

Abstract
Indoles are privileged heterocycles found in many biologically active pharmaceuticals and natural products. However, the selective functionalization of the benzenoid moiety in indoles in preference to the more reactive pyrrolic unit is a significant challenge. Herein we report that N‐acyl directing groups enable the C7‐selective C−H borylation of indoles using just BBr3. This transformation shows some functional‐group tolerance and notably proceeds with C6 substituted indoles. The directing group can be readily removed in situ and the products isolated as the pinacol boronate esters. Acyl‐directed electrophilic borylation can be extended to carbazoles and anilines with excellent ortho selectivity. 4‐amino‐indoles are amenable to this process, with acyl group installation and directed electrophilic C−H borylation enabling selective formation of C5‐BPin‐indoles.
Keywords: boranes, borenium, borylation, directing groups, electrophilic aromatic substitution
B‐directed: Acyl‐directed electrophilic C−H borylation provides access to novel C5 and C7 borylated indoles using just BBr3 as the borylating agent via the formation of six‐membered boracycles.
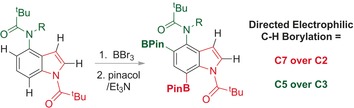
C−H borylation is a powerful methodology to form synthetically versatile C−B bonds.1 Numerous methods have been developed, with iridium‐catalysed C−H borylation one of the most notable.1 This method functionalises the pharmaceutically important heteroarene indole at the C2‐position.2 Alternative indole C−H borylation methods include electrophilic borylation (dominated by electronic effects)3 and C−H lithiation/borylation (controlled by C−H acidity).4 However, these also functionalise the pyrrole unit (at C3 and C2, respectively, Scheme 1 top left). Indole C−H borylation that occurs selectively on the less reactive benzenoid unit is desirable, including for accessing C5 and C7‐functionalised indoles which are motifs found in many biologically active natural products and pharmaceuticals (e.g. chloropeptin I, teleocidins, hippadine, tiplaxtinin).5 To date the selective C5−H/C7−H borylation of indoles in the presence of C2−H/C3−H requires prefunctionalised indoles (e.g. halide at C5/C7) or functionalisation of the more reactive C2−H/C3−H site prior to C5−H/C7−H borylation and then unmasking of the C2−H/C3−H.6 To the best of our knowledge, one example of directed iridium‐catalysed C−H borylation7 provides the only exception to these requirements (Scheme 1, middle left).8 This process while notable uses ruthenium and iridium catalysts and substrates containing C6 substituents are not viable (6,7‐disubstituted indoles are also bioactive motifs for example, indole isosteres of combrestatins).5, 6c, 9 Therefore a simple, precious metal free route for the C−H borylation of indoles that is selective for: (i) C7 (over C2), including for C6 substituted indoles, and (ii) C5 (over C3), would be highly notable particularly if using a readily removed directing group.
Scheme 1.
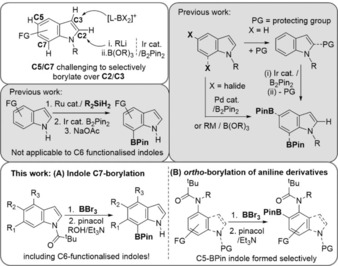
Select previous work on the borylation of indoles, specifically borylation reactions occurring at the C5 and C7 positions. Bottom inset, this work on acyl‐directed electrophilic C−H borylation at C5 and C7 using BBr3.
C−H borylation using BX3 (X=Cl or Br) is an attractive method to form organoboranes,3a, 3b, 10, 11 and directed borylation using BX3 has proved to be a powerful route to form B−C bonds for organic materials applications.12 Directed electrophilic C−H borylation is dominated by directing R2N‐ or N‐heterocycle groups with borylation generally forming six membered boracycles preferentially over other ring sizes.13 The extension of C−H borylation using BX3 to the C5/C7 positions of indoles would be highly attractive. However, this requires conditions that disfavour electrophilic C3−H borylation (which is relatively facile) and a directing group that: (i) is compatible with BX3; (ii) enables selective borylation at the desired position; (iii) is readily deprotected post C−H borylation. Transition metal‐catalysed C7−H indole functionalisation often uses bulky phosphinyl directing groups installed at N1 which are challenging to remove (requiring refluxing with LiAlH4),5, 6a, 14 however, in limited cases N‐acyl directing groups also have been used5, 15 and these are more readily removed. Herein we demonstrate that N‐acyl directing groups are compatible with BBr3 and lead to C7−H borylation of indoles generating useful C7‐BPin products on work up (Scheme 1, bottom). Notably, borylation is compatible with C6 substituted indoles in contrast to the iridium‐catalysed process. Furthermore, acyl directing groups also enable ortho C−H borylation of anilines using BBr3, including of 4‐amino indoles which affords C5‐BPin indoles.
To guide our selection of appropriate acyl directing groups initially we probed the thermodynamic outcome from indole borylation at C2 and C7 computationally. Notably, the C7 borylated isomer is calculated to be thermodynamically favoured over the C2 (Scheme 2) isomer in all cases, this is attributed to (i) the differing degrees of steric clash between R and the C7−H and C2−H hydrogens (as previously noted);16 (ii) the differing bond angles in 5 and 6‐membered boracycles, with the former leading to compressed O‐B‐C angles relative to the latter (which approaches the ideal for tetrahedral boron, Scheme 2). C7‐borylation is also calculated to be the kinetic outcome (for R=tBu) based on borylation proceeding via acyl→BBr3 formation, [acyl→BBr2]+ formation and then SEAr (see SI).
Scheme 2.
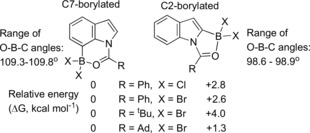
Relative energy of C2 and C7 borylated isomers calculated at the M06‐2X/6‐311G(d,p) level with a polarizable continuum model of DCM.
Based on these calculations the borylation of 1‐benzoyl‐indole, 1 a, and 1‐pivaloyl‐indole, 2 a, was targeted. To disfavour borenium cation formation and indole C3 borylation conditions were required avoiding coordinating exogenous base.3b, 17 For example, using reagents which lead to [(amine)BX2]+ cations (e.g. BBr3/ 2,6‐lutidine)11 led to the borylation of 2 a at C3 selectively (see SI) with no C2 or C7 borylation observed (Scheme 3). Therefore, BCl3 and BBr3 in the absence of base were utilised.
Scheme 3.
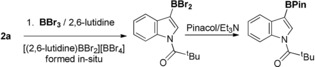
Borylation of 2 a under conditions that generate [(amine)BX2]+ borenium cations.
While BCl3 resulted in no borylation of 1 a and 2 a, with BBr3 C−B bond formation proceeded with both these indoles, forming products with δ 11B≈0 ppm (distinct to amide‐BBr3 adducts for which δ 11B is ca. −10 ppm). Subsequent addition of pinacol/Et3N led to formation of the pinacol boronate esters 3 a–5 a (Scheme 4). The disparity between BCl3/ BBr3 also has been observed in N‐heterocycle directed borylation and the origin of this has been examined previously.18 The regioselectivity of borylation using BBr3 was assessed by NMR spectroscopy in situ and post pinacol protection. This revealed that borylation of 1 a led to C7 and C2 borylation products (with 3 a and 4 a formed in a 4:1 ratio). Borylation of 2 a with BBr3 led to more selective C7 borylation, with compound 5 a‐BBr2 the major borylated product observed in situ (in ca. 85–90 % conversion, see SI). 5 a‐BBr2 and 6 a‐BBr2 are more soluble (than benzoyl congeners) enabling in situ reaction monitoring. Notably, while minor amounts of 6 a‐BBr2 were observed in situ no 6 a was observed after pinacol protection. To confirm regioselectivity ZnPh2 was added to the reaction mixture from 2 a/BBr3 to form predominantly 7 (right, Scheme 4) which has a δ 11B of 8.6 ppm indicating a four‐coordinate boron centre (in contrast 5 a has a broad δ 11B at 26 ppm consistent with a weaker PinB‐Opivaloyl interaction). 7 was isolated in 42 % yield and subsequently crystallised with X‐ray diffraction studies confirming the formulation as the C7‐borylated regioisomer. The solid state structure of 7 revealed a B−O distance of 1.610(2) Å and a O‐B‐C angle of 104.3(1)° that deviates from that calculated for 5 a‐BBr2 presumably due to the different steric demand of BPh2 vs. BBr2. The complete absence of C3‐borylation is consistent with the requirement for boranes more electrophilic than BBr3 (e.g. borenium salts) to effect intermolecular indole C3 borylation.3b, 17
Scheme 4.
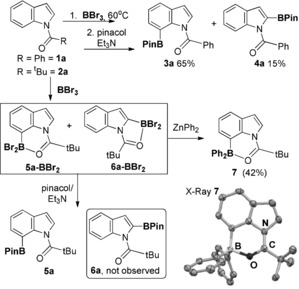
Borylation of 1 a and 2 a with BBr3 and subsequent protection with pinacol/Et3N or ZnPh2. Right, solid state structure of 7 (hydrogens omitted and ellipsoids at the 50 % probability level).24
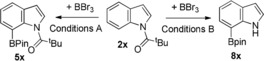
The substrate scope was explored next and notably C6 substituted N‐pivaloyl‐indoles were amenable to C−H borylation using BBr3 in moderate to good yields (e.g., 5 c and 5 d) (Table 1). The 6‐methoxy derivative 2 e was also a viable substrate, however, it underwent competitive ether cleavage with BBr3 producing two C7‐borylated products (5 e and 5 f) in varying amounts depending on the amount of BBr3 used. Conditions for one‐pot C−H borylation, pinacol protection and pivaloyl deprotection simply required the addition of methanol after BPin formation and heating to 60 °C. The removal of the pivaloyl group occurs without any observable C−B cleavage. This enables three steps to be achieved in one‐pot with no solvent switches with 8 a formed in 71 % isolated yield. These conditions were applicable to indoles substituted at C2, C3, C4, C5 and C6 (8 g–8 l), and containing electron withdrawing and donating groups. The reaction was performed on a 3 mmol scale to provide 0.82 g of 8 g in 86 % yield. However, 5‐SMe, 5‐NO2 and 4‐CN substituted indoles did not furnish isolable C‐BPin products, while attempts with a bulkier group at C6, 6‐(p‐tolyl)‐N‐pivaloyl‐ indole, led to C2 borylation dominating (35:65 C7:C2). Compounds 8 x are useful in Suzuki–Miyaura cross couplings, allylations and halogenations,8 and we note that 8 a readily undergoes oxidation with H2O2/NaOH to form 7‐hydroxy‐indole.
Table 1.
Substrate scope of pivaloyl‐directed C7‐borylation.
|
|
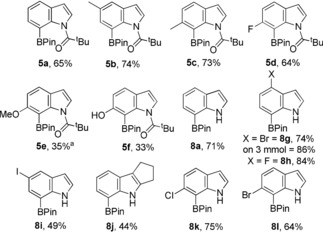
Conditions A=1. 2.2 equiv BBr3 in DCM. 2. +pinacol/Et3N. Conditions B=2.2 equiv BBr3 in DCM, 2. +pinacol/Et3N 3. +MeOH, 60 °C. Yields are of isolated products post column chromatography. [a]=using 1 equiv BBr3.
During substrate screening minor C2−BBr2 borylation (forming 6 x‐BBr2) often was observed. Attempts to form the C7−BBr2 products (5 x‐BBr2) selectively by heating (in sealed tubes so HBr does not leave the system) failed to change the C2:C7 ratio suggesting that C−H borylation of these indoles is irreversible under these conditions. However, it was observed that the ratio of C2:C7 BBr2 products was different to that of the C2:C7 BPin products (with C7‐BPin increasing). Furthermore, in a number of cases the amount of 5 x/8 x isolated was greater than that possible based on the observed 5 x‐BBr2:6 x‐BBr2 ratio (precluding C2‐selective protodeborylation during pinacol addition as the only origin of ratio changes). For example, substrate 2 k borylates to form a 5 k‐BBr2:6 k‐BBr2 ratio of ca. 55:45 (by 1H NMR spectroscopy), however, post work up 8 k was isolated in 75 % yield. This indicates that addition of pinacol enables C2−B protodeborylation and C7−H borylation. As the BBr2 products are stable to isomerisation in the presence of HBr this suggests that it is a C−B(OR)Br or C−B(OR)2 species that is undergoing protodeborylation and leading to more selective C7−H borylation.19 While the species undergoing C2→C7 isomerisation on pinacol addition is unknown Lewis/Brønsted acid initiated isomerisation of (RO)2B‐Aryl has been previously observed.17
To expand the utility of acyl‐directed electrophilic borylation other N‐heterocyclic frameworks were explored. However, N‐pivaloyl‐carbazole did not undergo C−H borylation using BBr3 (even on heating). This is attributed to steric crowding between the two proximal C−H units (at C1 and C8, Scheme 5, top left) and the pivaloyl tBu group that presumably results in large B‐O‐C‐N dihedral angles in the pivaloyl analogue of 10. Benzoyl contains a smaller R group (phenyl relative to tBu), therefore N‐benzoyl carbazole, 9, was combined with BBr3. This did not lead to C−H borylation at room temperature, instead the Lewis adduct, 10, was formed which was poorly soluble in DCM facilitating isolation and characterisation (including by X‐ray diffraction, Scheme 5).
Scheme 5.
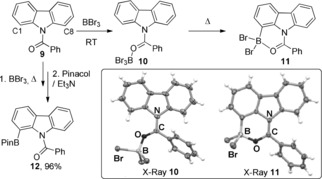
The directed borylation of N‐benzoyl carbazole using BBr3. Inset, the solid state structure of 10 and 11, ellipsoids at the 50 % probability level.24
Heating combinations of 9/ BBr3 led to high yielding C−H borylation at the C1 position. The C−H borylated product, 11, could be isolated (and structurally characterised by X‐ray diffraction studies) or protected at boron in situ to furnish the pinacol boronate ester 12 in excellent yield (96 %). For 10 and 11, the C=O (1.284(3) and 1.296(7) Å) and O−B distances (1.485(3) and 1.504(8) Å) reveal minimal difference, while the O‐B‐C angle in 11 (109.9(5)°) is comparable to that calculated for 5 a‐BBr2 and is close to ideal for four coordinate boron centres. Notably the B−O distance in 11 is significantly shorter than in 7 indicative of the greater Lewis acidity of the BBr2 moiety relative to BPh2.
We next explored the ortho borylation of anilines (Scheme 6). In previous work, borenium mediated electrophilic borylation of anilines proceeded at the para position.17 Ortho borylated anilines are accessible e.g., by directed lithiation of carbamate functionalised anilines,20 however, this approach has functional group limitations (e.g., C−Br). Both N‐pivaloyl and N‐benzoyl anilines were found to undergo selective ortho borylation using BBr3, with no para‐borylation observed.
Scheme 6.
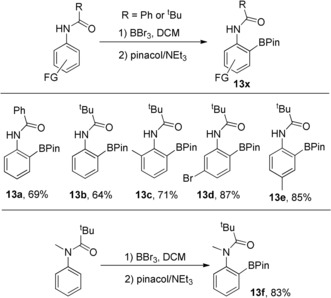
Directed ortho electrophilic borylation of N−H and N−Me anilines. Pivaloyl‐directed C−H borylation proceeds at 20 °C (over the course of 3–16 h), whereas as the benzoyl congener requires heating at 60 °C for 16 h.
This methodology was applicable to o‐, m‐ and p‐substituted anilines, forming 13 c–e in good yield, including for a bromo containing derivative (13 d). Directed borylation with BBr3 also can be applied to tertiary amides with the N‐Me derivative, 13 f, formed in good yield (83 %). Smith, Chattopadhyay and co‐workers have recently developed directed iridium‐catalysed ortho‐borylation of anilines using B2eg2 (eg=ethylene glycolate).21 This report is notable, but while excellent for N−H systems it is low yielding with N‐Me substituted anilines (<25 %),21 in contrast to the high yielding formation of 13 f using just commercially available DCM solutions of BBr3.
N‐Bn‐indol‐4‐yl‐2,2‐dimethylpropanamide, 14, next was investigated with it hypothesised that borylation would occur at C5 instead of C3 (the preferred site for SEAr in indoles) due to the preference for the formation of six membered boracycles over seven.13c Functionalisation of the C5−H of indoles is important for accessing pharmaceuticals such as C4‐amino‐C5‐functionalised indoles (e.g. Branebrutinib).5, 22 The thermodynamics of C5 vs. C3 borylation again was probed by DFT calculations which showed the C5 isomers 15A to be more stable than the C3 isomers 15B (inset, Scheme 7) for both halide and pinacol substituents. C5 borylation of 14 was achieved in high selectivity with the pinacol boronate ester 16 formed in moderate yield (77 % in situ and 40 % post purification). Attempts to monitor the borylation of 14 at the BBr2 stage were prevented by this intermediate being poorly soluble. Finally, the ability to perform a C5/C7 double C−H borylation using BBr3 was demonstrated using 17 (made in one step from 4‐amino‐indole). This formed 18 selectively post pinacol protection. Notably, in situ NMR spectra prior to pinacol addition show that the C3, C7 diborylated compound, 19, was formed as the major product and this does not isomerise on standing. However, addition of pinacol induces isomerisation of the C3−B moiety to form the thermodynamically favoured C5‐BPin unit and yield the desired C5/C7 product in good conversion (72 %).
Scheme 7.
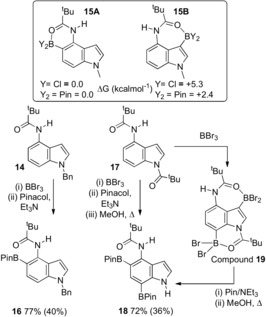
Top, relative energy of C3 and C5 borylated isomers at the M06‐2X/6‐311G(d,p) level, PCM (DCM). Bottom, borylation of 14 and 17. In situ yields versus an internal standard, isolated yields are provided in parentheses.23.
In summary, N‐pivaloyl is an effective and readily removed directing group enabling C7 borylation of indoles and ortho borylation of anilines simply using commercial solutions of BBr3. The process is complementary to borylation with [(amine)BBr2]+ and to iridium‐catalyzed directed borylation as C6‐substituted indoles are tolerated using BBr3, while it has complementary functional group tolerance to directed lithiation methods. Notably, in a number of cases pinacol induced isomerisation of the initial borylated regioisomer is essential to access the desired products containing C5−B and C7−B units. Due to the simplicity of this process and the many heterocycles containing N−H groups we believe acyl‐directed borylation with BBr3 will be applicable to many other systems.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge the Horizon 2020 Research and Innovation Program (Grant no. 769599) for financial support.
S. A. Iqbal, J. Cid, R. J. Procter, M. Uzelac, K. Yuan, M. J. Ingleson, Angew. Chem. Int. Ed. 2019, 58, 15381.
References
- 1.
- 1a. Boronic Acids: Preparation and Applications (Ed.: D. Hall), Wiley-VCH, Weinheim, 2011; [Google Scholar]
- 1b. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890; [DOI] [PubMed] [Google Scholar]
- 1c. Xu L., Wang G., Zhang S., Wang H., Wang L., Liu L., Jiao J., Li P., Tetrahedron 2017, 73, 7123. [Google Scholar]
- 2.For Ir-catalysed borylation of indoles see:
- 2a. Takagi J., Sato K., Hartwig J. F., Ishiyama T., Miyaura N., Tetrahedron Lett. 2002, 43, 5649; [Google Scholar]
- 2b.A recent analysis reported that 24 drugs currently on the market contain indole rings: Taylor R. D., MacCoss M., Lawson A. D. G., J. Med. Chem. 2014, 57, 5845. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a.For an early example see: Bujwid Z. J., Gerrard W., Lappert M. F., Chem. Ind. 1959, 1091; [Google Scholar]
- 3b.for reviews of early work see: Ingleson M. J., Synlett 2012, 23, 1411; and [Google Scholar]
- 3c. De Vries T. S., Prokofjevs A., Vedejs E., Chem. Rev. 2012, 112, 4246; For a more recent review see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Bähr S., Oestreich M., Pure Appl. Chem. 2018, 90, 723; For other select papers on electrophilic borylation see: [Google Scholar]
- 3e. Legare M. A., Courtemanche M.-A., Rochette E., Fontaine F.-G., Science 2015, 349, 513; [DOI] [PubMed] [Google Scholar]
- 3f. Yin Q., Klare H. F., Oestreich M., Angew. Chem. Int. Ed. 2017, 56, 3712; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3766; It should be noted in extremely limited examples C2 borylation has been reported under electrophilic conditions see: [Google Scholar]
- 3g. Zhong Q., Qin S., Yin Y., Hu J., Zhang H., Angew. Chem. Int. Ed. 2018, 57, 14891; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15107. [Google Scholar]
- 4. Rewcastle G. W., Katritzky A. R., Adv. Heterocycl. Chem. 1993, 56, 155. [Google Scholar]
- 5.
- 5a. Shah T. A., De P. B., Pradhan S., Punniyamurthy T., Chem. Commun. 2019, 55, 572; [DOI] [PubMed] [Google Scholar]
- 5b. Leitch J. A., Bhonoah Y., Frost C. G., ACS Catal. 2017, 7, 5618. [Google Scholar]
- 6.
- 6a. Hartung C. G., Fecher A., Chappell B., Snieckus V., Org. Lett. 2003, 5, 1899; [DOI] [PubMed] [Google Scholar]
- 6b. Paul S., Chotana G. A., Holmes D., Reichle R. C., Maleczka R. E., M. R. Smith III , J. Am. Chem. Soc. 2006, 128, 15552; [DOI] [PubMed] [Google Scholar]
- 6c. Loach R. P., Fenton O. S., Amaike K., Siegel D. S., Ozkal E., Movassaghi M. J., J. Org. Chem. 2014, 79, 11254; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Eastabrook A. S., Sperry J., Synthesis 2017, 49, 4731; [Google Scholar]
- 6e. Shen F., Tyagarajan S., Perera D., Krska S. W., Maligres P. E., M. R. Smith III , Maleczka R. E., Org. Lett. 2016, 18, 1544; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6f. Zhang S., Han Y., He J., Zhang Y., J. Org. Chem. 2018, 83, 1377. [DOI] [PubMed] [Google Scholar]
- 7.For a review on directed borylation see: Ros A., Fernandez R., Lassaletta J. M., Chem. Soc. Rev. 2014, 43, 3229. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Robbins D. W., Boebel T. A., Hartwig J. F., J. Am. Chem. Soc. 2010, 132, 4068; [DOI] [PubMed] [Google Scholar]
- 8b.One example of a 6-substituted indole subsequently was reported to undergo C−H borylation using this methodology, albeit in low yield: Eastabrook A. S., Wang C., Davison E. K., Sperry J., J. Org. Chem. 2015, 80, 1006.25525818 [Google Scholar]
- 9.For examples of important 6,7-disubstituted indoles see: Nakamura H., Yasui K., Kanda Y., Baran P. S., J. Am. Chem. Soc. 2019, 141, 1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For an example with arenes see: John A., Bolte M., Lerner H.-W., Wagner M., Angew. Chem. Int. Ed. 2017, 56, 5588; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5680. [Google Scholar]
- 11.For an example with alkenes see: Tanaka S., Saito Y., Yamamoto T., Hattori T., Org. Lett. 2018, 20, 1828. [DOI] [PubMed] [Google Scholar]
- 12.For select examples of N-directed electrophilic sp2C−H borylation see:
- 12a. Dewar M. J. S., Kubba V. P., Pettit R., J. Chem. Soc. 1958, 3073; [Google Scholar]
- 12b. Ishida N., Moriya T., Goya T., Murakami M., J. Org. Chem. 2010, 75, 8709; [DOI] [PubMed] [Google Scholar]
- 12c. Niu L., Yang H., Wang R., Fu H., Org. Lett. 2012, 14, 2618; [DOI] [PubMed] [Google Scholar]
- 12d. Crossley D. L., Cade I. A., Clark E. R., Escande A., Humphries M. J., King S. M., Vitorica-Yrezabal I., Ingleson M. J., Turner M. L., Chem. Sci. 2015, 6, 5144; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12e.For a recent example see: Liu K., Lalancette R. A., Jäkle F., J. Am. Chem. Soc. 2019, 141, 7453. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Letsinger R. L., Smith J. M., Gilpin J., MacLean D. B., J. Org. Chem. 1965, 30, 807; [Google Scholar]
- 13b. Kondrashov M., Provost D., Wendt O. F., Dalton Trans. 2016, 45, 525; [DOI] [PubMed] [Google Scholar]
- 13c. Escande A., Crossley D. L., Cid J., Cade I. A., Vitorica-Yrezabal I., Ingleson M. J., Dalton Trans. 2016, 45, 17160. [DOI] [PubMed] [Google Scholar]
- 14.For bulky phosphinyl-directed C7-arylation of indoles see: Yang Y., Qiu X., Zhao Y., Mu Y., Shi Z., J. Am. Chem. Soc. 2016, 138, 495. [DOI] [PubMed] [Google Scholar]
- 15.For some of the limited examples of pivaloyl-directed C7-functionalisation of indoles see:
- 15a. Xu L., Zhang C., He Y., Tan L., Ma D., Angew. Chem. Int. Ed. 2016, 55, 321; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 329; [Google Scholar]
- 15b. Kim Y., Park J., Chang S., Org. Lett. 2016, 18, 1892; [DOI] [PubMed] [Google Scholar]
- 15c. Kim Y., Park Y., Chang S., ACS Cent. Sci. 2018, 4, 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fukuda T., Maeda R., Iwao M., Tetrahedron 1999, 55, 9151. [Google Scholar]
- 17. Bagutski V., Del Grosso A., Ayuso Carrillo J., Cade I. A., Helm M. D., Lawson J. R., Singleton P. J., Solomon S. A., Marcelli T., Ingleson M. J., J. Am. Chem. Soc. 2013, 135, 474. [DOI] [PubMed] [Google Scholar]
- 18. Wang D. Y., Minami H., Wang C., Uchiyama M., Chem. Lett. 2015, 44, 1380. [Google Scholar]
- 19.Attempts to use B-bromo-catecholborane, targeting selective C7-borylation, did not lead to any borylation, even of the activated indole 2 b.
- 20.For an early example see: Stanetty P., Koller H., Mihovilovic M., J. Org. Chem. 1992, 57, 6833. [Google Scholar]
- 21. M. R. Smith III , Bisht R., Haldar C., Pandey G., Dannatt J. E., Ghaffari B., R. E. Maleczka, Jr. , Chattopadhyay B., ACS Catal. 2018, 8, 6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watterson S. H., et al., J. Med. Chem. 2019, 62, 3228. [DOI] [PubMed] [Google Scholar]
- 23.Compounds 16 and 19 are both prone to C5 protodeborylation on prolonged exposure to silica.
- 24.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201909786 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary