE based fluorescence imaging and ESI‐MS analysis (Figure 1 d; Supporting Information, Figures S2, S3). Labeling of proteins containing 1 or 2 was also specific towards the E. coli proteome as shown by selective labeling of over‐expressed intracellular and cell‐surface proteins bearing tetrazine amino acids with fluorophores 3 or 4 via iEDDAC (Figure 1 e; Supporting Information, Figures S4–S7).
Development of a Rapid, Photo‐induced iEDDAC
With tetrazine‐modified proteins at hand, we set out to develop photochemically triggered iEDDAC (photo‐iEDDAC) that confers temporal and spatial control to the labeling of target proteins.12a To achieve this goal we explored the photochemical generation of reactive BCN‐probes. Inspired by work from Popik, who described cyclopropenone‐caged dibenzocyclooctynes for photo‐SPAAC,18 we aimed at designing a cyclopropenone‐caged version of BCN that displays a tetrazine‐reactive triple bond upon photochemical decarbonylation (Scheme 1). As cyclopropenone compounds without aryl substituents are known to absorb and thereby photo‐decage at wavelengths below 250 nm,23 we decided to synthesize a cyclopropenone‐caged dibenzoannulated BCN probe (cyclopropenone‐caged di(methoxybenzo)bicyclo[6.1.0]nonyne, dubbed photo‐DMBO, Scheme 1), which should be amenable to photo‐decarbonylation to form DMBO by irradiation above 350 nm. Synthesis of photo‐9 (Figure 2 a) started with Sonogashira cross‐coupling of 3‐iodoanisol and 3‐ethynylanisol to give the symmetrical acetylene 5. Partial hydrogenation of 5 using Lindlar's catalyst generated Z‐alkene 6, which was subjected to cyclopropanation with ethyl diazoacetate to yield 7. The ester group in 7 was reduced to the corresponding hydroxyl group and protected by acetylation (8) before subsequent Friedel–Crafts alkylation with tetrachlorocyclopropene in the presence of AlCl3 to yield photo‐9 after in situ hydrolysis of the corresponding dichlorocyclopropene intermediate (Figure 2 a, Supporting Information).24 Despite its considerable ring strain, photo‐9 is stable in aqueous buffers at physiological pH over several days (Supporting Information, Figure S8 a). Upon 5 min of irradiation at 365 nm, photo‐9 decarbonylates quantitatively to form DMBO compound 9 (Figure 2 b). Photo‐decaging is effective in different organic solvents (MeOH, DMSO) and in aqueous buffers. To test reactivity of photo‐9 towards methyl‐tetrazine amino acid 2, we incubated a 100 μm solution of 2 with a five‐fold excess of photo‐9. Only upon UV irradiation of the mixture (5 min at 365 nm), amino acid 2 reacted quantitatively to the cycloaddition compound 10, while in the absence of UV irradiation no formation of product 10 could be observed even after prolonged incubation (Figure 2 c), confirming the exquisite photo‐inducibility of the cycloaddition reaction.
Figure 2.
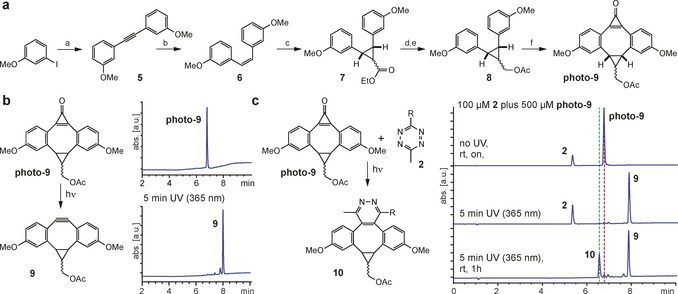
Establishing photo‐iEDDAC reactivity. a) Synthesis of photo‐9. Conditions: a) 1.2 equiv 3‐ethynylanisole, 3 equiv DIPEA, 0.1 equiv CuI, 0.05 equiv Pd(PPh3)4, in THF, reflux, overnight, (71 %); b) 20 % w/w Lindlar catalyst under H2 atmosphere, in hexane, 2 h, rt, (77 %); c) 2.5 equiv ethyl diazoacetate, 0.06 equiv CuSO4, in toluene, 75 °C, overnight, (18 %); d) 2 equiv LiAlH4, in Et2O, 0 °C–rt, 2 h, (68 %); e) 2.6 equiv Ac2O, 0.05 equiv DMAP, 4.9 equiv NEt3, in DCM, 0 °C–rt, 2 h, (70 %); f) 1 equiv tetrachlorocyclopropene, 3 equiv AlCl3, in DCM, −20 °C–rt, 4 h, aqueous workup (55 %). b) Short UV irradiation (5 min, 365 nm) converts photo‐9 quantitatively to 9. c) Photo‐9 reacts with amino acid 2 only when irradiated at 365 nm for 5 min to form iEDDAC product 10.
Rapid, Light‐Induced Protein Labeling via photo‐iEDDAC
We next set out to test if tetrazine‐modified proteins could be labeled in a photo‐induced and selective fashion with functionalized photo‐DMBO compounds. By decorating photo‐DMBO with a PEG‐linker and a primary amino group for further functionalization with fluorophores we generated water‐soluble photo‐11 (Figure 3 a) that is quantitatively decarbonylated to 11 by irradiation at 365 nm for 5 min (Figure S8 b). Incubation of a 10 μm solution of sfGFP‐2‐His6 with a 25‐fold excess of 11 led to quantitative sfGFP‐labeling within 5 min (Supporting Information, Figure S9). Similarly an irradiated solution (365 nm, 5 min) of sfGFP‐2‐His6 and photo‐11 yielded quantitative sfGFP‐modification within short incubation times (Figure 3 b; Supporting Information, Figure S9). Importantly, incubation in the absence of light did not yield any cycloaddition product. Furthermore, sfGFP bearing BocK at position 150 instead of mTetK did not result in any labeling, confirming the selectivity of photo‐iEDDAC (Supporting Information, Figure S9). Also, TetK‐modified proteins (sfGFP‐1‐His6) reacted selectively upon light‐activation with photo‐11, albeit much more slowly, and quantitative protein labeling with 25‐fold excess of 11 was only achieved after several hours of incubation (Supporting Information, Figure S10). To check how DMBO compounds performed in SPAAC reactions, we incubated an azide‐bearing sfGFP25 with photo‐11. Also this labeling took 2–3 hours to go to completion, highlighting the superior reactivity of DMBO in iEDDAC versus SPAAC (Supporting Information, Figure S11). Furthermore, azide‐modified amino acids are not completely stable towards reduction to the corresponding amine in the cytosol of E. coli and therefore our stable methyl‐tetrazine amino acids make for more reliable reporters in live E. coli. To benchmark photo‐DMBO reactivity against previously reported cyclopropenone‐caged cycloalkynes,18 we synthesized a cyclopropenone‐caged dibenzocyclooctyne (photo‐12) that contains no extra ring strain on the dibenzoannulated cyclooctyne26 and incubated it after light irradiation with sfGFP‐2‐His6 (Supporting Information, Figure S12). No cycloaddition product was observed even after overnight incubation, though decarbonylation to 12 had proceeded cleanly as proven by LC‐MS, highlighting that the extra ring strain in DMBO is needed for reaction with methyl‐tetrazines.2c, 27 Incubation of photo‐12 with azide‐bearing sfGFP under UV‐irradiation resulted in approximately 60 % SPAAC product upon overnight incubation, demonstrating that the extra strain in DMBO results also in higher reactivity towards azides (Supporting Information, Figures S11, S13). Interestingly, also a recently reported dibenzoannulated and cyclopropenone‐caged silicon‐containing cycloheptyne (photo‐13)28 failed in reacting with tetrazine‐ or azide‐modified proteins under identical conditions (Supporting Information, Figures S14, S15).
Figure 3.
