

Summary
Current treatments for systemic autoimmune diseases partially improve the health of patients displaying low pharmacological efficacy and systemic immunosuppression. Here, the therapeutic potential of transferring tolerogenic dendritic cells (tolDCs) generated with heme‐oxygenase inductor cobalt (III) protoporphyrin IX (CoPP), dexamethasone and rosiglitazone for the treatment of systemic autoimmunity was evaluated in two murine models of systemic lupus erythematosus (SLE), MRL‐Faslpr and NZM2410 mice. Dendritic cells treated ex vivo with these drugs showed a stable tolerogenic profile after lipopolysaccharide stimulation. Regular doses of tolDCs were administered to anti‐nuclear antibody‐positive mice throughout 60–70 days, and the clinical score was evaluated. Long‐term treatment with these tolDCs was well tolerated and effective to improve the clinical score on MRL‐Faslpr lupus‐prone mice. Additionally, decreased levels of anti‐nuclear antibodies in NZM2410 mice were observed. Although tolDC treatment increased regulatory T cells, no significant reduction of renal damage or glomerulonephritis could be found. In conclusion, these results suggest that the transfer of histone‐loaded tolDCs could improve only some SLE symptoms and reduced anti‐nuclear antibodies. This is the first study to evaluate antigen‐specific tolDC administration to treat SLE. Our report strengthens the clinical relevance of tolDC generation with CoPP, dexamethasone and rosiglitazone and the use of these modified cells as a therapy for systemic autoimmunity.
Keywords: autoimmunity, Heme‐oxygenase‐1, self‐tolerance, systemic lupus erythematosus, tolerogenic dendritic cells
Abbreviations
- ANA
anti‐nuclear antibodies
- APC
allophycocyanin
- BSA
bovine serum albumin
- CFSE
5,6‐carboxyfluorescein succinimidyl ester
- CoPP
cobalt (III) protoporphyrin IX chloride
- DCs
dendritic cells
- Dexa
dexamethasone
- FITC
fluorescein isothiocyanate
- HO‐1
heme‐oxygenase‐1
- HRP
horseradish peroxidase
- IFN‐γ
interferon‐γ
- IL‐10
interleukin‐10
- LPS
lipopolysaccharide
- MRL‐Faslpr
MRL/MpJ‐Faslpr/J
- NZM2410
New Zealand mixed 2410
- PE
phycoerythrin
- Rgz
rosiglitazone
- SLE
systemic lupus erythematosus
- tolDCs
tolerogenic dendritic cells
- Treg cells
regulatory T cells
Introduction
Dendritic cells (DCs) are specialized antigen‐presenting cells that play a central role in determining the induction of immunity or tolerance. Notably, the tolerogenic function of DCs has gained interest in recent years.1, 2 Tolerogenic dendritic cells (tolDCs) are characterized by reduced expression of co‐stimulatory molecules, the up‐regulation of inhibitory molecules and increased production of anti‐inflammatory cytokines.1 A stable tolDC phenotype can be genetic and pharmacologically generated in vitro by treatments that block or prevent DC maturation.1, 3, 4 These in vitro treatments reduce both the production of pro‐inflammatory cytokines and the expression of co‐stimulatory molecules, leading to a lower CD4+ T‐cell priming.3, 5 Hence, tolDCs pulsed with antigens can lead to antigen‐specific T‐cell hyporesponsiveness and as a result self‐antigen selection is a central issue for the appropriate design of immunotherapy by using antigen‐presenting cells in immune‐mediated diseases.1, 6, 7
Heme‐oxygenase‐1 (HO‐1) is the rate‐limiting enzyme in heme catabolism and can be induced by its natural substrate or by synthetic metalloporphyrins.8, 9 HO‐1 induction has been associated with injury protection, antioxidant properties and anti‐inflammatory responses.10 Although the expression of HO‐1 is drastically decreased during DC maturation in vitro,11 the induction of this enzyme has been reported to foster a tolerogenic phenotype on DCs.12
Due to their ability to promote immune tolerance, tolDCs have emerged as a promising antigen‐specific immunotherapy approach in several autoimmune conditions.7, 13, 14 Accordingly, these cells have been successfully used in murine models for type 1 diabetes, multiple sclerosis, inflammatory bowel disease and rheumatoid arthritis. In addition, favorable safety has been reported in clinical trials involving the transfer of tolDCs to patients with multiple sclerosis, arthritis and diabetes.15
Systemic lupus erythematosus (SLE) is a systemic autoimmune disorder of unknown etiology and high incidence.16 The disease is characterized by a broad spectrum of clinical manifestations, such as arthritis, skin lesions, glomerulonephritis, and psychiatric and neurological alterations.17 It is typically associated with overproduction of autoantibodies against nuclear constituents, as a result of the dysregulation of adaptive and innate immune cells.18, 19 Interestingly, the HO‐1 expression is down‐regulated in monocytes isolated from patients with SLE18, 20 and HO‐1 induction has been associated with disease amelioration in murine lupus.21 Finally, carbon monoxide, which is a product of HO‐1 enzymatic activity over heme group, decreases activated T‐cell numbers in kidneys and lungs, and decreases levels of anti‐nuclear antibodies (ANA) in lupus‐prone mice.22
In this study, we have evaluated the immunotherapeutic role of tolDCs generated by the HO‐1 inductor cobalt (III) protoporphyrin IX (CoPP) in combination with two known tolerogenic drugs: dexamethasone (Dexa) and rosiglitazone (Rgz) in autoimmune mice. Given the background summarized above, tolDCs loaded with nuclear antigens, such as histones, arise as a promising self‐antigen‐specific therapy for SLE. The present study aims to evaluate the therapeutic capacity of the adoptive transfer of histone‐loaded tolDCs in chronic and acute lupus‐prone murine models, NZM2410 and MRL‐Faslpr, respectively. We observed that tolDCs from NZM2410 and MRL‐Faslpr mice, generated by HO‐1 induction and Dexa‐Rgz treatment, have a stable tolerogenic profile in vitro. Furthermore, adoptive tolDC transfer into autoimmune MRL‐Faslpr recipient mice reduced their clinical scores. To our knowledge, this is the first study to evaluate the effect of antigen‐loaded tolDC transfer to reduce SLE severity in murine models.
Materials and methods
Animals
MRL/MpJ‐Faslpr/J (MRL‐Faslpr) mice and New Zealand mixed (B6.NZM‐Sle1NZM2410/Aeg Sle2NZM2410/Aeg Sle3NZM2410/Aeg/LmoJ also mentioned as NZM2410) lupus‐prone mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and kept under pathogen‐free conditions at the animal core facility of the Pontificia Universidad Católica de Chile. Mice were kept under specific pathogen‐free conditions and provided with environmental enrichment, sterile food and water ad libitum. Eight‐week‐old MRL‐Faslpr mice and 14‐week‐old NZM2410 mice (ANA‐positive) were intravenously transferred through the lateral tail vein with tolDCs under the supervision of a veterinarian and according to institutional guidelines. All efforts were made to minimize suffering or distress.
Ethics statement
All procedures were approved and supervised by the Scientific Ethics Committee for the Care of Animals and the Environment of the Pontificia Universidad Católica de Chile (reference number CBB‐170317008). The protocol is under the Chilean Law 20380 on Animal Protection (2009), the Terrestrial Animal Health Code of the World Organization for Animal Health, the European Directive 2010/63/EU and the Guide for the Care and Use of Experimental Animals.
Measurement of ANAs
Anti‐nuclear antibody levels in serum were determined with HEp‐2 cells on 12‐well slides (BioRad, Hercules, CA) and dsDNA Crithidia luciliae 12‐well slides (Inova Diagnostics, San Diego, CA). Both HEp‐2 cells and Crithidia luciliae slides were incubated for 30 min in a humid chamber with a 1 : 40 dilution of serum derived from control and treated mice, followed by staining for 30 min with 1 : 300 Alexa 488‐conjugated anti‐mouse immunoglobulin G (IgG) (Biolegend, San Diego, CA). The fluorescence patterns were evaluated immediately with an Olympus fluorescence microscope at 40×. Pictures were collected with Infinity camera and software.
Experimental design
To evaluate the effect of tolDCs generated with the HO‐1 inductor and Dexa‐Rgz treatment, lupus‐prone mice (from each murine model) received six or seven adoptive transfers of DCs for 3 months. Animals were followed up throughout the experiment and killed at the end of the experiment. Twenty‐eight lupus‐prone mice from MRL‐Faslpr or NZM2410 strains were randomly assigned to one of the experimental groups defined by the drugs used to generate tolDCs (Table 1).
Table 1.
Specification of the different treatments used to generate the tolerogenic profile
| Histones | CoPP | Dexamethasone | Rosiglitazone | |
|---|---|---|---|---|
| DCs | − | − | − | − |
| DCs + Hist | + | − | − | − |
| tolDCs1 | + | + | + | + |
| tolDCs2 | + | + | − | − |
| tolDCs3 | + | − | + | + |
Cobalt (III) protoporphyrin (CoPP) 50 μm was employed for 2 hr while rosiglitazone 10 μm and dexamethasone 1 μm for 24 hr. Finally, the cells were pulsed with histones 30 μg/ml for 24 hr. Non‐treated dendritic cells (DCs) were included as controls.
Each group received adoptive transfer of DCs treated as described in Table 1. Additionally, two control groups were included, a group given vehicle [phosphate‐buffered saline (PBS)] and an environmental control group.
Tolerogenic DC generation and evaluation
Bone‐marrow‐derived DCs were generated as previously described.23 Briefly, bone marrow from 6‐week‐old MRL‐Faslpr or NZM2410 mice was isolated by bone perfusion and, after ACK treatment, cells were counted and resuspended to a concentration of 1 × 106 cells/ml in RPMI‐1640 containing 10% fetal bovine serum (Hyclone Laboratories, Logan, UT) and supplemented with 10 ng/ml murine granulocyte–macrophage colony‐stimulating factor (PeproTech, Rocky Hill, NJ). At day 5, DCs were treated with CoPP (ChemCruz Biochemicals, TE Huissen, the Netherlands) 50 μm for 2 hr, carefully washed and treated with Rgz (Sigma‐Aldrich, St Louis, MO) 10 μm and Dexa (Sigma‐Aldrich) 1 μm for 24 hr. Finally, drugs were washed out and then DCs were pulsed with histones (Roche Diagnostics, Mannheim, Germany) 30 μg/ml for 24 hr. DC maturation was induced as a control, with 0·5 μg/ml lipopolysaccharide (LPS) (Sigma‐Aldrich) for 24 hr. Untreated control DCs and vehicle (PBS) ‐treated DCs were included in all experiments. Three experimental treatments were applied to DCs to induce the tolerogenic phenotype: DCs + Dexa + Rgz + CoPP (tolDCs1) DCs + CoPP (tolDCs2) and DCs + Dexa + Rgz (tolDCs3); all of them were loaded with histones. A control treatment was used with DCs loaded with histones but without drugs treatment (DCs + Hist) (Table 1). After LPS‐induced maturation, cells were evaluated for expression of surface markers CD86, CD80 and CD40 on CD11c+ MHC‐II+ cells on a Canto II flow cytometer (Becton Dickinson, San Jose, CA). To evaluate DC maturation, samples were multi‐stained with phycoerythrin‐Cychrome 7 (PE‐Cy7) ‐conjugated anti‐CD11c (clone HL3; BD Biosciences Pharmingen, San Diego, CA), fluorescein isothiocyanate (FITC) ‐conjugated anti‐MHCII (clone M5/114.15.2; BD Biosciences Pharmingen), allophycocyanin‐Cy7 (APC‐Cy7) ‐conjugated anti‐CD86 (clone GL1; BD Biosciences Pharmingen), PE‐conjugated anti‐CD40 (clone 3/23; BD Biosciences Pharmingen), APC‐conjugated anti‐CD80 (clone 16‐10A1; BD Biosciences Pharmingen), fixed in 1% paraformaldehyde in PBS, and analyzed by FACS. Viability of DCs after treatments was determined by staining cells with VS700 (BD Biosciences Pharmingen) for 15 min followed by FACS analysis. HO‐1 expression was determined by flow cytometry using an HO‐1‐specific primary antibody (Clone ab13248; Abcam, San Francisco, CA) and a secondary goat anti‐mouse (IgG H + L) APC‐conjugated antibody.
Adoptive transfer experiments
Cellular suspensions were obtained from bone‐marrow‐derived DC culture as described above, and concentrations were adjusted to a dose ranging from 2·5 × 105 to 1 × 106 cells per injection (100 μl in PBS). Intravenous administration into naive recipient MRL‐Faslpr or NZM2410 mice was done through the vein of the tail under anesthesia with isoflurane (MRL‐Faslpr mice) or under subjection without anesthesia (NZM2410 mice). Seven transfers were performed every 10 days, with the first transfer starting at 8 weeks of age in MRL‐Faslpr mice and 14 weeks of age in NZM2410 mice. Serum samples were obtained at the beginning of the experiment from the facial vein and at the end of the experiment (10 weeks after starting the treatment) by cardiac puncture. No additional blood samples were taken because several alterations in the coagulation system have been observed in these animals.24, 25
Assessment of urinary protein excretion, clinical score and splenic index
Proteinuria in fresh urine was measured with CentriVet sticks for urinalysis (ACON Laboratories Inc., San Diego, CA) using a scale of 0–5 as following: 0/trace, negative; 1, 15 mg/dl; 2, 30 mg/dl; 3, 100 mg/dl; 4, 300 mg/dl; and 5, 2000 mg/dl. Proteinuria scores > 3 were considered to represent moderate glomerulonephritis. Additionally, the renal function was evaluated to identify the occurrence of renal disease by measuring the levels of waste products and toxins, such as creatinine and blood urea nitrogen using the photometric Jaffe kinetic method and urea enzymatic urease method, respectively, by chemical autoanalyzer equipment MINDRAY BS300.
The clinical score was determined taking into consideration the following parameters: behavior and mobility (0, normal; 1, small changes; 2, difficult movement; 3, immobile), skin lesions (0, none; 1, one <0·5 cm2; 2, more than one <0·5 cm2; 3, 0·5–1·0 cm2; 4, >1·0 cm2), coat (0, normal; 1, bristly), alopecia (0, none; 1, <0·5 cm2; 2, 0·5–1·0 cm2; 3, >1·0 cm2), proteinuria (as was described above) and body weight loss (0, none; 1, <10%; 2, 10–20%; 3, >20%). The sum of the score in each parameter indicates the clinical score of each animal, which was determined three times per week. Finally, the splenic index was determined by the ratio between spleen weight (mg) and body weight (mg) per 100. These values were expressed as arbitrary units.
Enzyme‐linked immunosorbent assay
Indirect ELISA
Serum samples obtained before and after cell therapy from all experimental groups were evaluated to determine antibodies against nuclear antigens (DNA and histones). Total IgG antibodies against DNA (Invitrogen Corp., Carlsbad, CA) and histones (Sigma‐Aldrich) were quantified by enzyme‐linked immunosorbent assay (ELISA). Briefly, ELISA plates were coated overnight at 4° with 15 μg/ml DNA or 12 μg/ml histones in PBS, washed three times with PBS–Tween 0·05% and then blocked with 200 μl PBS/bovine serum albumin (BSA) 3% for 2 hr at room temperature with agitation. After washing three times, serum samples diluted in PBS/BSA 1%–Tween 0·05% at 1 : 250 (NZM2410 at initial time), 1 : 1000 (MRL‐Faslpr at initial time), 1 : 500 (NZM2410 at final time) or 1 : 2000 (MRL‐Faslpr at final time) were incubated for 2 hr at room temperature with agitation. Total IgG was detected with a goat anti‐mouse IgG antibody conjugated with horseradish peroxidase (HRP; Gibco by Life Technologies, Grand Island, NY) at 1/2000 dilution in PBS/BSA 1%‐Tween 0·05%. After washing three times, the HRP substrate 3,3′,5,5′‐tetramethylbenzidine (1 mg/ml) was added (BD OptEIA™, BD Biosciences Pharmingen) and to stop the reaction, 50 μl H2SO4 2 m was added. Optical density at 450 nm was measured on a microplate reader 800 TS BioTek.
Sandwich ELISA
For cytokine determination in bone‐marrow‐derived DC culture and sera samples, sandwich ELISAs were performed following the kits’ specifications. Interleukin‐10 (IL‐10), IL‐6, IL‐12p70 and interferon‐γ (IFN‐γ) determinations were performed in DC supernatant cultures with or without LPS stimulation (0·5 μg/ml by 24 hr) and IL‐10, IL‐6, 1L‐12p70 (BD OptEIA™; BD Biosciences, BD Biosciences Pharmingen), IL‐17 (BioLegend) and IL‐23 (R&D Systems, Minneapolis, MN) were measured in sera from the different groups at the end of the experiment using commercial kits. Briefly, 96‐well plates were coated overnight at 4° with 1·25 μg/well purified capture antibody and blocked with 200 μl PBS/BSA 3% for 2 hr at room temperature. Wells were washed, and 200 μl culture supernatant was added and incubated for 12 hr at 4°. Later, plates were washed and incubated with 0·85 μg/well biotinylated detection antibody for 1 hr at room temperature. For detection, the samples were incubated for 1 hr at room temperature with streptavidin‐HRP (dilution 1/1500) and revealed with 50 μl 3,3′,5,5′‐tetramethylbenzidine (1 mg/ml). To stop the reaction, 50 μl H2SO4 2 m was added, and the absorbance was measured at 450 nm in a microplate reader 800 TS BioTek. Data derived from measurements performed on cell culture supernatant and serum samples were expressed as cytokine concentration and the fold change was calculated relative to the respective unstimulated controls for each pharmacological treatment.
Histopathology
For histological evaluation, MRL‐Faslpr (17 weeks old) and NZM2410 (24 weeks old) mice at the end of the experiment were killed by sedation followed by cervical dislocation. Kidneys from the different groups of mice were harvested at the end of the therapy and fixed in 4% buffered paraformaldehyde for 24 hr. After that, formalin‐fixed tissues were embedded in paraffin, cut with a microtome in transverse sections into 5 μm‐thick sections, and stained with hematoxylin & eosin and Periodic acid–Schiff reagents. The obtained slides were evaluated by conventional light microscopy by a renal pathologist. The extension of glomerular, tubule, interstitial and vascular injury was evaluated using the current classification of lupus nephritis of the International Society of Nephrology/Renal Pathology Society 2003.26 Active lesions included glomerular hypercellularity (endocapillary and extracapillary), leukocyte infiltration, necrosis/karyorrhexis, wire‐loops and/or hyaline thrombi (subendothelial and/or intraluminal immune complexes, respectively), crescent formation, interstitial inflammation, membranous‐like glomerular lesions (subepithelial immune complexes), vasculitis and vascular immune complex deposition. Chronic lesions included glomerular sclerosis (segmental, global), fibrous adhesions and fibrous crescents. According to the microscopic alterations, the cases were classified as follows: Class 1, minimal mesangial lupus nephritis; Class 2, mesangial proliferative lupus nephritis; Class 3, focal lupus nephritis – active or inactive focal, segmental or global endocapillary or extracapillary glomerulonephritis involving < 50% of all glomeruli, typically with focal subendothelial immune deposits, with or without mesangial; Grade 4, diffuse lupus nephritis – active or inactive diffuse, segmental or global endocapillary or extracapillary glomerulonephritis involving > 50% of all glomeruli, typically with diffuse subendothelial immune deposits, with or without mesangial alterations.26
Flow cytometry
Spleen, kidney, mesenteric lymph nodes, skin and lung from MRL‐Faslpr and NZM2410 mice from different groups were harvested at the end of the experiment (70 days of therapy). Organs were weighed, minced with PBS supplemented with 10% fetal bovine serum and filtered through a 70‐μm cell strainer (BD Biosciences Pharmingen) until a homogeneous cell suspension was obtained. Cells were washed with PBS/BSA 1%, resuspended at 2 × 106 cells/ml and stained with BD Horizon™ Fixable Viability Stain 700 following the manufacturer's instructions. Consequently, samples were incubated with FITC‐, PE‐, Peridinin chlorophyll protein‐Cy5.5‐, APC‐Cy7‐, BV605‐, BV786‐, PE‐Cy7‐ and APC‐conjugated antibodies for 30 min at 4°. For Foxp3 intracellular staining, fixed cells were incubated for 1 hr with a PE‐conjugated anti‐FoxP3 antibody in BD Cytofix/Cytoperm™ (BD Biosciences Pharmingen) permeabilization buffer.27 Cells were washed with permeabilization buffer and acquired using a BD LSRFortessa X‐20 flow cytometer (BD Biosciences Pharmingen). To discriminate between CD45 single cells and doublets, and between live cells and cell debris, events were gated sequentially on side‐scatter (SSC)‐A and forward‐scatter (FSC)‐A, FSC‐H and FSC‐A, SSC‐A and Alexa 700, and SSC‐A and CD45‐APC plots. Flow cytometry data were analyzed using flowjo (TreeStar Inc., Ashland, OR).
CFSE tracking
For tracking experiments, DCs were labeled for 20 min at 37° in 5% CO2 with 5 μm/ml 5,6‐carboxyfluorescein succinimidyl ester (CFSE; Invitrogen) in PBS. After labeling, cells were washed in RPMI‐1640 plus 10% fetal bovine serum, rested for 5 min at 4° and washed with PBS. CFSE‐labeled DCs (4 × 106 cells) were injected intravenously into 10‐week‐old lupus mice. One day after the DC injection, skin, spleen, lungs, submandibular lymph nodes and kidneys of the mice were harvested. The skin was removed from the dorsal area. The lungs, skin and kidneys were cut into small pieces and treated for 30 min with collagenase type IV (Gibco by Life Technologies). The tissues were ground through a cell strainer to prepare single‐cell suspensions for subsequent flow cytometric analysis.
Quantitative real‐time RT‐PCR
Total RNA was isolated from DC cultures after treatment with Dexa, Rzg, CoPP and histones by using Trizol (Life Technologies, Invitrogen), according to the instructions provided by the manufacturer. cDNA synthesis from total RNA was performed using the ImProm‐II Reverse Transcription kit (Promega, Madison, WI). Quantitative RT‐PCR were carried out using a StepOne plus thermocycler (Applied Biosystems, Foster City, CA). The abundance of HO‐1 mRNAs was determined by relative expression to the respective housekeeping gene by the 2‐ΔΔ threshold cycle (ΔΔCt) method. The following primers were used: HO‐1 forward 5′‐CCTCTGACGAAGTGACGCC‐3′ and reverse 5′‐CAGCCCCACCAAGTTCAAA‐3′; β‐actin forward 5′‐ACCTTCTACAATGAGCTGCG‐3′ and reverse 5′‐CTGGATGGCTACGTACATGG‐3′.
Statistical analyses
Data and statistical analyses were performed using prism 7 software (Graph Pad Software, Inc., San Diego, CA). One‐way or two‐way analysis of variance tests and Bonferroni post‐tests were used. P‐values < 0·05 were considered statistically significant. Contingency table was performed to analyze non‐parametric data such as proteinuria.
Results
tolDC characterization
We evaluated the effectiveness of different pharmacological treatments to induce a tolerogenic profile in MRL‐Faslpr and NZM2410 DCs through the analyses of co‐stimulatory molecules expression by FACS. None of the treatments produced significant changes in the viability of DCs (see Supplementary material, Fig. S1). MRL‐Faslpr and NZM2410 bone marrow‐derived tolDCs generated ex vivo through culturing with CoPP, Dexa and Rgz, and further loaded with histones showed a reduced expression of CD40, CD80 and CD86 in comparison with non‐treated DCs, in response to LPS stimulation (Fig. 1). The maturation state of DCs following LPS stimulation can determine whether the tolerogenic phenotype achieved by pharmacological treatment is resistant to maturation. It is important to highlight that untreated DCs loaded with histones showed a higher maturation phenotype than unloaded DCs in response to LPS (Fig. 1b,d,e,f). Herein, tolDCs generated with the three drugs will be defined as tolDCs1; DCs treated with CoPP as tolDCs2 and DCs generated with Dexa and Rgz as tolDCs3. Interestingly, tolDCs1 showed a lower maturation status than untreated DCs loaded with histones. Similar results were observed in DCs generated with CoPP (tolDCs2) from both lupus‐prone mouse models (Fig. 1). It is important to highlight that tolDCs additionally exhibited a reduced expression of MHCII molecules, especially tolDCs1 (Fig. 1g,h,o,p) contributing most likely to diminishing their capacity to prime T cells.1 HO‐1 expression on DCs was evaluated after CoPP treatment by quantitative PCR and flow cytometry. A significant HO‐1 induction was observed for tolDCs1 and tolDCs2 but not under dexamethasone and rosiglitazone treatments (see Supplementary material, Fig. S2).
Figure 1.
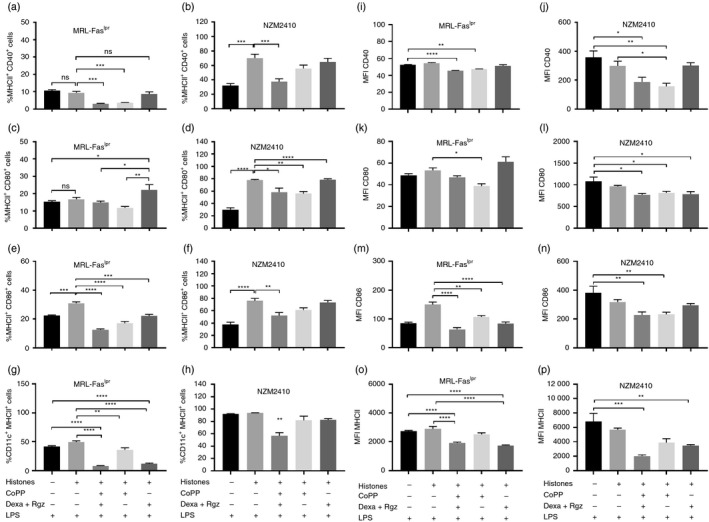
Dendritic cells (DCs) treated with cobalt (III) protoporphyrin IX (CoPP), dexamethasone (Dexa) and rosiglitazone (Rgz) display fewer co‐stimulatory molecules. Bone‐marrow‐derived DCs were treated with Dexa, Rgz and/or CoPP depending on the case. Untreated DCs and non‐treated DCs loaded with histones are included as controls. The frequency and expression of co‐stimulatory molecules are quantified by flow cytometry after lipopolysaccharide (LPS) stimulation (0·5 µg/ml). (a, b) CD40, (c, d) CD80 and (e, f) CD86 together with MHCII are plotted from MRL‐Faslpr (a, c and e) and NZM2410 mice DCs (b, d and f). In addition, (g, h) CD11c+ cells expressing MHCII are plotted for MRL‐Faslpr and NZM2410 mice; and mean fluorescence intensity for (i, j) CD40, (k, l) CD80, (m, n) CD89 and (o, p) MHCII is shown for MRL‐Faslpr and NZM2410 mice. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001 by one‐way analysis of variance. n = 4. Data are shown as the mean ± standard error of the mean.
Finally, pro‐ and anti‐inflammatory cytokine production by tolDCs after LPS stimulation was determined. A reduction in IL‐6 and IL‐12p70 release was observed for tolDCs1 compared with DCs + Hist (Fig. 2c,d,e). On the other hand, we could not detect significant changes in either IFN‐γ or IL‐10 production for tolDCs compared with untreated DCs (Fig. 2a,b,g,h). Although an important autocrine role of IFN‐γ involved in DC maturation has been reported, these cells produce only low levels of this cytokine after LPS stimulation.28 Moreover, DCs from lupus murine models have been shown to produce IFN‐γ.29
Figure 2.
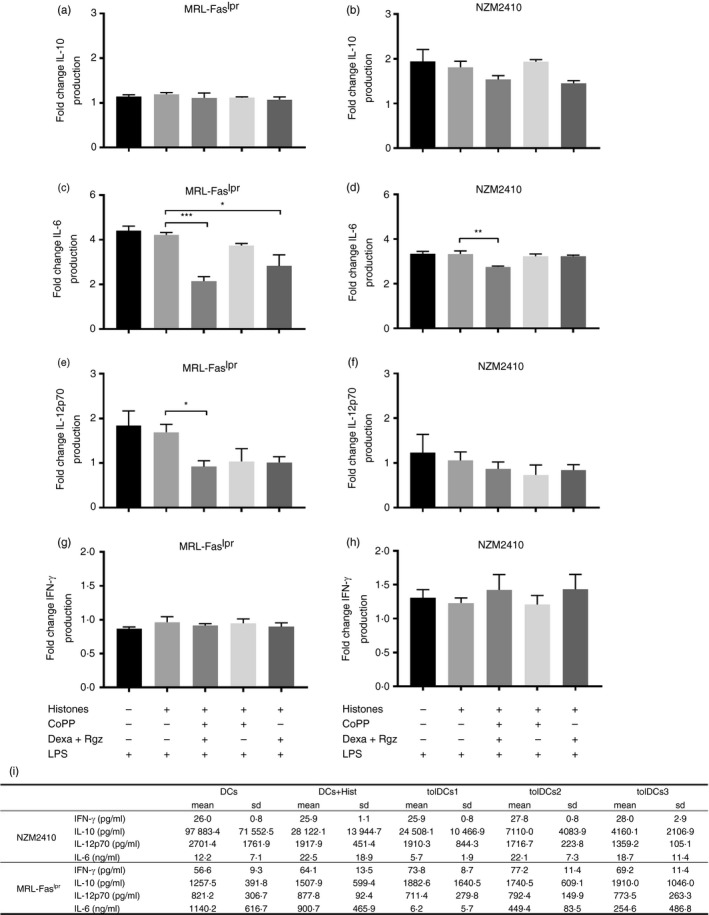
Dendritic cells (DCs) treated with cobalt (III) protoporphyrin IX (CoPP), dexamethasone (Dexa) and rosiglitazone (Rgz) produce less interleukin‐6 (IL‐6) and IL‐12p70 cytokines. Production of several cytokines after lipopolysaccharide (LPS) stimulation was evaluated after treatment with CoPP, Dexa and Rgz. Supernatants were harvested after 24 hr incubation with 0·5 µg/ml LPS, and cytokine levels were determined by ELISA. Graphs show (a, b) IL‐10, (c, d) IL‐6, (e, f) IL‐12p70 and (g, h) interferon‐γ (IFN‐γ) fold change from non‐stimulated DCs. DCs from MRL‐Faslpr mice (a, c, e and g) and NZM2410 mice (b, d, f and h) are shown. Finally, (i) absolute values of cytokines released after LPS stimulation are shown. *P < 0·05, **P < 0·01, ***P < 0·001 by one‐way analysis of variance. Data are shown as mean ± standard error (n = 4).
Adoptive transfer of antigen‐loaded tolDCs ameliorates clinical manifestations in MRL‐Faslpr mice
To assess whether tolDCs can improve the clinical score of murine lupus models, tolDCs were transferred to MRL‐Faslpr or NZM2410 mice (as described in Materials and methods). To evaluate disease severity, mice of all groups were evaluated by determining weight loss, proteinuria, autoantibodies and kidney histopathology. Throughout the experiment, the weight gain of transferred mice was similar to that observed in control groups, indicating the safety of the treatments (see Supplementary material, Fig. S3). In addition, the splenic index remained constant in mice treated with tolDCs compared with control treatments (see Supplementary material, Fig. S4).
Interestingly, MRL‐Faslpr mice treated with tolDCs1 and tolDCs3 improved their clinical scores compared with mice treated with PBS or with tolDCs2 at the end of the experiment (Fig. 3). Notably, mice treated with tolDCs1 and tolDCs3 showed reduced cutaneous lesions in comparison with control mice (PBS‐treated mice; see Supplementary material, Fig. S5). These differences were observed from the fourth cell transfer, and the effect was maintained up to the end of the experiment (Fig. 3a). In contrast, tolDC‐transferred NZM2410 mice did not show significant differences in their clinical score compared with PBS‐treated mice (Fig. 3b). It is important to note that untreated control mice were included in both experiments. These animals were only manipulated to evaluate their clinical score. Interestingly, the control mice group displayed the lowest clinical score between NZM2410 groups showing the sensitivity to manipulation of this murine lupus model.
Figure 3.
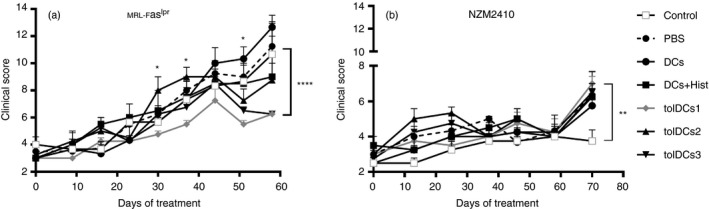
MRL‐Faslpr transferred with tolerogenic dendritic cells (tolDCs) show a reduction in their clinical score. Mice were monitored for 60–70 days from the beginning of the experiment. Clinical scores of mice from all experimental groups were recorded three times per week; the figure shows the average of the clinical score per group until 10 days after the last transfer. (a) MRL‐Faslpr and (b) NZM2410 mice transferred are shown. *P < 0·05, **P < 0·01, ***P < 0·001 by one‐way or two‐way analysis of variance test and Bonferroni post‐test. Data are shown as the mean ± standard error (for three or four animals per group depending on the case).
Although MRL‐Faslpr mice showed a reduced clinical score after treatment with tolDCs, no significant reduction in proteinuria levels was observed in these animals compared with mice treated with PBS (Fig. 4). Interestingly, after the fourth transfer, there was a tendency to reduce proteinuria in MRL‐Faslpr mice, although there were no statistically significant differences. However, at the end of the protocol, there were no differences in renal function between groups considering the serum measurements of creatinine and blood urea nitrogen levels (Fig. 4c–f).
Figure 4.
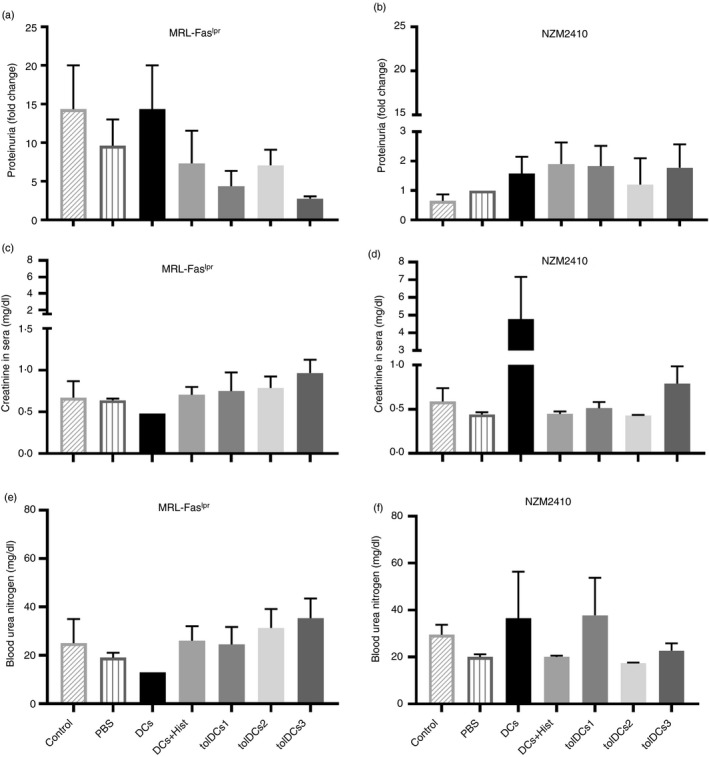
Adoptive transference of tolerogenic dendritic cells (tolDCs) does not decrease proteinuria. This parameter was calculated by the relation with proteinuria at the beginning of the experiment and at the end‐point, in comparison with control mice both in (a) MRL‐Faslpr and (b) NZM2410 mice, by a non‐parametric analysis. On the other hand, serum parameters related to renal failure such as creatinine and blood urea nitrogen were determined at the end of the treatment (c–f). Data are shown for (c, e) MRL‐Faslpr and (d, f) NZM mice. Data are shown as mean ± standard error (for three or four animals per group depending on the case).
In addition, we evaluated serum levels of anti‐DNA and anti‐histone antibodies at the end of the experiment by ELISA. There were no significant differences between groups derived from MRL‐Faslpr mice (Fig. 5a,c), but a reduction in anti‐DNA and anti‐histone antibodies fold change was observed in sera from NZM2410 mice treated with tolDCs1 and tolDCs3 (Fig. 5b,d). In addition, a nuclear staining pattern was determined in sera from control and tolDC‐treated mice, showing mainly a diffuse homogeneous pattern in MRL‐Faslpr mice and a cytoplasmic pattern in NZM2410 lupus‐prone mice (see Supplementary material, Fig. S5a,b). Similar results were obtained by staining Crithidia luciliae for detection of anti‐dsDNA IgG, showing a more intense fluorescence with MRL‐Faslpr sera (see Supplementary material, Fig. S6c,d). Although both SLE models showed elevated levels of autoantibodies, MRL‐Faslpr mice had a higher amount of these autoantibodies. On the other hand, NZM2410 mice presented lower autoantibody levels at the beginning of the protocol, showing a drastic increment at the end of the experiment in all treated groups (Fig. 5).
Figure 5.
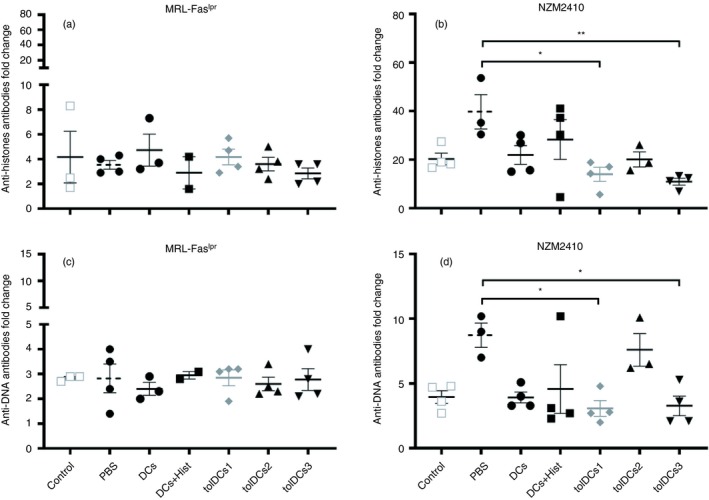
NZM2410 mice treated with tolerogenic dendritic cells (tolDCs) have less anti‐nuclear autoantibodies. Fold change of anti‐histone IgG antibodies was determined in sera from (a) MRL‐Faslpr and (b) NZM2410 mice. Furthermore, fold change of anti‐DNA IgG antibodies was determined in sera from (c) MRL‐Faslpr and (d) NZM2410 mice. The values were calculated by the ratio between antibodies at the end and the beginning of the experiment. **P < 001 and *P < 005 by two‐way analysis of variance test. Data are shown as the mean ± standard error of the mean (for three or four animals per group depending on the case).
Finally, T helper type 1 and type 17 cytokines were measured in sera and no significant differences were detected between groups for IL‐10, IL‐6, 1L‐12p70, IL‐17 or IL‐23 levels at the end of the experiment (see Supplementary material, Fig. S7).
Adoptive transfer of antigen‐loaded tolDCs alters leukocyte infiltration in the kidney
To assess whether tolDC transfer reduces inflammatory infiltration in target organs, leukocyte subsets present in kidneys, spleen and lungs were evaluated by flow cytometry. Although tolDCs can induce tolerance by deletion of autoreactive CD4+ T cells, we did not detect significant differences in CD4+ T‐cell amounts in kidneys and lungs from both murine models. In summary, adoptive transfer of tolDCs did not reduce CD4+ T‐cell numbers in kidneys or lungs from NZM2410 or MRL‐Faslpr mice (Fig. 6i–l).
Figure 6.
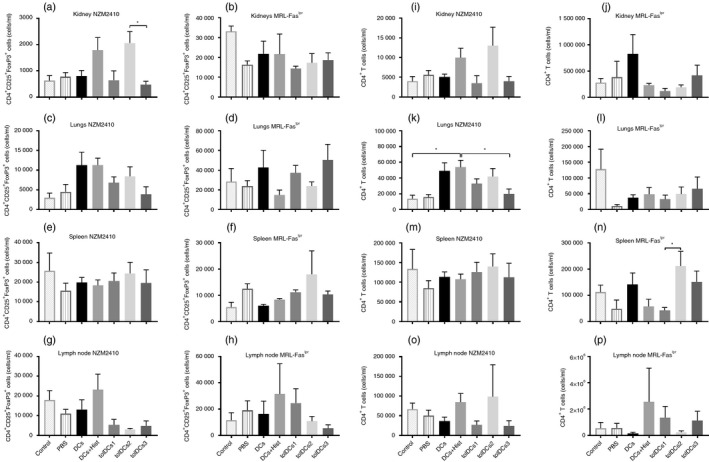
Numbers of regulatory T cells and CD4+ T cells were not altered after tolerogenic dendritic cell (tolDC) transference. Determination of CD4+ CD25+ Foxp3+ cells from (a, b) kidneys, (c, d) lungs, (e, f) spleen and (g, h) lymph node from (a, c, e and g) MRL‐Faslpr mice and (b, d, f and h) NZM2410 mice. CD4+ T cells from (i, j) kidneys, (k, l) lungs, (m, n) spleen and (o, p) lymph nodes from (i, k, m and o) MRL‐Faslpr mice and (j, l, n and p) NZM2410 mice. Counts are presented as cells ×106 per ml. *P < 005 by two‐way analysis of variance test. Data are shown as the mean ± standard error of the mean (for three or four animals per group depending on the case).
On the other hand, we decided to assess whether tolDC transfer induces regulatory T (Treg) cell expansion in any of the murine models of SLE; spleen, lymph nodes, lungs, skin and kidneys were evaluated by flow cytometry. No differences were detected in secondary lymphoid organs (Fig. 6e–h), but an increase in Treg cells was observed in lungs, although not statistically significant; a tendency for Treg cells to increase in this tissue was found in both models (Fig. 6c,d). When Treg cell infiltration was evaluated in the kidney, no differences were detected in MRL‐Faslpr mice. However, there was a tendency for Treg cell numbers to increasing in kidneys from tolDCs2 transferred to NZM2410 mice (Fig. 6a,b).
The presence of neutrophil infiltration in kidneys has been associated with severe inflammation and renal damage in patients with SLE.30, 31 Consequently, we evaluated these cells in the kidneys from both MRL‐Faslpr and NZM2410 mice. The mice treated with tolDCs showed a tendency for decreased neutrophil infiltration in tolDCs2‐ and tolDCs3‐treated mice (see Supplementary material, Fig. S8). Nevertheless, this tendency was not detected in NZM2410 mice treated with tolDCs1 (see Supplementary material, Fig. S8).
Finally, a phenotypic parameter evaluated in DCs was the expression of the co‐stimulatory molecule CD80. The levels of this molecule have been associated with maturation of DCs, and in vitro treatment of DCs with CoPP, Dexa and Rgz have proved to avoid DCs maturation (Figs 1 and 2). Interestingly, a tendency to reduce the expression of CD80 was detected in kidneys from both models whereas its expression in lung DCs was unaffected (see Supplementary material, Fig. S9).
Adoptive transfer of antigen‐loaded tolDCs alters leukocyte infiltration in skin from NZM2410 mice
Because lupus‐prone mice display an increased susceptibility to develop skin lesions in the medium‐ to long‐term, we decided to assess CD4+ T cells and DC infiltration in the skin from NZM2410 mice, which is a chronic model of lupus. The tolDCs1‐treated mice showed an increased proportion of CD4+ CD69+ T cells in the skin (Fig. 7a). Additionally, we detected a tendency to increase DC frequency in the skin from tolDCs1‐ and tolDCs2‐treated mice, nevertheless these DCs have reduced expression of the co‐stimulatory molecule CD80 (Fig. 7c), and there is an increase of Treg cell frequency in tolDCs1‐treated mice (Fig. 7d).
Figure 7.
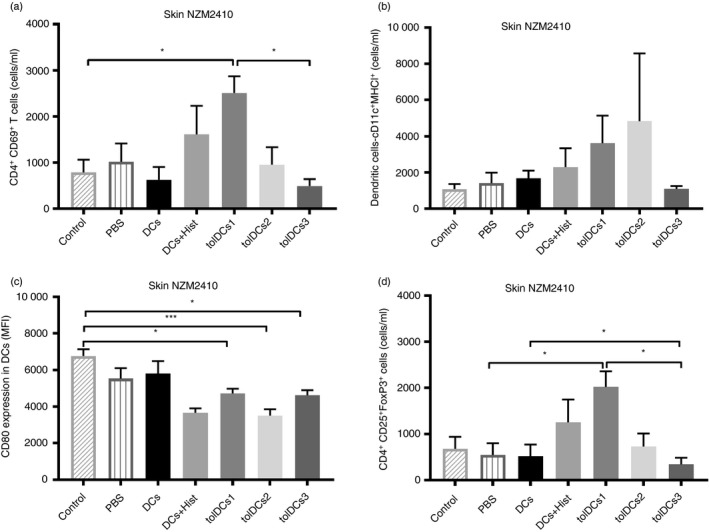
Immune cell infiltration into the skin from NZM2410 mice. (a) CD4+ CD69+ T cells, (b) dendritic cells (DCs), (c) CD80 expression as mean fluorescence intensity (MFI) in DCs and (d) CD4+ CD25+ FoxP3+ cells were determined from the skin in NZM2410 mice. Counts are presented as cells per ml. *P < 0·05 and ***P < 0·001 by two‐way analysis of variance test. Data are shown as mean and standard deviation of the mean (for three or four animals per group depending on the case).
Adoptive transfer of tolDCs does not reduce proliferative glomerulonephritis
To check whether tolDC transfer showed a protective effect on renal damage, kidney histopathological evaluation was performed taking into consideration the parameters mentioned in the Materials and methods.
Contrary to what was expected, kidneys from MRL‐Faslpr mice treated with tolDCs showed increased proliferative glomerulonephritis and greater infiltrating inflammatory cells that disrupted glomerular structure and function compared with PBS‐treated or control mice (Fig. 8a). Similar effects were reported in tolDC‐treated NZM2410 mice (Fig. 8b). Representative images of the different scores found are shown in Fig. 8(c–h).
Figure 8.
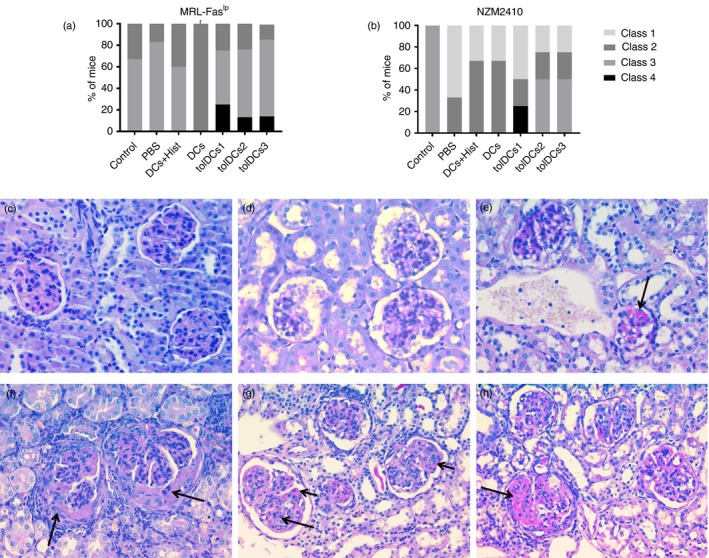
Histopathology of kidneys. The extension of glomerular, tubule interstitial and vascular injury was evaluated using the current classification for patients with lupus nephritis. The score for grading active lesions was a modification of the activity index used currently in renal diagnostic pathology. Grade of glomerulonephritis according to International Society of Nephrology/Renal Pathology Society 2003 classification of glomerulonephritis is shown for each animal group for (a) MRL‐Faslpr mice and (b) NZM2410 mice (for three or four animals per group depending on the case). Representative images for each class observed are shown at 40× with Periodic acid–Schiff stain (c–h). (c) Class I/Minimal mesangial lupus nephritis. High view with normal glomeruli by light microscopy. (d) Class II/Mesangial proliferative lupus nephritis. Light microscopy high view showing a slightly mesangial hypercellularity with focal mesangial matrix expansion. (e) Class III A/Active lesions: Focal (lesion involving < 50% of glomeruli) proliferative lupus nephritis. Arrow shows intraluminal immune aggregates (hyaline thrombi). (f) Class III (A/C)/Active and chronic lesions: Focal proliferative and sclerosing lupus nephritis. Arrows indicate fibrocellular crescents. (g) Class IV‐S (A/C)/Active and chronic lesions: Diffuse (lesion involving ≥ 50% glomeruli) segmental proliferative and sclerosing lupus nephritis. Active lesions are karyorrhexis (long arrow) and subendothelial deposits identifiable by light microscopy (wireloops, short arrow). (h) Class IV‐S (A/C)/Active and chronic lesions: Diffuse segmental proliferative and sclerosing lupus nephritis. Arrow shows segmental glomerular sclerosis.
In vivo tracking of antigen‐loaded tolDCs
The maturation state of DCs modifies the expression of surface homing molecules, which could increase the residence time in one tissue instead of another. Migration of transferred DCs generated by the three drugs (tolDCs1), untreated DCs loaded with histones (DCs + Hist) and unloaded DCs was evaluated in MRL‐Faslpr and NZM2410 mice (Fig. 9a–j). Although differences detected were not statistically significant, DCs loaded with histones migrated further to kidneys and lymph nodes in MRL‐Faslpr mice (Fig. 9a,i). On the other hand, unloaded DCs showed a tendency to migrate towards spleen, kidneys and skin, whereas tolDCs1 showed a higher propensity to migrate towards the lungs and lymph nodes in NZM2410 mice (Fig. 9f,j).
Figure 9.
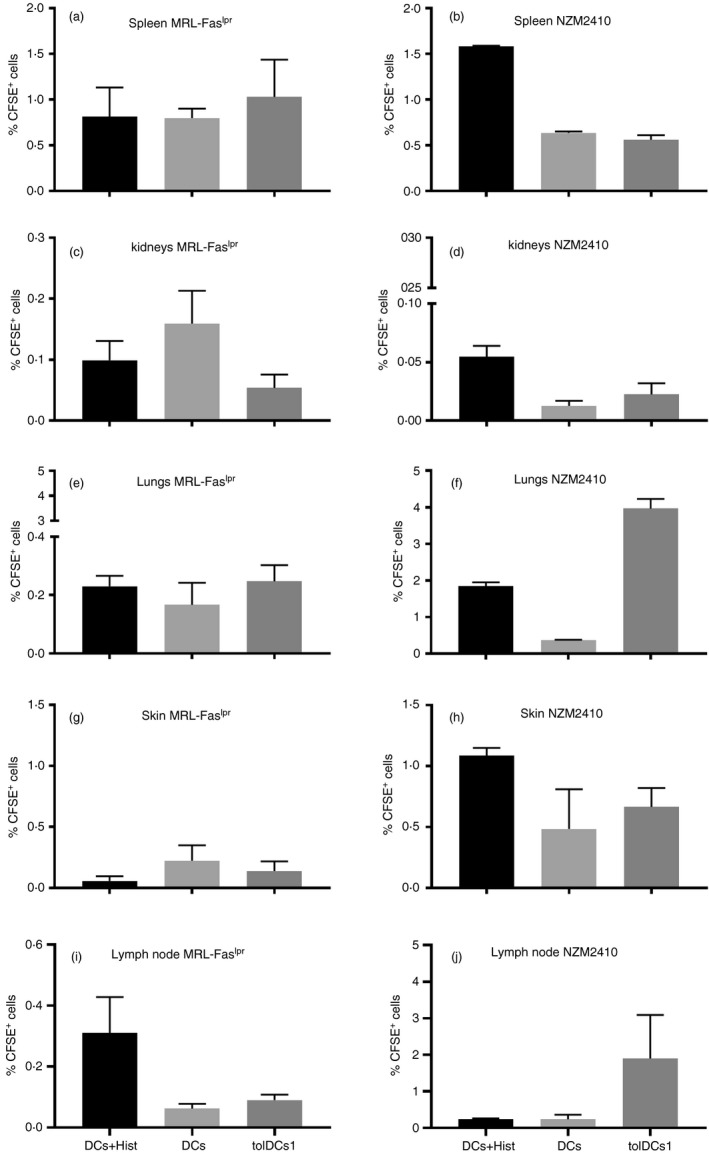
Tracking of transferred dendritic cells (DCs). Determination of DC migration towards (a, b) spleen, (c, d) kidneys, (e, f) lungs, (g, h) skin and (i, j) lymph nodes from (a, c, e, g and i) MRL‐Faslpr mice and (b, d, f, h and j) NZM2410 mice. The percentage of CFSE+ cells was determined in different organs. No differences were detected by one‐way analysis of variance test. Data are shown as mean and standard deviation of the mean (n = 4).
Discussion
To our knowledge, this is the first study evaluating the therapeutic effect of self‐antigen‐loaded tolDC transfer into two different SLE murine models.1 The pathogenic role of DCs has been evaluated in several autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis and SLE.7, 32 In fact, DCs from SLE patients display an increased expression of maturation markers (CD86/CD80) indicating an inflammatory profile in comparison to healthy donors.2, 32 Interestingly, a tolerogenic phenotype can be successfully induced in DCs from patients by pharmacological modulation with Dexa and Rgz.3 Tolerogenic DCs with similar properties have been loaded with autoantigens and employed in randomized clinical trials to induce tolerance in multiple sclerosis, rheumatoid arthritis and type I diabetes patients.1, 15
In addition to the increased expression of co‐stimulatory molecules, myeloid cells from SLE patients show reduced expression of the anti‐inflammatory enzyme HO‐1.20 Here, we evaluate whether HO‐1 induction in combination with Dexa and Rgz generates an improved tolerogenic profile. Our results showed that DCs under pharmacological modulation with CoPP, Dexa and Rgz (tolDCs1) reduced the frequency of DCs expressing maturation markers, such as CD80, CD86 and CD40 in comparison with DCs treated only with CoPP or Dexa and Rgz (Fig. 1). It is well known that mature DCs are immunogenic cells that can stimulate an inflammatory profile in T cells. In contrast, tolDCs characterized by low levels of maturation markers, and high anti‐inflammatory mediator and low pro‐inflammatory cytokine production promote regulatory properties in T and B cells.33 Although in our in vitro experiment, we did not detect an increase in IL‐10 production after LPS stimulation, a reduction in the release of pro‐inflammatory cytokines IL‐6 and IL‐12p70 was observed (Fig. 2). These features are essential when the adoptive transfer into an inflammatory context, such as systemic autoimmunity, is evaluated. The tolerogenic profile in DCs must be resistant to maturation in order to avoid adverse inflammatory effects in the recipient.1 Along these lines, tolDCs1 is suggested as the better option when considering co‐stimulatory molecule expression and cytokine production.
Furthermore, our results show that tolDCs1 (generated by pharmacological modulation with Dexa, Rgz and CoPP) and tolDCs3 (produced with Dexa and Rgz) have a beneficial therapeutic effect in MRL‐Faslpr lupus‐prone mice according to clinical score registry (Fig. 3a). Adoptive transfer of these tolDCs effectively reduces SLE severity and progression in comparison with vehicle (PBS) ‐treated mice and non‐treated DCs. As tolDCs were loaded with histones, the expected therapeutic effect must be antigen‐specific, whereas unpulsed DCs did not improve clinical score (Fig. 3a). Skin lesions, which are included in the determination of clinical scores, were especially reduced in MRL‐Faslpr mice treated with tolDCs compared with control mice (see Supplementary material, Fig. S5). This result is consistent with previous reports of DC accumulation in cutaneous lupus erythematosus lesions and reinforces the pathological importance of these cells in skin lesions.34 The selection of histones as target antigens to induce specific tolerance is sustained by the frequent association of anti‐histone antibodies and disease severity in SLE patients.35, 36 However, we could not rule out a non‐specific tolerogenic effect of self‐antigen‐loaded tolDCs, particularly when considering that tolDCs can induce Treg cells, which in turn could act through linked suppression. In other words, suppression may spread to different epitopes presented by DCs that are located in the lesion.37 Interestingly, NZM2410 lupus‐prone mice did not show any difference in the clinical score during the entire length of the experiment between treated groups (see Supplementary material, Fig. 3b). This could indicate the presence of pronounced differences in immunological mechanisms between the lupus‐prone murine models. Intriguingly, an increased proportion of CD4+ CD69+ T cells was detected in the skin from NZM2410 mice treated with tolDCs1 (Fig. 7a). Accordingly, CD69+ T cells have been associated with immune tolerance induction in tumor‐associated macrophages.38 Although a tendency to increase DC infiltration in the skin was observed, these DCs also showed a reduction in the expression of CD80 and MHCII molecules (Fig. 7b–d). This apparent incongruence could be to the result of a compensatory effect where DCs tend to reduce inflammation in the skin. Whether these DCs displayed a tolerogenic, inflammatory, or intermediate maturation phenotype remains unknown.
On the other hand, NZM2410 mice treated with tolDCs loaded with histones showed a significant reduction in total IgG anti‐histones and anti‐DNA antibodies (Fig. 5b). It is important to note that NZM2410 mice produce several kinds of ANAs (anti‐dsDNA, anti‐chromatin, anti‐histone)39 and although tolDCs reduce anti‐histones and anti‐DNA, other autoantibodies could be involved in the pathogenicity of the model. For example, although anti‐histone antibodies have been associated with neuropsychiatric lupus40 (also described in MRL‐Faslpr mice41) and nephritic lupus,35 the reduced histone‐reactive antibody levels induced in our study by histone‐loaded tolDCs were not able to suppress kidney pathology. Besides, although tolDCs were loaded with histones, NZM2410 mice also reduce anti‐DNA antibody levels. These data suggest the presence of a linked suppression mechanism.33, 37
Although CD4+ T‐cell deletion following anergy induction has been reported as a mechanism of action for tolDCs, we could not detect a reduction of CD4+ T cells in kidneys from either NZM2410 or MRL‐Faslpr mice (Fig. 6i–p). The lack of T‐cell depletion effect of tolDCs could be related to the intense systemic lymphoproliferation that occurs in both of these strains.42, 43, 44 Treg cell expansion has also been described as a mechanism associated with the modulatory function of tolDCs,45, 46, 47, 48 but we did not detect an expansion of Treg cell number in the kidney or spleen from MRL‐Faslpr mice (Fig. 6a,e). On the other hand, a tendency to increase Treg cell infiltration in the kidney was detected in NZM2410 mice treated with tolDCs2 (Fig. 6b,d,f).
Importantly, neutrophil activation is related to inflammation, the formation of neutrophil extracellular traps and ANA increment.49 However, we could not detect a reduction in neutrophil infiltration in kidneys (see Supplementary material, Fig. S8), suggesting a leading inflammatory context, which would be associated with a consequent increase of Treg cell number tending to reduce inflammation and damage. Accordingly, it has been reported that DCs infiltrating kidneys are involved in the development of chronic glomerulonephritis.50 However, we did not detect a significant reduction in DCs from kidney as well as their co‐stimulatory molecule expression (see Supplementary material, Fig. S9). For all the above mentioned, and because of the continuous transgression of the renal nephron, the normal function of the organ was affected, which was shown by the increment of proteinuria.
Strikingly, congruence was not detected between clinical score and renal condition. In other words, although MRL‐Faslpr mice showed a lower clinical score at the end of the treatment, glomerular damage remained unaffected after tolDC transfer (Fig. 8a). On the other hand, despite finding a reduction of fold change in anti‐DNA and anti‐histone antibodies in NZM2410 mice, tolDC‐treated mice did not show an improvement in clinical score (Figs 3 and 5) or renal damage (Fig. 8b). Moreover, concordant with these data, we did not detect a reduction in proteinuria of either of the two models after the adoptive transference of tolDCs in comparison with control mice (Fig. 4). On the other hand, one could speculate that peripheral tolerance induction might not be enough when the tissue inflammation and target‐organ damage are already established,51 especially in SLE, where already damaged tissue and dying cells prime and boost immune cells, which supports chronic inflammation. Moreover, even though autoantibodies can induce target organ inflammation, they will not necessarily be the leading cause of loss of function. Indeed, almost all SLE patients have immunocomplex deposits in their glomeruli, although only 40–60% of them develop glomerulonephritis.52
In consequence, our data suggest that tolDCs could be effective in reducing cutaneous manifestations instead of renal lesions. Indeed, skin evaluation showed a tendency to increase DC numbers with a reduced expression of maturation markers and increased Treg cell infiltration (Fig. 7). Cutaneous lesions (considered within the clinical score) are more evident in MRL‐Faslpr mice than the NZM2410 mice, which develop dermatitis only in advanced stages of the disease.53 In consequence, the improvement of the clinical score in MRL‐Faslpr is more evident than that in NZM2410 mice at the evaluated periods.
Our results support the notion that, since different mechanisms of action are associated with each drug, the pharmacological combination could result in additive and synergistic effects. Indeed, it has been demonstrated that treatment with Dexa blocks DC maturation54 and it has been previously used together with Rgz,3 which is a known nuclear factor‐κB inhibitor.55 HO‐1 is a critical enzyme involved in renal protection56 and its expression is reduced in SLE patients.20 Induction of this enzyme has been related to amelioration in lupus‐prone mice.21, 57 So, it could be expected that the combination of the three drugs could have a better effect in suppressing the broad spectrum of SLE manifestations. As a whole, our results indicate that the combined treatment (CoPP, Rgz, Dexa and histones) is more efficient at inducing tolerance in DCs than individual treatments.
The effect of tolDC transfer in our experiments was observed mainly in the skin, as shown by the reduction of cutaneous lesions, clinical score in MRL‐Faslpr mice and the reduction of the mature profile in DCs in NZM2410 mice (Fig. 7). It has been reported that immature DCs migrate differently from mature DCs,58 but although the selected antigen could determine the differential migration of loaded DCs,37 we did not detect such difference in MRL‐Faslpr mice (Fig. 9). Interestingly, tolDCs1 were more prone to migrate toward the lungs and unloaded DCs migrate to the kidneys in NZM2410 mice (Fig. 9). In this sense, immature DCs that go to this organ may be more susceptible to revert their profile toward an inflammatory phenotype that is detrimental for kidney function. Interestingly, a recent study has described that antigen‐loaded tolDCs have a reduced capacity to prevent autoimmunity in comparison with non‐loaded tolDCs.59 In the same way, histone‐loaded tolDCs could be effective in vitro but ineffective in reducing renal damage in vivo.
Conclusions
Here, we showed for the first time that transfer of tolDCs could improve clinical manifestations in the setting of inflammation associated with chronic SLE. Significant differences were observed between both murine models in terms of changing the clinical score and antibody titers. We found that DCs treated with the three drugs were more efficient than those treated only with CoPP or with Dexa and Rgz. However, tolDCs did not reduce glomerulonephritis in kidneys despite reducing cutaneous lesions in MRL‐Faslpr mice. These results suggest that loaded tolDCs would have an immunoregulatory profile; nevertheless, they could preferentially perform their suppressive function in tissues with greater tropism to specific tolDCs.
To conclude, tolDCs generated with CoPP, Dexa and Rgz formulation, and loaded with histones as an SLE‐specific self‐antigen, were not efficient as immunotherapy for reducing nephritic lupus severity and progression. Unknown antigens as well as the great diversity of reported self‐antigens in systemic autoimmune pathologies hinder the selection of the correct tissue‐specific antigens, especially considering that SLE is a heterogeneous disease composed of multiple clinical presentations. We suggest that tolDCs generated with CoPP, Dexa and Rgz have an efficient tolerogenic profile in vitro; however, a strategy employing multiple known self‐antigens to induce tolerance against several subtypes of SLE must be assayed.
Author Contributions
SCF, MR, FGS, MJAL and DRP carried out the experiments. SCF wrote the manuscript with support from MR and FGS. AV and JCR performed the histopathological evaluation of the renal samples. RPS and EJ consolidated the original project. AFF and DRZ performed the additional experiment required by reviewers. JPMO and AK supervised the work and performed critical revision of the manuscript. All authors revised and approved the manuscript.
Funding
This research was funded by Corporación de Fomento de la Producción (CORFO) grant No. 13CTI‐21526 and the Millennium Institute on Immunology and Immunotherapy (P09‐016‐F).
Disclosure
The authors declare no conflict of interest.
Supporting information
Figure S1. Treatment with Dexamethasone, Rosiglitazone and cobalt (III) protoporphyrin IX do not reduce viability in dendritic cells.
Figure S2. Cobalt (III) protoporphyrin IX induces heme‐oxygenase‐1 expression in DCs.
Figure S3. Body weight variations.
Figure S4. Splenic index determination.
Figure S5. Skin lesions in MRL‐Faslpr mice.
Figure S6. Anti‐nuclear antibody determination.
Figure S7. Cytokine determination in sera.
Figure S8. Neutrophil determination in kidney.
Figure S9. CD80 expression in dendritic cells.
References
- 1. Funes SC, Manrique de Lara A, Altamirano‐Lagos MJ, Mackern‐Oberti JP, Escobar‐Vera J, Kalergis AM. Immune checkpoints and the regulation of tolerogenicity in dendritic cells: implications for autoimmunity and immunotherapy. Autoimmun Rev 2019; 18:359–68. [DOI] [PubMed] [Google Scholar]
- 2. Schäfer C, Ascui G, Ribeiro CH, López M, Prados‐Rosales R, González PA et al Innate immune cells for immunotherapy of autoimmune and cancer disorders. Int Rev Immunol 2017; 36:315–37. [DOI] [PubMed] [Google Scholar]
- 3. Obreque J, Vega F, Torres A, Cuitino L, Mackern‐Oberti JP, Viviani P et al Autologous tolerogenic dendritic cells derived from monocytes of systemic lupus erythematosus patients and healthy donors show a stable and immunosuppressive phenotype. Immunology 2017; 152:648–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adorini L, Giarratana N, Penna G. Pharmacological induction of tolerogenic dendritic cells and regulatory T cells. Semin Immunol 2004; 16:127–34. [DOI] [PubMed] [Google Scholar]
- 5. Carreño LJ, González PA, Bueno SM, Riedel CA, Kalergis AM. Modulation of the dendritic cell–T‐cell synapse to promote pathogen immunity and prevent autoimmunity. Immunotherapy 2011; 3:6–11. [DOI] [PubMed] [Google Scholar]
- 6. Mackern‐Oberti JP, Riquelme SA, Llanos C, Schmidt CB, Simon T, Anegon I et al Heme oxygenase‐1 as a target for the design of gene and pharmaceutical therapies for autoimmune diseases. Curr Gene Ther 2014; 14:218–35. [DOI] [PubMed] [Google Scholar]
- 7. Mackern‐Oberti JP, Llanos C, Vega F, Salazar‐Onfray F, Riedel CA, Bueno SM et al Role of dendritic cells in the initiation, progress and modulation of systemic autoimmune diseases. Autoimmun Rev 2015; 14:127–39. [DOI] [PubMed] [Google Scholar]
- 8. Alam J, Shibahara S, Smith A. Transcriptional activation of the heme oxygenase gene by heme and cadmium in mouse hepatoma cells. J Biol Chem 1989; 264:6371–5. [PubMed] [Google Scholar]
- 9. Ryter SW, Alam J, Choi AM. Heme oxygenase‐1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 2006; 86:583–650. [DOI] [PubMed] [Google Scholar]
- 10. Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev 2008; 60:79–127. [DOI] [PubMed] [Google Scholar]
- 11. Moreau A, Hill M, Thebault P, Deschamps J, Chiffoleau E, Chauveau C et al Tolerogenic dendritic cells actively inhibit T cells through heme oxygenase‐1 in rodents and in nonhuman primates. FASEB J 2009; 23:3070–7. [DOI] [PubMed] [Google Scholar]
- 12. Chauveau C, Rémy S, Royer PJ, Hill M, Tanguy‐Royer S, Hubert F‐X et al Heme oxygenase‐1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL‐10 expression. Blood 2005; 106:1694–702. [DOI] [PubMed] [Google Scholar]
- 13. Mackern‐Oberti JP, Vega F, Llanos C, Bueno SM, Kalergis AM. Targeting dendritic cell function during systemic autoimmunity to restore tolerance. Int J Mol Sci 2014; 15:16381–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Llanos C, Mackern‐Oberti JP, Vega F, Jacobelli SH, Kalergis AM. Tolerogenic dendritic cells as a therapy for treating lupus. Clin Immunol 2013; 148:237–45. [DOI] [PubMed] [Google Scholar]
- 15. Kim S‐H, Jung H‐H, Lee C‐K. Generation, characteristics and clinical trials of ex vivo generated tolerogenic dendritic cells. Yonsei Med J 2018; 59:807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rees F, Doherty M, Grainge MJ, Lanyon P, Zhang W. The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatology 2017; 56:1945–61. [DOI] [PubMed] [Google Scholar]
- 17. Agmon‐Levin N, Mosca M, Petri M, Shoenfeld Y. Systemic lupus erythematosus one disease or many? Autoimmun Rev 2012; 11:593–5. [DOI] [PubMed] [Google Scholar]
- 18. Mackern‐Oberti JP, Llanos C, Riedel CA, Bueno SM, Kalergis AM. Contribution of dendritic cells to the autoimmune pathology of systemic lupus erythematosus. Immunology 2015; 146:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol 2016; 12:716. [DOI] [PubMed] [Google Scholar]
- 20. Herrada AA, Llanos C, Mackern‐Oberti JP, Carreño LJ, Henriquez C, Gómez RS et al Haem oxygenase 1 expression is altered in monocytes from patients with systemic lupus erythematosus. Immunology 2012; 136:414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mackern‐Oberti JP, Llanos C, Carreño LJ, Riquelme SA, Jacobelli SH, Anegon I et al Carbon monoxide exposure improves immune function in lupus‐prone mice. Immunology 2013; 140:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mackern‐Oberti JP, Obreque J, Méndez GP, Llanos C, Kalergis AM. Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus. Clin Exp Immunol 2015; 182:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S et al Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony‐stimulating factor. J Exp Med 1992; 176:1693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moratz C, Robbins R, Eickhoff J, Edison J, Lui H, Peng SJCI. Regulation of systemic tissue injury by coagulation inhibitors in B6.MRL/lpr autoimmune mice. Clin Immunol 2018; 197:169–78. [DOI] [PubMed] [Google Scholar]
- 25. Lichtnekert J, Rupanagudi KV, Kulkarni OP, Darisipudi MN, Allam R, Anders H‐J. Activated protein C attenuates systemic lupus erythematosus and lupus nephritis in MRL‐Fas (lpr) mice J Immunol 2011; 187:3413–21. [DOI] [PubMed] [Google Scholar]
- 26. Weening JJ, D'agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB et al The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int 2004; 65:521–30. [DOI] [PubMed] [Google Scholar]
- 27. Dzangué‐Tchoupou G, Corneau A, Blanc C, Benveniste O. Analysis of cell surface and intranuclear markers on non‐stimulated human PBMC using mass cytometry. PLoS ONE 2018; 13:e0194593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pan J, Zhang M, Wang J, Wang Q, Xia D, Sun W et al Interferon‐γ is an autocrine mediator for dendritic cell maturation. Immunol Lett 2004; 94:141–51. [DOI] [PubMed] [Google Scholar]
- 29. Sang A, Zheng Y‐Y, Yin Y, Dozmorov I, Li H, Hsu H‐C et al Dysregulated cytokine production by dendritic cells modulates B cell responses in the NZM2410 mouse model of lupus. PLoS ONE 2014; 9:e102151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM et al Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 2011; 187:538–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Flores‐Mendoza G, Sansón SP, Rodríguez‐Castro S, Crispín JC, Rosetti F. Mechanisms of tissue injury in lupus nephritis. Trends Mol Med 2018; 24:364–78. [DOI] [PubMed] [Google Scholar]
- 32. Carreño LJ, Pacheco R, Gutierrez MA, Jacobelli S, Kalergis AM. Disease activity in systemic lupus erythematosus is associated with an altered expression of low‐affinity Fcγ receptors and costimulatory molecules on dendritic cells. Immunology 2009; 128:334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. García‐González P, Ubilla‐Olguín G, Catalán D, Schinnerling K, Aguillón JC. Tolerogenic dendritic cells for reprogramming of lymphocyte responses in autoimmune diseases. Autoimmun Rev 2016; 15:1071–80. [DOI] [PubMed] [Google Scholar]
- 34. Farkas L, Beiske K, Lund‐Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon‐α/β‐producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol 2001; 159:237–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dieker J, Berden JH, Bakker M, Briand J‐P, Muller S, Voll R et al Autoantibodies against modified histone peptides in SLE patients are associated with disease activity and lupus nephritis. PLoS ONE 2016; 11:e0165373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jimenez AL, Mowrey WB, Putterman C, Buyon J, Goilav B, Broder A. Tubulointerstitial damage in lupus nephritis: a comparison of the factors associated with tubulointerstitial inflammation and renal scarring. Arthritis Rheumatol 2018; 70:1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Suwandi JS, Nikolic T, Roep BO. Translating mechanism of regulatory action of tolerogenic dendritic cells to monitoring endpoints in clinical trials. Front Immunol 2017; 8:1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao Q, Kuang D‐M, Wu Y, Xiao X, Li X‐F, Li T‐J et al Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor‐associated macrophages. J Immunol 2012; 188:1117–24. [DOI] [PubMed] [Google Scholar]
- 39. Vyse TJ, Rozzo SJ, Drake CG, Izui S, Kotzin BL. Control of multiple autoantibodies linked with a lupus nephritis susceptibility locus in New Zealand black mice. J Immunol 1997; 158:5566–74. [PubMed] [Google Scholar]
- 40. Sun X‐Y, Shi J, Han L, Su Y, Li Z‐G. Anti‐histones antibodies in systemic lupus erythematosus: prevalence and frequency in neuropsychiatric lupus. J Clin Lab Anal 2008; 22:271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gulinello M, Putterman C. The MRL/lpr mouse strain as a model for neuropsychiatric systemic lupus erythematosus. Biomed Res Int 2011; 2011:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rieux‐Laucat F, Magérus‐Chatinet A, Neven B. The autoimmune lymphoproliferative syndrome with defective FAS or FAS‐ligand functions. J Clin Immunol 2018; 38:558–68. [DOI] [PubMed] [Google Scholar]
- 43. Shi X, Xie C, Kreska D, Richardson JA, Mohan C. Genetic dissection of SLE: SLE1 and FAS impact alternate pathways leading to lymphoproliferative autoimmunity. J Exp Med 2002; 196:281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li W, Titov AA, Morel L. An update on lupus animal models. Curr Opin Rheumatol 2017; 29:434–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dáňová K, Grohová A, Strnadová P, Funda DP, Šumník Z, Lebl J et al Tolerogenic dendritic cells from poorly compensated type 1 diabetes patients have decreased ability to induce stable antigen‐specific T cell hyporesponsiveness and generation of suppressive regulatory T cells. J Immunol 2016; 198:729–40. [DOI] [PubMed] [Google Scholar]
- 46. Raker VK, Domogalla MP, Steinbrink K. Tolerogenic dendritic cells for regulatory T cell induction in man. Front Immunol 2015; 6:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Volchenkov R, Karlsen M, Jonsson R, Appel S. Type 1 regulatory T cells and regulatory B cells induced by tolerogenic dendritic cells. Scand J Immunol 2013; 77:246–54. [DOI] [PubMed] [Google Scholar]
- 48. Mansilla MJ, Sellès‐Moreno C, Fàbregas‐Puig S, Amoedo J, Navarro‐Barriuso J, Teniente‐Serra A et al Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci Ther 2015; 21:222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van der Linden M, van den Hoogen LL, Westerlaken GH, Fritsch‐Stork RD, van Roon JA, Radstake TR et al Neutrophil extracellular trap release is associated with antinuclear antibodies in systemic lupus erythematosus and anti‐phospholipid syndrome. Rheumatology 2018; 57:1228–34. [DOI] [PubMed] [Google Scholar]
- 50. Hochheiser K, Tittel A, Kurts C. Kidney dendritic cells in acute and chronic renal disease. Int J Exp Pathol 2011; 92:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tabas I, Glass CK. Anti‐inflammatory therapy in chronic disease: challenges and opportunities. Science 2013; 339:166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hanly JG, O'keeffe AG, Su L, Urowitz MB, Romero‐Diaz J, Gordon C et al The frequency and outcome of lupus nephritis: results from an international inception cohort study. Rheumatology 2015; 55:252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Richard ML, Gilkeson G. Mouse models of lupus: what they tell us and what they don't. Lupus Sci Med 2018; 5:e000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matyszak MK, Citterio S, Rescigno M, Ricciardi‐Castagnoli P. Differential effects of corticosteroids during different stages of dendritic cell maturation. Eur J Immunol 2000; 30:1233–42. [DOI] [PubMed] [Google Scholar]
- 55. Kalergis AM, Iruretagoyena MI, Barrientos MJ, González PA, Herrada AA, Leiva ED et al Modulation of nuclear factor‐κB activity can influence the susceptibility to systemic lupus erythematosus. Immunology 2009; 128:e306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kawashima A, Oda Y, Yachie A, Koizumi S, Nakanishi I. Heme oxygenase‐1 deficiency: the first autopsy case. Hum Pathol 2002; 33:125–30. [DOI] [PubMed] [Google Scholar]
- 57. Takeda Y, Takeno M, Iwasaki M, Kobayashi H, Kirino Y, Ueda A et al Chemical induction of HO‐1 suppresses lupus nephritis by reducing local iNOS expression and synthesis of anti‐dsDNA antibody. Clin Exp Immunol 2004; 138:237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. de Vries IJM, Krooshoop DJEB, Scharenborg NM, Lesterhuis WJ, Diepstra JHS, van Muijen GNP et al Effective migration of antigen‐pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res 2003; 63:12–7. [PubMed] [Google Scholar]
- 59. Funda DP, Goliáš J, Hudcovic T, Kozáková H, Špíšek R, Palová‐Jelínková L. Antigen loading (e.g. glutamic acid decarboxylase 65) of tolerogenic DCs (tolDCs) reduces their capacity to prevent diabetes in the non‐obese diabetes (NOD)‐severe combined immunodeficiency model of adoptive cotransfer of diabetes as well as in NOD mice. Front Immunol 2018; 9:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Treatment with Dexamethasone, Rosiglitazone and cobalt (III) protoporphyrin IX do not reduce viability in dendritic cells.
Figure S2. Cobalt (III) protoporphyrin IX induces heme‐oxygenase‐1 expression in DCs.
Figure S3. Body weight variations.
Figure S4. Splenic index determination.
Figure S5. Skin lesions in MRL‐Faslpr mice.
Figure S6. Anti‐nuclear antibody determination.
Figure S7. Cytokine determination in sera.
Figure S8. Neutrophil determination in kidney.
Figure S9. CD80 expression in dendritic cells.