

Abstract
Premise
Spore‐bearing plants are capable of dispersing very long distances. However, it is not known if gene flow can prevent genetic divergence in widely distributed taxa. Here we address this issue, and examine systematic relationships at a global geographic scale for the fern genus Pteridium.
Methods
We sampled plants from 100 localities worldwide, and generated nucleotide data from four nuclear genes and two plastid regions. We also examined 2801 single nucleotide polymorphisms detected by a restriction site‐associated DNA approach.
Results
We found evidence for two distinct diploid species and two allotetraploids between them. The “northern” species (Pteridium aquilinum) has distinct groups at the continental scale (Europe, Asia, Africa, and North America). The northern European subspecies pinetorum appears to involve admixture among all of these. A sample from the Hawaiian Islands contained elements of both North American and Asian P. aquilinum. The “southern” species, P. esculentum, shows little genetic differentiation between South American and Australian samples. Components of African genotypes are detected on all continents.
Conclusions
We find evidence of distinct continental‐scale genetic differentiation in Pteridium. However, on top of this is a clear signal of recent hybridization. Thus, spore‐bearing plants are clearly capable of extensive long‐distance gene flow; yet appear to have differentiated genetically at the continental scale. Either gene flow in the past was at a reduced level, or vicariance is possible even in the face of long‐distance gene flow.
Keywords: allotetraploid, fern, hybrids, introgression, long distance gene flow, spore‐bearing plants
Spore‐bearing plants are dispersed by microscopic haploid propagules that can travel vast distances in wind currents. Thus, many species of ferns, lycophytes, fungi, and other such groups are theoretically capable of extensive long‐distance dispersal, some of which may lead to establishment. How much intercontinental gene flow occurs? Is this sufficient to prevent genetic divergence? And at what scale are we able to detect these processes in spore‐bearing plants? Here we address these questions in one of the most widely distributed genera on the planet: Pteridium (bracken). Pteridium Gled. ex Scop. (Dennstaedtiaceae) is a fern genus found in most temperate to tropical regions from almost 70 degrees north (Alaska, Scandinavia, Siberia) to almost 50 degrees south (Argentina, Tasmania, New Zealand) and from sea level to above 3000 m (GBIF Secretariat, 2018). Bracken is a noxious weed in many parts of the world, often invading grazing land and outcompeting forage species (Holm et al., 1997). Furthermore, Pteridium is toxic to humans and other animals; consumption can result in teratogenesis, carcinogenesis, thiamine deficiency, retinal degeneration, and acute poisoning (Alonso‐Amelot and Avendano, 2002; Gil da Costa et al., 2012). Nevertheless, bracken has been used as fuel, thatch, litter, compost, food, medicine, and in the production of potash for the glass, soap, and bleaching industries (Rymer, 1976; McGlone et al., 2005).
Following a landmark revision by Tryon (1941), Pteridium has generally been treated as monospecific, although previously many more species had been described at regional levels (e.g., Ching, 1940). In a later review, Page (1976) noted that more systematic work is needed at a global scale to understand the number of distinct entities within the genus worldwide, and this is still the case. The majority of brackens are diploid (2n = 104: summarized by Page, 1976; Marrs and Watt, 2006). Current treatments have converged to recognize two diploid species (Thomson, 2012; Zhou et al., 2014; Wolf et al., 2015; Schwartsburd et al., 2018). Pteridium aquilinum (L.) Kuhn is predominant in the northern hemisphere whereas P. esculentum (G. Forst.) Cockayne is predominantly southern hemisphere in distribution. Additional to the two widespread diploid species, we recognize two allotetraploid taxa resulting from hybridization of these two species: (1) P. caudatum (L.) Maxon from Central and South America (and parts of North America); and (2) P. semihastatum (Wall. ex J. Agardh) S.B. Andrews from southeastern Asia and northern Australia. Other allopolyploids and/or introgressants may also exist locally where the northern and southern diploid species are, or have been, in contact (Brownsey, 1989; Wolf et al., 2015).
Studies on any taxon at a worldwide scale face the challenge of sampling populations in such a way as to represent diversity effectively, and this is inherently difficult at a global scale. In Pteridium, the problem is exacerbated by inconsistencies in morphological variation and what appears to be phenotypic plasticity for many traits (Page, 1976). In P. aquilinum (Boodle, 1903; Bright, 1928; Tryon, 1941) and in P. esculentum (O'Brien, 1963; Schwartsburd et al., 2014, 2018), environmentally labile characters include features regarded as critical for taxonomic assignment, as well as occasional paedomorphy in the latter species. Furthermore, because of uncertainties in taxonomic delimitations, and because fern spores are so mobile, it is not unexpected to find intermediate phenotypes and apparent hybrids. Plants collected for the present study were subsequently grown at a single site in Sydney, Australia under standardized garden culture in raised tubs preventing rhizome cross‐contamination, to facilitate taxonomic diagnosis and morphometric comparisons. This included 72 accessions (Thomson, 2000) used in both the present study and earlier work by Der et al. (2009) for a global phylogeny based on chloroplast DNA.
The number and rankings of sub‐specific taxa within the diploid brackens are still contentious. In Pteridium aquilinum, both morphometric analysis and molecular evidence from phenetic cluster analysis using the arbitrarily‐primed polymerase chain reaction (A‐P PCR) and inter simple sequence repeats (ISSR) suggest four subspecies in North America, two in Europe and North Africa, two in sub‐Saharan Africa, two in Asia, and one in the Hawaiian Islands (Thomson et al., 2005, 2008; Thomson, 2008). All eleven subspecies of P. aquilinum are represented in the present analysis. Strikingly, each of these taxa falls into one or the other of two major phenotypic groups separated on a set of morphological, phenological, and physiological criteria delimiting the “latiusculum” and “aquilinum” morphotypes respectively (Thomson et al., 2005, 2008). These are features likely to affect growth rate, frond size, laminal subdivision, light penetration, and litter breakdown in relation to adaptation to local climate (Thomson et al., 2005; Thomson, 2008). All four North American diploid brackens have the “latiusculum” morphotype while one subspecies of each morphotype is represented in Europe, sub‐Saharan Africa, and Asia respectively (Thomson et al., 2005). Intermediates consistent with hybridization have been observed in zones of contact or parapatry between the two morphotypes (e.g., Rumsey et al., 1991; Verdcourt, 2000; Karlsson, 2001). Nuclear genomic comparisons using A‐P PCR showed that sequence similarity for contrasting morphotypes from the same geographical region is generally higher than for morphotypes of the same kind from other geographical regions, providing strong evidence of local gene flow between them (Thomson, 2008). The genetic basis of these morphotypes remains unclear pending detailed comparative nuclear genomic analysis, although simple allelic differences do correlate with smaller‐scale polymorphisms in other ferns (Thomson, 2008).
Many of the main groupings discussed above were identified in a global scale analysis of molecular variation focused on chloroplast genes (Der et al., 2009). The results again highlighted the divergence between northern hemisphere (Pteridium aquilinum) and southern hemisphere (P. esculentum) clades, a pattern further corroborated in the work of Zhou and colleagues (Zhou et al., 2014). At the subspecies level, two results obtained by the latter authors are particularly noteworthy. As already noted by Der et al. (2009), chloroplast sequences from P. aquilinum subsp. japonicum (Nakai) A. Love and D. Love and P. aquilinum subsp. pinetorum (C.N. Page and R.R. Mill) J.A. Thomson were not distinguishable, suggesting their synonymy. In contrast, genomic comparison using AP‐PCR and ISSR sequence data clearly separate these two forms (see Fig. 6 in Thomson et al., 2005). Secondly, the chloroplast sequence analysis presented by Zhou et al. (2014) did not separate the two sub‐Saharan forms from each other or from P. aquilinum subsp. aquilinum from western Europe. Additional nuclear genome studies are particularly desirable as chloroplast DNA is maternally inherited in ferns that have been examined (Gastony and Yatskievych, 1992; Vogel et al., 2014; Kuo et al., 2018). Thus, hybrid and polyploid samples usually carry only one chloroplast haplotype and so only the maternal parent can be inferred. Conversely, recent hybrids usually retain nuclear DNA markers from both parents. Here, we addressed the issues of Pteridium systematics and hybridization at a global scale, using data from two distinct approaches. We used Polymerase Chain Reaction (PCR) to amplify chloroplast and nuclear genomic regions. We also prepared a genomic library with double‐digest restriction site‐associated DNA markers (ddRADseq). The objectives of our study are: (1) to infer distinct genetic entities in the genus and their geographic distributions; (2) to infer the number of diploid species; (3) to infer origins of tetraploid samples; and (4) to explore cases of genetic admixture resulting from dispersal between geographic regions.