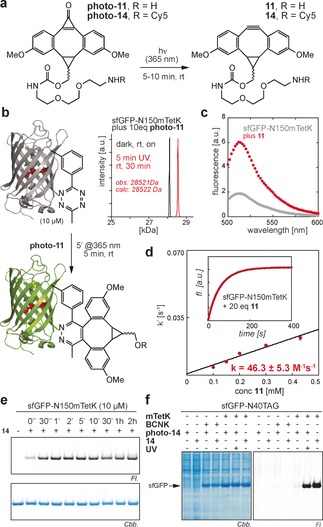
Characterization of photo‐iEDDAC on tetrazine‐bearing proteins. a) Structures of water‐soluble photo‐DMBO conjugates. Both photo‐11 and the Cy5‐conjugate photo‐14 are decarbonylated by short irradiation at 365 nm to quantitatively form 11 and 14, respectively. b) Quantitative and rapid labeling of sfGFP‐N150mTetK with photo‐11 analyzed by LC‐MS. c) Excitation at 488 nm produces quenched fluorescence for sfGFP‐N150mTetK, which is restored after reaction with 11. d) Determination of second‐order rate constant k 2 of sfGFP‐N150mTetK and 11. Inset shows fluorescence increase (508 nm) of sfGFP‐N150mTetK upon addition of a 20‐fold excess of 11 over time. e) Rapid protein labeling with fluorophore‐conjugate 14. f) Selective and light‐induced labeling of mTetK‐bearing sfGFP with photo‐14 towards the E. coli proteome.
Encouraged by the short incubation times needed for quantitative iEDDAC labeling between DMBO compounds and sfGFP‐2‐His6, we set out to determine on‐protein rate constants in a more quantitative way. As previously observed and exploited for determination of on‐protein kinetics in vitro and in cellulo, site‐specific incorporation of tetrazine amino acids in vicinity to the sfGFP chromophore (N150) lead to sfGFP‐fluorescence quenching that is restored after iEDDAC of the tetrazine moiety with a strained trans‐cyclooctene label.11 Similarly, also genetic encoding of flexible mTetK into sfGFP‐N150TAG led to sfGFP chromophore quenching and fluorescence recuperated after reaction with 11 (Figure 3 c).
By following the exponential increase over time in sfGFP‐fluorescence, we were able to quantify second order on‐protein rate constants under pseudo first‐order conditions. We determined a rate constant of about 50 m −1 s−1 (Figure 3 d; Supporting Information, Figure S16), which is nearly twice as fast as iEDDAC between mTetK and BCN−OH and 50 or 1000 times faster than the corresponding reactions with 1,3‐disubstituted cyclopropene or norbornenol (Supporting Information, Figures S17–S19), alkene reporters that are routinely used for biomolecule labeling.4, 5, 29 Reaction between sfGFP‐2‐His6 and 11 is also significantly faster than reaction between 11 and a sfGFP mutant bearing a previously described phenylalanine based tetrazine amino acid (TetF),11b which is presumably due to higher flexibility and on‐protein accessibility of mTetK versus TetF (Supporting Information, Figure S20). The fast kinetic rate constants enable photo‐triggered labeling of mTetK‐modified proteins with photo‐11 within minutes at low μm or nm concentrations and put photo‐iEDDAC reactions between DMBO and tetrazines among the fastest photo‐induced bioorthogonal reactions, several orders of magnitude faster than recently described approaches with caged cyclopropenes (k2≈10−2–10−4 m −1 s−1),17 and considerably faster than most photo‐SPAAC reactions with strained dibenzocyclooctynes (k2≈10−1–10−2 m −1 s−1),30 and on par with the recently described and so far fastest photo‐SPAAC reaction employing photo‐oxa‐dibenzocylooctyne (k2≈40 m −1 s−1).31
