

Abstract
The terminal diarsene HAs=AsH ligand attracts special interest concerning its bonding relation in comparison to its isolable relative, ethene. Herein, by the methanolysis of [{Fe(CO)4}As(SiMe3)3] (1) the synthesis of [{Fe(CO)4}(η2‐As2H2)] (2) is reported, containing a parent diarsene as unprecedented side‐on coordinated ligand. Following this synthetic route, also the D‐labeled complex [{Fe(CO)4}(η2‐As2D2)] (2D) could be isolated. The electronic structure and bonding situation of 2 was elucidated by DFT calculations revealing that 2 is best described as an olefin‐like complex. Moreover, the reactivity of 2 towards the Lewis acids [{M(CO)5}(thf)] (M=Cr, W) was investigated, leading to the complexes [Fe(CO)4AsHW(CO)5]2 (3) and [{Fe(CO)4}2AsH{Cr(CO)5}] (4), respectively.
Keywords: arsenic, chromium, diarsene, iron, tungsten
The elusive HAs=AsH, the heavy analogue of ethylene, is side‐on coordinated by a 3d transition metal complex without using kinetic stabilisation. By methanolysis of [{Fe(CO)4}As(SiMe3)3], the complex [{Fe(CO)4}{η2‐As2H2}] was synthesized. DFT calculations show that the bonding mode can be best described by the Dewar–Chatt–Duncanson model. Subsequent reactivity studies reveal that it reacts as an AsH transfer reagent.
The first ethylene complex K[PtCl3(C2H4)], reported by Zeise in 1827, represents the first organometallic transition metal compound ever synthesized.1 Since then, this type of complexes has enormously gained in importance in chemistry as they are for example, intermediates in a broad range of industrial catalytic processes such as hydrogenation, dehydrogenation, or hydrosilylation.2 Simultaneously, they represent fundamental examples of classic bonding modes. Concerning bonding relations, [{Fe(CO)4}(η2‐C2H4)] is a classic prototype of an ethene complex, which, some decades ago, was synthesized by a high pressure synthesis3a or by matrix isolation techniques3b because of its instability at ambient conditions. Analyses showed that its bonding behavior was predominantly the Dewar–Chatt–Duncanson one.3c, 3d In view of the isolobal relationships, it is of principal interest to look at the parent non‐carbon analogues of ethene, for example, the dipnictenes of the general formulae HE=EH (E=N, P, As, Sb, Bi), with respect to their properties as ligands. The study of such parent HE=EH compounds can clarify the true reactivity and structural behavior of these systems without the distortions induced by sterically demanding substituents in RE=ER, which are usually required in order to stabilize the E=E double bonds.4
The diazene HN=NH, which, to the best of our knowledge, is the only isolated hydrogen‐substituted dipnictene known, is stable, though only at very low temperatures (< −165 °C; ΔH f 298=212 kJ mol−1), but well accessible as a ligand in metal complexes such as for example, [μ‐N2H2{Fe(NHS4)}2] (NHS4=2,2‐bis(2‐mercaptophenylthio)diethylamine2−).4c Complexes containing a diphosphene HP=PH ligand are very rare and only three complexes are known so far.5 The parent diarsene HAs=AsH was first mentioned by Davy in 1810 as a product of the reaction of potassium and arsenic in a hydrogen atmosphere.6 However, its existence could not be unequivocally proved until the synthesis and characterization of [(TrenTIPSU)2(As2H2)] (A) (TrenTIPS=N(CH2CH2NSiPri 3)3) was reported containing the diarsene HAs=AsH as bridging ligand between two bulky metal fragments of uranium.7 However, the surroundings of the sterically demanding ligand have a pronounced influence on the properties and geometry of the diarsene ligand. Therefore, the search for synthetic pathways to achieve a simple and mononuclear complex of the diarsene free of any steric restrictions is still ongoing, also to shed light onto the bonding situation of this ethene‐like ligand.
Recently, we investigated the usage of single‐source precursors for the synthesis of transition metal phosphide nanoparticles and could show that complexes containing only labile CO ligands and hydrogen substituents on phosphorus are suitable precursors for the synthesis of size‐ and stoichiometry‐controlled nanoparticles.8 This was demonstrated by the synthesis of FeP nanoparticles with a precise stoichiometry control by starting from the single‐source precursor [Fe(CO)4(PH3)] or [{Fe(CO)3}2(μ‐PH2)]2.8b, 9 Targeting the corresponding As‐containing complexes, we realized that the synthetic pathway for this kind of compounds goes totally different ways and, to our surprise, we achieved the synthesis of the unprecedented diarsene iron carbonyl complex [Fe(CO)4(η2‐As2H2)] (2)—a complex without sterically demanding ligands—in which the diarsene ligand is only stabilized in the coordination sphere of one transition metal. Additionally, reactivity studies towards Lewis acids show that 2 can serve as a source of AsH units, resulting in novel complexes, such as [{Fe(CO)4}AsH{W(CO)5}]2 (3) and [{Fe2(CO)8}AsH{Cr(CO)5}] (4).
Due to the high toxicity and difficulties in handling AsH3 gas, we chose a synthetic strategy in order to avoid the usage of AsH3 gas. The irradiation of As(SiMe3)3 and Fe(CO)5 in n‐pentane affords [{Fe(CO)4}As(SiMe3)3] (1) in 81 % yield (Scheme 1). Solid 1 is air‐sensitive but can be stored at −30 °C under an inert atmosphere for several months, while, at room temperature, it decomposes to an unidentified brown solid.
Scheme 1.
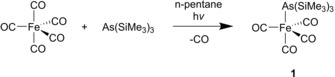
Synthesis of 1 from Fe(CO)5 and As(SiMe3)3.
In the mass spectrum of 1, the molecular ion peak was found. The 1H NMR spectrum of 1 shows a singlet at 0.30 ppm with the corresponding 29Si satellites (2 J Si‐H=6.67 Hz). The infrared spectrum of 1 reveals three CO stretches at 2025, 1940 and 1899 cm−1, being in agreement with an axially coordinating As(SiMe3)3 ligand. According to DFT calculations, the isomer of 1 with the As(SiMe3)3 ligand, in equatorial position, is, with 18.37 kJ mol−1, less stable. Similarly, [{Fe(CO)4}Sb(SiMe3)3] shows three CO stretching modes in the IR spectrum.10 In contrast, for [{Fe(CO)4}P(SiMe3)3], four CO bands were reported and, therefore, an equatorially coordinating ligand was proposed,11 although, in the solid state structure, the P(SiMe3)3 ligand takes the axial position.12 The molecular structure of 1 (Figure 1) shows a trigonal bipyramidally coordinated iron center with the As(SiMe3)3 ligand in axial position as also found for the corresponding Sb compound [{Fe(CO)4}Sb(SiMe3)3].13
Figure 1.
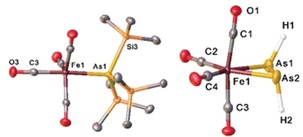
Molecular structure of 1 (left, hydrogen atoms are omitted for clarity) and 2 (right) with ellipsoids set at 50 % probability. Selected distances [Å] and angles [°] of 1: As1−Fe1 2.4406(8), As1−Si1 2.3823(11), As1−Si2 2.3717(12), As1−Si3 2.3780(12) and 2 (major part) [Å]: As1−As2 2.3680(5), As1−Fe1 2.5004(5), As2−Fe1 2.4907(5); C2−Fe1−C4 104.71(12), As1−Fe1−As2 56.645(14).
The methanolysis of 1 in diethylether at low temperatures results in the formation of yellow crystals of 2 in a very low yield, which are surrounded by a large amount of unidentified brown solid (Scheme 2). If the reaction is performed at −80 °C and all volatiles are removed in vacuum at a maximum temperature of −50 °C, and while warming up to room temperature, the residue is sublimated into a cooled Schlenk tube (−80 °C) as a yellow solid of [{Fe(CO)4}{η2‐As2H2}] (2) obtained in 86 % yield.14 Compound 2 is extremely air‐ and moisture‐sensitive and decomposes rapidly at temperatures above −20 °C. Interestingly, under similar reaction conditions, the methanolysis of [{Fe(CO)4}P(SiMe3)3] leads to [{Fe(CO)4}(PH3)] in good yields. Thermolysis or photolysis of [{Fe(CO)4}(PH3)] lead to the formation of the dinuclear complex [{Fe(CO)3}2(μ‐PH2)]2,8b indicating that the hypothetical complex [Fe(CO)4(AsH3)] might form as an intermediate, which, however, is unstable and decomposes to 2 and other unidentified products.