MATERIALS AND METHODS
We sampled Pteridium from most named taxa (Fig. 1, Appendix 1) except for some recently described forms of P. esculentum from South and Central America (Schwartsburd et al., 2018). We used 101 samples for ddRADseq and 96 for amplicon analysis. Seven samples in ddRADseq samples were not in the amplicon samples (141PWLT, 321KISA, 293CPHW, MALW, CP73, 040CONT, AZ01) and two samples in the amplicon set were not used for ddRADseq set (Wolf_1006 and Wolf_1007). Total genomic DNA was extracted from tissue that had been silica‐dried, freeze‐dried, or pickled in CTAB/NaCl/ascorbate following Thomson (2002), or we extracted from silica‐dried material using the DNeasy Plant Mini kit (QIAGEN, Germantown, Maryland, USA), following the manufacturer's protocol.
Figure 1.
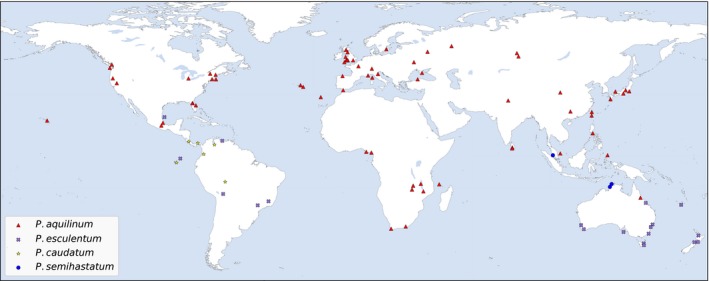
Map showing location of samples used in this study. See also Appendix 1.
Amplicon sequencing
We selected two plastid genes (rpl16 and rps4) and four nuclear encoded genes (ApPEFP_C, CRY2, GapCp, and SQD) based on previous studies on Pteridium (Der et al., 2009; Wolf et al., 2015) and other ferns (Schuettpelz et al., 2008; Rothfels et al., 2013). PCR primer sequences were modified slightly (Table 1). Note that most of the plastid gene sequences are available on GenBank from an earlier study (Der et al., 2009); these were included for quality checking. Because sequence reads are shorter than the gene regions, we divided the genes into pieces (except for the targeted portions of CRY2 and GapCp which are <600 bp; Fig. 2) for a total of 12 amplicons (Table 1). Because amplicons were to be processed for sequencing on the Illumina platform, primers also included sequences that corresponded to the Illumina sequencing primers. Thus, all forward primers also included TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG at the 5′ end and reverse primers also included GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG at the 5′ end. We used a high‐fidelity DNA polymerase (Amplitaq Gold, Thermofisher Scientific, Waltham, Massachusetts, USA) to amplify the above 12 regions. Amplicons from all primer sets were then pooled for each individual and indexed during the standard library preparation. DNA sequencing was performed on a single run of the MiSeq platform with 2 × 300 bp paired‐end reads.
Table 1.
Primers used for amplicon sequencing of Pteridium samples, based on previous studies (Schuettpelz et al., 2008; Der et al., 2009; Wolf et al., 2015). All forward primers included TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG at the 5′ end and all reverse primers included GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG at the 5′ end.
| Marker name | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| rpl16a | ACGCTTAGTGTGCGACTCGTT | GTTCAGATATTACTTGGTGC |
| rpl16b | GGTTAACCACTCAACTCCCG | TCCSCNATGTTGYTTACGAAAT |
| rps4a | ATGTCSCGTTAYCGAGGACCT | GGCTAGCAATGCGATTCACT |
| rps4b | TCTCTTTTGGGGGAAAAGATACC | AGAAGAGCGAAAGGGTTC |
| rps4c | CAGATTACTGAAAAACTAGC | TTACCGAGGGTTCGAATCCCTC |
| SQDa | GCAAGGGTACHAAGGTHATGATCATAGG | GCGTGARTCRTGCACTTTGCTRAGATG |
| SQDb | TGTGGGTGATATATGTGATTTTGAG | CCTTTGCCATAAACTGTAAGGGGGTG |
| ApPEFPa | ATACAGCTGCGGAAAATGCT | GCAATGCTGAAACAACCACA |
| ApPEFPb | ACACACACTATTGGGCACTGA | GAAGTGCAAAATACTAGAGCAGAAAC |
| ApPEFPc | ACTAGTTTGACAGAAACTCAGTACACA | TCAAAGGTGCGAGCTAACAA |
| CRY | AGGATGARYTGGAGAAAGGYAGCAATG | GTRTCCCAGAAATAYTTCATACCCC |
| GAPC | ATYCCAAGYTCAACTGGTGCTGC | GTAACCCAGAATGCCCTTCA |
Figure 2.
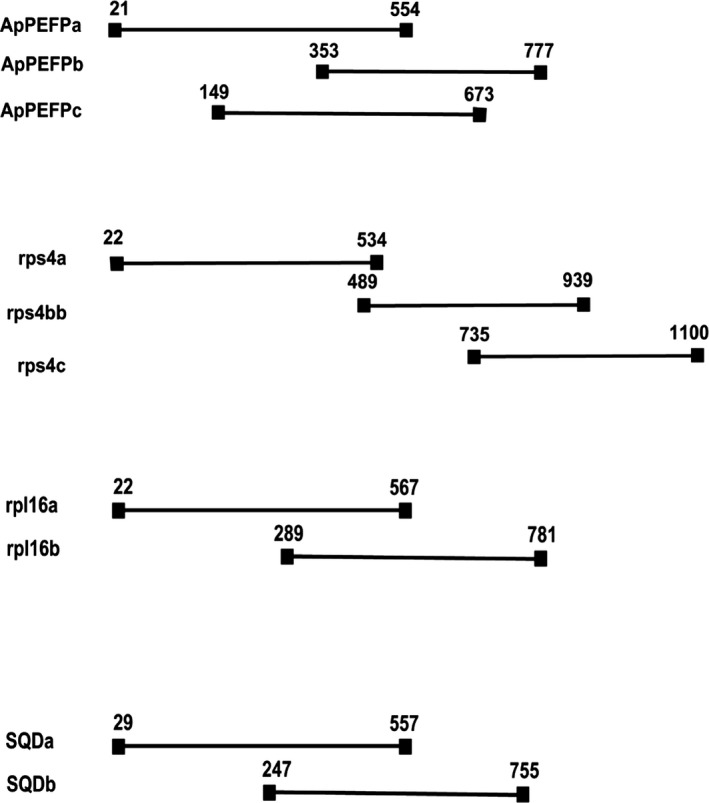
Relative locations of amplicons for each gene.
Amplicon analysis
Paired‐end sequences were first demultiplexed using the sample‐specific index. Sequences within samples were then paired and sorted by amplicon using PANDAseq with a strict quality threshold of 0.9 (Masella et al., 2012). We next used VSEARCH version 2.4.2 (Rognes et al., 2016) to cluster identical sequences within samples, while accounting for variation in read length. To remove possible sequence errors and PCR chimeras we used the usearch unoise algorithm (Edgar, 2016 [preprint]) to produce zero‐radius OTUs (zotus). This resulted in 595 amplicons with sequences. The remaining amplicons were clustered at 99%, or 98% identity using VSEARCH to retrieve sequences from amplicon‐sample combinations not in the zotus set. We estimated haplotype networks using the TCS method (Templeton et al., 1992), implemented in POPART (Leigh and Bryant, 2015). We attempted to phase haplotypes (Patterson et al., 2015) across overlapping amplicons, but could not achieve this for most samples due to a lack of single nucleotide polymorphisms (SNPs) within the regions of overlap, and a higher than expected number of haplotypes in some samples (see results).
ddRADseq
We generated a genomic library following a double‐digestion restriction site‐associated DNA sequencing (ddRADseq) protocol (Gompert et al., 2012; Parchman et al., 2012). We used EcoR1 and Mse1 to generate fragments of genomic DNA that were ligated to indexed oligonucleotides (with an index unique to each individual) on the EcoRI ends of the fragments. A standard, non‐barcoded oligonucleotide was ligated to the MseI ends of the fragmented DNA. Samples were then PCR‐amplified using iproof high‐fidelity DNA polymerase (New England Biolabs, Ipswich, Massachusetts, USA) with primers that overlap the ligated oligonucleotides. To reduce stochastic variation in PCR amplification, all fragments were first mixed with one other individual, which were further amplified in duplicate. The library was then reduced to fragments in the size range of 350‐450 bp using a Blue Pippin (Sage Science, Beverly, Massachusetts, USA). Quality and quantity was verified using TapeStation 2200 (Agilent Technologies, Santa Clara, California, USA). The size‐selected, multiplexed samples were run on a single lane of Illumina HiSeq 4000 with 100 bp single‐end sequencing at Genomic Sequencing and Analysis Facility at the University of Texas at Austin, Texas, USA (GSAF).
ddRADseq data processing
Processing the raw Illumina data was conducted in roughly three steps: (1) creation of a pseudo‐reference genome; (2) alignment of reads and variant calling; and (3) ENTROPY admixture analysis. Raw Illumina reads were demultiplexed and split by individuals using custom Perl scripts (Gompert et al., 2014). Since there is no reference genome available for Pteridium, we constructed a pseudo‐reference using the diploid taxa. We chose to use only the diploid taxa for the reference because they contain the majority of the sequence variation present in the tetraploids (known to be hybrid taxa between the diploids). Our pseudo‐reference was created by first clustering similar sequences for each diploid taxon separately using VSEARCH version 2.4.2 (Rognes et al., 2016); clustering was done at 92% similarity to create centroids for further clustering. This first stage attempts to combine different alleles from a locus, based on high levels of sequence similarity. However, similar paralogs within a diploid genome also tend to get included. Potential paralogs are removed, by eliminating loci that combine at a second clustering phase of at 84% sequence similarity. We then combined loci from the diploid species by selecting the intersection of all sequences from each diploid. After combining, we clustered using VSEARCH again at 84% similarity; the resulting sequences were then used as our pseudo‐reference genome.
Alignment of reads & variant calling
Before calling single nucleotide polymorphism (SNP) variants for all of the included species, we first had to index the pseudo‐reference genome from the previous step, which gives the sequence position points for the alignment. This was done using the INDEX function of BWA (Burrow‐Wheeler Aligner, version 0.7.10; Li and Durbin, 2009). Next we used PicardTools (website http://broadinstitute.github.io/picard/) to create a sequence dictionary; SAMTOOLS (Li et al., 2009) version 1.5 INDEX function was used to create a FASTA index file. For the alignment of reads we used BWA ALN to align all individuals to the pseudo‐reference. We then used the SAMTOOLS functions VIEW, SORT, and INDEX. To call variants we used the GATK (Van der Auwera et al., 2013) HaplotypeCaller (version 3.8.0) because it allows the user to specify ploidy. Because of this option, we called variants separately on the diploids and tetraploids. We used a custom Perl script to filter the resulting variant call format (VCF) files. We filtered for four stringency variables: (1) minimum coverage; (2) minimum number of sequences with an alternative allele; (3) loci fixed for the alternative allele; and (4) minimum mapping quality. Before combining the variants for the diploids and tetraploids we used custom Perl scripts to find the intersection of variants and then combine those intersecting variants from both files. Raw sequence data and details of amplicon analyses are available at website https://digitalcommons.usu.edu/all_datasets/67/.
ENTROPY analysis
We used the population genetics program ENTROPY (Gompert et al., 2014), which is very similar to STRUCTURE (Pritchard et al., 2000). Both are Bayesian, model‐based approaches to examine population structure, and both assume each individual's genome is a mosaic of loci inherited from K source populations. A key difference between them is that STRUCTURE assumes that individual genotypes are known, whereas ENTROPY uses genotype likelihoods calculated from raw sequence data and quality estimates. These genotype likelihoods are then used as an input for ENTROPY, whereas STRUCTURE calculates them repeatedly at each Markov chain Monte Carlo (MCMC) step from prior assignments (Gompert et al., 2014). The output graphic displays the population structure of samples at a range of source population numbers (K), with each sample coded by the proportion of the genome from each source. The first step of this analysis was to convert our VCF variant file to a Genotype Likelihood (GL) file format using a custom Perl script (see below). A second Perl script was used to convert the GL file to a matrix for input to R version 3.4.4 (R Core Team, 2008. We used the R package ADEGENET version 2.1.1 (Jombart, 2008) to perform a discriminant analysis of principal components to find the most likely source population for each individual. This analysis is similar to ENTROPY, but lacks much of the complexity. It is used to seed ENTROPY with starting values, which helps eliminate label swapping, and enables the Bayesian model to converge on the posterior more quickly. We followed the DAPC vignette (website http://adegenet.r-forge.r-project.org/files/tutorial-dapc.pdf), and chose a starting K value of 2.
Using our starting values, we ran ENTROPY for K = 2 through K = 15. Runs were 50,000 iterations with a burn‐in of 10,000 iterations and three independent chains per run. We chose to investigate those K values because K = 2 would be our most parsimonious explanation for the two diploid taxa contributing to the tetraploids; K = 15 would represent each sub‐species and tetraploid as its own separate population. We used a custom R script and functions to visualize our ENTROPY output, and chose K = 5 as the most informative. An R Markdown file with all custom scripts is located at website https://github.com/sylviakinosian/Pteridium_DDRAD-SEQ_Pipeline.
RESULTS
Amplicon haplotypes
We detected 1028 of the possible 1200 amplicons (i.e., 100 samples, each with a maximum of 12 possible amplicons). On average, we detected 1.57 haplotypes per plastid amplicon per sample. For nuclear amplicons we detected fewer haplotypes for presumed diploid samples (2.93 for Pteridium aquilinum, 2.64 for P. esculentum) than tetraploids (7.89 for P. caudatum, and 11.46 for P. semihastatum). However, a few amplicons had very high read depth and a corresponding high number of haplotypes, the highest of which was 49 haplotypes for GapCp in a sample of the tetraploid P. semihastatum (278KMAL). Thus, it is likely that at least some of retained haplotypes included both PCR and sequencing error. Removing sequencing and PCR error requires a tradeoff in losing possible true variants. Our approach has been more to the variant retention end of the spectrum, because we want to avoid losing variants associated with polyploidy and admixture. Broad scale relationships among haplotypes are illustrated by ApPEFP_C and SQD (Figs. 3 and 4), with remaining amplicon haplotype networks in supplementary data. In general, for each amplicon there are two dominant haplotypes, i.e., one from P. esculentum and one from P. aquilinum. All minor haplotypes emerge by a few steps (nucleotide substitutions) from these. Most of the presumed allotetraploids P. semihastatum and P. caudatum combine haplotypes of the two diploid species, or haplotypes that are within one or two steps (Figs. 3 and 4); however, some of the haplotypes found in the allotetraploids were unique to individual samples, and these may represent either haplotypes present in diploids that we failed to sample, or possible PCR chimeras.
Figure 3.
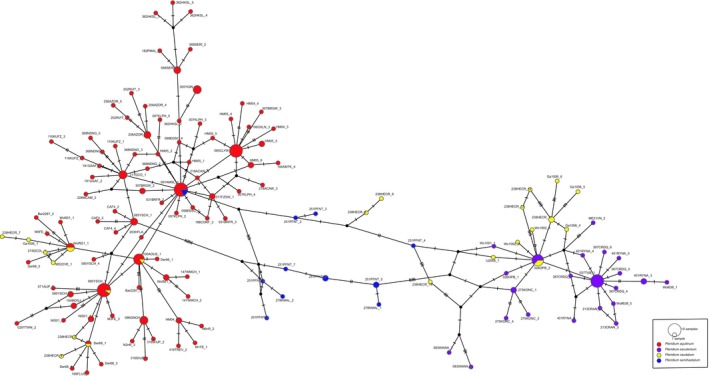
Haplotype network for SQDa.
Figure 4.
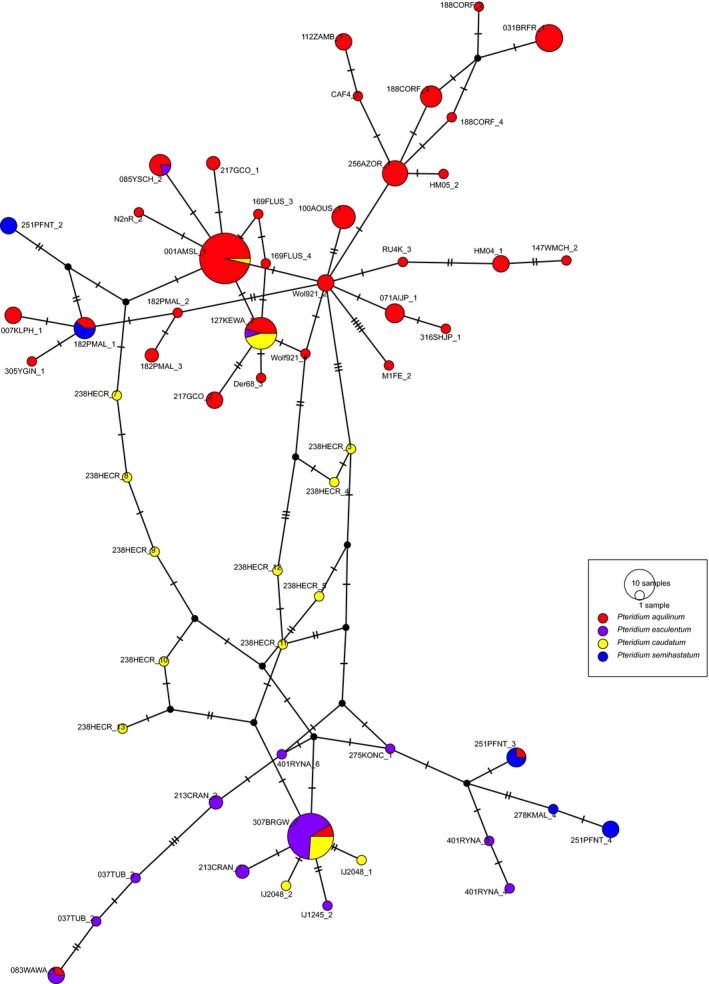
Haplotype network for ApPEFPb.
ENTROPY analysis of ddRADseq genotypes
We independently examined relationships among samples based on ddRADseq data. We detected 2801 SNP loci across diploid samples. With a few notable exceptions, at K = 2 the two species segregate and most presumed allotetraploid samples include contributions from each of the two diploids (Fig. 5). The exceptions include 040CONT (Pteridium esculentum from Tasmania), which grouped with P. aquilinum, and Wolf 921 (P. aquilinum from Connecticut, USA), which had some apparent contribution from P. esculentum at K = 2 (although at K = 5 this sample grouped with remaining North American P. aquilinum). Four of the six P. caudatum samples from Central and South America had equal contributions from P. aquilinum and P. esculentum. One sample from Bolivia (ij_2048) had mostly P. aquilinum at K = 2 (but see below at K = 5). The sample Wolf 1002 (P. caudatum from Galapagos) appeared to be P. aquilinum at K = 2 but a mix of P. aquilinum and P. esculentum at K = 5. All three P. semihastatum samples were an equal mix of P. aquilinum and P. esculentum.
Figure 5.
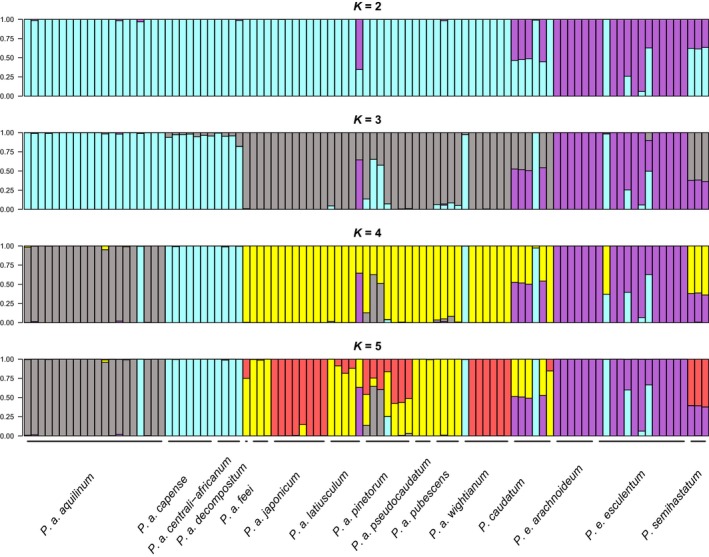
Entropy patterns of genetic cluster contributions for K = 2 through K = 5 .
We detected more geographic and taxonomic resolution at K = 5 (Fig. 6), which appears to have the optimal signal, i.e., K > 5 did not appear to add any signal to the analysis. At K = 5. Pteridium aquilinum splits approximately into continents (Europe, Asia, North America, and Africa). However, P. esculentum in Australia and South America remain part of the same group, not splitting, even at high values of K. This result is consistent with treatment of P. esculentum as one species (Thomson, 2012). Within P. aquilinum, some subspecies appeared to involve admixture. Pteridium aquilinum subsp. decompositum (Gaudich.) Lamoureux ex J.A. Thomson appeared to include contributions from both North America and Asia, consistent with its location between the two, on the Hawaiian Islands. Likewise, P. a. subsp. pinetorum appeared to be a complex admixture involving contributions from Europe, Asia, North America, and even Africa. This is also somewhat consistent with morphology, distribution, and taxonomic treatments (Thomson, 2004). The most widely scattered genomic elements appear to be those of African P. aquilinum (subspecies centrali‐africanum Hieron. ex R.E. Fr. and capense (Thunb.) C. Chr.), components of which were detected in Europe, Australia, and one sample of P. caudatum (ij_2048) from South America.
Figure 6.
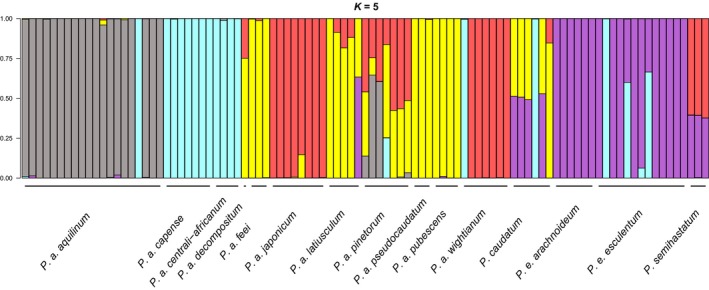
Entropy patterns for K = 5.
DISCUSSION
Choosing the best approach for studying a group of closely related species and their derivatives is often challenging. Here we chose to use two very different data sets in an attempt to detect complementary signals. Amplicon sequencing exploits previously known phylogenetic markers to examine relationships among haplotypes that differ in nucleotide sequence, whereas ddRADseq samples many more genomic regions, in the form of single nucleotide polymorphisms. Filtering PCR error is challenging because an incorrect nucleotide incorporated at an early PCR cycle can result in a haplotype with high read depth (Andrews and Luikart, 2014). Furthermore, because polyploids possess extra gene copies, selection for maintaining extra functional copies may be reduced. This could lead to divergence, adding substantial noise to the analysis. We have been conservative with our filtering scheme, opting to risk retaining false haplotypes rather than eliminate possible real ones. However, we suspect that our amplicon data contain significant errors. Therefore, we have not submitted our amplicon sequences to GenBank, but instead have made them available from Digital Commons: website https://doi.org/10.26078/p64t-9h61. Despite extensive exploration, we are unable to determine the source of additional unexpected haplotypes, which may result from PCR error, despite our attempts to avoid it. We do not think that amplicon sequencing is an altogether ineffective method, although we suggest that longer read lengths, which eliminate the need for assembly and haplotype phasing, may result in higher success (Rothfels et al., 2017). Furthermore, various forms of target sequence capture of nuclear genes have been used very successfully at higher evolutionary levels (e.g., Uribe‐Convers et al., 2016; Dauphin et al., 2018). Here, we detected patterns consistent with previous findings in Pteridium, and the highest depth chloroplast haplotypes were identical to those obtained in earlier studies (Der et al., 2009; Wolf et al., 2015) using Sanger sequencing approaches. Conversely, ddRADseq has not been used previously for studies on Pteridium. We found that the ENTROPY approach revealed patterns than enabled a simpler interpretation of relationships among entities. Overall, we detected consistent patterns between the two approaches. Although both approaches entail similar levels of data analysis, ddRADseq requires considerably less lab work than amplicon sequencing. However, the latter can be streamlined by using alternative approaches to obtaining the data, such as using a target sequence capture protocol (Wolf et al., 2018).
Pteridium aquilinum versus P. esculentum
The strongest signal of genetic subdivision observed in both data sets globally distinguish P. aquilinum and P. esculentum. The same pattern has been observed with chloroplast‐only markers (Der et al., 2009) and on a more focused study on South American Pteridium (Wolf et al., 2015). The paradox surrounding this observation is that given the apparent high levels of admixture at the global scale and the fact that allopolyploids between these taxa have formed, how was it possible for P. aquilinum and P. esculentum to diverge in the first place. The division between the main Pteridium taxa is roughly that of a Laurasian P. aquilinum and a Gondwanan P. esculentum. We posit that these northern and southern groups were able to diverge because of reduced gene flow between them. Prevailing winds in equatorial regions are mostly easterly and westerly (Oort and Yienger, 1996), whereas air flows across the equator are relatively rare (Oort and Yienger, 1996; Wright et al., 2001). For example, the inter‐tropical convergence zone could enable spores to cross the equator, resulting in hybridization and subsequent allopolyploid formation in South America and in Australia/Southeast Asia (Thomson and Alonso‐Amelot, 2002). Another possibility is related to the conditions needed for spore production in Pteridium. In general, Pteridium sporulates more readily in dry conditions. If the climate was moist in the area where Pteridum was growing during the period of separation, then this could have contributed to accelerated divergence (via reduced gene flow) between P. aquilinum and P. esculentum.
Subspecific taxa
Two major features are apparent from the ENTROPY analysis of the ddRADseq data (Figs. 5 and 6). First, genetic distinctions within Pteridium aquilinum that reflect geographic distribution on a global scale are clearly evident at the subspecies level. Second, in agreement with previous studies (summarized by Thomson, 2008 and see Fig. 6 in Thomson et al., 2008), contrasting morphotypes from each continent are more similar genetically to each other than to the same morphotype from other continents.
In Asia, taxa of “aquilinum” and “latiusculum” morphotype are represented respectively by Pteridium aquilinum subsp. wightianum (J. Agardh) W.C. Shieh and japonicum which are seen as genetically close (Figs. 5 and 6 at K = 5,6), as are the sub‐Saharan African forms subsp. capense and centrali‐africanum. In western Europe, subsp. aquilinum is, with one exception, uniform genetically, i.e., all but one sample share a distinct genetic source. In contrast, subsp. pinetorum (“latiusculum” morphotype) shows elements of genetic similarity with subspecies latiusculum (Desv.) Hultén sensu Thomson, aquilinum, and japonicum. The ddRAD‐SEQ analysis supports distinction between the two sub‐Saharan African brackens and subsp. aquilinum (contra Zhou et al., 2014). In North and Central America, subsp. feei (W. Schaffn. ex Fée) J.A. Thomson, Mickel & K. Mehltreter, latiusculum, pubescens (Underw.) J.A. Thomson, Mickel & K. Mehltreter, and pseudocaudatum (Clute) Hultén all have a “latiusculum” phenotype. The close genetic similarity of the subspecies in each continental zone appears consistent with past interbreeding leading to reports of intermediate forms (e.g., Tryon, 1941; Page, 1976; Speer et al., 1999; Mickel and Smith, 2004).
In contrast to the genetic subdivisions observed in Pteridium aquilinum, we detected no clear differences between Australian and South American P. esculentum. This finding is more consistent with the treatment as a single species (Thomson, 2012; Zhou et al., 2014; Schwartsburd et al., 2018) rather than as separate taxa with P. esculentum in Australia and P. arachnoideum in South America (see e.g., Schwartsburd et al., 2014).
Origins of tetraploid taxa
The two tetraploid species, Pteridium caudatum and P. semihastatum, clearly show signs of admixture from the two diploid species in both our amplicon and ddRAD‐SEQ analyses. The only exception was one sample of P. caudatum from Bolivia that has a genetic composition more similar to African Pteridium. Otherwise, the presumed tetraploids were composed of approximately equal contributions from each of the two diploids. Several questions remain regarding the allotetraploid taxa: (1) How many independent origins have occurred? (2) Is allotetraploid formation ongoing? (3) Are tetraploids generated via sterile triploids? and (4) Can such triploids be detected? Evidence from P. caudatum suggests several origins and even possible backcrosses to parents (Wolf et al., 2015). Multiple origins of allotetraploid species is not an uncommon observation (see e.g., Soltis and Soltis, 1991; Zou et al., 2015). Several lines of theory and data suggest that tetraploids often arise in nature via a sterile triploid bridge in seed plants (Harlan and deWet, 1975; Bretagnolle and Thompson, 1995) and in ferns (Gastony, 1986). Initially, an unreduced spore is produced and the resulting diploid gametophyte is most likely to combine with a more common haploid gametophyte. The resulting triploid would be sterile except when it produces an unreduced (3x) spore that again, is more likely to combine with a common haploid gametophyte resulting in a tetraploid, which restores fertility so long as the combining genomes are balanced. To date, only a presumed autotriploid Pteridium has been observed (Sheffield et al., 1993), but allotriploids are likely to be common. The problem is that they are generally difficult to detect without cytological analysis. Future studies would benefit from sampling with a goal of detecting variation in genome size by flow cytometry, from which ploidy may be inferred.
Current patterns of admixture
Genetic groupings in Pteridium, even within P. aquilinum, were distinct at the continental scale. Furthermore, with a few notable exceptions, most samples within named taxa were genetically similar. However, we did detect several cases of possible admixture. One such case was the single sample of P. aquilinum subsp. decompositum from the Hawaiian Islands. The ENTROPY analysis of this sample indicated approximately three‐quarters contribution from North America, and one quarter from Asia, consistent with the location of the islands between these source continents. A similar situation is seen in P. aquilinum subsp. pinetorum, which is distributed in northern parts of Europe. Samples of this taxon have apparent contributions from Western Europe, Asia, and North America. This taxon has always been taxonomically challenging (Thomson, 2004). However, the proximity of P. aquilinum subsp. pinetorum (if looking from above the arctic) to all three source populations is consistent with the genetic results.
One of the major exceptions to consistency in the genetic results was the distribution of sub‐Saharan African genotypes. Samples from Pteridium aquilinum subsp. capense and subsp. centrali‐africanum grouped as a single source in the ENTROPY results at K = 5 (blue samples in Fig. 5). However, this genetic component was also detected in South America, Europe, and Australia. One possibility is that the African genotypes represent a relict southern entity. However, if this were the case then we would expect that admixture would have reduced the distinctness of the genotypes. Alternatively, the occurrence of African genotypes outside Africa more likely represents recent dispersal. If plants in Africa produce spores more readily (because of a drier climate) then it is not unlikely for them to enter airflows both above and below the equator. From there, reaching South America and Australia is not unreasonable. A previous study has detected North American genotypes in western Europe (Rumsey et al., 1991), providing evidence for recent trans‐oceanic dispersal.
Our findings are consistent with those of a chloroplast‐only study of many of the same samples (Der et al., 2009). However, inclusion of nuclear DNA markers enables us to see a more complete picture, including possible patterns of introgression and the origins of allotetraploid taxa. One possible issue that can confound analyses and make interpretations more challenging is possible variation in ploidy. In this study we assumed that taxa were either diploid or allotetraploid. In addition to alternative cytotypes, it is known that recent polyploids can have disrupted meiosis (e.g., Lim et al., 2008; Szadkowski et al., 2010; Chester et al., 2012). This can result in variation in the number of gene copies, a possible explanation for the high number of haplotypes observed in the amplicon data from some samples. In general, the results of the ddRADseq study were easier to summarize and interpret. Nevertheless, combining long sequences with phylogenetic signal, with large numbers of SNP data enables more insight into the evolutionary history of this group of ferns. We find evidence for two distinct (presumably diploid) clades with allotetraploids between them. Samples of the northern Pteridium aquilinum group approximately by continent, whereas the southern clade, P. esculentum, appears to be more homogeneous. Overlaid on this distinct geographic pattern is evidence for several cases of admixture between genetic (and geographic) groups. This is a similar pattern to that seen in scaly tree ferns (Korall and Pryer, 2013). We think that the treatment of Pteridium as two diploid and two allotetraploid taxa is warranted. Future studies would benefit from denser sampling in key geographic areas, especially near the equator and on oceanic islands. Screening for possible triploids, and other cytotypes, using flow cytometry would also provide useful information on the evolution of this ubiquitous group of plants.
ACKNOWLEDGMENTS
This research was supported by the Mary Gunson Memorial Bequest. SPK was supported by a U. S. National Science Foundation Graduate Research Fellowship. JPD was partially supported by faculty start‐up funds from California State University, Fullerton. We thank Elizabeth Robertson for assistance with lab work, and Katherine Downs (NSW) for specimen curation. Thanks also to the University of Utah Center for High‐Performance Computing, particularly Anita Orendt, for providing computational resources for data analyses. We also thank many colleagues for material used in this study (see Appendix 1). Access to specimens collected by A. Dolling, E. A. Ershova, V. V. Korzhenevsky, H. McHaffie, C. N. Page, A. I. Shmakov, and N. I. Shorina for a collaborative taxonomic study of European brackens, a work still in progress, is also gratefully acknowledged. We thank two anonymous reviewers who provided constructive feedback that resulted in a greatly improved manuscript.
Appendix 1. Taxa, sample IDs, collectors, vouchers, and localities for specimens included in this study. Voucher specimens are deposited in the following herbaria: NSW = National Herbarium of New South Wales, Sydney, Australia; UTC = Intermountain Herbarium, Utah State University, Logan, Utah, USA.
Format: Taxon – IDs, Collector_and_Number, Voucher, Country, Latitude, Longitude
Pteridium aquilinum (L.) Kuhn subsp. aquilinum – 017FZSW, T. Reichstein, NSW420392, Switzerland, 47.12, 9.1; 031BRFR, R. Prelli, NSW420391, France, 48.38, 2.27; 065CLYW, M.E. Gillham, NSW420404, United Kingdom, 51.65, ‐3.8; 099EDSC, A.F. Dyer, NSW429393, United Kingdom, 55.5, ‐3.25; 103ASSP, R. Viane, NSW420405, Spain, 43.37, ‐5.83; 106OXLN, M.E. Gillham, NSW420396, United Kingdom, 51.38, ‐0.42; 188CORF, F. Piccoli, NSW420390, France, 42.57, 9.3; 194AMTK, E. Sheffield, NSW420403, Turkey, 41.73, 32.4; 202RVIT, F. Piccoli, NSW420394, Italy, 44.42, 12.2; 218ACAW, C.N. Page, NSW420408, United Kingdom, 53.13, ‐4.27; 226LBSP, B. Capezedo & A.E.Salvo, NSW420374, Spain, 36.18, ‐5.5; 256AZOR, A.C. Jermy 20209, NSW420397, Portugal (Azores), 38.7, ‐27.2; 293CPHW, M.E. Gillham, NSW420380, United Kingdom, 51.35, ‐3.12; 307BRGW, M.E. Gillham, NSW420399, United Kingdom, 51.52, ‐3.58; 336RAMD, R. Viane, NSW420388, Portugal (Madeira), 32.65, ‐16.88; 350NIFR, R. Prelli, NSW420376, France, 43.67, 7.15; AZ01, C.A. Pinto 011, NSW619625, Portugal (Azores), 37.85, ‐25.84; CP73, C.N. Page 37073, NSW619615, United Kingdom, 50.42, ‐4.89; HM05, H.McHaffie 05, NSW617824, United Kingdom, 56.71, ‐4.02; TAUR, V. Korzhenevsky, NSW627463, Ukraine, 44.98, 34.62; Pteridium aquilinum subsp. capense (Thunb.) C. Chr.– 110KUFZ, C.S. McMaster, NSW617745, Zambia, ‐12.45, 30.13; 191GSAF, V. Rashbrook, NSW420332, Republic of South Africa, ‐33.19, 26.32; 217GCOM, V. Rashbrook, NSW420330, Comoros Islands, ‐11.72, 43.37; 228MCAM, Watling, NSW420334, Cameroon, 4.42, 8.87; 353BBSA, P.J. Myerscough, NSW420333, Republic of South Africa, ‐34.35, 18.9; 368NDNG, B.E. Okoli, NSW420328, Nigeria, 4.83, 6.25; 503CAPM_ZOMA, A.C. Chikuni 503, NSW617743, Malawi, ‐15.3, 35.32; Pteridium aquilinum subsp. centrali‐africanum Hieron. ex R.E. Fr.– 112ZAMB_CH3Z, C.S. McMaster 03, NSW617746, Zambia, ‐14.38, 29.53; AKFZ, C.S. McMaster, NSW617747, Zambia, ‐12.45, 30.13; CAF4, C.S. McMaster 04, NSW617757, Zambia, ‐14.38, 29.53; MALW, A.C. Chikuni 500, NSW617739, Malawi, ‐11.46, 34.02; Pteridium aquilinum subsp. decompositum (Gaudich.) Lamoureux ex J.A. Thomson – 292MAHI, C.W. Smith, NSW420357, USA, 20.72, ‐156.155; Pteridium aquilinum subsp . feei (W. Schaffn. ex Fée) J.A. Thomson, Mickel & K. Mehltreter, M1FE, K. Mehltreter 1064, NSW704194, Mexico, 19.55; M3FE, K. Mehltreter 1066, NSW705070, Mexico, 19.55; M5FE, K. Mehltreter1068, NSW705073, Mexico, 18.17; Pteridium aquilinum subsp. japonicum (Nakai) A. Löve & D. Löve – 029TTWN, Shu‐Miaw Chaw, NSW420347, Taiwan, 25.07, 121.02; 071AIJP, R. Hirose, NSW420335, Japan, 34.49, 137.12; 085YSCH, K.U. Kramer, NSW420340, China, 25.35, 110.18; 096GNCH, Yao‐Jia Zhang, NSW420336, China, 35.0, 105.0; 104MOGJ, R. Hirose, NSW420351, Japan, 36.17, 138.25; 113YOJP, T. Kumashiro & H. Tagawa, NSW420356, Japan, 31.58, 130.55; 280CHJP, R. Hirose, NSW420348, Japan, 35.6, 140.1; 316SHJP, I. Miyata, NSW420349, Japan, 35.47, 133.03; Pteridium aquilinum subsp. latiusculum (Desv.) Hultén sensu Thomson (2004)– 143YCCM, E. Klekowski, NSW420315, USA, 41.71, ‐70.23; 147WMCH, W.H. Wagner Jr, NSW420310, USA, 42.15, ‐84.24; 148BRMN, D.S. Barrington, NSW420311, USA, 44.04, ‐70.42; Barr2287, D.S Barrington 2287, UTC247719, USA, 44.583, ‐73.2; Wolf921, P.G. Wolf 921, UTC249671, USA, 41.792, ‐72.217; Pteridium aquilinum subsp. pinetorum (C.N. Page & R.R. Mill) J.A. Thomson – 164KUKR, V. Bogatyr, NSW420402, Ukraine, 50.43, 30.5; AD02, A. Dolling 02, NSW807719, Sweden, 57.04, 16.44; HM04, H. McHaffie 04, NSW617809, United Kingdom, 56.71, ‐4.02; N2nR, E.A. Ershova 02 & A.I. Schmakov, NSW807731, Russia, 53.35, 83.73; N6nR, E.A. Ershova 06 and A.I. Schmakov, NSW807726, Russia, 55.03, 82.92; NIS01, N.I. Shorina, NSW807707, Russia, 55.73, 37.43; RU4K, O.N. Perestoronina, NSW807736, Russia, 58.6, 49.65; Pteridium aquilinum subsp. pseudocaudatum (Clute) Hultén – 169FLUS, E. Sheffield 103, NSW420317, USA, 28.47, ‐80.47; 203HFLA, P.G. Wolf 55 & M.D. Windham, NSW420316, USA, 29.55, ‐82.08; Der68, P. Soltis, UTC249632, USA, 29.519, ‐82.298; Pteridium aquilinum subsp. pubescens (Underw.) J.A. Thomson, Mickel & K. Mehltreter – 100AOUS, D. Barrington, NSW419173, USA, 42.23, ‐122.73; 325OWUS, 325OWUS, J. Schneller, NSW420312, USA, 47.55, ‐124.24; Der66, J.P. Der 66, UTC249629, Canada, 49.264, ‐123.26; Der67, J.P. Der 67, UTC247718, USA, 39.739, ‐120.713; Pteridium aquilinum subsp. wightianum (J. Agardh) W.C. Shieh – 001AMSL, R.D.E. Jayesekara, NSW419562, Sri Lanka, 7.03, 80.59; 007KLPH, J.V. Pancho, NSW420371, Philippines, 14.25, 121.5; 068SERI, T. Partomihardjo, NSW419534, Indonesia, 3.1, 129.05; 182PMAL, R. Kiew, NSW420251, Malaysia, 4.0, 105.0; 305YGIN, S.P. Khullar, NSW419570, India, 30.92, 78.47; 354WFNQ, J.A. Thomson 354, NSW420257, Australia, ‐18.55, 145.8; 362HKSL, M.D. Dassayanake, NSW419571, Sri Lanka, 6.55, 80.48; 416TREV, S.J. Moore, NSW705077, Taiwan, 23.27, 120.96; Pteridium caudatum (L.) Maxon – 238HECR, J. Villalobos‐Salazar & M.Firenczi, NSW420297, Costa Rica, 10.0, ‐84.08; 274QCOL, J.Lenne, NSW420295, Colombia, 3.6, ‐76.48; 328CJPN, G. McPherson, NSW420294, Panama, 9.23, ‐79.5; IJ_2048, I. Jimenez 2048, LPB, Bolivia, ‐10.483, ‐65.567; MD2_2VENZ, M.E. Alonso‐Amelot, NSW505770, Venezuela, 8.42, ‐71.03; Wolf_1002, P. G. Wolf 1002, UTC, Ecuador, ‐0.66375, ‐90.396; Pteridium esculentum subsp. arachnoideum (Kaulf.) J.A. Thomson – 026OPBR, J. Badini, NSW420300, Brazil, ‐20.38, ‐43.5; 144RMEX, B. Perez‐Garcia, NSW420308, Mexico, 20.48, ‐98.44; 317SPBR, M.G.E. Noronha & P.G. Windisch, NSW420303, Brazil, ‐22.42, ‐49.0; IJ_1245, I. Jimenez 1245, LPB, Bolivia, ‐16.65, ‐66.617; ME2_1VNZA, M.E. Alonso‐Amelot, NSW505771, Venezuela, 8.42, ‐69.03; Wolf_1001, UTC, Ecuador, ‐0.626, ‐90.385; Wolf_1006, UTC, Ecuador,‐0.6688,‐90.32385; Wolf_1007, UTC, Ecuador, ‐0.815, ‐91.089; Pteridium esculentum (G.Forst.) Cockayne subsp. esculentum– 037TUBN, J.A. Thomson 037, NSW420871, Australia, ‐36.86, 149.92; 040CONT, J.A. Thomson 040, NSW420278, Australia, ‐41.83, 147.44; 083WAWA, J.A. Thomson 083, NSW420273, Australia, ‐32.51, 115.59; 127KEWA, J.A. Thomson 127, NSW420274, Australia, ‐34.57, 117.01; 141PWLT, J.A. Thomson 141, NSW420880, Australia, ‐42.73, 147.59; 213CRAN, J.A. Thomson 213, NSW420848, Australia, ‐32.15, 151.95; 275KONC, J.A. Thomson 275, NSW420264, New Caledonia, ‐22.17, 166.5; 321KISA, N.A. Walker, NSW420279, Australia, ‐35.98, 137.18; 324HNNZ, C. Surman, NSW420267, New Zealand, ‐41.18, 173.17; 332SVNZ, C. Surman, NSW420289, New Zealand, ‐41.07, 175.08; 387CRDQ, J.A. Thomson 387, NSW420879, Australia, ‐21.2, 148.53; 401RYNA, J.A. Thomson 401, NSW420269, Australia, ‐33.475, 151.045; Wolf638, P.G. Wolf 638, UTC250545, New Zealand, ‐37.776, 175.309; Pteridium semihastatum (Wall. ex J. Agardh) S.B. Andrews – 251PFNT, P. Brocklehurst & G. Wightman, NSW420865, Australia, ‐13.06, 130.24; 278KMAL, Mujamil & T.J. Ho, NSW419554, Malaysia, 3.07, 101.24; 281MLNT, C.R. Dunlop, NSW420423, Australia, ‐11.58, 131.12.
Wolf, P. G. , Rowe C. A., Kinosian S. P., Der J. P., Lockhart P. J., Shepherd L. D., McLenachan P. A., and Thomson J. A.. 2019. Worldwide relationships in the fern genus Pteridium (bracken) based on nuclear genome markers. American Journal of Botany 106(10): 1365–1376.
DATA AVAILABILITY
Raw DNA sequence data and details of amplicon analyses are available at website https://digitalcommons.usu.edu/all_datasets/67/.
An R Markdown file with all custom scripts for the ENTROPY analysis: website https://github.com/sylviakinosian/Pteridium_DDRAD-SEQ_Pipeline.
LITERATURE CITED
- Alonso‐Amelot, M. E. , and Avendano M.. 2002. Human carcinogenesis and bracken fern: A review of the evidence. Current Medicinal Chemistry 9: 675–686. [DOI] [PubMed] [Google Scholar]
- Andrews, K. R. , and Luikart G.. 2014. Recent novel approaches for population genomics data analysis. Molecular Ecology 23: 1661–1667. [DOI] [PubMed] [Google Scholar]
- Boodle, L. A. 1903. The structure of the leaves of the bracken (Pteris aquilina, Linn.) in relation to environment. Botanical Journal of the Linnean Society. Linnean Society of London 35: 659–669. [Google Scholar]
- Bretagnolle, F. , and Thompson J. D.. 1995. Gametes with the somatic chromosome number: Mechanisms of their formation and role in the evolution of autopolyploid plants. New Phytologist 129: 1–22. [DOI] [PubMed] [Google Scholar]
- Bright, D. N. E. 1928. The effects of exposure upon the structure of certain heath‐plants. Journal of Ecology 16: 323–365. [Google Scholar]
- Brownsey, P. J. 1989. The taxonomy of bracken (Pteridium: Dennstaedtiaceae) in Australia. Australian Systematic Botany 2: 113–128. [Google Scholar]
- Chester, M. , Gallagher J. P., Symonds V. V., da Silva A. V. C., Mavrodiev E. V., Leitch A. R., Soltis P. S., and Soltis D. E.. 2012. Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). Proceedings of the National Academy of Sciences, USA 109: 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching, R. C. 1940. On natural classification of the family “Polypodiaceae”. Sunyatsenia 5: 201–268. [Google Scholar]
- Dauphin, B. , Grant J. R., Farrar D. R., and Rothfels C. J.. 2018. Rapid allopolyploid radiation of moonwort ferns (Botrychium; Ophioglossaceae) revealed by PacBio sequencing of homologous and homeologous nuclear regions. Molecular Phylogenetics and Evolution 120: 342–353. [DOI] [PubMed] [Google Scholar]
- Der, J. P. , Thomson J. A., Stratford J. K., and Wolf P. G.. 2009. Global chloroplast phylogeny and biogeography of bracken (Pteridium; Dennstaedtiaceae). American Journal of Botany 96: 1041–1049. [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. 2016. UNOISE2: Improved error‐correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016: 10.1101/081257 (Preprint). [DOI] [Google Scholar]
- Gastony, G. J. 1986. Electrophoretic evidence for the origin of fern species by unreduced spores. American Journal of Botany 73: 1563–1569. [Google Scholar]
- Gastony, G. J. , and Yatskievych G.. 1992. Maternal inheritance of the chloroplast and mitochondrial genomes in Cheilanthoid ferns. American Journal of Botany 79: 716–722. [Google Scholar]
- GBIF Secretariat . 2018. GBIF Backbone Taxonomy. Website 10.15468/39omei, accessed via https://www.gbif.org/species/9744831 on 10 December 2018. [DOI]
- Gil da Costa, R. M. , Bastos M. M. S. M., Oliveira P. A., and Lopes C.. 2012. Bracken‐associated human and animal health hazards: Chemical, biological and pathological evidence. Journal of Hazardous Materials 203–204: 1–12. [DOI] [PubMed] [Google Scholar]
- Gompert, Z. , Lucas L. K., Nice C. C., Fordyce J. A., Forister M. L., and Buerkle C. A.. 2012. Genomic regions with a history of divergent selection affect fitness of hybrids between two butterfly species. Evolution 66: 2167–2181. [DOI] [PubMed] [Google Scholar]
- Gompert, Z. , Lucas L. K., Buerkle C. A., Forister M. L., Fordyce J. A., and Nice C. C.. 2014. Admixture and the organization of genetic diversity in a butterfly species complex revealed through common and rare genetic variants. Molecular Ecology 23: 4555–4573. [DOI] [PubMed] [Google Scholar]
- Harlan, J. R. , and deWet J. M. J.. 1975. On Ö. Winge and prayer: The origins of polyploidy. Botanical Review 41: 361–390. [Google Scholar]
- Holm, L. , Doll J., Holm E., Pancho J., and Herberger J.. 1997. World weeds: Natural histories and distributions. Wiley, New York, New York, USA. [Google Scholar]
- Jombart, T. 2008. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24: 1403–1405. [DOI] [PubMed] [Google Scholar]
- Karlsson, T. 2001. Pteridium In Jonsell B. [ed.], Flora Nordica, vol. 1, 43–47. Royal Swedish Academy of Sciences, Stockholm, Sweden. [Google Scholar]
- Korall, P. , and Pryer K. M.. 2013. Global biogeography of scaly tree ferns (Cyatheaceae): Evidence for Gondwanan vicariance and limited transoceanic dispersal. Journal of Biogeography 41: 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, L.‐Y. , Tang T.‐Y., Li F.‐W., Su H.‐J., Chiou W.‐L., Huang Y.‐M., and Wang C.‐N.. 2018. Organelle genome inheritance in Deparia ferns (Athyriaceae, Aspleniineae, Polypodiales). Frontiers in Plant Science 9: 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh, J. , and Bryant D.. 2015. PopART: Full‐feature software for haplotype network construction. Methods in Ecology and Evolution 6: 1110–1116. [Google Scholar]
- Li, H. , and Durbin R.. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K. Y. , Soltis D. E., Soltis P. S., Tate J., Matyasek R., Srubarova H., Kovarik A., et al. 2008. Rapid chromosome evolution in recently formed polyploids in Tragopogon (Asteraceae). PLoS One 3: e3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs, R. H. , and Watt A. S.. 2006. Biological flora of the British Isles: Pteridium aquilinum (L.) Kuhn. Journal of Ecology 94: 1272–1321. [Google Scholar]
- Masella, A. P. , Bartram A. K., Truszkowski J. M., Brown D. G., and Neufeld J. D.. 2012. PANDAseq: Paired‐end assembler for illumina sequences. BMC Bioinformatics 13: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlone, M. S. , Wilmshurst J. M., and Leach H. M.. 2005. An ecological and historical review of bracken (Pteridium esculentum) in New Zealand, and its cultural significance. New Zealand Journal of Ecology 29: 165–184. [Google Scholar]
- Mickel, J. T. , and Smith A. R.. 2004. The pteridophytes of Mexico. Memoirs of the New York Botanical Garden 88: 1–1055. [Google Scholar]
- O'Brien, T. P. 1963. The morphology and growth of Pteridium aquilinum var. esculentum (Forst.) Kuhn. Annals of Botany 27: 253–267. [Google Scholar]
- Oort, A. H. , and Yienger J. J.. 1996. Observed interannual variability in the Hadley circulation and its connection to ENSO. Journal of Climate 9: 2751–2767. [Google Scholar]
- Page, C. N. 1976. The taxonomy and phytogeography of bracken– a review. Botanical Journal of the Linnean Society. Linnean Society of London 73: 1–34. [Google Scholar]
- Parchman, T. L. , Gompert Z., Mudge J., Schilkey F. D., Benkman C. W., and Buerkle C. A.. 2012. Genome‐wide association genetics of an adaptive trait in lodgepole pine. Molecular Ecology 21: 2991–3005. [DOI] [PubMed] [Google Scholar]
- Patterson, M. , Marschall T., Pisanti N., van Iersel L., Stougie L., Klau G., and Schönhuth A.. 2015. WhatsHap: Weighted haplotype assembly for future‐generation sequencing reads. Journal of Computational Biology 22: 498–509. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens M., and Donnelly P.. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2008. R: A language and environment for Ssatistical computing. R Foundaton for Statistical Computing, Vienna, Austria: Website http://www.R-project.org. [Google Scholar]
- Rognes, T. , Flouri T., Nichols B., Quince C., and Mahé F.. 2016. VSEARCH: A versatile open source tool for metagenomics. PeerJ 4: e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfels, C. J. , Larsson A., Li F. W., Sigel E. M., Huiet L., Burge D. O., Ruhsam M., et al. 2013. Transcriptome‐mining for single‐copy nuclear markers in ferns. PLoS One 8: e76957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfels, C. J. , Pryer K. M., and Li F.‐W.. 2017. Next‐generation polyploid phylogenetics: Rapid resolution of hybrid polyploid complexes using PacBio single‐molecule sequencing. New Phytologist 213: 413–429. [DOI] [PubMed] [Google Scholar]
- Rumsey, F. J. , Sheffield E., and Haufler C. H.. 1991. A re‐assessment of Pteridium aquilinum (L.) Kuhn in Britain. Watsonia 18: 297–301. [Google Scholar]
- Rymer, L. 1976. The history and ethnobotany of bracken. Botanical Journal of the Linnean Society. Linnean Society of London 73: 151–176. [Google Scholar]
- Schuettpelz, E. , Grusz A. L., Windham M. D., and Pryer K. M.. 2008. The utility of nuclear gapCp in resolving polyploid fern origins. Systematic Botany 33: 621–629. [Google Scholar]
- Schwartsburd, P. B. , De Moraes P. L. R., and Lopes‐Mattos K. L. B.. 2014. Recognition of two morpho‐types in eastern South American brackens (Pteridium — Dennstaedtiaceae — Polypodiopsida). Phytotaxa 170: 15. [Google Scholar]
- Schwartsburd, P. B. , Yanez A., and Prado J.. 2018. Formal recognition of six subordinate taxa within the South American bracken fern, Pteridium esculentum (P. esculentum subsp. arachnoideum s.l. — Dennstaedtiaceae), based on morphology and geography. Phytotaxa 333: 22. [Google Scholar]
- Sheffield, E. , Wolf P. G., Rumsey F. J., Robson D. J., Ranker T. A., and Challinor S. M.. 1993. Spatial distribution and reproductive behaviour of a triploid bracken (Pteridium aquilinum) clone in Britain. Annals of Botany 72: 231–237. [Google Scholar]
- Soltis, P. S. , and Soltis D. E.. 1991. Multiple origins of the allotetraploid Tragopogon mirus (Compositae) ‐ rDNA evidence. Systematic Botany 16: 407–413. [Google Scholar]
- Speer, W. D. , Werth C. R., and Hilu K. W.. 1999. Relationships between two infraspecific taxa of Pteridium aquilinum (Dennstaedtiaceae). II. Isozyme evidence. Systematic Botany 23: 313–325. [Google Scholar]
- Szadkowski, E. , Eber F., Huteau V., Lodé M., Huneau C., Belcram H., Coriton O., et al. 2010. The first meiosis of resynthesized Brassica napus, a genome blender. New Phytologist 186: 102–112. [DOI] [PubMed] [Google Scholar]
- Templeton, A. R. , Crandall K. A., and Sing C. F.. 1992. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132: 619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, J. A. 2000. Morphological and genomic diversity in the genus Pteridium (Dennstaedtiaceae). Annals of Botany 85: 77–99. [Google Scholar]
- Thomson, J. A. 2002. An improved non‐cryogenic transport and storage preservative facilitating DNA extraction from ‘difficult’ plants collected at remote sites. Telopea 9: 755–760. [Google Scholar]
- Thomson, J. A. 2004. Towards a taxonomic revision of Pteridium (Dennstaedtiaceae). Telopea 10: 793–803. [Google Scholar]
- Thomson, J. A. 2008. Morphotype and conflicting taxonomies in Pteridium (Dennstaedtiaceae: Pteridophyta). Fern Gazette 18: 101–109. [Google Scholar]
- Thomson, J. A. 2012. Taxonomic status of diploid southern hemisphere brackens (Pteridium: Dennstaedtiaceae). Telopea 14: 43–48. [Google Scholar]
- Thomson, J. A. , and Alonso‐Amelot M. E.. 2002. Clarification of the taxonomic status and relationships of Pteridium caudatum (Dennstaedtiaceae) in Central and South America. Botanical Journal of the Linnean Society. Linnean Society of London 140: 237–248. [Google Scholar]
- Thomson, J. A. , Chikuni A. C., and McMaster C. S.. 2005. The taxonomic status and relationship of bracken ferns (Pteridium: Dennstaedtiaceae) from sub‐Saharan Africa. Botanical Journal of the Linnean Societ. Linnean Society of London 148: 311–321. [Google Scholar]
- Thomson, J. A. , Mickel J. T., and Mehltreter K.. 2008. Taxonomic status and relationships of bracken ferns (Pteridium: Dennstaedtiaceae) of Laurasian affinity in Central and North America. Botanical Journal of the Linnean Society. Linnean Society of London 157: 1–17. [Google Scholar]
- Tryon, R. M. 1941. A revision of the genus Pteridium . Rhodora 43: 1–67. [Google Scholar]
- Uribe‐Convers, S. , Settles M. L., and Tank D. C.. 2016. A phylogenomic approach based on PCR target enrichment and high throughput sequencing: Resolving the diversity within the South American Species of Bartsia L. (Orobanchaceae). PLoS One 11: e0148203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera, G. A. , Carneiro M. O., Hartl C., Poplin R., Del Angel G., Levy‐Moonshine A., Jordan T., et al. 2013. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Current Protocols in Bioinformatics 43: 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdcourt, B. 2000. Pteridium In Beentje H. J. [ed.], Flora of tropical East Africa, Dennstaedtiaceae, 5–8. A.A. Balkema, Rotterdam, Netherlands. [Google Scholar]
- Vogel, J. C. , Russell S. J., Rumsey F. J., Barrett J. A., and Gibby M.. 2014. Evidence for maternal transmission of chloroplast DNA in the genus Asplenium (Aspleniaceae, Pteridophyta). Botanica Acta 111: 247–249. [Google Scholar]
- Wolf, P. G. , Rowe C. A., Der J. P., Schilling M. P., Visger C. J., and Thomson J.. 2015. Origins and diversity of a cosmopolitan fern genus on an island archipelago. AoB Plants 7: plv118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, P. G. , Robison T. A., Johnson M. G., Sundue M. A., Testo W. L., and Rothfels C. J.. 2018. Target sequence capture of nuclear‐encoded genes for phylogenetic analysis in ferns. Applications in Plant Sciences 6: e01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. D. , Yong C. G., Wichman S. R., Dawson J. W., and Gardner R. C.. 2001. Stepping stones to Hawaii: A trans‐equatorial dispersal pathway for Metrosideros (Myrtaceae) inferred from nrDNA (ITS plus ETS). Journal of Biogeography 28: 769–774. [Google Scholar]
- Zhou, S. , Dong W., Chen X., Zhang X.‐C., Wen J., and Schneider H.. 2014. How many species of bracken (Pteridium) are there? Assessing the Chinese brackens using molecular evidence. Taxon 63: 509–521. [Google Scholar]
- Zou, X.‐H. , Du Y.‐S., Tang L., Xu X.‐W., Doyle J. J., Sang T., and Ge S.. 2015. Multiple origins of BBCC allopolyploid species in the rice genus (Oryza). Scientific Reports 5: 14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw DNA sequence data and details of amplicon analyses are available at website https://digitalcommons.usu.edu/all_datasets/67/.
An R Markdown file with all custom scripts for the ENTROPY analysis: website https://github.com/sylviakinosian/Pteridium_DDRAD-SEQ_Pipeline.