Photo‐iEDDAC Confers Spatio‐temporal Control to Protein Labeling on Living Cells
To create fluorescent photo‐DMBO probes, the primary amino group of photo‐11 was coupled to succinimidyl esters of Cy5 and tetramethylrhodamine (TAMRA) dyes, creating photo‐14 (Figure 3 a; Supporting Information, Figure S21) and photo‐15 (Supporting Information, Figure S22). Photo‐DMBO fluorophores react rapidly, specifically and quantitatively with recombinant proteins bearing site‐specifically incorporated mTetK upon irradiation at 365 nm, as confirmed by LC‐MS and SDS‐PAGE‐based fluorescence imaging (Figure 3 e; Supporting Information, Figures S21, S22). Light‐induced labeling with photo‐14 and photo‐15 is also distinct towards cellular proteins of the E. coli proteome, as shown by specific and selective labeling of mTetK‐bearing proteins in E. coli lysates with minimal background. Importantly, efficient labeling is reliant on irradiation at 365 nm (Figure 3 f). Encouraged by specific modification of mTetK‐bearing proteins in cell lysates we set out to use photo‐iEDDAC for temporally controlled labeling of living E. coli. We over‐expressed outer membrane protein OmpC‐Y232mTetK on the surface of E. coli cells, washed cells and treated them with photo‐14 either in the presence or absence of UV light. Labeling was specific for mTetK‐bearing cells that were irradiated at 365 nm (Figure 4 a; Supporting Information, Figure S23). As photo‐DMBO reactivity is strictly controlled by light and remains dormant until activated by UV irradiation, photo‐iEDDAC can be used in one‐pot reactions with iEDDAC for sequential dual labeling of proteins.32 To show this mutual orthogonality, we constructed a heterobifunctional crosslinker, which unifies two moieties displaying comparable iEDDAC reactivities, namely BCN and DMBO, in one molecule for sequential one‐pot labeling (Figure 4 b). Photo‐16 displays both BCN‐ and photo‐DMBO reactivity and permits controlled assembly of different methyl‐tetrazine tagged conjugates. E. coli cells expressing mTetK‐bearing OmpC on their cell surface were treated with photo‐16, allowing covalent attachment of photo‐16 via its BCN moiety. The photo‐DMBO functionality in photo‐16 is refractory to tetrazine ligation prior to UV irradiation and cells are efficiently labeled with a methyl‐tetrazine–fluorophore conjugate (Tet‐Cy5) only after light activation (Figure 4 c; Supporting Information, Figure S24), conferring mutual orthogonality to iEDDA cycloadditions with tetrazines and providing in principle control over protein‐labeling both in time and space.32a, 33
Figure 4.
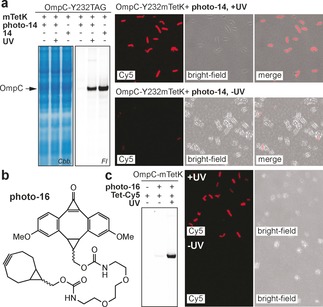
Photo‐iEDDAC labeling on living cells. a) In gel fluorescence imaging and fluorescence microscopy show efficient and light‐induced labeling of a cell‐surface protein in living E. coli. b) Structure of bifunctional linker photo‐16, displaying a BCN and a photo‐DMBO moiety. c) The DMBO moiety in photo‐16 becomes only available upon UV‐irradiation and thereby allows sequential labeling of live E. coli with temporal and spatial control.
Conclusion
We have reported a novel light‐induced iEDDAC between tetrazines and a cyclopropenone‐caged dibenzoannulated BCN probe (photo‐DMBO). We have demonstrated the efficient site‐specific incorporation of methyl‐tetrazine modified amino acids 1 and 2 into proteins in E. coli and their efficient, specific, and light‐triggered labeling with photo‐DMBO fluorophore conjugates. Photo‐iEDDAC proceeds with on‐protein rate‐constants of about 50 m −1 s−1, on par with the fastest bioorthogonal photo‐induced reactions, enabling protein labeling within minutes at low μm concentrations. These reactions are 2–4 orders of magnitude faster than iEDDAC using cyclopropene or norbornene as dienophiles and nearly twice as fast as cycloadditions between BCN and metabolically stable methyl‐tetrazines. While we have demonstrated the advantages of our approach in vitro and in living E. coli, the ability to incorporate UAAs in mammalian cells and C. elegans,34 zebrafish,35 and D. melanogaster 36 using engineered PylRS/tRNACUA pairs suggests that it may be possible to extend the photo‐induced labeling approach described here to site‐specifically label proteins in animals in a photo‐induced fashion, which might be especially attractive in combination with multi‐photon activation of photo‐DMBO compounds.37 Exploitation of photo‐iEDDAC and genetically encodable tetrazine reporters to study receptor activation is focus of current studies in our lab. We furthermore expect that other fields such as the fabrication of microarrays,38 biosensors, and the preparation of multifunctional material may benefit from this photo‐triggered and rapid iEDDAC reaction.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Excellence Initiative CIPSM and the DFG through the following programs: SFB1035, GRK1721 and SPP1623 (to K.L). K.L. is a Mössbauer Professor at TUM‐IAS and as such acknowledges funding by the Excellence Initiative and the EU Marie Curie COFUND Program. We thank Benedikt Buchmann for help with microscopy.
S. V. Mayer, A. Murnauer, M.-K. von Wrisberg, M.-L. Jokisch, K. Lang, Angew. Chem. Int. Ed. 2019, 58, 15876.
References
- 1.
- 1a. Lang K., Chin J. W., Chem. Rev. 2014, 114, 4764–4806; [DOI] [PubMed] [Google Scholar]
- 1b. Sletten E. M., Bertozzi C. R., Angew. Chem. Int. Ed. 2009, 48, 6974–6998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7108–7133; [Google Scholar]
- 1c. Patterson D. M., Nazarova L. A., Prescher J. A., ACS Chem. Biol. 2014, 9, 592–605. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. McKay C. S., Finn M. G., Chem. Biol. 2014, 21, 1075–1101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Jewett J. C., Bertozzi C. R., Chem. Soc. Rev. 2010, 39, 1272–1279; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Patterson D. M., Prescher J. A., Curr. Opin. Chem. Biol. 2015, 28, 141–149; [DOI] [PubMed] [Google Scholar]
- 2d. Shih H. W., Kamber D. N., Prescher J. A., Curr. Opin. Chem. Biol. 2014, 21, 103–111; [DOI] [PubMed] [Google Scholar]
- 2e. Ji X., Pan Z., Yu B., De La Cruz L. K., Zheng Y., Ke B., Wang B., Chem. Soc. Rev. 2019, 48, 1077–1094; [DOI] [PubMed] [Google Scholar]
- 2f. Azagarsamy M. A., Anseth K. S., ACS Macro Lett. 2013, 2, 5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Mayer S., Lang K., Synthesis 2017, 49, 830–848; [Google Scholar]
- 3b. Oliveira B. L., Guo Z., Bernardes G. J. L., Chem. Soc. Rev. 2017, 46, 4895–4950; [DOI] [PubMed] [Google Scholar]
- 3c. Blackman M. L., Royzen M., Fox J. M., J. Am. Chem. Soc. 2008, 130, 13518–13519; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Kozma E., Demeter O., Kele P., ChemBioChem 2017, 18, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Lang K., Davis L., Torres-Kolbus J., Chou C., Deiters A., Chin J. W., Nat. Chem. 2012, 4, 298–304; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Kaya E., Vrabel M., Deiml C., Prill S., Fluxa V. S., Carell T., Angew. Chem. Int. Ed. 2012, 51, 4466–4469; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4542–4545. [Google Scholar]
- 5. Elliott T. S., Townsley F. M., Bianco A., Ernst R. J., Sachdeva A., Elsasser S. J., Davis L., Lang K., Pisa R., Greiss S., Lilley K. S., Chin J. W., Nat. Biotechnol. 2014, 32, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Lang K., Davis L., Wallace S., Mahesh M., Cox D. J., Blackman M. L., Fox J. M., Chin J. W., J. Am. Chem. Soc. 2012, 134, 10317–10320; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Borrmann A., Milles S., Plass T., Dommerholt J., Verkade J. M., Wiessler M., Schultz C., van Hest J. C., van Delft F. L., Lemke E. A., ChemBioChem 2012, 13, 2094–2099. [DOI] [PubMed] [Google Scholar]
- 7. Plass T., Milles S., Koehler C., Szymanski J., Mueller R., Wiessler M., Schultz C., Lemke E. A., Angew. Chem. Int. Ed. 2012, 51, 4166–4170; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4242–4246. [Google Scholar]
- 8.
- 8a. Nikić I., Plass T., Schraidt O., Szymański J., Briggs J. A., Schultz C., Lemke E. A., Angew. Chem. Int. Ed. 2014, 53, 2245–2249; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2278–2282; [Google Scholar]
- 8b. Uttamapinant C., Howe J. D., Lang K., Beranek V., Davis L., Mahesh M., Barry N. P., Chin J. W., J. Am. Chem. Soc. 2015, 137, 4602–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsai Y. H., Essig S., James J. R., Lang K., Chin J. W., Nat. Chem. 2015, 7, 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li J., Jia S., Chen P. R., Nat. Chem. Biol. 2014, 10, 1003–1005. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Seitchik J. L., Peeler J. C., Taylor M. T., Blackman M. L., Rhoads T. W., Cooley R. B., Refakis C., Fox J. M., Mehl R. A., J. Am. Chem. Soc. 2012, 134, 2898–2901; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Blizzard R. J., Backus D. R., Brown W., Bazewicz C. G., Li Y., Mehl R. A., J. Am. Chem. Soc. 2015, 137, 10044–10047. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Kumar P., Laughlin S. T., Methods Enzymol. 2019, 622, 153–182; [DOI] [PubMed] [Google Scholar]
- 12b. Tasdelen M. A., Yagci Y., Angew. Chem. Int. Ed. 2013, 52, 5930–5938; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6044–6053. [Google Scholar]
- 13. Ramil C. P., Lin Q., Curr. Opin. Chem. Biol. 2014, 21, 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J., Kong H., Huang L., Cheng B., Qin K., Zheng M., Yan Z., Zhang Y., J. Am. Chem. Soc. 2018, 140, 14542–14546. [DOI] [PubMed] [Google Scholar]
- 15. Arumugam S., Orski S. V., Mbua N. E., McNitt C., Boons G. J., Locklin J., Popik V. V., Pure Appl. Chem. 2013, 85, 1499–1513. [Google Scholar]
- 16. Zhang H., Trout W. S., Liu S., Andrade G. A., Hudson D. A., Scinto S. L., Dicker K. T., Li Y., Lazouski N., Rosenthal J., Thorpe C., Jia X., Fox J. M., J. Am. Chem. Soc. 2016, 138, 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Kumar P., Jiang T., Li S., Zainul O., Laughlin S. T., Org. Biomol. Chem. 2018, 16, 4081–4085; [DOI] [PubMed] [Google Scholar]
- 17b. Kumar P., Zainul O., Camarda F. M., Jiang T., Mannone J. A., Huang W., Laughlin S. T., Org. Lett. 2019, 21, 3721–3725. [DOI] [PubMed] [Google Scholar]
- 18. Poloukhtine A. A., Mbua N. E., Wolfert M. A., Boons G. J., Popik V. V., J. Am. Chem. Soc. 2009, 131, 15769–15776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Baalmann M., Ziegler M. J., Werther P., Wilhelm J., Wombacher R., Bioconjugate Chem. 2019, 30, 1405–1414; [DOI] [PubMed] [Google Scholar]
- 19b. Karver M. R., Weissleder R., Hilderbrand S. A., Bioconjugate Chem. 2011, 22, 2263–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang J., Karver M. R., Li W., Sahu S., Devaraj N. K., Angew. Chem. Int. Ed. 2012, 51, 5222–5225; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5312–5315. [Google Scholar]
- 21.
- 21a. Liu C. C., Schultz P. G., Annu. Rev. Biochem. 2010, 79, 413–444; [DOI] [PubMed] [Google Scholar]
- 21b. Dumas A., Lercher L., Spicer C. D., Davis B. G., Chem. Sci. 2015, 6, 50–69; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Wan W., Tharp J. M., Liu W. R., Biochim. Biophys. Acta Proteins Proteomics 2014, 1844, 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kavran J. M., Gundllapalli S., O'Donoghue P., Englert M., Soll D., Steitz T. A., Proc. Natl. Acad. Sci. USA 2007, 104, 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poloukhtine A., Popik V. V., J. Org. Chem. 2003, 68, 7833–7840. [DOI] [PubMed] [Google Scholar]
- 24. Friscourt F., Fahrni C. J., Boons G. J., J. Am. Chem. Soc. 2012, 134, 18809–18815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fottner M., Brunner A. D., Bittl V., Horn-Ghetko D., Jussupow A., Kaila V. R. I., Bremm A., Lang K., Nat. Chem. Biol. 2019, 15, 276–284. [DOI] [PubMed] [Google Scholar]
- 26. Ning X., Guo J., Wolfert M. A., Boons G. J., Angew. Chem. Int. Ed. 2008, 47, 2253–2255; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2285–2287. [Google Scholar]
- 27. Karver M. R., Weissleder R., Hilderbrand S. A., Angew. Chem. Int. Ed. 2012, 51, 920–922; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 944–946. [Google Scholar]
- 28. Martínek M., Filipova L., Galeta J., Ludvikova L., Klan P., Org. Lett. 2016, 18, 4892–4895. [DOI] [PubMed] [Google Scholar]
- 29. Patterson D. M., Nazarova L. A., Xie B., Kamber D. N., Prescher J. A., J. Am. Chem. Soc. 2012, 134, 18638–18643. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Gordon C. G., Mackey J. L., Jewett J. C., Sletten E. M., Houk K. N., Bertozzi C. R., J. Am. Chem. Soc. 2012, 134, 9199–9208; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b. Jewett J. C., Sletten E. M., Bertozzi C. R., J. Am. Chem. Soc. 2010, 132, 3688–3690; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30c. Debets M. F., van Berkel S. S., Schoffelen S., Rutjes F. P., van Hest J. C., van Delft F. L., Chem. Commun. 2010, 46, 97–99. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. McNitt C. D., Popik V. V., Org. Biomol. Chem. 2012, 10, 8200–8202; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31b. Bjerknes M., Cheng H., McNitt C. D., Popik V. V., Bioconjugate Chem. 2017, 28, 1560–1565; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31c. Nainar S., Kubota M., McNitt C., Tran C., Popik V. V., Spitale R. C., J. Am. Chem. Soc. 2017, 139, 8090–8093. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Arumugam S., Popik V. V., J. Org. Chem. 2014, 79, 2702–2708; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32b. Kele P., Mezo G., Achatz D., Wolfbeis O. S., Angew. Chem. Int. Ed. 2009, 48, 344–347; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 350–353. [Google Scholar]
- 33.
- 33a. Sutton D. A., Yu S. H., Steet R., Popik V. V., Chem. Commun. 2016, 52, 553–556; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33b. Sutton D. A., Popik V. V., J. Org. Chem. 2016, 81, 8850–8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Greiss S., Chin J. W., J. Am. Chem. Soc. 2011, 133, 14196–14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu J., Hemphill J., Samanta S., Tsang M., Deiters A., J. Am. Chem. Soc. 2017, 139, 9100–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bianco A., Townsley F. M., Greiss S., Lang K., Chin J. W., Nat. Chem. Biol. 2012, 8, 748–750. [DOI] [PubMed] [Google Scholar]
- 37. McNitt C. D., Cheng H., Ullrich S., Popik V. V., Bjerknes M., J. Am. Chem. Soc. 2017, 139, 14029–14032. [DOI] [PubMed] [Google Scholar]
- 38. Orski S. V., Poloukhtine A. A., Arumugam S., Mao L., Popik V. V., Locklin J., J. Am. Chem. Soc. 2010, 132, 11024–11026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary