

Abstract
Mature T helper (Th) effector cells originate following antigen recognition by naive T precursors. The maturation process is accompanied by the acquisition of specific effector functions that distinguish at least three different T helper subsets: Th1, Th2 and Th17. In general, maturation of somatic cells is accompanied by terminal differentiation. However, accumulating evidence shows that effector T cells retain a certain degree of plasticity. This is especially true for Th17 cells, which have been shown to converge towards other phenotypes in response to specific microenvironmental pressure. In this review we will discuss the experimental evidence that supports the hypothesis of Th17 plasticity, with particular emphasis on the generation of Th17‐derived ‘non‐classic’ Th1 cells, and the molecular networks that control it. Moreover, we will consider why Th17 plasticity is important for host protection, but also why it can have pathogenic functions during chronic inflammation. Regarding the last point, we will discuss a possible role for biological drugs in the control of Th17 plasticity and disease course.
Keywords: inflammation, T helper 1, T helper 17
Abbreviations
- AhR
aryl hydrocarbon receptor
- CD
cluster of differentiation
- GM‐CSF
granulocyte–macrophage colony‐stimulating factor
- IFN
interferon
- IL
interleukin
- MOG
myelin oligodendrocyte glycoprotein
- mTORC1
mammalian target of rapamycin complex 1
- RAR‐α
retinoic acid receptor‐α
- ROR‐γt
RAR‐related orphan receptor‐γt
- STAT
signal transducer and activator of transcription
- TGF‐β
transforming growth factor β
- Th
T helper
- TNF‐α
tumor necrosis factor α
Introduction
The immune system has evolved to mount specific and specialized immune responses against invading pathogens. In this context, CD4+ T helper (Th) cells are crucial regulators of adaptive immunity. Upon recognition of the specific antigen presented by professional antigen‐presenting cells, naive Th cells differentiate towards one of multiple possible lineages. In this way, CD4 Th cells guarantee the optimal immune response to achieve pathogen clearance. The original model of Th cell differentiation, described in the early 1990s by Mosmann and Coffman in mouse and by Romagnani in humans, contemplated the existence of two alternative differentiation programs, Th1 and Th2.1, 2 Th1 cells protect mainly from intracellular pathogens through the secretion of interferon‐γ (IFN‐γ), a cytokine that potentiates the phagocytic capacity of macrophages. In addition, Th1 cells promote the IgG class‐switch of antigen‐specific B lymphocytes.3 Th2 cells protect from extracellular parasites by the activation of mast cells, basophils and eosinophils, the main characters of the so‐called non‐phagocytic response.3 Indeed, parasites are usually large and cannot be phagocytosed. Th2 cells secrete interleukin‐4 (IL‐4) and IL‐13, which promote the production of antigen‐specific IgE, and IL‐5, which promotes the differentiation of eosinophils in the bone marrow and their survival in the periphery. IgE‐opsonized parasites can be recognized by eosinophils by their low‐affinity IgE receptors.3 This leads to the release of molecules that can damage the parasite’s robust tegument and to the secretion of histamine and other mediators that promote contraction of intestinal or bronchoalveolar districts to favor parasite expulsion.4 The differentiation of naive cells towards these alternative pathways is induced by different instructive cytokines that are released by antigen‐presenting cells in the local microenvironment at the immune synapse. Indeed, Th1 cell development is favored by the presence of IFN‐γ and IL‐12, whereas Th2 cells are induced by the presence of IL‐4.5 The role of instructive cytokines is to promote intracellular signaling pathways that activate lineage‐specific transcription factors, that in turn promote chromatin remodeling and the stabilization of a specific Th cell program. In particular, the transcription factors signal transducer and activator of transcription 1 (STAT1), STAT4 and Tbet are crucial in Th1 differentiation, while STAT6 and GATA3 are involved in Th2 cell development.5
More recently Harrington and Park with their colleagues defined the existence of a third, distinct, lineage of Th cells producing IL‐17, named Th17.6, 7 Since this discovery, most efforts have been focused on understanding how these cells originate, what is their biological function and their possible involvement in autoimmunity. Th17 responses are induced against extracellular bacteria and fungi, and through IL‐17 promote the secretion of IL‐8 by tissue‐resident cells, so favoring neutrophil recruitment at sites of infection. Th17 cells also produce IL‐22, which has tissue protective functions.8 In contrast to Th1 and Th2 polarization processes, there is still no consensus in the literature regarding how Th17 responses are induced. Indeed, multiple cytokines have been proposed in various combinations to be the most effective in driving Th17 polarization. To further complicate this scenario, significant differences in mouse and human polarization settings have been proposed. The paper by Langrish et al. demonstrated that the development of IL‐17‐producing cells is strictly dependent on the activity of IL‐23, which signals through STAT3.9 Moreover, these cells have a highly pathogenic potential in a mouse model of experimental autoimmune encephalomyelitis. Subsequent studies instead proposed IL‐6 and transforming growth factor‐β (TGF‐β) as crucial components of the murine Th17 polarization cocktail.10, 11 IL‐6 turns on the STAT3 signaling pathway, while TGF‐β activates transcription factors of the Smad family. However, as already stated, there is still no consensus on the reciprocal role of these cytokines. In particular, the role of TGF‐β is debated. Some studies in humans confirmed the importance of this cytokine.12, 13, 14 Instead, other studies denied the importance of TGF‐β in Th17 development.15 Indeed, IL‐1β and IL‐616 or IL‐1β and IL‐2317 were found to be effective in generating Th17 cells. In our experience, TGF‐β plays an indirect role in promoting Th17 cell development. Indeed, TGF‐β is an immunoregulatory cytokine and is more effective in suppressing Th1 rather than Th17 cell responses,18 so indirectly promoting the accumulation of IL‐17‐secreting cells. Moreover, we have demonstrated that human Th17 cells preferentially develop from a naive CD4+ CD161+ precursor that can be detected in the umbilical cord blood and thymus, under the combined activity of IL‐1β and IL‐23.19 It has recently been shown that multiple redundant pathways converge during the course of Th17 differentiation in vivo. Patients with IL‐6 receptor (IL‐6R) inactivating mutations have normal levels of circulating Th17 cells, but their development in vitro from naive precursors is affected.20 Hence, IL‐6 signaling deficiency alone in vivo can be substituted by other redundant factors. In agreement with this, the frequency of peripheral CCR4+ CCR6+ Th17 cells is normal in patients with autosomal recessive IL23R deficiency, but they do not differentiate properly in vitro. 21 A critical step during the differentiation process is the activation of the transcription factor STAT3, which controls the expression of several genes associated with the Th17 program.22 The importance of STAT3 in this process is demonstrated by patients affected by the hyper‐IgE syndrome, a genetic disease caused by loss of function mutations in the STAT3 locus, that display absence of Th17 cells.23 The final differentiation step is the expression of the transcription factor retinoic acid‐related orphan receptor‐γt (ROR‐γt),24 which defines Th17 as a distinct CD4 Th cell lineage. ROR‐γt activates a transcriptional program typical of Th17 cells that includes the expression of typical surface molecules such as CD161, the chemokine receptor CCR6 and IL‐23R. Indeed, patients with RORC bi‐allelic inactivating mutations display absence of circulating Th17 cells.25
Despite CD4, Th effector cells are crucial players in the fight against microbial pathogens, their activity must be tightly controlled to avoid exaggerated and unwanted responses. Indeed, inappropriate responses of Th1 and Th17 are responsible for autoimmune disease development, and Th2 cells sustain allergic disorders.26 In this view, regulatory T (Treg) cells have a crucial role in maintaining tissue homeostasis through preventing inappropriate immune responses against self or innocuous antigens. Treg cells express the transcription factor Foxp3 and can develop either in the thymus or in the periphery (iTreg). A subset of Foxp3– Treg cells has also been identified, type 1 regulatory cells (Tr1).27 The Tr1 cells originate in the periphery, produce high levels of IL‐10 and express CD49b and LAG3 on their surface.28
Plasticity of Th17 cells
The original model of Th cell differentiation considered polarized cells as terminally differentiated and unable to acquire phenotypical properties typical of other subsets. This concept originated mainly from experiments that suggested the existence of a transcriptional program that is reinforced by epigenetic changes and that antagonizes the expression of a gene signature from alternative subsets. Indeed, it was demonstrated that recently in vitro polarized Th1 or Th2 cells could be redirected towards the opposite lineage if cultured in the presence of the appropriate cytokine cocktail. Instead, long‐term polarized cells maintained their original phenotype even if stimulated with cytokines promoting the development of the opposite lineage.29 To further reinforce this hypothesis, several data showing the reciprocal counter‐regulation of Th1 and Th2 differentiation programs were published. Indeed, it has been shown that Tbet is induced downstream of the IFN‐γ–STAT1 signaling pathway30 and promotes Runx3 expression. Tbet and Runx3 then cooperate to increase IFNG gene transcription and IL4 gene silencing.31 On the other hand, IL‐4 represses the Th1 differentiation program by inhibiting the expression of the IL‐12Rβ2 chain, so impeding IL‐12 sensitivity.32 Counter‐regulation of alternative Th phenotypes was demonstrated even after the discovery of the Th17 subset. Indeed, IL‐27, a cytokine that belongs to the IL‐12 family, was demonstrated to trigger Th1 cell development through the up‐regulation of Tbet and IL‐12Rβ2, while inhibiting the Th17 program.33, 34 Similar evidence was produced regarding the ability of Th2‐associated molecular machinery to impede Th17 differentiation.35, 36
Despite all these findings, several others confuted the idea that polarized Th cells are terminally differentiated. Indeed, it was demonstrated that allergen‐specific Th2 cells can be redirected towards IFN‐γ production37, 38 and that either Th1 or Th2 cells can express alternative cytokine genes when stimulated under opposite polarizing conditions.39 The concept of Th cell plasticity became even more evident for the Th17 subset. Our group demonstrated that cells simultaneously producing IL‐17 and IFN‐γ (named Th17/Th1) were detectable in the gut of patients with Crohn’s disease. Th17/Th1 cells exhibited features of Th17 cells, such as expression of the surface markers CCR6 and IL‐23R and of the transcription factor ROR‐γt.40 These data suggested the possibility that Th17/Th1 cells may originate from Th17 upon appropriate cytokine stimulation. Indeed, culturing of Th17 cells in the presence of IL‐12 resulted in an up‐regulation of Tbet expression and an increase of IFN‐γ production.40 Moreover, our group provided evidence that the phenotypical conversion of Th17 cells can further progress to a complete trans‐differentiation to a Th1 phenotype with loss of IL‐17 secretion. This process is induced by IL‐12 and occurs at sites of chronic inflammation such as the affected joints of children with juvenile idiopathic arthritis.41, 42 Th1 cells derived from the shifting of Th17 precursors were defined as non‐classic Th1. Non‐classic Th1 cells are phenotypically distinct from classic Th1, that instead do not derive from Th17. Indeed, non‐classic Th1 cells express surface markers of the Th17 phenotype such as CCR6, CD161 and IL‐23R and the transcription factor ROR‐γt, although at lower levels than Th17. In parallel, non‐classic Th1 cells acquire Th1 features such as expression of the chemokine receptor CXCR3 and of the transcription factor Tbet.43 Non‐classic Th1 cells produce IFN‐γ and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) as classic Th1 cells do.44 More recently we showed that also tumor necrosis factor‐α (TNF‐α) can promote the development of non‐classic Th1 cells from Th17 cells.45 In addition to these data demonstrating the plasticity of human Th17 cells, similar findings have been obtained in mouse. Lee et al. 46 demonstrated that in vitro generated Th17 cells could be redirected towards a Th1‐like phenotype when exposed to IL‐12 or IL‐23 in a STAT4‐ and Tbet‐dependent manner. The same phenotypic transition was observed even in vivo in a model of colitis when reconstituting Rag1–/– mice with in vitro generated Th17 cells.46 However, most murine data were generated using in vitro polarized Th17 cells, thus raising some concerns about their real phenotypic stability. Supporting this possibility, Lexberg et al. 47 demonstrated that murine in vitro polarized Th17 cells can be shifted towards Th1 or Th2 if exposed to IL‐12 or IL‐4, respectively, while in vivo polarized Th17 cells are instead phenotypically stable. However, the same authors provided evidence in a subsequent publication.48 that in vivo generated murine Th17 cells are also plastic. Indeed, these cells do not express the IL‐12Rβ2 chain, so are not sensitive to IL‐12 stimulation. However, the authors demonstrated that IFN‐γ up‐regulates the expression of the IL‐12Rβ2 chain by Th17 cells, which become Th17/Th1 following IL‐12 exposure. It has also been demonstrated that Th17 cell plasticity is strictly dependent on their metabolic activity because strong mTORC1 signaling and anabolic metabolism are required for their trans‐differentiation into Th1 cells.49
Differentiation routes of Th17 cells towards different phenotypes other than Th1 have been demonstrated. Our group has shown the existence of Th17/Th2 cells, producing both IL‐17 and IL‐4, that originate from Th17 upon exposure to IL‐4.50 Th17/Th2 cells are enriched in the blood of patients with chronic asthma.50 Of note, an accumulation of this subset in the bronchoalveolar lavage of individuals with asthma is associated with steroid‐resistant forms of the disease, the so‐called ‘difficult to treat asthma’.51 In mice it has been demonstrated that Th17 cells accumulate in intestinal Peyer’s patches where they acquire T follicular helper cell properties, leading to the promotion of IgA production.52 Finally, it has been shown that murine intestinal Th17 cells can also differentiate into Type 1 regulatory cells through a mechanism that includes TGF‐β signaling and the transcription factor aryl hydrocarbon receptor (AhR).53 Collectively these data further support plasticity of the Th17 phenotype and highlight the possibility that these cells may behave as pro‐ or anti‐inflammatory depending on the specific microenvironmental context. Finding out how to redirect Th17 responses may have clinical implications as it may be relevant to modulate the course of not only chronic inflammatory diseases but also tumoral diseases.
Plasticity towards the Th17 phenotype is more debated. Th1 cells are traditionally considered a highly stable cell subset. Indeed, we and others have shown that Th1 cells (either classic or non‐classic) are resistant to the activity of pro‐Th17 cytokines.44, 54 Only ROR‐γt forced expression by way of lentiviral vector can trigger IL‐17 production by Th1 cells.44, 54 Supporting this notion, it has been shown that several molecular mechanisms stabilize the Th1 phenotype and restrain the expression of Th17 genes. Retinoic acid via its receptor RARα has a critical role in maintaining Th1 cell lineage as it promotes STAT‐4, Tbet and IFN‐γ while simultaneously inhibiting the expression of Th17‐associated genes.55 In agreement with this, we have shown that human Th1 cells express the transcription factor Eomes, which selectively inhibits the expression of RORC2 and IL17A. Of note, Eomes is expressed also by non‐classic Th1 cells, which suggests that the differentiation of Th17 cells into Th1 is irreversible, at least until the molecular machinery that stabilizes the Th1 phenotype is expressed.44 For this reason, the development of non‐classic Th1 cells from Th17 is tightly controlled. Indeed, we have shown that acquisition of Eomes expression by Th17 cells is strongly regulated and requires proper IL‐2 sensitivity.44 It is known that Th17 cells have reduced IL‐2 production56, 57 and sensitivity58, thus indirectly controlling Eomes expression.
Another report showed instead that during intestinal inflammation in mice the TGF‐β–Runx1 axis can differentiate Th1 cells into Th17.59 However, as all experiments were performed with in vitro polarized cells, the possibility that it may happen in vivo is still debated. Regarding plasticity of Treg cells towards Th17, it has been shown the existence of a population of regulatory cells co‐expressing Foxp3 and ROR‐γt that potently suppresses inflammation in a T‐cell‐dependent colitis model.60 These cells originate mainly from Foxp3+ ROR‐γt– precursors61 and acquire IL‐17 production thanks to ROR‐γt expression.62 Hence, Foxp3+ ROR‐γt+ cells are considered to be bifunctional because they exhibit both pro‐ and anti‐inflammatory properties.63
Epigenetic control of Th17 cells plasticity
T helper cell polarization is accompanied by chromatin remodeling to sustain the expression of a specific transcriptional program. Architectural changes of chromatin structure are achieved through the addition of epigenetic marks that include mainly histone modifications and DNA methylation. Wei et al. 64 demonstrated that murine naive, Th1, Th2 and Th17 cells display distinct histone H3 lysine 4 (H3K4) and lysine 27 (H3K27) trimethylation at cytokine gene loci, confirming the lineage commitment of these subsets. However, they also provided evidence that transcription factor genes display instead bivalent histone marks, suggesting that this might be crucial for Th cell plasticity. Mukasa et al. confirmed that the IL‐12‐driven plasticity of murine in vitro polarized Th17 is sustained by epigenetic remodeling. Indeed, upon IL‐12 treatment, Th17 cells underwent a rapid reorganization of histone modifications, with the accumulation of permissive marks at Tbx21 and Ifng genes and the concurrent appearance of inhibitory marks at Rorc2 and Il17 loci.65 Of note, similar data were generated also in a subsequent study performed on in vivo polarized Th17 cells.66 This paper highlighted also the presence of repressive histone marks at the Il12rβ2 gene, which correlates with the reduced sensitivity of in vivo generated Th17 cells to IL‐12.48 Cell activation lead to the removal of non‐permissive histone modifications and to the expression of the IL‐12Rβ2 chain.66 The first data on epigenetic plasticity of human Th17 cells were provided by a study performed on in vivo generated Th1 and Th17 cells. The authors showed that human Th1 and Th17 cells have a distinct profile of histone modifications and DNA methylation at cytokine (IL17, IFNG) and transcription factor (RORC2, TBX21) genes.54 When cultured in the presence of IL‐12 in vitro, Th17 cells acquired the capacity to produce IFN‐γ, but this was not mirrored by the appearance of epigenetic permissive modifications. Hence, the authors concluded that, despite the possible transient expression of non‐signature cytokines, human T cells are epigenetically stable and DNA methylation has a crucial role in maintaining their phenotypic identity. However, these conclusions were drawn from the exposure of ex vivo‐recovered cells to IL‐12 in vitro, which may not recapitulate what really happens in vivo. Indeed, in a subsequent study we took advantage of a combination of surface markers and cytokine production capabilities to sort in vivo generated, Th17‐derived, non‐classic‐Th1 cells. DNA methylation analysis showed that non‐classic Th1 cells have reduced CpG methylation at IL17A and RORC2 gene loci, which confirms their Th17 origin. Moreover, these cells exhibited complete demethylation of TBX21 and IFNG genes, that instead are fully methylated in Th17 cells.67 Hence, in vivo plasticity of human Th17 cells towards Th1 is sustained at an epigenetic level.
Th17 plasticity and host defense
Despite Th17 plasticity having been clearly demonstrated and characterized at molecular level, the biological significance of this phenomenon is less clear. Transition to the non‐classic Th1 phenotype may be associated with the acquisition of peculiar antimicrobial properties that may be fundamental for pathogen clearance. Th17 responses are classically associated with neutrophilic inflammation, which is commonly present during the initial phases of bacterial infections. However, exaggerated and sustained neutrophil activity may also lead to tissue damage, hence the shifting to a macrophage‐based (Th1‐triggered) response may be important in chronic infections. It is also worth mentioning that non‐classic Th1 cells have a unique pattern of chemokine receptors, i.e. CCR6 and CXCR3 co‐expression, that allows migration to tissues expressing either Th17 (CCL20) or Th1 (CXCL9, CXCL10, CXCL11) ‐recruiting chemokines.43 Regarding host protection, it has been demonstrated that Mycobacterium tuberculosis‐specific T cells are multifunctional (IFN‐γ +, TNF‐α +, IL‐2+) and are contained in the CXCR3+ CCR6+ subset.68, 69 Further supporting these data, patients with bi‐allelic RORC loss of function mutations present not only with chronic candidiasis due to the absence of IL‐17‐producing cells, but also mycobacteriosis. Indeed, these patients displayed a defective IFN‐γ production by CD4+ CXCR3+ CCR6+ cells in response to the weak pathogen bacillus Calmette–Guérin.25 Non‐classic Th1 cells cooperate also with classic Th1 cells in the response against influenza. 69 Hence, non‐classic Th1 cells provide a specific line of defense against certain pathogens. In addition, it has been shown that CCR6+ CXCR3+ Th17/Th1 cells responded specifically to extracellular bacteria such as Streptococcus pneumoniae, Escherichia coli and Lactobacillus rhamnosus. 70 The observation that different pathogens evoke distinct multifunctional Th17 responses further supports the concept that T‐cell plasticity is aimed to provide the optimal response to the specific invading pathogen. Indeed, Candida‐albicans‐specific Th17 cells were shown to produce IL‐17 and IFN‐γ but no IL‐10, whereas Staphylococcus‐aureus‐specific Th17 cells produced IL‐17 and no IFN‐γ but could also secrete IL‐10 upon restimulation71 (Fig. 1).
Figure 1.
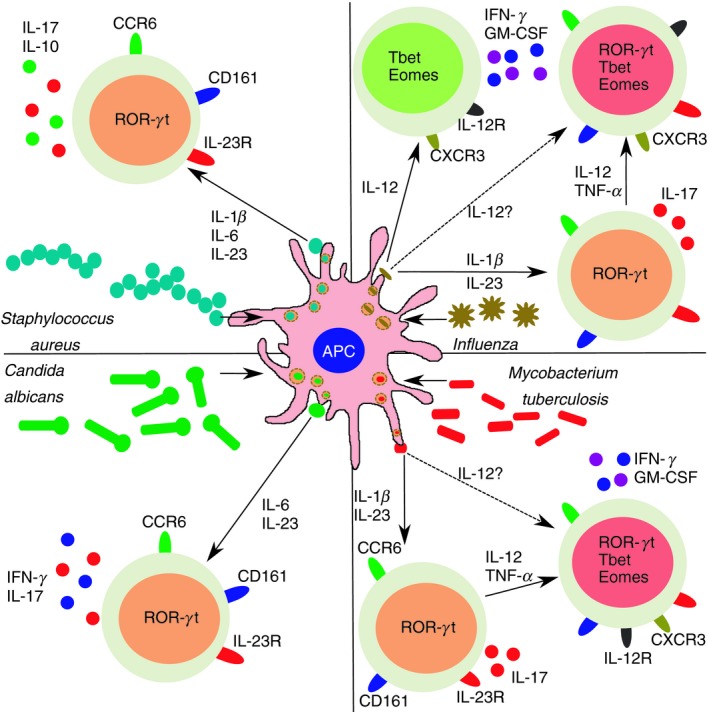
T helper type 17 (Th17) cell plasticity and protection from invading pathogens. Th17 cells can produce different combinations of cytokine to promote pathogen clearance. Following Staphylococcus aureus infection, antigen‐presenting cells produce interleukin‐1β (IL‐1β), IL‐6 and IL‐23 that promote the development of Th17 cells co‐secreting IL‐17 and IL‐10. In response to Candida albicans infection instead, IL‐6 and IL‐23 favor the appearance of Th17 cells producing IL‐17 and interferon‐γ (IFN‐γ). Response to Mycobacterium tuberculosis is driven by non‐classic Th1 cells, which originate from Th17 following exposure to pro‐Th1 cytokines such as IL‐12 and tumor necrosis factor‐α (TNF‐α). It is presently unknown if non‐classic Th1 cells can directly originate from naive precursors (dotted arrow). Influenza‐specific T cells can be found either in the classic or in the non‐classic Th1 subsets. Also in this case it is still to demonstrate if non‐classic Th1 cells generate entirely from Th17 or whether a direct polarization route exists. Orange cells: Th17; red cells: non‐classic Th1; green cells: classic Th1.
Th17 to Th1 cell plasticity and disease
Chronic inflammatory disorders were originally considered to be the result of uncontrolled autoreactive Th1 responses. However, with the discovery of the Th17 subset, it has been proposed that these cells may play a critical pathogenic role. Contrasting data have been proposed on the reciprocal role of Th1 and Th17 cells in autoimmunity.72 A solution to this debate came with the discovery of Th17 plasticity and the finding that cells simultaneously producing IL‐17 and IFN‐γ were detectable at sites of inflammation. Since that observation data, both in mice and humans, have been reported regarding the crucial role of the Th17 to Th1 trans‐differentiation in sustaining chronic inflammation in different organs (Fig. 2). It has been demonstrated that Th17 cells can induce pancreatic inflammation and type 1 diabetes in mice only after their transition to a Th1 phenotype.73 Diabetes development could be prevented through the administration of a neutralizing anti‐IFN‐γ antibody but not by an anti‐IL‐17 antibody.74 In agreement with this, Th17 cells from children with type 1 diabetes exhibited enhanced plasticity, and the degree of Th17/Th1 plasticity was correlated with the impaired glucose control.75 A significant amount of data strongly suggests a pathogenic role for Th17 plasticity in inflammatory bowel diseases. Cells simultaneously producing IL‐17 and IFN‐γ were detected in the gut of patients with Crohn’s disease.40 In a colitis model induced by transfer of in vitro polarized Th17 cells into Rag1–/– recipient mice, it was demonstrated that transition of Th17 cells to Th1 occurs in vivo. Transition was IL‐12‐ and IL‐23‐dependent and was correlated with colitis development as neutralization of these cytokines significantly ameliorated disease score.46 This hypothesis was further confirmed by a subsequent study showing that acquisition of IFN‐γ production is crucial for colitis development following transfer of in vitro polarized Th17 cells into Rag1–/– mice. Of note, Th17 to Th1 transition was largely STAT4‐dependent and required Tbet activity.76 Regulatory T cells have an important role in this process by preventing the development of colitogenic non‐classic Th1 cells, thus favoring an accumulation of Th17 and Th17/Th1 cells, which are non‐pathogenic.77 Th17 plasticity was observed even in a model of colitis induced by experimental infection with Helicobacter hepaticus. 78 Similar observations have been produced in experimental autoimmune encephalomyelitis, the mouse model of multiple sclerosis. Indeed, it has been shown that IFN‐γ + IL‐17+ cells infiltrating the central nervous system (CNS) are enriched in cells specific for the myelin antigen MOG35–55. Development of IFN‐γ‐producing Th17 cells was Tbet‐dependent, as Tbet–/– mice showed higher levels of IL‐17+ IFN‐γ – cells. Moreover, Tbet–/– mice developed a milder disease than wild‐type controls.79, 80 In humans, Th17/Th1 cells were enriched in the peripheral blood and CNS of individuals with relapsing–remitting multiple sclerosis and produced IL‐17, IFN‐γ and GM‐CSF. More importantly, Th17/Th1 cells derived from CNS produced effector cytokines in response to myelin‐derived peptides, but Th1 cells did not.81 A pathogenic role for non‐classic Th1 cells has been proposed also in arthritis. We and others have demonstrated that non‐classic Th1 cells develop in the inflamed joints in patients with juvenile idiopathic arthritis under the activity of IL‐12.41, 42 Of note, the frequency of non‐classic Th1 cells in the synovial fluid was directly correlated with parameters of disease activity.42 Synovial fluid‐infiltrating non‐classic Th1 cells were also found to produce the pro‐inflammatory cytokine GM‐CSF, and the frequency of GM‐CSF‐producing cells was strictly correlated with the erythrocyte sedimentation rate, further supporting their pathogenicity.82 In addition, non‐classic Th1 cells, as well as classic Th1, produce TNF‐α, which directly induces CD106 expression by synovial fibroblasts, so promoting leukocyte retention in the inflamed joints.83 Pathogenicity of Th17 to Th1 trans‐differentiation has been demonstrated also in the context of graft‐versus‐host disease84 while, on the contrary, Th17 cells seem phenotypically stable in the autoimmune kidney diseases.85 The last finding suggests that the microenvironment directs Th17 plasticity. In this view, the microbiome can play a crucial role because bacteria or their products interact with T cells at mucosal surfaces. Hence, Th17 cells may polarize or not depending on their location (a sterile organ or mucosal surfaces with direct contact with the external environment). Moreover, even at mucosal surfaces different microbiome composition may affect Th17 cell fate. However, Th17 cells have a different ability to polarize also in different ‘sterile’ organs, such as pancreas, joints or kidney. Hence, the microbiome alone cannot be entirely responsible for the polarization capacity of Th17 cells. Understanding which factors promote plasticity or stabilization of the Th17 phenotype is therefore a matter of interest.
Figure 2.
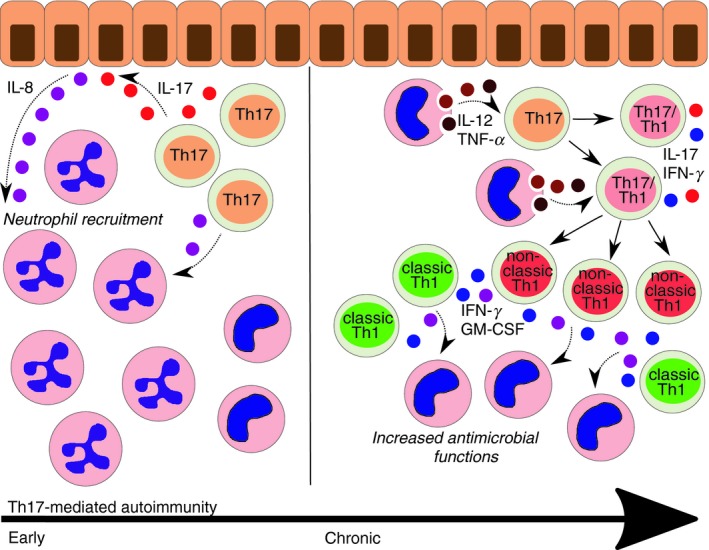
Different phases of T helper type 17 (Th17) ‐mediated autoimmunity. In early phases of Th17‐mediated inflammation, production of interleukin‐17 (IL‐17) by T cells induces the release of IL‐8 by local epithelial cells. Th17 cells are also a source of IL‐8. IL‐8 is a chemotactic molecule which attracts neutrophils in the tissue. Indeed, Th17 responses are commonly associated with neutrophil infiltrate. If inflammation becomes chronic, macrophage‐derived IL‐12 and tumor necrosis factor‐α (TNF‐α) act on activated Th17 cells, which acquire interferon‐γ (IFN‐γ) production and a Th17/Th1 phenotype. If inflammation persists, pro‐Th1 cytokines can convert Th17/Th1 into non‐classic Th1 cells, which are commonly found at later stages of inflammation together with classic Th1 cells. Both subsets of Th1 cells produce IFN‐γ and granulocyte–macrophage colony‐stimulating factor (GM‐CSF), which increase the antimicrobial functions of macrophages.
Targeting Th17 plasticity with biologicals
Accumulating data on inflammatory disorders in humans and experimental mouse models show that chronic inflammation can promote Th17 plasticity. Development of Th17‐derived non‐classic Th1 cells is strictly correlated with the severity and quality of inflammation and may be considered a selective target for biological treatment. The scenario that appears from experimental evidence suggests that IL‐17 may be the ideal target during initial phases of Th17‐mediated inflammation and in those cases where Th17 cells are present also during the chronic phase of the disease. In this view, the anti‐IL‐17 antibody Secukinumab is already available and successfully used in psoriasis.86, 87 However, in those cases where non‐classic Th1 cells are the most pathogenic subset, targeting IL‐17 may become less beneficial. Targeting IL‐17 was less efficacious in rheumatoid arthritis and uveitis than in psoriasis and even ineffective in Crohn’s disease.86, 88 Indeed, IL‐17 has a crucial role for the maintenance of the homeostasis at mucosal surfaces, so its targeting during the course of intestinal inflammation may have even worse effects, possibly due to an altered protection to microbial invasion. For this reason, targeting those axes that promote Th17 phenotypic shift may be beneficial. In this view, the anti‐TNF‐α monoclonal antibody etanercept has been shown to favor an accumulation of Th17 cells and a reduction of non‐classic Th1 cells in vivo. In agreement with this, etanercept prevented non‐classic Th1 cell development from Th17 in vitro. 45 Anti‐TNF‐α treatment is approved for the treatment of several immune‐mediated diseases including Crohn’s disease, rheumatoid arthritis, psoriasis and spondyloarthritis.89, 90, 91, 92 Abatacept, the CTLA‐4–IgG fusion protein that prevents T‐cell activation by binding to CD80 CD86 on antigen‐presenting cells, has been shown to reduce IFN‐γ and TNF‐α production by anti‐CD3 stimulated cells, so indirectly preventing possible Th17 trans‐differentiation.93 Other biologicals targeting IL‐12 or IL‐23 may also be useful as these cytokines are involved in non‐classic Th1 cell generation. Indeed ustekinumab, a monoclonal antibody that selectively targets the p40 chain shared by IL‐12 and IL‐23, improves symptoms in Crohn’s disease and psoriatic patients.94, 95, 96 Finally, antibodies directly targeting effector cytokines produced by both classic and non‐classic Th1 cells such as IFN‐γ and GM‐CSF have shown promising effects97, 98 (Fig. 3).
Figure 3.
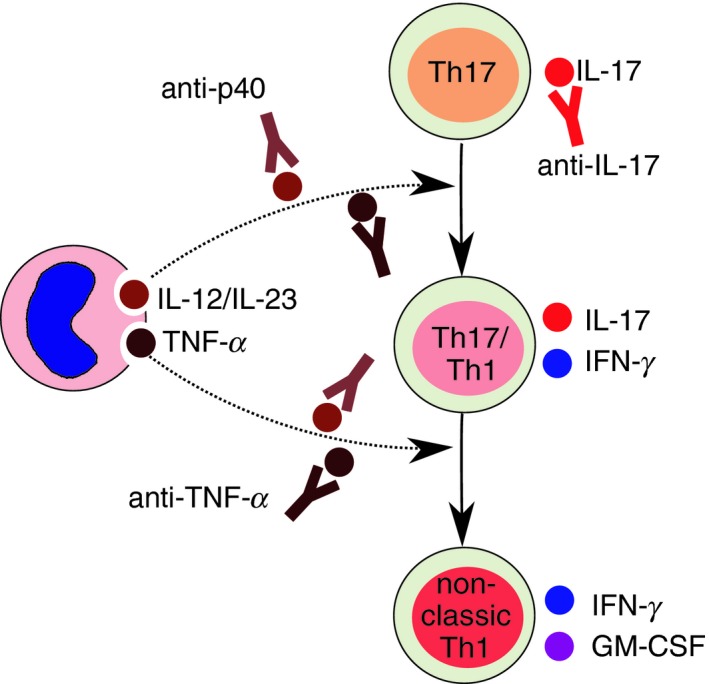
Targeting T helper type 17 (Th17) plasticity with biologicals. Th17‐mediated inflammation can be targeted with neutralizing anti‐interleukin‐17 (IL‐17) monoclonal antibodies (mAbs). To prevent the acquisition of interferon‐γ (IFN‐γ) production by Th17 cells and also the development of non‐classic Th1 cells can be used either anti‐tumor necrosis factor‐α (TNF‐α) or anti‐p40 mAbs. p40 is a subunit shared by IL‐12 and IL‐23 cytokines, which have both Th1 polarizing effects on Th17 cells. Targeting p40 allows simultaneous inhibition of IL‐12 and IL‐23.
Conclusions
Development of Th17‐derived non‐classic Th1 cells is strictly controlled at molecular level and sustained by epigenetic and transcriptional events. Non‐classic Th1 cells are crucial players in host protection – they cooperate with other Th subsets in microbial fight and provide a line of defense against specific pathogens. However, they are also responsible for chronic inflammation when their activity becomes deregulated. Targeting Th17 plasticity with biologicals may provide a valuable tool for the treatment of chronic inflammatory disorders.
Disclosures
The authors declare that they have no competing interests.
References
- 1. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989; 7:145–73. [DOI] [PubMed] [Google Scholar]
- 2. Romagnani S. Human TH1 and TH2 subsets: doubt no more. Immunol Today. 1991; 12:256–7. [DOI] [PubMed] [Google Scholar]
- 3. Romagnani S. T‐cell subsets (Th1 versus Th2). Ann Allergy Asthma Immunol. 2000; 85:9–18. [DOI] [PubMed] [Google Scholar]
- 4. Klion AD, Nutman TB. The role of eosinophils in host defense against helminth parasites. J Allergy Clin Immunol. 2004; 113:30–7. [DOI] [PubMed] [Google Scholar]
- 5. Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H et al Signaling and transcription in T helper development. Annu Rev Immunol. 2000; 18:451–94. [DOI] [PubMed] [Google Scholar]
- 6. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM et al Interleukin 17‐producing CD4 effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005; 6:1123–32. [DOI] [PubMed] [Google Scholar]
- 7. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y‐H et al A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005; 6:1133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liang SC, Tan X‐Y, Luxenberg DP, Karim R, Dunussi‐Joannopoulos K, Collins M et al Interleukin (IL)‐22 and IL‐17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006; 203:2271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD et al IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005; 201:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO et al Transforming growth factor‐β induces development of the TH17 lineage. Nature 2006; 441:231–4. [DOI] [PubMed] [Google Scholar]
- 11. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL‐17‐producing T cells. Immunity 2006; 24:179–89. [DOI] [PubMed] [Google Scholar]
- 12. Manel N, Unutmaz D, Littman DR. The differentiation of human Th17 cells requires transforming growth factor‐β and induction of the nuclear receptor RORγt. Nat. Immunol. 2008; 9:641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E et al. A critical function for transforming growth factor‐β, interleukin‐23 and proinflammatory cytokines in driving and modulating human Th17 responses. Nat. Immunol. 2008; 9:650–7. [DOI] [PubMed] [Google Scholar]
- 14. Yang L, Anderson DE, Baecher‐Allan C, Hastings WD, Bettelli E, Oukka M et al. IL‐21 and TGF‐β are required for differentiation of human TH17 cells. Nature 2008; 454:350–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE et al. Generation of pathogenic TH17 cells in the absence of TGF‐β signalling. Nature 2010; 467:967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Acosta‐Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F Interleukins 1b and 6 but not transforming growth factor‐β are essential for the differentiation of interleukin 17‐producing human T helper cells. Nat. Immunol. 2007; 8:942–9. [DOI] [PubMed] [Google Scholar]
- 17. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD et al. Development, cytokine profile and function of human interleukin 17‐producing helper T cells. Nat Immunol. 2007; 8:950–7. [DOI] [PubMed] [Google Scholar]
- 18. Santarlasci V, Maggi L, Capone M, Frosali F, Querci V, De Palma R et al. TGF‐β indirectly favors the development of human Th17 cells by inhibiting Th1 cells. Eur J Immunol. 2009; 39:207–15. [DOI] [PubMed] [Google Scholar]
- 19. Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F et al. Human interleukin‐17‐producing cells originate from a CD161+ CD4+ T‐cell precursor. J. Exp. Med. 2008; 205:1903–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spencer S, Köstel Bal S, Egner W, Lango Allen H, Raza SI, Ma CA et al. Loss of the interleukin‐6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med. 2019; 216:1986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martínez‐Barricarte R, Markle JG, Ma CS, Deenick EK, Ramírez‐Alejo N, Mele F et al. Human IFN‐γ immunity to mycobacteria is governed by both IL‐12 and IL‐23. Sci Immunol. 2018; 3:eaau6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010; 32:605–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM et al. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper‐IgE syndrome. Nature 2008; 452:773–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL‐17 T helper cells. Cell 2006; 126:1121–33. [DOI] [PubMed] [Google Scholar]
- 25. Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M et al. Immunodeficiencies. Impairment of immunity to Candida and Mycobacterium in humans with bi‐allelic RORC mutations. Science 2015; 349:606–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell‐mediated effector immunity. J Allergy Clin Immunol. 2015; 135:626–35. [DOI] [PubMed] [Google Scholar]
- 27. Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010; 140:845–58. [DOI] [PubMed] [Google Scholar]
- 28. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona‐Limon P et al. Coexpression of CD49b and LAG‐3 identifies human and mouse T regulatory type 1 cells. Nat Med. 2013; 19:739–46. [DOI] [PubMed] [Google Scholar]
- 29. Murphy E, Shibuya K, Hosken N, Openshaw P, Maino V, Davis K et al. Reversibility of T helper 1 and 2 populations is lost after long‐term stimulation. J Exp Med. 1996; 183:901–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mullen AC, High FA, Hutchins AS. Role of T‐bet in commitment of TH1 cells before IL‐12‐dependent selection. Science 2001; 292:1907–10. [DOI] [PubMed] [Google Scholar]
- 31. Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM Transcription factors T‐bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat. Immunol. 2007; 8:145–53. [DOI] [PubMed] [Google Scholar]
- 32. Szabo SJ, Dighe AS, Gubler U, Murphy KM Regulation of the interleukin (IL)‐12Rβ2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J. Exp. Med. 1997; 185:817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Takeda A, Hamano S, Yamanaka A, Hanada T, Ishibashi T, Mak TW et al. Cutting edge: role of IL‐27/WSX‐1 signaling for induction of T‐bet through activation of STAT1 during initial Th1 commitment. J. Immunol. 2003; 170:4886–90. [DOI] [PubMed] [Google Scholar]
- 34. Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17‐producing T cells. Nat. Immunol. 2006; 7:929–36. [DOI] [PubMed] [Google Scholar]
- 35. Zhu J, Guo L, Min B, Watson CJ, Hu‐Li J, Young HA et al. Growth factor independent‐1 induced by IL‐4 regulates Th2 cell proliferation. Immunity 2002; 16:733–44. [DOI] [PubMed] [Google Scholar]
- 36. Zhu J, Davidson TS, Wei G, Jankovic D, Cui K, Schones DE et al. Down‐regulation of Gfi‐1 expression by TGF‐β is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J. Exp. Med. 2009; 206:329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Annunziato F, Cosmi L, Manetti R, Brugnolo F, Parronchi P, Maggi E et al. Reversal of human allergen‐specific CRTH2+ TH2 cells by IL‐12 or the PS‐DSP30 oligodeoxynucleotide. J Allergy Clin Immunol. 2001; 108:815–21. [DOI] [PubMed] [Google Scholar]
- 38. Brugnolo F, Sampognaro S, Liotta F, Cosmi L, Annunziato F, Manuelli C et al. The novel synthetic immune response modifier R‐848 (Resiquimod) shifts human allergen‐specific CD4+ TH2 lymphocytes into IFN‐γ‐producing cells. J Allergy Clin Immunol. 2003; 111:380–8. [DOI] [PubMed] [Google Scholar]
- 39. Messi M, Giacchetto I, Nagata K, Lanzavecchia A, Natoli G, Sallusto F Memory and flexibility of cytokine gene expression as separable properties of human TH1 and TH2 lymphocytes. Nat Immunol. 2003; 4:78–86. [DOI] [PubMed] [Google Scholar]
- 40. Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007; 204:1849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W et al Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U S A. 2010; 107:14751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cosmi L, Cimaz R, Maggi L, Santarlasci V, Capone M, Borriello F et al. Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum. 2011; 63:2504–15. [DOI] [PubMed] [Google Scholar]
- 43. Maggi L, Santarlasci V, Capone M, Rossi MC, Querci V, Mazzoni A et al. Distinctive features of classic and nonclassic (Th17 derived) human Th1 cells. Eur J Immunol. 2012; 42:3180–8. [DOI] [PubMed] [Google Scholar]
- 44. Mazzoni A, Maggi L, Siracusa F, Ramazzotti M, Rossi MC, Santarlasci V et al. Eomes controls the development of Th17‐derived (non‐classic) Th1 cells during chronic inflammation. Eur J Immunol. 2019; 49:79–95. [DOI] [PubMed] [Google Scholar]
- 45. Maggi L, Cimaz R, Capone M, Santarlasci V, Querci V, Simonini G et al. Brief report: etanercept inhibits the tumor necrosis factor α‐driven shift of Th17 lymphocytes toward a nonclassic Th1 phenotype in juvenile idiopathic arthritis. Arthritis Rheumatol. 2014; 66:1372–7. [DOI] [PubMed] [Google Scholar]
- 46. Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO et al. Late developmental plasticity in the T helper 17 lineage. Immunity 2009; 30:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lexberg MH, Taubner A, Förster A, Albrecht I, Richter A, Kamradt T et al. Th memory for interleukin‐17 expression is stable in vivo. Eur J Immunol. 2008; 38:2654–64. [DOI] [PubMed] [Google Scholar]
- 48. Lexberg MH, Taubner A, Albrecht I, Lepenies I, Richter A, Kamradt T et al. IFN‐γ and IL‐12 synergize to convert in vivo generated Th17 into Th1/Th17 cells. Eur J Immunol. 2010; 40:3017–27. [DOI] [PubMed] [Google Scholar]
- 49. Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature 2019; 565:101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F et al. Identification of a novel subset of human circulating memory CD4+ T cells that produce both IL‐17A and IL‐4. J Allergy Clin Immunol. 2010; 125:222–30.e1‐4. [DOI] [PubMed] [Google Scholar]
- 51. Irvin C, Zafar I, Good J, Rollins D, Christianson C, Gorska MM et al. Increased frequency of dual‐positive TH2/TH17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J Allergy Clin Immunol. 2014; 134:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM et al. Plasticity of Th17 cells in Peyer's patches is responsible for the induction of T cell‐dependent IgA responses. Nat Immunol. 2013; 14:372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015; 523:221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cohen CJ, Crome SQ, MacDonald KG, Dai EL, Mager DL, Levings MK Human Th1 and Th17 cells exhibit epigenetic stability at signature cytokine and transcription factor loci. J Immunol. 2011; 187:5615–26. [DOI] [PubMed] [Google Scholar]
- 55. Brown CC, Esterhazy D, Sarde A, London M, Pullabhatla V, Osma‐Garcia I et al. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 2015; 42:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Santarlasci V, Maggi L, Capone M, Querci V, Beltrame L, Cavalieri D et al Rarity of human T helper 17 cells is due to retinoic acid orphan receptor‐dependent mechanisms that limit their expansion. Immunity 2012; 36:201–14. [DOI] [PubMed] [Google Scholar]
- 57. Santarlasci V, Maggi L, Mazzoni A, Capone M, Querci V, Rossi MC et al IL‐4‐induced gene 1 maintains high Tob1 expression that contributes to TCR unresponsiveness in human T helper 17 cells. Eur J Immunol. 2014; 44:654–61. [DOI] [PubMed] [Google Scholar]
- 58. Santarlasci V, Mazzoni A, Capone M, Rossi MC, Maggi L, Montaini G et al . Musculin inhibits human T‐helper 17 cell response to interleukin 2 by controlling STAT5B activity. Eur J Immunol. 2017; 47:1427–1442. [DOI] [PubMed] [Google Scholar]
- 59. Liu HP, Cao AT, Feng T, Li Q, Zhang W, Yao S et al. TGF‐β converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur J Immunol. 2015; 45:1010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M et al. Foxp3+ T cells expressing RORγt represent a stable regulatory T‐cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. 2016; 9:444–57. [DOI] [PubMed] [Google Scholar]
- 61. Solomon BD, Hsieh CS. Antigen‐specific development of mucosal Foxp3+RORγt+ T cells from regulatory T cell precursors. J Immunol. 2016; 197:3512–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Osorio F, LeibundGut‐Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G et al . DC activated via dectin‐1 convert Treg into IL‐17 producers. Eur J Immunol. 2008; 38:3274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kluger MA, Meyer MC, Nosko A, Goerke B, Luig M, Wegscheid C et al. RORγt+Foxp3+ Cells are an independent bifunctional regulatory T cell lineage and mediate crescentic GN. J Am Soc Nephrol. 2016; 27:454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wei G, Wei L, Zhu J, Zang C, Hu‐Li J, Yao Z et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009; 30:155–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mukasa R, Balasubramani A, Lee YK, Whitley SK, Weaver BT, Shibata Y et al. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 2010; 32:616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bending D, Newland S, Krejcí A, Phillips JM, Bray S, Cooke A Epigenetic changes at Il12rb2 and Tbx21 in relation to plasticity behavior of Th17 cells. J Immunol. 2011;186:3373–82. [DOI] [PubMed] [Google Scholar]
- 67. Mazzoni A, Santarlasci V, Maggi L, Capone M, Rossi MC, Querci V et al. Demethylation of the RORC2 and IL17A in human CD4+ T lymphocytes defines Th17 origin of nonclassic Th1 cells. J Immunol. 2015; 194:3116–26. [DOI] [PubMed] [Google Scholar]
- 68. Acosta‐Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A et al. Surface phenotype and antigenic specificity of human interleukin 17‐producing T helper memory cells. Nat Immunol. 2007; 8:639–46. [DOI] [PubMed] [Google Scholar]
- 69. Lindestam Arlehamn CS, Gerasimova A, Mele F, Henderson R, Swann J, Greenbaum JA et al. Memory T cells in latent Mycobacterium tuberculosis infection are directed against three antigenic islands and largely contained in a CXCR3+CCR6+ Th1 subset. PLoS Pathog. 2013; 9:e1003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Duhen T, Campbell DJ. IL‐1β promotes the differentiation of polyfunctional human CCR6+CXCR3+ Th1/17 cells that are specific for pathogenic and commensal microbes. J Immunol. 2014; 193:120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M et al. Pathogen‐induced human TH17 cells produce IFN‐γ or IL‐10 and are regulated by IL‐1β . Nature 2012; 484:514–8. [DOI] [PubMed] [Google Scholar]
- 72. Dardalhon V, Korn T, Kuchroo VK, Anderson AC Role of Th1 and Th17 cells in organ‐specific autoimmunity. J Autoimmun. 2008; 31:252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Martin‐Orozco N, Chung Y, Chang SH, Wang YH, Dong C Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol. 2009; 39:216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bending D, De la Peña H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B et al. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1‐like cells in NOD/SCID recipient mice. J Clin Invest. 2009; 119:565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reinert‐Hartwall L, Honkanen J, Salo HM, Nieminen JK, Luopajärvi K, Härkönen T et al. Th1/Th17 plasticity is a marker of advanced β cell autoimmunity and impaired glucose tolerance in humans. J Immunol. 2015; 194:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci U S A. 2015; 112:7061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sujino T, Kanai T, Ono Y, Mikami Y, Hayashi A, Doi T et al. Regulatory T cells suppress development of colitis, blocking differentiation of T‐helper 17 into alternative T‐helper 1 cells. Gastroenterology 2011; 141:1014–23. [DOI] [PubMed] [Google Scholar]
- 78. Morrison PJ, Bending D, Fouser LA, Wright JF, Stockinger B, Cooke A et al. Th17‐cell plasticity in Helicobacter hepaticus‐induced intestinal inflammation. Mucosal Immunol. 2013; 6:1143–56. [DOI] [PubMed] [Google Scholar]
- 79. Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E Cutting edge: the pathogenicity of IFN‐γ‐producing Th17 cells is independent of T‐bet. J Immunol. 2013; 190:4478–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Grifka‐Walk HM, Lalor SJ, Segal BM. Highly polarized Th17 cells induce EAE via a T‐bet independent mechanism. Eur J Immunol. 2013; 43:2824–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Paroni M, Maltese V, De Simone M, Ranzani V, Larghi P, Fenoglio C et al. Recognition of viral and self‐antigens by TH1 and TH1/TH17 central memory cells in patients with multiple sclerosis reveals distinct roles in immune surveillance and relapses. J Allergy Clin Immunol. 2017; 140:797–808. [DOI] [PubMed] [Google Scholar]
- 82. Piper C, Pesenacker AM, Bending D, Thirugnanabalan B, Varsani H, Wedderburn LR et al. T cell expression of granulocyte‐macrophage colony‐stimulating factor in juvenile arthritis is contingent upon Th17 plasticity. Arthritis Rheumatol. 2014; 66:1955–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Maggi L, Margheri F, Luciani C, Capone M, Rossi MC, Chillà A et al. Th1‐induced CD106 expression mediates leukocytes adhesion on synovial fibroblasts from juvenile idiopathic arthritis patients. PLoS One 2016; 11:e0154422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gartlan KH, Varelias A, Koyama M, Robb RJ, Markey KA, Chang K et al. Th17 plasticity and transition toward a pathogenic cytokine signature are regulated by cyclosporine after allogeneic SCT. Blood Adv. 2017; 1:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Krebs CF, Turner JE, Paust HJ, Kapffer S, Koyro T, Krohn S et al. Plasticity of Th17 cells in autoimmune kidney diseases. J Immunol. 2016; 197:449–57. [DOI] [PubMed] [Google Scholar]
- 86. Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G et al. Effects of AIN457, a fully human antibody to interleukin‐17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010; 2:52ra72. [DOI] [PubMed] [Google Scholar]
- 87. Frieder J, Kivelevitch D, Menter A. Secukinumab: a review of the anti‐IL‐17A biologic for the treatment of psoriasis. Ther Adv Chronic Dis. 2018; 9:5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD et al. Secukinumab, a human anti‐IL‐17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double‐blind placebo‐controlled trial. Gut 2012; 61:1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Haraoui B, Bykerk V. Etanercept in the treatment of rheumatoid arthritis. Ther Clin Risk Manag. 2007; 3:99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Levin AD, Wildenberg ME, van den Brink GR. Mechanism of action of anti‐TNF therapy in inflammatory bowel disease. J Crohns Colitis. 2016; 10:989–97. [DOI] [PubMed] [Google Scholar]
- 91. Adegbola SO, Sahnan K, Warusavitarne J, Hart A, Tozer P Anti‐TNF therapy in Crohn's disease. Int J Mol Sci. 2018; 19:E2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Menegatti S, Bianchi E, Rogge L. Anti‐TNF therapy in spondyloarthritis and related diseases, impact on the immune system and prediction of treatment responses. Front Immunol. 2019;10:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Maggi L, Cimaz R, Capone M, Santarlasci V, Rossi MC, Mazzoni A et al. Immunosuppressive activity of abatacept on circulating T helper lymphocytes from juvenile idiopathic arthritis patients. Int Arch Allergy Immunol. 2016; 171:45–53. [DOI] [PubMed] [Google Scholar]
- 94. Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y et al. A human interleukin‐12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med. 2007; 356:580–92. [DOI] [PubMed] [Google Scholar]
- 95. Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S et al. A randomized trial of Ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with moderate‐to‐severe Crohn's disease. Gastroenterology 2008; 135:1130–41. [DOI] [PubMed] [Google Scholar]
- 96. Thibodaux RJ, Triche MW, Espinoza LR. Ustekinumab for the treatment of psoriasis and psoriatic arthritis: a drug evaluation and literature review. Expert Opin Biol Ther. 2018; 18:821–827. [DOI] [PubMed] [Google Scholar]
- 97. Sigidin YA, Loukina GV, Skurkovich B, Skurkovich S Randomized, double‐blind trial of anti‐interferon‐γ antibodies in rheumatoid arthritis. Scand J Rheumatol. 2001; 30:203–7. [DOI] [PubMed] [Google Scholar]
- 98. Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis. 2013; 72:1445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]