

Summary
Siglecs are cell surface lectins that recognize sialic acids and are primarily expressed in hematopoietic cells. Previous studies showed that some Siglecs regulate macrophage function. In the present study, we examined the induction and putative roles of mouse Siglec‐F in bone‐marrow‐derived macrophages in mice. A quantitative RT‐PCR analysis showed that the basal expression of Siglec‐F was weak in bone‐marrow‐derived macrophages differentiated by macrophage colony‐stimulating factor. However, a 24‐hr stimulation with granulocyte–macrophage colony‐stimulating factor (GM‐CSF) enhanced Siglec‐F expression. GM‐CSF also enhanced Siglec‐F expression in thioglycollate‐induced peritoneal macrophages. The inhibition of signal transducer and activator of transcription 5 (STAT5), but not that of phosphoinositide 3‐kinase or mitogen‐activated protein kinase kinase, significantly reduced the induction of Siglec‐F. Interleukin‐3, which uses a common β‐chain shared with the GM‐CSF receptor to stimulate the STAT5 pathway, also enhanced Siglec‐F expression. The knockdown of Siglec‐F by a specific small interfering RNA enhanced GM‐CSF‐induced STAT5 phosphorylation, suggesting that Siglec‐F down‐regulates its own expression upon prolonged GM‐CSF stimulation. Furthermore, the knockdown of Siglec‐F reduced the STAT6 phosphorylation and expression of arginase‐1 in interleukin‐4‐stimulated macrophages. These results suggest that Siglec‐F fine‐tunes the immune responses of macrophages.
Keywords: arginase, granulocyte–macrophage colony‐stimulating factor, interleukin‐4, macrophage, Siglec‐F, signal transducer and activator of transcription 5
Abbreviations
- APC
allophycocyanin
- Arg1
arginase‐1
- BMDM
bone‐marrow‐derived macrophage
- FITC
fluorescein isothiocyanate
- GM‐CSF
granulocyte–macrophage colony‐stimulating factor
- HPRT
hypoxanthine‐guanosine phosphoribosyl transferase
- IFN
interferon
- IgG
immunoglobulin G
- iNOS
inducible NO synthase
- Jak2
Janus kinase 2
- L929‐BMDM
L929 supernatant‐differentiated BMDM
- LPS
lipopolysaccharide
- M‐BMDM
M‐CSF‐differentiated BMDM
- M-CSF
macrophage colony-stimulating factor
- PBS
phosphate-buffered saline
- qRT‐PCR
quantitative reverse transcription-polymerase chain reaction
- siRNA
small interfering RNA
- SOCS1
suppressor of cytokine signaling 1
- STAT5
signal transducer and activator of transcription 5
- STAT6
signal transducer and activator of transcription 6
Introduction
Macrophages are central players in inflammation and host defenses, and show considerable phenotype diversity and plasticity in response to environmental stimuli.1, 2 In many cases, macrophages have been categorized into two broad types – classical and alternatively activated states – as the extreme states of phenotypes. Experimentally, macrophages may polarize to classically activated macrophages (M1) in response to Toll‐like receptor ligands and interferon‐γ (IFN‐γ), and to alternatively activated macrophages (M2) by interleukin‐4 (IL‐4) and IL‐13. The former phenotype is characterized, for example, by the production of pro‐inflammatory cytokines and inducible nitric oxide synthase (iNOS), and the latter by the production of anti‐inflammatory cytokines and arginase‐1 (Arg1).1, 2
Siglecs are sialic acid‐recognizing immunoglobulin‐like lectins primarily expressed in immune cells.3, 4, 5 Five and 11 CD33‐related Siglecs have been identified in mice and humans, respectively. Most Siglecs have immunoreceptor tyrosine‐based inhibitory motifs in the cytoplasmic region of the protein, to which the phosphotyrosine phosphatases SHP1/2 bind. Siglecs exhibit potent immune‐modulating activities including inflammation.6, 7, 8 The expression patterns of Siglecs have been described,3, 4, 5 and some Siglecs are conveniently used as cell‐specific markers, including Siglec‐H as a plasmacytoid dendritic cell marker9 and Siglec‐F as an eosinophil marker.10 On the other hand, mouse macrophages have been reported to express several Siglecs upon activation. Siglec‐G, originally reported as a specific inhibitor of B1 cells, is induced by an infection by the RNA virus, such as vesicular stomatitis virus.11 Induced Siglec‐G inhibits anti‐virus responses by degrading the cytosolic pathogen sensor RIG‐I.11 Furthermore, the expression of Siglec‐E is induced by several Toll‐like receptor ligands such as lipopolysaccharide (LPS) or CpG DNA in bone‐marrow‐derived macrophages (BMDMs).7 Siglec‐E inhibits the production of pro‐inflammatory cytokines in vitro in macrophages stimulated by Toll‐like receptor ligands or group B Streptococcus, 7, 12 and in microglia stimulated by neural debris.13 Whether multiple Siglecs are regulated simultaneously on various stimuli has not been reported.
Siglec‐F is a functional paralog of human Siglec‐8, which is predominantly expressed in eosinophils.10 Cross‐linking of Siglec‐F or Siglec‐8 by antibodies results in the apoptosis of eosinophils through caspase activation.14, 15 Siglec‐F−/− mice exhibited enhanced lung inflammation in an allergic model due to an increased number of eosinophils.16 Hence, Siglec‐F appears to reduce eosinophilic inflammation. However, the changes in expression level and the physiological role of Siglec‐F in macrophages have not been clarified in detail, except that alveolar macrophages constitutively express a high level of Siglec‐F.17
In the present study, we examined the inducible expression of CD33‐related Siglecs by several stimuli in mouse BMDMs as a model system and found that Siglec‐F was induced by granulocyte–macrophage colony‐stimulating factor (GM‐CSF) through the activation of signal transducer and activator of transcription 5 (STAT5). The knockdown experiments showed that Siglec‐F reduced STAT5 signaling following GM‐CSF stimulation, suggesting the existence of a regulatory loop of Siglec‐F expression in which Siglec‐F negatively controls its own expression through the inhibition of STAT5. We also showed that Siglec‐F positively regulated the STAT6 signaling pathway as well as the expression of Arg1 in IL‐4‐stimulated macrophages. These results suggest that Siglec‐F is induced by GM‐CSF and fine‐tunes macrophage responses.
Materials and methods
Mice
Eight‐week‐old male C57BL/6 mice were purchased from Slc Japan (Shizuoka, Japan). All animal experiments were approved by the Institutional Animal Experiment Committee of Nagoya University and conducted in accordance with the regulations on animal experiments in Nagoya University and the Guidelines for the Proper Conduct of Animal Experiments (Science Council of Japan).
Reagents
Mouse macrophage colony‐stimulating factor (M‐CSF), GM‐CSF, IL‐4, IFN‐γ, IL‐3, and IL‐5 were purchased from PeproTech (Rocky Hill, NJ). LPS (Escherichia coli 0111:B4) was purchased from Sigma‐Aldrich (St. Louis, MO). Thioglycollate was obtained from Eiken Chemical (Tokyo, Japan). The mitogen‐activated protein kinase kinase (MEK) inhibitor PD0325901 and phosphoinositide 3‐kinase (PI3K) inhibitor wortmannin were purchased from Wako Chemicals (Osaka Japan) and Cayman Chemical (Ann Arbor, MI), respectively. The STAT5 inhibitor (CAS 285986‐31‐4) was obtained from Calbiochem (Darmstadt, Germany). The following antibodies were used in the analysis and purification of cells. A phycoerythrin‐labeled rat anti‐Siglec‐F (E50‐2440) antibody and an unlabeled rat anti‐Siglec‐F (238023) antibody were obtained from BD Biosciences (San Jose, CA) and R&D Systems (Minneapolis, MN), respectively. Fluorescein isothiocyanate (FITC) ‐labeled rat anti‐F4/80 (BM8.1) and allophycocyanin (APC) ‐labeled rat anti‐CD11b (M1/70) antibodies were from TONBO Biosciences (San Diego, CA). Fc block (anti‐CD16/CD32 (Fc receptor) antibody, 2.4G2, 553141) was from BD Biosciences. Phycoerythrin‐labeled control rat immunoglobulin G2a (IgG2a) (RTK2758) was obtained from Biolegend (San Diego, CA). Antibodies were used at 1 μg/ml in the flow cytometric analysis. Antibodies towards phospho‐p44/42 (Thr202/Tyr204) mitogen‐activated protein kinase [phospho‐extracellular signal‐regulated kinase (pERK), #9101], phospho‐Akt (Ser473, #9271), and phospho‐STAT5 (Y694, C11C5, #9359) were purchased from Cell Signaling Technology (Danvers, MA). Rabbit anti‐phospho‐STAT6 (Tyr641, sc‐11762) and anti‐IκB‐α (C21, sc‐371) antibodies were from Santa Cruz Biotechnology (Dallas, TX). A rabbit anti‐Arg1 (GTX109242) antibody was from GeneTex (Irvine, CA). Rabbit anti‐ERK (51068‐1‐AP) and anti‐Akt (10176‐2‐AP) antibodies were from Proteintech (Rosemont, IL). Rabbit anti‐STAT5 (AF2168) and anti‐STAT6 (HPA001861) antibodies were obtained from R&D Systems and Sigma‐Aldrich, respectively. A mouse anti‐β‐actin antibody (6D1) was from Medical and Biological Laboratories (MBL, Nagoya, Japan). Peroxidase‐labeled goat anti‐mouse IgG (#330) and anti‐rabbit IgG (#458) antibodies were from MBL.
Preparation of macrophages
L929 cells were obtained from the Riken Cell Bank (Tsukuba, Japan), and maintained in RPMI‐1640 medium supplemented with 10% fetal calf serum (Biological Industries, Kibbutz Beit Haemek, Israel), 100 U/ml penicillin, and 100 μg/ml streptomycin. L929 cells were cultured for 7 days, and the supernatant containing M‐CSF was harvested and filtered (0·45 μm), and aliquots were stored at −80°C until used.
Bone marrow cells were recovered from the femurs and tibias of the sacrificed mice by flushing out the bone cavities with phosphate‐buffered saline (PBS). Cells were treated for 3 min with ACK solution (155 mm NH4Cl, 10 mm KHCO3, and 0·1 mm EDTA) to lyze erythrocytes. To obtain L929‐BMDMs, bone marrow cells were cultured for 7–9 days in the presence of 20% L929 supernatant, and to obtain M‐CSF‐differentiated (M‐BMDMs), bone marrow cells were cultured in the presence of 10 ng/ml M‐CSF. After non‐adherent cells had been removed, macrophages were harvested. The purity of cells was routinely >90%, which was confirmed by staining with anti‐F4/80 or anti‐CD11b antibodies (data not shown). A quantitative reverse transcription polymerase chain reaction (qRT‐PCR) analysis revealed that the expression levels of eosinophil‐specific genes (IL‐5 receptor α chain and GATA118) were lower than 0·2% that of eosinophil freshly isolated from peritoneal cavity (data not shown). In some experiments, macrophages were purified based on the expression of CD11b and F4/80 using FACSJazz (BD Biosciences) as follows. Harvested M‐BMDMs were initially incubated with Fc block, and stained with APC‐labeled anti‐CD11b and FITC‐labeled anti‐F4/80 antibodies. CD11b+ F4/80+ cells were sorted as macrophages (see Supplementary material, Fig. S1a).
In order to obtain thioglycollate‐elicited peritoneal cavity‐derived macrophages, C57BL/6 mice were injected intraperitoneally with 2·5 ml of 3% thioglycollate. After 72 hr, peritoneal cells were harvested by lavage with 0·67% EDTA/PBS. Cells were incubated with Fc block, and then stained with phycoerythrin‐labeled anti‐Siglec‐F, APC‐labeled anti‐CD11b, and FITC‐labeled anti‐F4/80 antibodies. Siglec Flow CD11b+ F4/80+ cells were sorted as macrophages (see Supplementary material, Fig. S1b). In some experiments, Siglec‐Fhigh cells were sorted as eosinophils to compare the expression of Siglec‐F.
Cell stimulation
Macrophages (4·0 × 105) were seeded in RPMI‐1640 medium supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin on 24‐well plates and cultured for 2–4 hr to allow attachment. Medium was replaced with fresh medium to remove non‐adherent cells. Cells were then stimulated in the absence of M‐CSF. Unless otherwise stated, 10 ng/ml GM‐CSF, 20 ng/ml IL‐4, 10 ng/ml LPS, 10 ng/ml IFN‐γ, and 20 ng/ml IL‐3 were used.
Inhibitors were added at a predetermined concentration (ref.19 and data not shown). As a control, the corresponding concentration of vehicle (dimethyl sulfoxide) was added to the culture. For MEK inhibition, cells were preincubated with PD0325901 (1 μm) for 30 min, washed with PBS, and then stimulated with GM‐CSF in the absence of the inhibitor. To inhibit PI3K, cells were preincubated with wortmannin (100 nm) for 30 min, washed, and stimulated with GM‐CSF in the absence of the inhibitor, and to inhibit STAT5, cells were pretreated with the STAT5 inhibitor (500 μm) for 24 hr and stimulated with GM‐CSF in the presence of the inhibitor. In the case of the STAT5 inhibitor, the GM‐CSF stimulation was performed for 8 hr to reduce cell damage, and gene expression was analyzed by qRT‐PCR. For detection of the phosphorylation of ERK, Akt, and STAT5 by a Western blot analysis, cells were stimulated for 30 min.
qRT‐PCR
Unless otherwise stated, cells were stimulated for 24 hr. Total RNA was extracted by Isogen II (Nippon Gene, Tokyo, Japan) and reverse‐transcribed by ReverTra Ace (Toyobo, Osaka, Japan), as reported previously.20 To obtain the cDNA of eosinophils, total RNA was extracted by the ReliaPrep RNA Cell Miniprep System (Promega, Madison, WI) and reverse‐transcribed by the ReverTra Ace qPCR RT Master Mix with the gDNA Remover (Toyobo). The qPCR was performed using the Thunderbird SYBR qPCR mix (Toyobo) using Light Cycler 96 (Roche Diagnostics, Basel, Switzerland) at 95°C for 60 seconds, followed by 45 cycles at 95°C for 5 seconds, at 58°C for 10 seconds, and at 72°C for 20 seconds. Data were normalized by hypoxanthine‐guanosine phosphoribosyl transferase (HPRT) gene expression. To compare the expression levels of Siglecs, a purified plasmid DNA in which PCR fragments for the Siglecs and HPRT were inserted was used as standard after linearization. Normalization for other genes was by the ΔΔC t method. The primers used are listed in the Supplementary material (Table S1).
Knockdown by small interfering RNA (siRNA)
Stealth small siRNAs were purchased from Invitrogen (Carlsbad, CA). Stealth RNAi™ siRNA‐negative control med GC (Invitrogen) was used as control siRNA. The siRNAs were transfected to BMDMs by Lipofectamine 3000 (Invitrogen) or INTERFERin (Polyplus Transfection, Illkirch, France) according to the supplier's recommendations. In the knockdown of Siglec‐F, a mixture of equal amounts of the two siRNAs (#1 and #2) or each siRNA (#1, #2 or #3) was used in experiments. BMDMs (2·0 × 105) were seeded on 24‐well plates and knocked down for 48 hr in the presence of M‐CSF (10 ng/ml), followed by a gentle wash with PBS, and were then stimulated in the absence of M‐CSF. In some experiments, GM‐CSF was used in the place of M‐CSF. In the knockdown of STAT5, the siRNAs that targeted STAT5A and STAT5B were used. The sequences of siRNAs used are shown in the Supplementary material (Table S2).
Western blotting
Cells were washed with PBS and lyzed with sodium dodecyl sulfate sample buffer and subjected to Western blotting, as reported previously.19 Band intensities were assessed using imagej software,21 and compared after normalization by the band intensity of actin.
Statistical analysis
Data are presented as the mean ± standard error of at least three independent experiments. The significance of differences was analyzed by Student's t‐test or a one‐way analysis of variance followed by Tukey's post hoc test. A P value < 0·05 was considered to be significant.
Results
Up‐regulation of Siglec‐F by GM‐CSF in macrophages
The expression of CD33‐related Siglecs in BMDMs activated by various stimuli was analyzed. Mouse bone marrow cells were cultured with M‐CSF to induce M‐BMDMs. These cells were confirmed to express iNOS (M1 marker) and Arg1 (M2 marker) mRNA by LPS plus IFN‐γ and IL‐4, respectively, as judged by qRT‐PCR (data not shown). Hence, these cells were used as macrophages in subsequent analyses. Cells were challenged by several stimuli, such as LPS, IL‐4, and GM‐CSF, and the expression levels of Siglecs were analyzed by qRT‐PCR (Fig. 1). The expression of CD33 and Siglec‐G was induced by GM‐CSF (3·4‐fold and 4·5‐fold, respectively). The expression of CD33 was slightly increased by LPS. Siglec‐E was enhanced by LPS (8·3‐fold), but not by LPS plus IFN‐γ. Siglec‐F was induced by GM‐CSF (5‐fold), but appeared to be repressed by LPS or LPS plus IFN‐γ. Siglec‐H was not induced by any of the stimuli tested. After the stimulation with GM‐CSF, the expression level of Siglec‐F appeared to be markedly higher than those of CD33 and Siglec‐E, ‐G, and ‐H with all of the stimuli tested. In contrast to Siglec‐E (Fig. 1 and ref. 7), Siglec‐F was not induced by LPS, suggesting that the expression of each Siglec is regulated in a different manner. To confirm the induced expression of Siglec‐F by GM‐CSF, the cell surface expression of Siglec‐F was investigated using a flow cytometric analysis. A Siglec‐F signal was detected by two different anti‐Siglec‐F antibodies in the absence of stimulation, whereas GM‐CSF stimulation for 24 hr increased cell surface Siglec‐F levels (Fig. 2a, b; see Supplementary material, Fig. S2).
Figure 1.
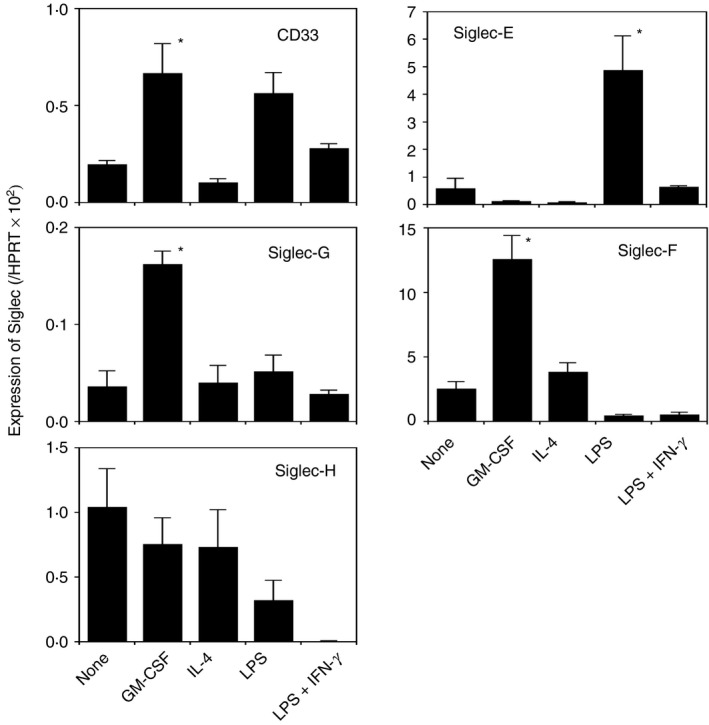
Induced expression of CD33‐related Siglecs in macrophage colony‐stimulating factor‐differentiated bone‐marrow‐derived macrophages (M‐BMDMs) by various stimuli. M‐BMDMs were seeded on 24‐well plates and stimulated as indicated for 24 hr. The expression of each Siglec was measured by qRT‐PCR. Data are the mean ± SE of three to nine independent experiments. *P < 0·05 versus none by a one‐way analysis of variance followed by Tukey's post hoc test.
Figure 2.
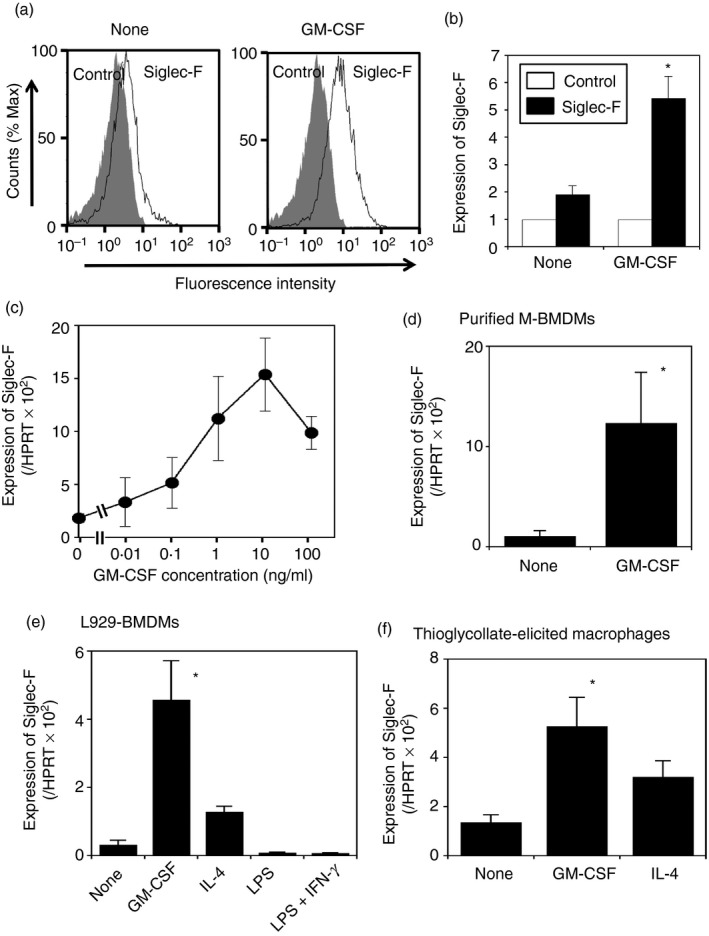
The expression of Siglec‐F is enhanced by granulocyte–macrophage colony‐stimulating factor (GM‐CSF) on macrophages. (a) A flow cytometric analysis of Siglec‐F on macrophages. Macrophage colony‐stimulating factor‐differentiated bone‐marrow‐derived macrophages (M‐BMDMs) were cultured for 24 hr with or without GM‐CSF, and stained by phycoerythrin‐labeled anti‐Siglec‐F (BD Biosciences) or control antibodies. The black lines and shaded images show anti‐Siglec‐F and control staining, respectively. A representative result of three independent experiments is shown. (b) Quantification of the flow cytometric analysis. Relative expression was calculated by mean fluorescence intensity (control staining = 1). White and black bars indicate staining with control and anti‐Siglec‐F antibodies, respectively. Data are the mean ± SE of three independent experiments. *P < 0·05 versus none by Student's t‐test. (c) Dose dependency of Siglec‐F expression induced by GM‐CSF. M‐BMDMs were stimulated with various concentrations of GM‐CSF for 24 hr and the expression of Siglec‐F was measured by qRT‐PCR. Data are the mean ± SE of three to five independent experiments. (d) GM‐CSF enhances Siglec‐F expression in purified BMDMs. Sorted M‐BMDMs were stimulated by GM‐CSF for 24 hr and Siglec‐F expression was examined by qRT‐PCR. Data are the mean ± SE of three independent experiments. *P < 0·05 versus none by Student's t‐test. (e) Induced expression of Siglec‐F in L929‐BMDMs. L929‐BMDMs were seeded on 24‐well plates and stimulated as indicated for 24 hr. The expression of Siglec‐F was measured by qRT‐PCR. Data are the mean ± SE of three independent experiments. *P < 0·05 versus none by a one‐way analysis of variance followed by Tukey's post hoc test. (f) Induced expression of Siglec‐F in thioglycollate‐elicited macrophages. Purified macrophages were seeded on 24‐well plates and stimulated as indicated for 24 hr, and Siglec‐F expression was then examined by qRT‐PCR. Data are the mean ± SE of four independent experiments. *P < 0·05 versus none by a one‐way analysis of variance followed by Tukey's post hoc test.
We then assessed the dose dependency of GM‐CSF to induce Siglec‐F. M‐BMDMs were stimulated with various concentrations of GM‐CSF for 24 hr because the maximal induction of Siglec‐F was observed after a 24‐hr stimulation (data not shown). The expression of Siglec‐F was up‐regulated by GM‐CSF in a dose‐dependent manner (Fig. 2c), and maximal expression levels were achieved at approximately 10 ng/ml GM‐CSF, which was similar to the concentration used in the in vitro culture to differentiate bone marrow cells to macrophages or dendritic cells by GM‐CSF22 and to stimulate eosinophils.23
To exclude the possibility that the up‐regulation of Siglec‐F was attributable to contaminated cells, we purified macrophages from M‐BMDMs using a cell sorter (see Supplementary material, Fig. S1a). Although eosinophils express high levels of Siglec‐F,10, 24 Siglec‐Fhigh cells were not detected in the M‐BMDM preparations. Therefore, CD11b+ and F4/80+ cells were collected without the removal of Siglec‐Fhigh cells. The up‐regulation of Siglec‐F by GM‐CSF was observed in purified M‐BMDMs (Fig. 2d). The extent of the induction by GM‐CSF (11·7‐fold) was higher than that in M‐BMDMs before purification (Fig. 1).
As the expression of Siglec‐F was up‐regulated by GM‐CSF stimulation in M‐BMDMs, we applied other conditions for macrophage differentiation. Mouse bone marrow cells were treated with an L929‐cell culture supernatant (L929‐BMDMs), which contains M‐CSF and has been used for the differentiation of macrophages from bone marrow cells.7 L929‐BMDMs expressed less Siglec‐F than M‐BMDMs without stimulation (compare Fig. 1 with Fig. 2e); however, the expression of Siglec‐F was induced by GM‐CSF (approximately 14‐fold) (Fig. 2e). Siglec‐F was slightly induced by IL‐4, but at a weaker magnitude than that by GM‐CSF (Fig. 2e). When IL‐4 was added in the presence of GM‐CSF, further enhancements were not observed in the expression of Siglec‐F (data not shown).
We then investigated whether similar up‐regulation occurs in thioglycollate‐elicited peritoneal cavity‐derived macrophages. In thioglycollate‐elicited peritoneal cavity cells, certain amounts of Siglec‐Fhigh cells were detected by flow cytometry (see Supplementary material, Fig. S1b), which possibly corresponded to eosinophils.24 Hence, macrophages were sorted as F4/80+ and CD11b+ cells after excluding Siglec‐Fhigh cells from peritoneal cavity cells using a cell sorter (see Supplementary material, Fig. S1b). Basal Siglec‐F mRNA levels were slightly lower in thioglycollate‐elicited macrophages than in M‐BMDMs (approximately 60–70%, compare Fig. 2f with Fig. 1), and a 3·9‐fold increase in Siglec‐F with GM‐CSF and a 2·4‐fold increase with IL‐4 were observed in the qRT‐PCR analysis of thioglycollate‐elicited macrophages (Fig. 2f).
Siglec‐F expression levels in GM‐CSF‐stimulated M‐BMDMs were 0·12‐fold to 0·15‐fold of HPRT (Figs 1 and 2), whereas those in freshly isolated eosinophils were 2·5‐fold of HPRT (data not shown): expression levels were approximately 17‐fold to 21‐fold higher in eosinophils than in GM‐CSF‐stimulated M‐BMDMs.
Identification of the downstream signal of GM‐CSF that drives Siglec‐F expression
The binding of GM‐CSF to its receptor activates Janus kinase 2 (JAK2),25 which results in the activation of three signaling pathways: the STAT5, mitogen‐activated protein kinase and PI3K pathways.26 To clarify which pathway is involved in the up‐regulation of Siglec‐F by GM‐CSF, downstream signaling pathways were blocked by their inhibitors. The MEK inhibitor PD0325901 did not affect GM‐CSF‐driven Siglec‐F expression (Fig. 3a), although the phosphorylated form of ERK was reduced. The PI3K inhibitor wortmannin inhibited Akt phosphorylation (Fig. 3b). Wortmannin slightly reduced Siglec‐F levels. In contrast, the STAT5 inhibitor strongly blocked GM‐CSF‐induced Siglec‐F expression (Fig. 3c). Furthermore, STAT5 siRNA, by which the mRNA level of STAT5 decreased to 60%, reduced the induced expression of Siglec‐F (Fig. 3d). These results indicate that the JAK2–STAT5 pathway was involved in Siglec‐F expression induced by GM‐CSF in macrophages.
Figure 3.
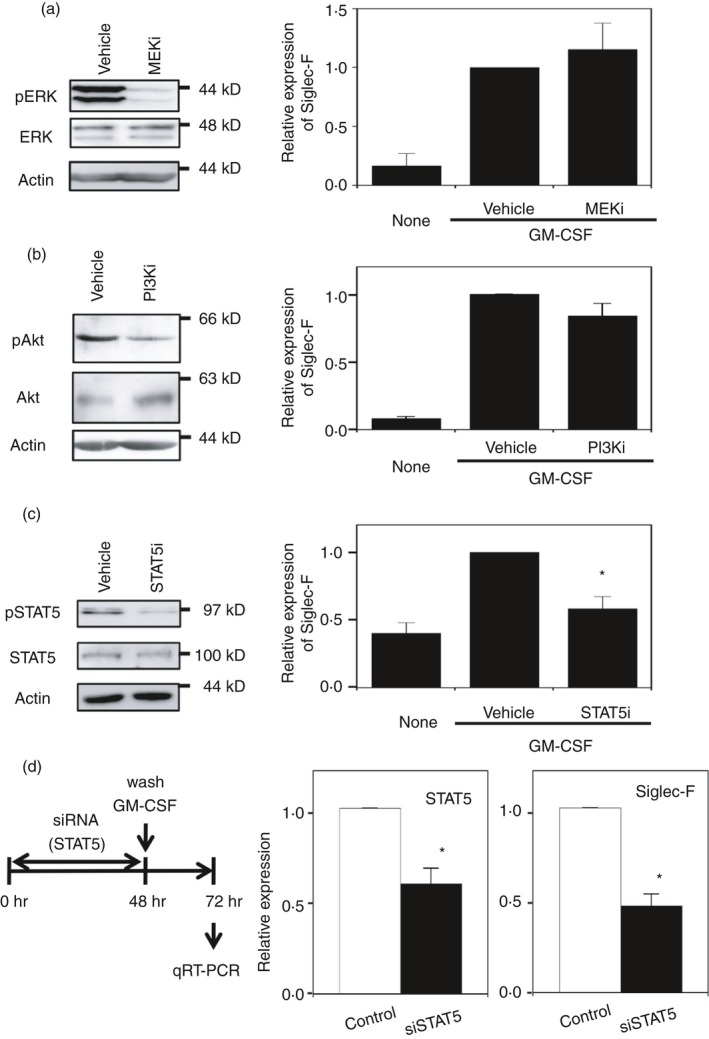
Enhancement of Siglec‐F expression by granulocyte–macrophage colony‐stimulating factor (GM‐CSF) depends on the signal transducer and activator of transcription 5 (STAT5) pathway. (a) Effects of the mitogen‐activated protein kinase kinase (MEK) inhibitor (PD0325901). (Left) Confirmation of MEK inhibition by a reduction in the phosphorylation of extracellular signal‐regulated kinase (ERK). Macrophage colony‐stimulating factor‐differentiated bone‐marrow‐derived macrophages (M‐BMDMs) were stimulated with GM‐CSF for 30 min. A representative result of two independent experiments is shown. (Right) The lack of inhibition of Siglec‐F expression by the MEK inhibitor. Siglec‐F expression was measured by qRT‐PCR after a 24‐hr stimulation. The vehicle control [dimethyl sulfoxide (DMSO) 0·1%] was regarded as 1. Data are the mean ± SE of three independent experiments. (b) Effects of the phosphoinositide 3‐kinase (PI3K) inhibitor (wortmannin). (Left) Confirmation of PI3K inhibition by a reduction in the phosphorylation of Akt. Cells were stimulated with GM‐CSF for 30 min. A representative result of three independent experiments is shown. (Right) The lack of inhibition of Siglec‐F expression by the PI3K inhibitor. Siglec‐F expression was measured by qRT‐PCR after a 24‐hr stimulation. The vehicle control (DMSO 0·1%) was regarded as 1. Data are the mean ± SE of five independent experiments. (c) Effects of the STAT5 inhibitor. (Left) Confirmation of STAT5 inhibition by a reduction in phosphorylation. Cells were stimulated with GM‐CSF for 30 min. A representative result of two independent experiments is shown. (Right) The inhibition of Siglec‐F expression by the STAT5 inhibitor. Siglec‐F expression was measured by qRT‐PCR after an 8‐hr stimulation. The vehicle control (DMSO 0·5%) was regarded as 1. Data are the mean ± SE of three independent experiments. *P < 0·05 versus the vehicle by Student's t‐test. (d) Enhancing effects of GM‐CSF on Siglec‐F expression are inhibited by the knockdown of STAT5. (Left) Schematic presentation of the experiment. M‐BMDMs were transfected with STAT5 or control small interfering RNA (siRNA). Cells were washed after a 48‐hr culture and stimulated with GM‐CSF for an additional 24 hr. The expression of STAT5 and Siglec‐F was measured by qRT‐PCR. (Middle) Knockdown efficiency of STAT5. (Right) Inhibition of Siglec‐F expression by STAT5 siRNA. The mRNA level in cells transfected with control siRNA was regarded as 1. Data are the mean ± SE of three independent experiments. *P < 0·05 versus control siRNA by Student's t‐test.
The receptors for IL‐3 and IL‐5 share a common βc subunit with the GM‐CSF receptor, and hence induce JAK2‐dependent downstream signaling including STAT5.27, 28 When M‐BMDMs were stimulated with IL‐3, Siglec‐F expression was induced (approximately sevenfold, see Supplementary material, Fig. S3). Consistent with the lack of the expression of IL‐5 receptor α chain in macrophages (data not shown), IL‐5 did not induce Siglec‐F expression in M‐BMDMs (data not shown).
Siglec‐F enhances Arg1 expression and STAT6 phosphorylation in IL‐4‐stimulated macrophages
To address the physiological roles of Siglec‐F, the induction of Arg1 by IL‐4 stimulation was analyzed in M‐BMDMs after the knockdown of Siglec‐F. Cells were simultaneously treated with siRNAs and GM‐CSF for 48 hr, and the Arg1 induction by IL‐4 was examined by Western blotting (Fig. 4a). By transfection of three different siRNAs against Siglec‐F, Siglec‐F expression decreased to approximately 25–50% of the control even after 24‐hr stimulation with IL‐4 (Fig. 4b). When the cells were stimulated with IL‐4 for 24 hr, two isoforms of Arg1 were induced in both control‐ and Siglec‐F‐knockdown M‐BMDMs (Fig. 4c), as reported previously.29 However, the expression level was reduced to 50–60% of the control by the knockdown (Fig. 4c, d). The results suggest that Siglec‐F enhanced Arg1 expression when macrophages were stimulated with IL‐4.
Figure 4.
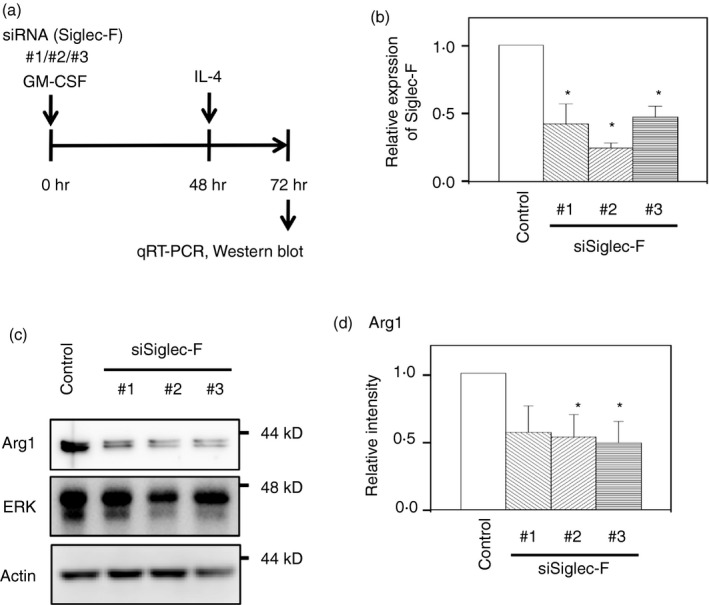
Siglec‐F knockdown reduces arginase‐1 (Arg1) expression induced by interleukin‐4 (IL‐4) in macrophage colony‐stimulating factor‐differentiated bone‐marrow‐derived macrophages (M‐BMDMs). (a) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control small interfering RNA (siRNA) in the presence of GM‐CSF. Forty‐eight hours later, cells were washed and stimulated with IL‐4 for an additional 24 hr. The expression of Siglec‐F and Arg1 was measured by qRT‐PCR and Western blotting, respectively. (b) Confirmation of Siglec‐F knockdown. Data are the mean ± SE of three independent experiments. *P < 0·05 versus the control by Student's t‐test. (c) Siglec‐F knockdown reduced IL‐4‐induced Arg1 expression. Total extracellular signal‐regulated kinase (ERK) and actin were measured as controls. A representative result of three independent experiments is shown. (d) Quantification of band intensity. The band intensity of Arg1 was normalized to that of actin. The band intensity of control siRNA‐transfected cells was regarded as 1. Data are the mean ± SE of three independent experiments. *P < 0·05 versus the control by Student's t‐test.
As IL‐4 responses require the activation of STAT6, the tyrosine phosphorylation of STAT6 was analyzed just after the IL‐4 stimulation. We failed to detect short‐term activation of STAT6 by IL‐4 in M‐BMDM that had been stimulated with GM‐CSF (data not shown), possibly because of the interference of IL‐4 signaling by prolonged GM‐CSF activation including cross‐activation of STAT6 by GM‐CSF.30 Therefore, Siglec‐F in M‐BMDMs was knocked down by the siRNA treatment for 48 hr in the absence of GM‐CSF, and cells were then stimulated with IL‐4 (Fig. 5a). Quantitative RT‐PCR confirmed that Siglec‐F mRNA was reduced to approximately one‐seventh that of the control by transfection of two‐siRNA mixture (Fig. 5b). This knockdown did not affect STAT6 protein levels (Fig. 5c). The phosphorylation of STAT6 was observed from 15 min after the addition of IL‐4 and the knockdown of Siglec‐F decreased the phosphorylation of STAT6 30 min after the stimulation (Fig. 5c, d). To confirm the effect of Siglec‐F knockdown on STAT6 phosphorylation, three siRNAs were separately transfected and cells were stimulated with IL‐4 (see Supplementary material, Fig. S4a). Knockdown efficiencies were 50–70% (see Supplementary material, Fig. S4b); slightly less than that by the mixture of siRNA#1 and #2. The knockdown of Siglec‐F reduced the STAT6 phosphorylation induced by IL‐4 (see Supplementary material, Fig. S4c, d). Similar results were obtained with L929‐BMDMs (see Supplementary material, Fig. S5). The changes in phosphorylation of ERK were not evident in these experiments (data not shown). The phosphorylation of Akt was not observed (data not shown), suggesting that PI3K activation was weak under these conditions, as reported previously.31 These results suggested that Siglec‐F enhanced IL‐4‐induced phosphorylation of STAT6.
Figure 5.
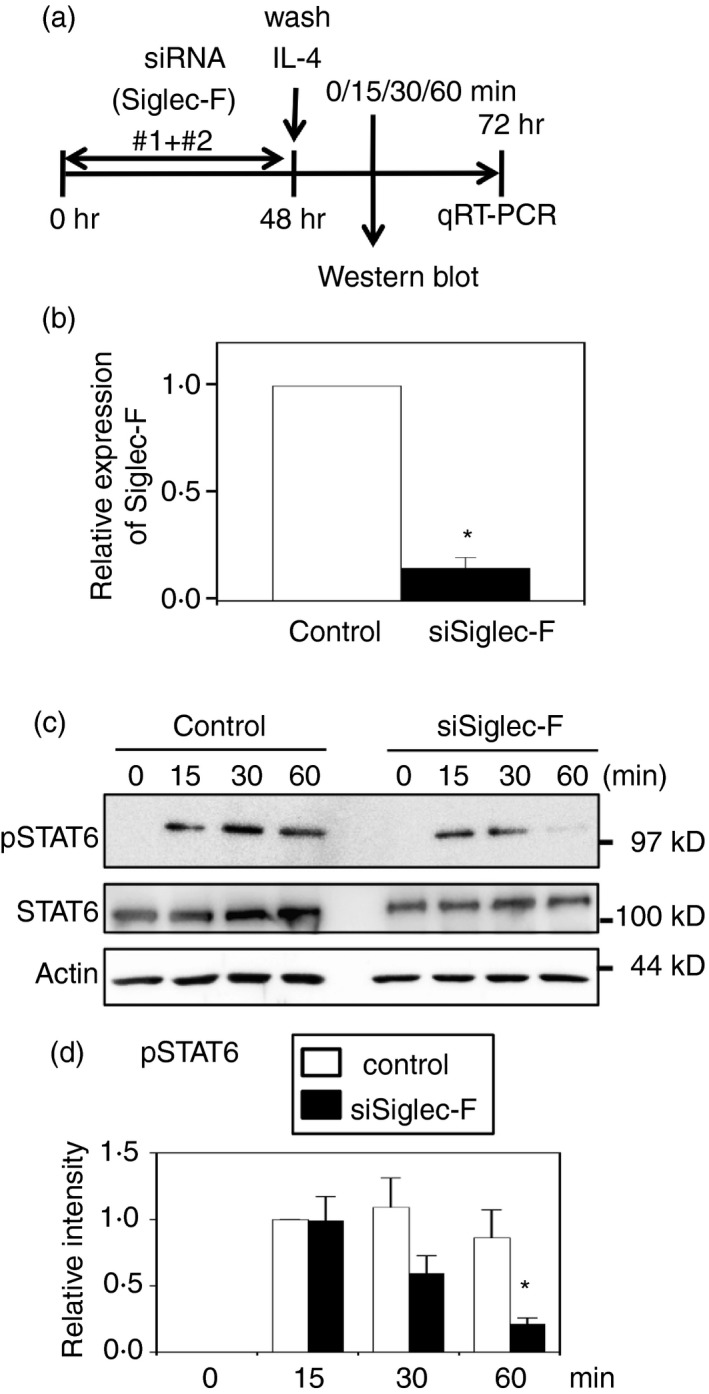
Siglec‐F knockdown reduces signal transducer and activator of transcription 6 (STAT6) phosphorylation induced by interleukin‐4 (IL‐4) in macrophage colony‐stimulating factor‐differentiated bone‐marrow‐derived macrophages (M‐BMDMs). (a) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control small interfering RNA (siRNA). Cells were washed after a 48‐hr culture and stimulated with IL‐4 for the indicated periods. The phosphorylation of STAT6 was examined by Western blotting. (b) Confirmation of knockdown. Total RNA was collected 24 hr after the stimulation and subjected to qRT‐PCR. Data are the mean ± SE of six independent experiments. *P < 0·05 versus the control by Student's t‐test. (c) Siglec‐F knockdown reduced the phosphorylation of STAT6. Total and the phosphorylated form of STAT6 were examined. Actin was measured as a control. A representative result of six independent experiments is shown. (d) Quantification of band intensity. The band intensity of pSTAT6 was normalized to that of actin. The band intensity of control siRNA at 15 min was regarded as 1. White and black bars indicate control and Siglec‐F siRNAs, respectively. Data are the mean ± SE of six independent experiments. *P < 0·05 versus the control at the same time point by Student's t‐test.
Siglec‐F does not affect nuclear factor‐κB activation induced by LPS plus IFN‐γ
We then investigated whether the knockdown of Siglec‐F modulates the responses induced by LPS plus IFN‐γ. Cells were simultaneously treated with siRNAs and GM‐CSF, and then washed and stimulated with LPS plus IFN‐γ (see Supplementary material, Fig. S6a). The 4‐hr stimulation strongly induced the expression of iNOS, IL‐10 and tumor necrosis factor‐α, which was not significantly changed by the knockdown of Siglec‐F (see Supplementary material, Fig. S6b, c). The activation of nuclear factor‐κB, which was inhibited by Siglec‐E,7 was then examined as the degradation of IκB. IκB was similarly reduced in control‐ and Siglec‐F‐siRNA‐treated macrophages after the LPS plus IFN‐γ stimulation for 1 hr (see Supplementary material, Fig. S6d–f). These results suggest that Siglec‐F may not directly affect inflammation signals by LPS plus IFN‐γ under these conditions.
Siglec‐F reduces the STAT5 phosphorylation induced by GM‐CSF
We also investigated whether Siglec‐F affected GM‐CSF responses in macrophages. M‐BMDMs were transfected with Siglec‐F siRNA, and cells were stimulated after 48 hr with GM‐CSF (Fig. 6a; see Supplementary material, Fig. S7a). The amount of total STAT5 was not changed by the knockdown of Siglec‐F (Fig. 6b; see Supplementary material, Fig. S7b), whereas the phosphorylation of STAT5 was enhanced (Fig. 6b, c; see Supplementary material, Fig. S7b, c). These results suggest that Siglec‐F at least partly blocked the STAT5 activation in the GM‐CSF stimulation. As the expression of class II MHC is controlled by GM‐CSF,32 the effect of the knockdown on the expression of this gene was then analyzed. M‐BMDMs were transfected with three different siRNAs against Siglec‐F and cultured for 48 hr, then stimulated for 24 hr with GM‐CSF. Quantitative RT‐PCR analysis indicated that the expression of class II MHC was induced by GM‐CSF (approximately 10‐fold), which was not changed by the knockdown of Siglec‐F (data not shown).
Figure 6.
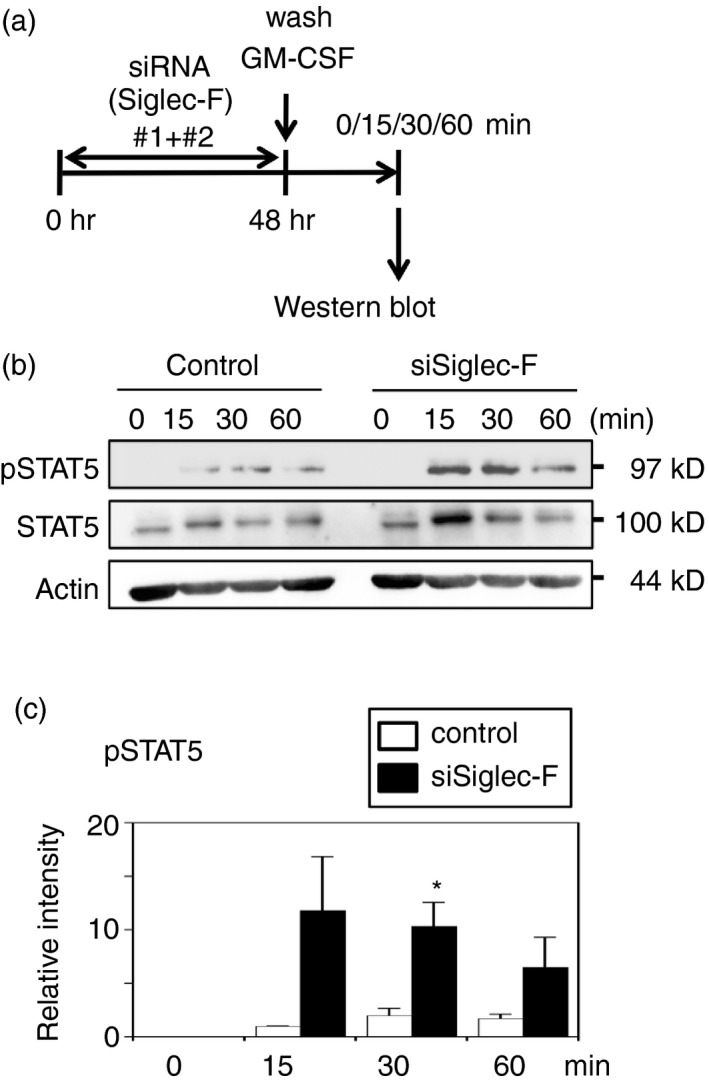
Siglec‐F knockdown enhances signal transducer and activator of transcription 5 (STAT5) phosphorylation induced by granulocyte–macrophage colony‐stimulating factor (GM‐CSF) in macrophage colony‐stimulating factor‐differentiated bone‐marrow‐derived macrophages (M‐BMDMs). (a) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control small interfering RNA (siRNA). Cells were washed after a 48‐hr culture and stimulated with GM‐CSF for the indicated periods. Phosphorylation was assessed by Western blotting. (b) Siglec‐F knockdown enhanced the phosphorylation of STAT5. Total and the phosphorylated form of STAT5 were examined. Actin was measured as a control. A representative result of three independent experiments is shown. (c) Quantification of band intensity. The band intensities of pSTAT5 were normalized to that of actin. The band intensity of control siRNA at 15 min was regarded as 1. White and black bars indicate control and Siglec‐F siRNAs, respectively. Data are the mean ± SE of three independent experiments. *P < 0·05 versus the control at the same time point by Student's t‐test.
Discussion
In the present study, we examined Siglec expression in BMDMs (Fig. 1). Siglec‐E was strongly induced by LPS, but other CD33‐related Siglecs were not, suggesting different regulatory mechanisms for the expression of each Siglec. Furthermore, we found that several Siglecs, such as CD33 and Siglec‐G and ‐F, were induced by GM‐CSF simultaneously (Fig. 1). Among the Siglecs, we analyzed GM‐CSF‐driven Siglec‐F expression in more detail because the results of the qRT‐PCR analysis suggested that Siglec‐F is a major Siglec expressed in BMDMs. Although macrophages show diversity, GM‐CSF enhanced Siglec‐F expression in two different BMDM preparations as well as thioglycollate‐elicited macrophages (Figs 1 and 2). As GM‐CSF is essential for the proper differentiation of alveolar macrophages,33, 34 the strong expression of Siglec‐F in alveolar macrophages17, 35 may be related to the activity of GM‐CSF.
Originally identified as a hematopoietic growth factor, GM‐CSF is now known to be a potent cytokine that induces the proliferation, differentiation, and activation of macrophages.36, 37, 38, 39 Although the basal concentration of GM‐CSF is low, its levels are often elevated during inflammatory reactions.37, 38 A number of different cell types including T lymphocytes, macrophages, and cancer cells produce GM‐CSF.37, 40 In most cases, inflammatory stimuli induce the production of GM‐CSF, including typical inflammatory mediators/inducers, such as tumor necrosis factor‐α,41 IL‐1,42 and LPS.42 In addition to GM‐CSF, IL‐3 enhanced Siglec‐F expression (see Supplementary material, Fig. S3). IL‐3 is a major hematopoietic cytokine, and has been reported to play important roles in inflammation. A large amount of IL‐3 was shown to be produced by BMDMs following stimulation with LPS plus IFN‐γ.43 Furthermore, GM‐CSF and IL‐3 were simultaneously produced in septic shock induced by LPS or cecal ligation and puncture.44 These findings suggest that Siglec‐F is indirectly induced by a number of inflammatory stimuli.
In an attempt to clarify the physiological significance of Siglec‐F in BMDMs, we investigated whether Siglec‐F modulates the activation of two JAK/STAT signaling pathways: STAT6 induced by IL‐4 and STAT5 by GM‐CSF. Siglec‐F facilitated IL‐4‐induced STAT6 phosphorylation (Fig. 5; see Supplementary material, Figs S4 and S5), but inhibited GM‐CSF‐induced STAT5 phosphorylation (Fig. 6; see Supplementary material, Fig. S7). The latter result suggests the existence of a feedback loop for the regulation of Siglec‐F expression. Siglec‐F, which is induced by GM‐CSF (Figs 1, 2; see Supplementary material, Fig. S2), inhibited the downstream signaling of GM‐CSF, suggesting that Siglec‐F restricted its own expression through the inhibition of STAT5 when cells were continuously stimulated by GM‐CSF.
The mechanisms underlying Siglec‐F activity to modulate signaling have not yet been elucidated in detail, but may be due to the binding proteins of immunoreceptor tyrosine‐based inhibitory motifs in Siglec‐F. One possibility is that immunoreceptor tyrosine‐based inhibitory motifs of Siglec‐F sequester cytosolic regulators from the receptors for GM‐CSF or IL‐4. As the common βc requires SHP‐2 to initiate downstream signaling,45 sequestration of SHP‐2 from GM‐CSF receptor by Siglec‐F may reduce STAT5 signaling. Alternatively, sequestration of inducible suppressor by Siglec‐F from IL‐4 receptor may affect IL‐4 signaling: when BMDMs that had been knocked down for Siglec‐F were stimulated by IL‐4, the phosphorylation of STAT6 was observed just after the stimulation, but rapidly became weaker than that observed in control cells (Fig. 5; see Supplementary material, Fig. S5). Suppressor of cytokine signaling 1 (SOCS1), which is induced by IL‐4 and suppresses STAT6 phosphorylation,46 may be such a candidate, although binding between SOCS and Siglecs is not reported so far except for SOCS3 and Siglec‐7 or CD33.47, 48 These possibilities remain to be investigated.
Although the knockdown of Siglec‐F did not affect inflammatory gene expression or nuclear factor‐κB activation induced by LPS plus IFN‐γ (see Supplementary material, Fig. S6), Siglec‐F knockdown reduced the expression of Arg1 in IL‐4‐stimulated macrophages (Fig. 4). On the other hand, the results of the qRT‐PCR analysis suggested that other M2‐specific genes, including Fizz1 or Mrc1, were not reduced by this knockdown (data not shown), indicating that Siglec‐F specifically enhances Arg1 rather than generally promoting M2 phenotypes. Arg1 is known to participate in the inhibition of iNOS activity and in the production of polyamine and proline, which are required for tissue repair in inflammation,49 while enhancing the growth of Leishmania parasites.50 Our results imply the possibility that Siglec‐F may participate in the regulation of macrophage activity by fine‐tuning of Arg1 expression. Further studies, such as those using Siglec‐F‐knockout mice, are needed to clarify the exact role of Siglecs in the physiology of macrophages.
Author Contribution
HT, YM, HH, HK, and YI designed and performed experiments. SI and KN conceived the study and wrote the manuscript. All authors analyzed the results and approved the final version of the manuscript.
Disclosures
The authors declare no financial conflicts of interest.
Supporting information
Figure S1. Sorting strategy for macrophage purification. (a) Procedure for the purification of BMDMs (associated with Fig. 1d). M‐BMDMs were sorted as F4/80+ CD11b+ cells. R1, sorted region. A representative result of 3 independent experiments is shown. (b) Procedure for the purification of thioglycollate‐elicited macrophages (associated with Fig. 1f). Five days after the thioglycollate injection, macrophages were purified from peritoneal cavity cells as F4/80+ CD11b+ cells after excluding Siglec‐Fhigh cells. R1, sorted macrophage fraction. Siglec‐Fhigh cells were sorted as eosinophils for comparison. A representative result of 4 independent experiments is shown.
Figure S2. GM‐CSF induces Siglec‐F expression on M‐BMDMs. Cells were stimulated for 24 h and cell surface expression of Siglec‐F was examined by indirect staining. Anti‐Siglec‐F antibody (R&D Systems) and control rat IgG (Santa Cruz Biotechnology) were used for primary antibody. A representative result of 3 independent experiments is shown.
Figure S3. IL‐3 enhances Siglec‐F expression in BMDMs. M‐BMDMs were stimulated with IL‐3 for 24 h and Siglec‐F expression was examined by qRT‐PCR. Data are the mean ± SE of 5 independent experiments. *P < 0·05 versus none by the Student’s t‐test.
Figure S4. Siglec‐F knockdown reduces STAT6 phosphorylation induced by IL‐4 in M‐BMDMs. (a) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control siRNA. Cells were washed after a 48‐h culture, and stimulated with IL‐4 for 60 min. The phosphorylation of STAT6 was examined by Western blotting. (b) Confirmation of knockdown. Total RNA was collected 24 h after the stimulation and subjected to qRT‐PCR. Data are the mean ± SE of 3 independent experiments. *P < 0·05 versus the control by the Student’s t‐test. (c) Siglec‐F knockdown reduced the phosphorylation of STAT6. Total and the phosphorylated form of STAT6 were examined. Actin was measured as a control. A representative result of 7 independent experiments is shown. (d) Quantification of band intensity. The band intensity of pSTAT6 was normalized to that of actin. The band intensity of control siRNA was regarded as 1. Data are the mean ± SE of 7 independent experiments. *P < 0·05 versus the control by the Student’s t‐test.
Figure S5. Siglec‐F knockdown reduces STAT6 phosphorylation induced by IL‐4 in L929‐BMDMs. (a) Schematic presentation of the knockdown experiment. L929‐BMDMs were transfected with Siglec‐F or control siRNA. Cells were washed after a 48‐h culture, and stimulated with IL‐4 for the indicated periods. The phosphorylation of STAT6 was examined by Western blotting. (b) Siglec‐F knockdown reduced the phosphorylation of STAT6. Total and the phosphorylated form of STAT6 were examined. Actin was measured as a control. A representative result of 3 independent experiments is shown. (c) Quantification of band intensity. The band intensity of pSTAT6 was normalized to that of actin. The band intensity of control siRNA at 15 min was regarded as 1. White and black bars indicate control and Siglec‐F siRNAs, respectively. Data are the mean ± SE of 3 independent experiments. *P < 0·05 versus the control at the same time point by the Student’s t‐test.
Figure S6. Effects of Siglec‐F knockdown on the stimulation of LPS plus IFN‐γ in M‐BMDMs. (a‐c) Effects of Siglec‐F knockdown on gene expression induced by LPS plus IFN‐γ. (a) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control siRNA in the presence of GM‐CSF. Forty‐eight hours later, cells were washed and stimulated with LPS plus IFN‐γ for an additional 4 h. The expression of Siglec‐F, iNOS, IL‐10, and TNF‐α was measured by qRT‐PCR. (b) Confirmation of Siglec‐F knockdown at 52 h. (c) Effect of Siglec‐F knockdown on the gene expression. Data are the mean ± SE. N=3 (iNOS), 4 (IL‐10), 3 (TNF‐α). *P < 0·05 versus the control by the Student’s t‐test. (d‐f) The effects of Siglec‐F knockdown on IκBα degradation induced by LPS plus IFN‐γ. (d) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control siRNA in the presence of GM‐CSF. Cells were washed after a 48‐h culture, and stimulated with LPS plus IFN‐γ for 60 min. The amount of IκBα was assessed by Western blotting. (e) Siglec‐F knockdown did not affect IκBα degradation. A representative result of 3 independent experiments is shown. (f) Quantification of band intensity. The band intensity of IκBα was normalized by that of actin. The band intensity of control siRNA without a stimulation was regarded as 1. White and black bars indicate control and Siglec‐F siRNAs, respectively. Data are the mean ± SE of 3 independent experiments.
Figure S7. Siglec‐F knockdown enhances the STAT5 phosphorylation induced by GM‐CSF in M‐BMDMs. (a) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control siRNA. Cells were washed after a 48‐h culture, and stimulated with GM‐CSF for 60 min. (b) Phosphorylation was assessed by Western blotting. A representative result of 7 independent experiments is shown. Total and the phosphorylated form of STAT5 were examined. Actin was measured as a control. (c) Quantification of band intensity. The band intensities of pSTAT5 were normalized to that of actin. The band intensity of control siRNA was regarded as 1. Data are the mean ± SE of 7 independent experiments. *P < 0·05 versus the control by the Student’s t‐test.
Table S1. Primers used in this study.
Table S2. Small interfering RNAs used in this study.
Acknowledgements
We thank Dr. Y. Yoshida (Innovative Research Center for Preventive Medical Engineering, Nagoya University) for the use of FACSJazz. This work is partly supported by Mizutani Foundation for Glycoscience to KN.
References
- 1. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008; 8:958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 122:787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cao H, Crocker PR. Evolution of CD33‐related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology 2010; 132:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pillai S, Netravali IA, Cariappa A, Mattoo H. Siglecs and immune regulation. Annu Rev Immunol 2012; 30:357–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol 2007; 7:255–66. [DOI] [PubMed] [Google Scholar]
- 6. McMillan SJ, Sharma RS, McKenzie EJ, Richards HE, Zhang J, Prescott A et al Siglec‐E is a negative regulator of acute pulmonary neutrophil inflammation and suppresses CD11b β2‐integrin‐dependent signaling. Blood 2013; 121:2084–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boyd CR, Orr SJ, Spence S, Burrows JF, Elliott J, Carroll HP et al Siglec‐E is up‐regulated and phosphorylated following lipopolysaccharide stimulation in order to limit TLR‐driven cytokine production. J Immunol 2009; 183:7703–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ando M, Tu W, Nishijima K, Iijima S. Siglec‐9 enhances IL‐10 production in macrophages via tyrosine‐based motifs. Biochem Biophys Res Commun 2008; 369:878–83. [DOI] [PubMed] [Google Scholar]
- 9. Zhang JQ, Raper A, Sugita N, Hingorani R, Salio M, Palmowski MJ et al Characterization of Siglec‐H as a novel endocytic receptor expressed on murine plasmacytoid dendritic cell precursors. Blood 2006; 107:3600–8. [DOI] [PubMed] [Google Scholar]
- 10. Zhang JQ, Biedermann B, Nitschke L, Crocker PR. The murine inhibitory receptor mSiglec‐E is expressed broadly on cells of the innate immune system whereas mSiglec‐F is restricted to eosinophils. Eur J Immunol 2004; 34:1175–84. [DOI] [PubMed] [Google Scholar]
- 11. Chen WL, Han CF, Xie B, Hu X, Yu Q, Shi LY et al Induction of Siglec‐G by RNA viruses inhibits the innate immune response by promoting RIG‐I degradation. Cell 2013; 152:467–78. [DOI] [PubMed] [Google Scholar]
- 12. Chang YC, Olson J, Beasley FC, Tung C, Zhang J, Crocker PR et al Group B Streptococcus engages an inhibitory Siglec through sialic acid mimicry to blunt innate immune and inflammatory responses in vivo. PLoS Pathog 2014; 10:e1003846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Claude J, Linnartz‐Gerlach B, Kudin AP, Kunz WS, Neumann H. Microglial CD33‐related Siglec‐E inhibits neurotoxicity by preventing the phagocytosis‐associated oxidative burst. J Neurosci 2013; 33:18270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nutku E, Aizawa H, Hudson SA, Bochner BS. Ligation of Siglec‐8: a selective mechanism for induction of human eosinophil apoptosis. Blood 2003; 101:5014–20. [DOI] [PubMed] [Google Scholar]
- 15. Song DJ, Cho JY, Lee SY, Miller M, Rosenthal P, Soroosh P et al Anti‐Siglec‐F antibody reduces allergen‐induced eosinophilic inflammation and airway remodeling. J Immunol 2009; 183:5333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang M, Angata T, Cho JY, Miller M, Broide DH, Varki A. Defining the in vivo function of Siglec‐F, a CD33‐related Siglec expressed on mouse eosinophils. Blood 2007; 109:4280–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S et al Alveolar macrophages develop from fetal monocytes that differentiate into long‐lived cells in the first week of life via GM‐CSF. J Exp Med 2013; 210:1977–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drissen R, Buza‐Vidas N, Woll P, Thongjuea S, Gambardella A, Giustacchini A et al Distinct myeloid progenitor‐differentiation pathways identified through single‐cell RNA sequencing. Nat Immunol 2016; 17:666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Higuchi H, Shoji T, Murase Y, Iijima S, Nishijima K. Siglec‐9 modulated IL‐4 responses in the macrophage cell line RAW264. Biosci Biotechnol Biochem 2016; 80:501–9. [DOI] [PubMed] [Google Scholar]
- 20. Kidani S, Okuzaki Y, Kaneoka H, Asai S, Murakami S, Murase Y et al Expression of interferon‐inducible transmembrane proteins in the chicken and possible role in prevention of viral infections. Cytotechnology 2017; 69:477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miah MA, Yoon CH, Kim J, Jang J, Seong YR, Bae YS. CISH is induced during DC development and regulates DC‐mediated CTL activation. Eur J Immunol 2012; 42:58–68. [DOI] [PubMed] [Google Scholar]
- 23. Griseri T, Arnold IC, Pearson C, Krausgruber T, Schiering C, Franchini F et al Granulocyte macrophage colony‐stimulating factor‐activated eosinophils promote interleukin‐23 driven chronic colitis. Immunity 2015; 43:187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Misharin AV, Saber R, Perlman H. Eosinophil contamination of thioglycollate‐elicited peritoneal macrophage cultures skews the functional readouts of in vitro assays. J Leukoc Biol 2012; 92:325–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hansen G, Hercus TR, McClure BJ, Stomski FC, Dottore M, Powell J et al The structure of the GM‐CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell 2008; 134:496–507. [DOI] [PubMed] [Google Scholar]
- 26. Itoh T, Liu R, Yokota T, Arai K, Watanabe S. Definition of the role of tyrosine residues of the common β subunit regulating multiple signaling pathways of granulocyte‐macrophage colony‐stimulating factor receptor. Mol Cell Biol 1998; 18:742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hercus TR, Dhagat U, Kan WL, Broughton SE, Nero TL, Perugini M et al Signalling by the βc family of cytokines. Cytokine Growth Factor Rev 2013; 24:189–201. [DOI] [PubMed] [Google Scholar]
- 28. Broughton SE, Nero TL, Dhagat U, Kan WL, Hercus TR, Tvorogov D et al The betac receptor family – structural insights and their functional implications. Cytokine 2015; 74:247–58. [DOI] [PubMed] [Google Scholar]
- 29. Munder M, Eichmann K, Morán J, Centeno F, Soler G, Modolell M. Th1/Th2‐regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol 1999; 163:3771–7. [PubMed] [Google Scholar]
- 30. Welte T, Koch F, Schuler G, Lechner J, Doppler W, Heufler C. Granulocyte‐macrophage colony‐stimulating factor induces a unique set of STAT factors in murine dendritic cells. Eur J Immunol 1997; 27:2737–40. [DOI] [PubMed] [Google Scholar]
- 31. Heller NM, Qi X, Junttila IS, Shirey KA, Vogel SN, Paul WE et al Type I IL‐4Rs selectively activate IRS‐2 to induce target gene expression in macrophages. Sci Signal 2008; 1:ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Willman CL, Stewart CC, Miller V, Yi T‐L, Tomasi TB. Regulation of MHC class II gene expression in macrophages by hematopoietic colony‐stimulating factors (CSF). Induction by granulocyte/macrophage CSF and inhibition by CSF‐1. J Exp Med 1989; 170:1559–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA et al Granulocyte/macrophage colony‐stimulating factor‐deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA 1994; 91:5592–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM‐CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 2001; 15:557–67. [DOI] [PubMed] [Google Scholar]
- 35. Misharin AV, Morales‐Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 2013; 49:503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y et al Biology and action of colony‐stimulating factor‐1. Mol Reprod Dev 1997; 46:4–10. [DOI] [PubMed] [Google Scholar]
- 37. Barreda DR, Hanington PC, Belosevic M. Regulation of myeloid development and function by colony stimulating factors. Dev Comp Immunol 2004; 28:509–54. [DOI] [PubMed] [Google Scholar]
- 38. Hamilton JA. Colony‐stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 2008; 8:533–44. [DOI] [PubMed] [Google Scholar]
- 39. Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol 2013; 34:81–9. [DOI] [PubMed] [Google Scholar]
- 40. Shiomi A, Usui T. Pivotal roles of GM‐CSF in autoimmunity and inflammation. Mediators Inflamm 2015; 2015:568543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Broudy VC, Kaushansky K, Segal GM, Harlan JM, Adamson JW. Tumor necrosis factor type α stimulates human endothelial cells to produce granulocyte/macrophage colony‐stimulating factor. Proc Natl Acad Sci USA 1986; 83:7467–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rennick D, Yang G, Gemmell L, Lee F. Control of hemopoiesis by a bone marrow stromal cell clone: lipopolysaccharide‐ and interleukin‐1‐inducible production of colony‐stimulating factors. Blood 1987; 69:682–91. [PubMed] [Google Scholar]
- 43. Melton DW, McManus LM, Gelfond JA, Shireman PK. Temporal phenotypic features distinguish polarized macrophages in vitro. Autoimmunity 2015; 48:161–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A et al Interleukin‐3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science 2015; 347:1260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu WM, Hawley TS, Hawley RG, Qu CK. Catalytic‐dependent and ‐independent roles of SHP‐2 tyrosine phosphatase in interleukin‐3 signaling. Oncogene 2003; 22:5995–6004. [DOI] [PubMed] [Google Scholar]
- 46. Dickensheets H, Vazquez N, Sheikh F, Gingras S, Murray PJ, Ryan JJ et al Suppressor of cytokine signaling‐1 is an IL‐4‐inducible gene in macrophages and feedback inhibits IL‐4 signaling. Genes Immun 2007; 8:21–7. [DOI] [PubMed] [Google Scholar]
- 47. Orr SJ, Morgan NM, Buick RJ, Boyd CR, Elliott J, Burrows JF et al SOCS3 targets Siglec 7 for proteasomal degradation and blocks Siglec 7‐mediated responses. J Biol Chem 2007; 282:3418–22. [DOI] [PubMed] [Google Scholar]
- 48. Orr SJ, Morgan NM, Elliott J, Burrows JF, Scott CJ, McVicar DW et al CD33 responses are blocked by SOCS3 through accelerated proteasomal‐mediated turnover. Blood 2007; 109:1061–8. [DOI] [PubMed] [Google Scholar]
- 49. Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol 2014; 5:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Muylder G, Daulouede S, Lecordier L, Uzureau P, Morias Y, Van Den Abbeele J et al A Trypanosoma brucei kinesin heavy chain promotes parasite growth by triggering host arginase activity. PLoS Pathog 2013; 9:e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sorting strategy for macrophage purification. (a) Procedure for the purification of BMDMs (associated with Fig. 1d). M‐BMDMs were sorted as F4/80+ CD11b+ cells. R1, sorted region. A representative result of 3 independent experiments is shown. (b) Procedure for the purification of thioglycollate‐elicited macrophages (associated with Fig. 1f). Five days after the thioglycollate injection, macrophages were purified from peritoneal cavity cells as F4/80+ CD11b+ cells after excluding Siglec‐Fhigh cells. R1, sorted macrophage fraction. Siglec‐Fhigh cells were sorted as eosinophils for comparison. A representative result of 4 independent experiments is shown.
Figure S2. GM‐CSF induces Siglec‐F expression on M‐BMDMs. Cells were stimulated for 24 h and cell surface expression of Siglec‐F was examined by indirect staining. Anti‐Siglec‐F antibody (R&D Systems) and control rat IgG (Santa Cruz Biotechnology) were used for primary antibody. A representative result of 3 independent experiments is shown.
Figure S3. IL‐3 enhances Siglec‐F expression in BMDMs. M‐BMDMs were stimulated with IL‐3 for 24 h and Siglec‐F expression was examined by qRT‐PCR. Data are the mean ± SE of 5 independent experiments. *P < 0·05 versus none by the Student’s t‐test.
Figure S4. Siglec‐F knockdown reduces STAT6 phosphorylation induced by IL‐4 in M‐BMDMs. (a) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control siRNA. Cells were washed after a 48‐h culture, and stimulated with IL‐4 for 60 min. The phosphorylation of STAT6 was examined by Western blotting. (b) Confirmation of knockdown. Total RNA was collected 24 h after the stimulation and subjected to qRT‐PCR. Data are the mean ± SE of 3 independent experiments. *P < 0·05 versus the control by the Student’s t‐test. (c) Siglec‐F knockdown reduced the phosphorylation of STAT6. Total and the phosphorylated form of STAT6 were examined. Actin was measured as a control. A representative result of 7 independent experiments is shown. (d) Quantification of band intensity. The band intensity of pSTAT6 was normalized to that of actin. The band intensity of control siRNA was regarded as 1. Data are the mean ± SE of 7 independent experiments. *P < 0·05 versus the control by the Student’s t‐test.
Figure S5. Siglec‐F knockdown reduces STAT6 phosphorylation induced by IL‐4 in L929‐BMDMs. (a) Schematic presentation of the knockdown experiment. L929‐BMDMs were transfected with Siglec‐F or control siRNA. Cells were washed after a 48‐h culture, and stimulated with IL‐4 for the indicated periods. The phosphorylation of STAT6 was examined by Western blotting. (b) Siglec‐F knockdown reduced the phosphorylation of STAT6. Total and the phosphorylated form of STAT6 were examined. Actin was measured as a control. A representative result of 3 independent experiments is shown. (c) Quantification of band intensity. The band intensity of pSTAT6 was normalized to that of actin. The band intensity of control siRNA at 15 min was regarded as 1. White and black bars indicate control and Siglec‐F siRNAs, respectively. Data are the mean ± SE of 3 independent experiments. *P < 0·05 versus the control at the same time point by the Student’s t‐test.
Figure S6. Effects of Siglec‐F knockdown on the stimulation of LPS plus IFN‐γ in M‐BMDMs. (a‐c) Effects of Siglec‐F knockdown on gene expression induced by LPS plus IFN‐γ. (a) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control siRNA in the presence of GM‐CSF. Forty‐eight hours later, cells were washed and stimulated with LPS plus IFN‐γ for an additional 4 h. The expression of Siglec‐F, iNOS, IL‐10, and TNF‐α was measured by qRT‐PCR. (b) Confirmation of Siglec‐F knockdown at 52 h. (c) Effect of Siglec‐F knockdown on the gene expression. Data are the mean ± SE. N=3 (iNOS), 4 (IL‐10), 3 (TNF‐α). *P < 0·05 versus the control by the Student’s t‐test. (d‐f) The effects of Siglec‐F knockdown on IκBα degradation induced by LPS plus IFN‐γ. (d) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control siRNA in the presence of GM‐CSF. Cells were washed after a 48‐h culture, and stimulated with LPS plus IFN‐γ for 60 min. The amount of IκBα was assessed by Western blotting. (e) Siglec‐F knockdown did not affect IκBα degradation. A representative result of 3 independent experiments is shown. (f) Quantification of band intensity. The band intensity of IκBα was normalized by that of actin. The band intensity of control siRNA without a stimulation was regarded as 1. White and black bars indicate control and Siglec‐F siRNAs, respectively. Data are the mean ± SE of 3 independent experiments.
Figure S7. Siglec‐F knockdown enhances the STAT5 phosphorylation induced by GM‐CSF in M‐BMDMs. (a) Schematic presentation of the knockdown experiment. M‐BMDMs were transfected with Siglec‐F or control siRNA. Cells were washed after a 48‐h culture, and stimulated with GM‐CSF for 60 min. (b) Phosphorylation was assessed by Western blotting. A representative result of 7 independent experiments is shown. Total and the phosphorylated form of STAT5 were examined. Actin was measured as a control. (c) Quantification of band intensity. The band intensities of pSTAT5 were normalized to that of actin. The band intensity of control siRNA was regarded as 1. Data are the mean ± SE of 7 independent experiments. *P < 0·05 versus the control by the Student’s t‐test.
Table S1. Primers used in this study.
Table S2. Small interfering RNAs used in this study.