Scheme 2.
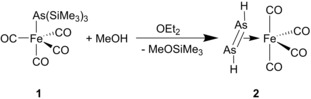
Synthesis of 2 by methanolysis of 1.
Besides the bridging complex [{U(TrenTIPS)}2(μ‐η2:η2‐As2H2)] (A),7 compound 2 is the only known complex containing a side‐on coordinated parent diarsene ligand and here for the first time in a terminal fashion. Complexes containing substituted diarsene ligands such as phenyl groups in [{Fe(CO)4}{η2‐As2Ph2}] (B),15 were reported.16 In A, the diarsene ligand is stabilized between two bulky [U(TrenTIPS)] fragments. Due to the strong back donation of the U(TrenTIPS) fragments, the As−As distance in A is strongly elongated (As−As 2.4102(13) Å) and corresponds to an As−As single bond.7 Hence, 2 is the first complex containing a HAs=AsH ligand stabilized by only one sterically not demanding organometallic fragment.
The solid state structure of 2 (Figure 1) shows a trans‐diarsene HAs=AsH ligand, which coordinates to an Fe(CO)4 fragment in equatorial position (Figure 1). The As−As distances in 2 (two independent molecules 2.3680(5), 2.3683(8) Å) is between an As−As single bond (2.42 Å) and an As=As double bond (2.28 Å) and can be considered as being an elongated double bond.17 This slight elongation, which is due to the back bonding from the Fe(CO)4 moiety,4a is much shorter than in the dinuclear U complex A. The positions of the hydrogen atoms bonded to arsenic were located from the difference Fourier map and subsequently refined with fixed coordinates.18 According to this, the trans isomer of As2H2 is present in the complex and only a minor amount of the cis isomer could be refined in the crystal structure of 2 (cf. Supporting Information). This is in line with the results of DFT calculations showing that the trans isomer of 2 is thermodynamically preferred by 7.60 kJ mol−1 over the cis isomer.19 Similarly, at the same level of theory, the parent trans‐diarsene HAs=AsH is with 11.25 kJ mol−1 more stable than the cis‐isomer.20
The mass spectrum of 2 shows the molecular ion peak at m/z=319.77 and fragments according to the subsequent loss of CO and/or H. According to DFT calculations, the symmetric and asymmetric As−H stretches in the IR spectra are situated at 2136 cm−1 and 2149 cm−1, respectively, and hence are obscured by the CO absorption bands. In order to unambiguously attribute the As−H stretch in the IR spectrum of 2, we prepared the deuterium‐labeled derivative [{Fe(CO)4}{η2‐As2D2}] (2D) by the reaction of 1 with deuterated methanol. The infrared spectrum of 2D shows four signals for the CO ligands at 1912, 1999, 2053, and 2124 cm−1, which are in agreement with the calculated values and, additionally, a new absorption at 1473 cm−1, which is attributed to the As−D vibration and in line with the expected shift due to the mass difference between hydrogen and deuterium (νH/νD=1.41). This is also in agreement with the observations for A, in which the As−H stretches appear at 2030 cm−1 and the As−D ones at 1410–1490 cm−1.7
Due to the high sensitivity of 2, a partial decomposition takes place when it is dissolved in deuterated solvents and, in the 1H NMR spectrum, among the signals corresponding to 2, further signals for unidentified decomposition products are present. The partial decomposition of 2 when being dissolved in organic solvents cannot be suppressed even when being dissolved at low temperatures. 2H NMR spectroscopic investigations of 2D show a main resonance signal at δ=1.33 ppm, which is attributed to the As2D2 unit. For A, no resonance signal for the hydrogen atoms attached to the arsenic atoms could be detected, due to the paramagnetic nature of the complex.
In order to clarify the electronic structure of 2, DFT calculations at the B3LYP/def2‐TZVP level of theory were performed. The orbital interaction diagram (Figure 2) shows that the π orbitals of the As2H2 ligand are stabilized while the As−As σ bond is slightly destabilized upon coordination. The overlap of the Highest Occupied Molecular Orbital (HOMO) of the As2H2 ligand with the Lowest Unoccupied Molecular Orbital (LUMO) of the Fe(CO)4 fragment represents the π‐donor coordination mode. The Fe‐As backbonding becomes obvious by the overlap of the HOMO orbital of the Fe(CO)4 fragment with the LUMO orbital (which is the As=As π* orbital) of the As2H2 ligand, which leads to the HOMO−1 orbital in 2. Considering the main fragment contribution to the HOMO−1 and HOMO−2 orbitals, complex 2 can be best described as an olefin‐like complex. The NBO analysis21 shows NBO orbits corresponding to an As−As bond, which is realized by almost pure p orbitals (sp11 hybrid orbital) and two Fe−As bonds. The occupancy of the latter two is 1.75e and polarized toward iron. The lone pair of the arsenic atoms are of high s character (sp0.3 hybrid orbitals). The Wiberg Bond Indices show an As−As bond order of 1.18, which indicates the multiple bond character and a bond order of 0.73 for the Fe‐As bonds. For comparison, the WBI of the As−Fe bond in 1 is 0.58, while the WBI of the As−As bond in the free trans‐As2H2 is 2.05. Further, the electronic structure of 2 was investigated by the Bader's theory of atoms in molecules (AIM).22 Bond Critical Points (BCPs) could be located corresponding to the As−As and two As−Fe bonds with electron densities of 0.090 e a.u.−3 and 0.057 e a.u.−3, respectively. The Laplacian at the BCP corresponding to the As−As bond is negative (∇2 ρ=−0.046 e a.u.−5), while the one at the BCPs corresponding to the As−Fe bond is positive (∇2 ρ=0.046 e a.u.−5). The positive Laplacian at the BCPs of the Fe−As bond and the negative energy density values (−0.273 hartee a.u.−3) indicate a moderately polar Fe−As bond. The pronounced ellipticity at these BCPs of 0.24 and 0.28 for the BCPs corresponding to the As−As as well as the As−Fe bonds, respectively, indicates the concentration of the electron density in the FeAs2 plane. Based on the WBIs, complex 2 can be described as a diarsa‐metallacyclopropane while, based on the fragment contribution to the molecular orbitals, complex 2 can be better described with a Dewar–Chatt–Duncanson model. The latter is also confirmed by the torsion angles of the As2H2 unit being close to 90°.3c, 3d, 18
Figure 2.
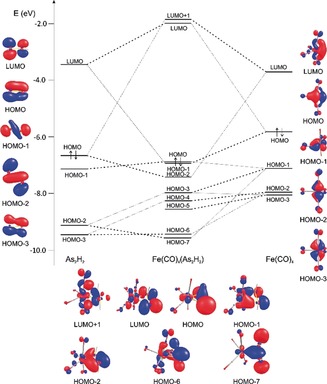
Orbital interaction diagram as well as molecular and fragment orbitals in 2, calculated at the B3LYP/def2‐TZVP level. The lines drawn in bold represent the main character of the corresponding molecular orbital.
To investigate the reactivity of 2, it was reacted with the Lewis acids [M(CO)5(thf)] (M=Cr, W) at −80 °C in THF. From the reaction with [W(CO)5(thf)], [{Fe(CO)4}AsH{W(CO)5}]2 (3) (Figure 3) was isolated in 43 % yield. 3 represents the first compound with a planar As2Fe2 ring that has been structurally characterized. A related substituted compound [{(C5H5)Fe(CO)}{As(CH3)2}]2, has only been characterized by spectroscopic methods (1H NMR and IR) and elemental analysis, but not by single crystal X‐ray diffractions.23 A compound with a butterfly‐like structure, containing an Fe−Fe bond, was reported for [{Fe2(CO)6}{μ‐As(CH3)2}2].24 Performing the reaction of [W(CO)5(thf)] with 2D leads to [{Fe(CO)4}AsD{W(CO)5}]2 (3D). The 2H NMR spectrum of 3D shows a singlet at 3.05 ppm, corresponding to the As−D unit. In the 1H NMR spectrum of 3, the signal of the As−H unit appears at 3.12 ppm (note that the spectra were recorded in different solvents). In the EI mass spectrum of 3D, the molecular ion peak could not be detected, but several peaks corresponding to fragments such as {W(CO)5}AsDFe2(CO)7 + and {W(CO)5}AsDFe+. The infrared spectra of 3 show four CO stretches at 1940, 2048, 2067 and 2095 cm−1. A single As−D stretch appears at 1422 cm−1 for 3D, whereas, in the IR spectrum of 3, the As−H stretch is obscured by the CO absorption bands.
Figure 3.
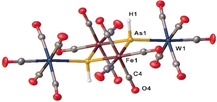
Molecular structure of 3, with ellipsoids at 50 % probability. Selected distances [Å]: As1−Fe1 2.4728(7), As1−Fe1′ 2.4787(8), As1−W1 2.6380(5).
The molecular structure of 3 (Figure 3) shows a planar As2Fe2 ring. A hydrogen atom is attached to each arsenic atom, which additionally coordinates to a W(CO)5 fragment. The W(CO)5 fragments are tilted by 30° out of the plane of the Fe2As2 ring. The Fe−As distances in 3 (As1‐Fe1 2.4728(7) and As1−Fe1′ 2.4787(8) Å) are slightly shorter than the As−Fe distances in 2 (As1‐Fe1 2.5004(5), As2−Fe1 2.4907(5) Å) but slightly longer than the Fe−As distance in 1 (2.4406(8) Å).
For the reaction of [{Fe(CO)4}(η2‐As2H2}] (2) with [{Cr(CO)5}(thf)], an isostructural compound to 3 was expected. Instead, however, the new compound [{Fe2(CO)8}AsH{Cr(CO)5}] (4) (Scheme 3) is obtained in 20 % yield after column chromatographic workup of the reaction mixture. Besides crystals of 4, very few crystals of [Fe3(CO)9{μ3‐AsCr(CO)5}2] (5) were obtained and identified by single crystal X‐ray diffractions and by mass spectrometry. The structure of the latter compound 5 was already reported.25
Scheme 3.
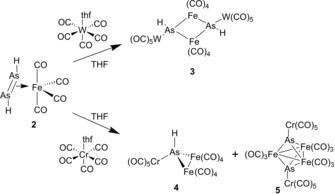
Reactivity of 2 towards the Lewis acids [M(CO)5thf] (M=Cr, W).
The infrared spectrum of 4 shows 5 stretches at 1940, 2034, 2054, 2065, and 2114 cm−1 for the CO and As−H vibrations. In the 1H NMR spectrum of 4, a singlet at 3.9 ppm can be attributed to the H atom of the AsH unit. In order to clearly attribute the As−H stretches and hence to prove the presence of the AsH unit, we performed the reaction of 2D with [Cr(CO)5(thf)]. To our surprise, the same reaction product 4 was isolated instead of the expected deuterated complex [{Fe2(CO)8}AsD{Cr(CO)5}]. 2H NMR and IR spectroscopic investigations indicate that the H/D exchange occurs during the reaction and not during the column chromatographic workup, since the resonance signal corresponding to [{Fe2(CO)8}AsD{Cr(CO)5}] is not present in the 2H NMR spectrum of the reaction mixture. In the EI mass spectrum of 4, the molecular ion peak could be detected at m/z=603.68 as well as peaks corresponding to the consecutive loss of all carbonyl groups, the hydrogen and the arsenic atom.
In the solid state structure of 4 (Figure 4), an AsH ligand coordinates to a Cr(CO)5 and an Fe2(CO)8 fragment. The coordination geometry around the arsenic atom is tetrahedrally distorted. A phenyl‐substituted complex [{Fe2(CO)8]AsPh{Cr(CO)5}] (C) with a related structural motif was reported.26 Moreover, in the literature, there are only a few complexes described containing AsH ligands in which the arsenic is tetrahedrally coordinated such as [{HOs2(CO)7}AsH{HOs(CO)4}] or [Et4N][HAs{Fe2(CO)6(μ‐CO)(μ‐H)}{Fe(CO)4}].27
Figure 4.
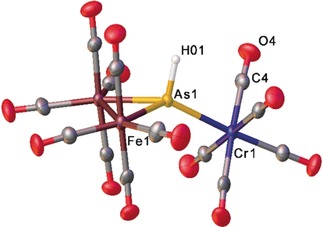
Molecular structure of 4 with ellipsoids set at 50 % probability. Selected distances [Å]: As1−Fe1 2.3833(5), As1−Fe2 2.3873(5), As1−Cr1 2.4855(5).
In summary, we were able to isolate and characterize the very sensitive complex 2, containing the unprecedented parent diarsene HAs=AsH as side‐on coordinating ligand, which is stabilized in the coordination sphere of a mononuclear Fe(CO)4 fragment without further stabilization by bulky organic substituents. Although 2 is highly sensitive and only stable at low temperatures, it could be comprehensively characterized. DFT calculations show that 2 is best described as an olefin‐like complex in which the As=As double bond character is preserved. Further, the initial reactivity of 2 towards Lewis acids was investigated leading to new products containing As−H ligands. Among others, complex 3 is the first structurally proved representative containing a planar Fe2As2 unit.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the project Sche 384/32‐2.
R. Rund, G. Balázs, M. Bodensteiner, M. Scheer, Angew. Chem. Int. Ed. 2019, 58, 16092.
Dedicated to Professor H. Schmidbaur on the occasion of his 85th birthday
Contributor Information
Reinhard Rund, https://www.uni-regensburg.de/chemistry-pharmacy/inorganic-chemistry-scheer/index.html.
Prof. Dr. Manfred Scheer, Email: manfred.scheer@ur.de.
References
- 1. Zeise W. C., Ann. Phys. 1831, 97, 497–541. [Google Scholar]
- 2.
- 2a. Koga N., Daniel C., Han J., Fu X. Y., Morokuma K., J. Am. Chem. Soc. 1987, 109, 3455–3456; [Google Scholar]
- 2b. Niu S., Hall M. B., J. Am. Chem. Soc. 1999, 121, 3992–3999; [Google Scholar]
- 2c. Bergens S. H., Noheda P., Whelan J., Bosnich B., J. Am. Chem. Soc. 1992, 114, 2128–2135; [Google Scholar]
- 2d. Weissermel K., Arpe H.-J., Industrial Organic Chemistry, Vol. 3, VCH, Weinheim, 1997. [Google Scholar]
- 3.
- 3a. Murdoch H. D., Weiss E., Helv. Chim. Acta 1963, 46, 1588–1594; [Google Scholar]
- 3b. Yamada Y., Tominaga T., J. Radioanal. Nucl. Chem. 1988, 126, 455–466; [Google Scholar]
- 3c. Dewar M. J. S., Bull. Soc. Chim. Fr. 1951, 18, C71–C79; [Google Scholar]
- 3d. Chatt J., Duncanson L. A., J. Chem. Soc. 1953, 2939–2947. [Google Scholar]
- 4.
- 4a. Guo J. D., Liptrot D. J., Nagase S., Power P. P., Chem. Sci. 2015, 6, 6235–6244; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Fischer R. C., Power P. P., Chem. Rev. 2010, 110, 3877–3923; [DOI] [PubMed] [Google Scholar]
- 4c. Power P. P., Chem. Rev. 1999, 99, 3463–3504; [DOI] [PubMed] [Google Scholar]
- 4d. Weber L., Chem. Rev. 1992, 92, 1839; [Google Scholar]
- 4e. Rosengren K., Pimentel G. C., J. Chem. Phys. 1965, 43, 507–516; [Google Scholar]
- 4f. Sellmann D., Soglowek W., Knoch F., Moll M., Angew. Chem. Int. Ed. Engl. 1989, 28, 1271–1272; [Google Scholar]; Angew. Chem. 1989, 101, 1244–1245. [Google Scholar]
- 5.
- 5a. Etkin N., Benson M. T., Courtenay S., McGlinchey M. J., Bain A. D., Stephan D. W., Organometallics 1997, 16, 3504–3510; [Google Scholar]
- 5b. Green J. C., Green M. L. H., Morris G. E., J. Chem. Soc. Chem. Commun. 1974, 212–213; [Google Scholar]
- 5c. Fermin M. C., Ho J., Stephan D. W., Organometallics 1995, 14, 4247–4256. [Google Scholar]
- 6. Davy H., Philos. Trans. R. Soc. London 1810, 100, 16–74. [Google Scholar]
- 7. Gardner B. M., Balázs G., Scheer M., Wooles A. J., Tuna F., McInnes E. J. L., McMaster J., Lewis W., Blake A. J., Liddle S. T., Angew. Chem. Int. Ed. 2015, 54, 15250–15254; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15465–15469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Bauer S., Hunger C., Bodensteiner M., Ojo W.-S., Cros-Gagneux A., Chaudret B., Nayral C., Delpech F., Scheer M., Inorg. Chem. 2014, 53, 11438–11446; [DOI] [PubMed] [Google Scholar]
- 8b. Hunger C., Ojo W.-S., Bauer S., Xu S., Zabel M., Chaudret B., Lacroix L.-M., Scheer M., Nayral C., Delpech F., Chem. Commun. 2013, 49, 11788–11790. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Schäfer H., Leske W., Z. Anorg. Allg. Chem. 1987, 552, 50–68; [Google Scholar]
- 9b. Dreher C., Zabel M., Bodensteiner M., Scheer M., Organometallics 2010, 29, 5187–5191. [Google Scholar]
- 10. Breunig H. J., Polyhedron 1984, 3, 757–758. [Google Scholar]
- 11. Schumann H., Stelzer O., J. Organomet. Chem. 1968, 13, 25–27. [Google Scholar]
- 12. Barron A. R., Cowley A. H., Nunn C. M., Acta Crystallogr. Sect. C 1988, 44, 750–751. [Google Scholar]
- 13.The synthesis was reported: see ref [10]. A new synthesis and the crystal structure analysis are reported in the Supporting Information.
- 14.Yield related to arsenic. Due to low stability and the formation of byproducts, this yield can vary between 50 and 86 %.
- 15. Jacob M., Weiss E., J. Organomet. Chem. 1978, 153, 31–38. [Google Scholar]
- 16.For selected examples see:
- 16a. Fenske D., Merzweiler K., Angew. Chem. Int. Ed. Engl. 1984, 23, 635–637; [Google Scholar]; Angew. Chem. 1984, 96, 600–602; [Google Scholar]
- 16b. Grobe J., Karst A., Krebs B., Läge M., Würthwein E.-U., Z. Anorg. Allg. Chem. 2006, 632, 599–608. [Google Scholar]
- 17.
- 17a. Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 186–197; [DOI] [PubMed] [Google Scholar]
- 17b. Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 12770–12779. [DOI] [PubMed] [Google Scholar]
- 18.Crystallographic details, including details about the location of the hydrogen atoms and their refinement are given in the Supporting Information.
- 19.For details see Supporting Information.
- 20.According to calculations at different levels of theory, the trans isomer of the parent diarsene HAs=AsH is more stable than the cis isomer. See for example:
- 20a. Schöller W. W., Begemann C., Tubbesing U., Strutwolf J., J. Chem. Soc. Faraday Trans. 1997, 93, 2957; [Google Scholar]
- 20b. Nagase S., Suzuki S., Kurakake T., J. Chem. Soc. Chem. Commun. 1990, 1724–1726; [Google Scholar]
- 20c. Lai C.-H., Su M.-D., J. Comput. Chem. 2008, 29, 2487–2499. [DOI] [PubMed] [Google Scholar]
- 21.NBO 6.0.: E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis, F. Weinhold (Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013); http://nbo6.chem.wisc.edu/.
- 22.
- 22a. Bader R. F. W., Atoms in Molecules: A Quantum Theory, Oxford, Clarendon Press, 1990; [Google Scholar]
- 22b. Bader R. F. W., Acc. Chem. Res. 1985, 18–19; [Google Scholar]
- 22c. Bader R. F. W., Chem. Rev. 1991, 91, 893–928. [Google Scholar]
- 23. Hayter R. G., J. Am. Chem. Soc. 1963, 85, 3120–3124. [Google Scholar]
- 24. Keller E., Vahrenkamp H., Chem. Ber. 1977, 110, 430–440. [Google Scholar]
- 25. Collins B. E., Koide Y., Schauer C. K., White P. S., Inorg. Chem. 1997, 36, 6172–6183. [Google Scholar]
- 26. Huttner G., Mohr G., Friedrich P., Schmid H. G., J. Organomet. Chem. 1978, 160, 59–66. [Google Scholar]
- 27.
- 27a. Guldner K., Johnson B. F. G., Lewis J., J. Organomet. Chem. 1988, 355, 419–425; [Google Scholar]
- 27b. Schipper D. E., Young B. E., Whitmire K. H., Organometallics 2016, 35, 471–483. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary