

Abstract

In the field of supramolecular (co)polymerizations, the ability to predict and control the composition and length of the supramolecular (co)polymers is a topic of great interest. In this work, we elucidate the mechanism that controls the polymer length in a two-component cooperative supramolecular polymerization and unveil the role of the second component in the system. We focus on the supramolecular copolymerization between two derivatives of benzene-1,3,5-tricarboxamide (BTA) monomers: a-BTA and Nle-BTA. As a single component, a-BTA cooperatively polymerizes into long supramolecular polymers, whereas Nle-BTA only forms dimers. By mixing a-BTA and Nle-BTA in different ratios, two-component systems are obtained, which are analyzed in-depth by combining spectroscopy and light-scattering techniques with theoretical modeling. The results show that the length of the supramolecular polymers formed by a-BTA is controlled by competitive sequestration of a-BTA monomers by Nle-BTA, while the obvious alternative Nle-BTA acts as a chain-capper is not operative. This sequestration of a-BTA leads to short, stable species coexisting with long cooperative aggregates. The analysis of the experimental data by theoretical modeling elucidates the thermodynamic parameters of the copolymerization, the distributions of the various species, and the composition and length of the supramolecular polymers at various mixing ratios of a-BTA and Nle-BTA. Moreover, the model was used to generalize our results and to predict the impact of adding a chain-capper or a competitor on the length of the cooperative supramolecular polymers under thermodynamic control. Overall, this work unveils comprehensive guidelines to master the nature of supramolecular (co)polymers and brings the field one step closer to applications.
Introduction
Since the seminal work of Staudinger, controlling the chain length and the molecular weight distribution of macromolecules has been a topic of major interest.1,2 In the synthesis of covalent polymers, the ability to make a product with a desired molecular weight offers remarkable control over the material properties.1,2 Methods to tune the chain length in step-growth polymerizations are well-established and generally involve stoichiometric imbalance or the addition of a small amount of monofunctional monomer, which is often referred to as a chain-stopper. This chain-stopper inhibits further polymerization because the chain-ends lack reactive groups and, thus, become unable to grow further.3
In the field of supramolecular polymers, isodesmic supramolecular polymerization is the noncovalent counterpart of step-growth polymerization, as the reactivity of the chain-end is independent of the chain length.4 Therefore, next to stoichiometric imbalance and impurities, the control over chain length in isodesmic polymerizations has also been achieved by the use of chain-stoppers.5 In fact, many elegant examples have been reported in which the chain-stopper is a monotopic derivative of a ditopic monomer with two electronically uncoupled functional groups.5−10 This is simple to design because the association at one of the two binding sites does not affect the second association at the other binding site. In addition, the effect of the chain-stopper on the molecular weight is simple to compute because the association constant between chain-stopper and chain-end is as strong as the interaction between the ditopic monomers.5 As a result, the reduction in chain length is very efficient, even in the presence of small amounts of chain-stopper.5−10 The impact of chain-stoppers on the decrease of the molecular weights of the isodesmic supramolecular polymers and on their property changes have been studied in great detail.11
Similarly, the control of polymer chain length in cooperative supramolecular polymerizations has attracted a great deal of interest as well.12 In this case, the reactivities of the two functional groups of a ditopic monomer are electronically coupled, because the binding on one side of the molecule influences the binding affinity on the other side of the molecule. As a result, monomers are more likely to react on the active site of the polymer than on another monomer, resulting in the coexistence of long polymers and monomers. In chain-growth polymerizations, the covalent counterpart of cooperative supramolecular polymerization, the degree of polymerization depends not only on the rates of propagation and initiation steps but also on termination by disproportionation, by recombination and the possibility of chain transfer. Different strategies have been developed to control the polymer length such as the addition of chain transfer agents (thiols in radical polymerization or antioxidant in PVC polymerization) and the development of living polymerization to control the molecular weight of the final polymer.1,2 This last strategy has been successfully applied to cooperative supramolecular polymerizations under kinetic control, with the use of metastable monomers and tailored initiators.13−16 A conformationally dormant monomer was used to initiate the polymerization, and as a result, supramolecular polymers with controlled chain growth and narrow dispersity have been obtained.
However, these strategies have not been applied to dynamic cooperative supramolecular polymerization under thermodynamic control. To steer polymer chain length in cooperative supramolecular polymerization, the term chain-stopper or end-capper has been used,17−22 although it is not clear how this additive controls the length or deactivates the chain growth. Hence, this second component is not necessarily present at the end of the chain. Then, the question is how this second component interacts with the polymer, with the monomer, or with both and whether it copolymerizes by intercalation or by chain-capping. An extreme case is the addition of a good solvent, which depolymerizes the supramolecular chains without participating in the polymer sequence.23,24 A similar mechanism explains the denaturation of proteins by urea, which unfolds the protein by stabilizing the unfolded protein.25 In such cases, the second component is a competitor that preferentially stabilizes the monomers and, therefore, pushes the thermodynamic equilibrium to depolymerization in accordance with Le Chatelier’s principle.26 With these scenarios in mind, the synthesis of a chain-capper, a component that interacts with the chain-end and inhibits further growth, requires a rational design that encompasses a subtle balance of reactivity with the monomer and the polymer chain. In a number of studies, the chain-capper reported seems monofunctional (similar to the methodology in step-growth polymerizations) or has a very specific interaction with the end of the chain.17−22
Here we delineate a general experimental approach supported by theoretical modeling to elucidate the mechanism directing the chain length in two-component thermodynamically controlled cooperative supramolecular polymerizations. Extensive studies on the assembly of benzene-1,3,5-tricarboxamides (BTAs) derivatives have demonstrated that the nature of the side chains dramatically influences the structure, length, and mechanism of formation of the aggregates.27−30 Monomers of BTA decorated with achiral alkyl chains (a-BTA) self-assemble cooperatively into one-dimensional (1D) aggregates stabilized by threefold helical intermolecular hydrogen bonding. In contrast, the aggregation of BTAs decorated with bulky α-esters side chains, derived from norleucine, exhibits exclusive formation of dimers with intermolecular hydrogen bond formation between the N–H protons and the ester carbonyls (Nle-BTA).31,32 A question that we are discussing here is how BTAs with either alkyl or bulky α-esters side chains would interact with one another in solution (Scheme 1). The elucidation of this question brought us to unravel the mechanisms of supramolecular copolymerization of a monomer with an intercalator, a chain-capper, or a competitor and its impact on the polymer’s length under thermodynamic control. The results show that the copolymerization processes of chain-capping and competitive aggregation pathways are challenging to characterize and subtle to differentiate.
Scheme 1. Chemical Structures of Benzene-1,3,5-tricarboxamides a-BTA and S-Nle-BTA (A); Schematic Molecular Structure of the Helical Stack Formed by a-BTA (B) and the Dimeric Hydrogen-Bonded Structure Formed by S-Nle-BTA (C).

Results
Gelation Study in Mixtures of a-BTA and S-Nle-BTA
It is well-known that BTAs comprising alkyl side chain form gels in apolar solvents.33−36 We started our investigation with an initial qualitative study based on a gel of a-BTA in methylcyclohexane (MCH) (Figure S1). Interestingly, the addition of only 10 mol % S-Nle-BTA to this gel resulted in an immediate loss of the self-supporting properties of the gel. This loss of gel property is indicative of a strong interaction between the two monomers a-BTA and S-Nle-BTA with a possible shortening of the polymers. In the interaction with a-BTA, S-Nle-BTA is a hydrogen bond acceptor via the amide or via the ester. When all monomers interact only via the amides of each monomer, this results in threefold helical hydrogen bonding between the amide groups of S-Nle-BTA and a-BTA, and a growth into 1D helical polymers. However, when S-Nle-BTA interacts via the ester with a-BTA, the amides of S-Nle-BTA remain inactivated at the polymer end, which should decrease the cooperative character of the chain and reduce its length. This hypothesis is supported by the qualitative gel study. To elucidate the origin of this strong effect, we conduct a series of experiments combining “Sergeant and Soldiers” experiments37−40 between chiral S-Nle-BTA and achiral a-BTA analyzed by UV–vis and circular dichroism (CD) spectroscopy and by static light scattering (SLS).
Homoaggregation of a-BTA and S-Nle-BTA
We first analyzed the thermodynamics of dimerization of the chiral S-Nle-BTA monomer using temperature- and concentration-dependent CD measurements (Figure S2). The experimental cooling curves were fitted, and the thermodynamic parameters obtained show that the Gibbs free energy for the dimer (ΔG° = −40.0 kJ mol–1) is lower than that for the elongation of a-BTA cooperative aggregates (ΔG°e = −38.3 kJ mol–1) at 298 K,41 confirming that S-Nle-BTA will form a dimer rather than 1D 3-fold hydrogen-bonded aggregates. These S-Nle-BTA dimers have been shown to be stabilized by six hydrogen bonds between the N–H amides and C=O esters.31,32 As a result, unbound C=O groups are pointing outward, which prevents a dimer from forming hydrogen bonds with another dimer. The thermodynamic parameters of the a-BTA homopolymerization were previously reported.41
Supramolecular Copolymerization of a-BTA and S-Nle-BTA in Dilute Solutions Studied by Spectroscopy
We then continued with “Sergeant and Soldiers” experiments where S-Nle-BTA and a-BTA play the role of sergeants and soldiers, respectively. We analyzed the copolymerization by UV–vis and CD spectroscopy in dilute solutions. Hereto, we gradually increased the fraction of the S-Nle-BTA sergeant in a mixture with a-BTA soldiers, keeping the total monomer concentration constant at 50 μM in MCH and monitored the changes in the molar circular dichroism (Δε). An amount of 4 mol % of the chiral S-Nle-BTA successfully biases the helical preference of the achiral a-BTA as illustrated by the appearance of a CD signal for the mixture (Figure 1A). This transfer of chirality underlines the fact that a-BTA and S-Nle-BTA coassemble. However, the molar circular dichroism Δε for the mixture (Δε = 28 L·mol–1·cm–1) is lower than values previously observed when mixing chiral, nonracemic BTAs with a-BTA (Δε = 40–45 L·mol–1·cm–1).28 In addition, two distinct patterns in the shape of the CD signal can be discerned for mixtures of S-Nle-BTA and a-BTA (Figure 1A): at fractions of sergeant up to ∼25 mol %, the CD spectrum shows a double Cotton effect with a maximum at 220 nm and a shoulder at ∼245 nm, similar to the shape obtained with helical columnar aggregates of a-BTA. However, at higher fractions of sergeant (>50 mol %), the CD spectrum changes gradually to the spectrum of pure sergeant, with a single maximum at 226 nm, indicative of the formation of dimers.31 No isodichroic point is observed, which indicates the existence of more than two optically active species. Since the contribution of S-Nle-BTA dimers to the CD signal is about double (Δε = 90 L·mol–1·cm–1) compared to the contribution of helical columnar aggregates of BTAs (Δε = 40–45 L·mol–1·cm–1), the large CD increase suggests that, at a higher fraction of sergeants, dimers of S-Nle-BTA coexist in solution with a-BTA dominated aggregates and contribute strongly to the CD signal.
Figure 1.

(A) CD spectra of the mixture a-BTA and S-Nle-BTA at ctot = 50 μM in MCH at 20 °C. (B–C) Temperature-dependent UV (B) and CD (C) spectra of solutions containing different ratios of S-Nle-BTA/a-BTA probed at λmax with ctot = 50 μM in MCH (cooling rate = 2 K·min–1). (D) Normalized CD and UV cooling curves probed at λmax with ctot = 50 μM in MCH at the ratio of S-Nle-BTA/a-BTA 1/9.
To further understand the mixing of S-Nle-BTA and a-BTA, variable-temperature UV and CD experiments were carried out on these mixtures (Figure 1B–1D). Interestingly, two different trends in the UV and CD cooling curves are observed. Up to 25 mol % of S-Nle-BTA, all the UV cooling curves have a similar nonsigmoidal shape indicating a cooperative mechanism of copolymerization as observed for the a-BTA homopolymer (Figure 1B). However, despite this similarity, a drop in the elongation temperature Te (about 13 °C) is observed upon addition of sergeants. This is indicative of either a decrease in the stability of the copolymers formed or a lower quantity of monomers participating in these cooperative aggregates. Interestingly, this drop of Te observed is much higher than the variation expected by the equivalent 25% dilution of a-BTA monomers (Figure 1B, green dotted trace). Above the threshold of 50 mol % of S-Nle-BTA, the cooling curves gradually become similar to the pure dimer curve with a sigmoidal shape (Figure 1C), indicating a change toward the predominance of dimers.
Theoretical Modeling of the Two-Component Copolymerization
To understand the origin of this strong decrease of Te, we devised a series of theoretical mass-balance models43−45 for different reaction schemes. With these models, we calculate the corresponding temperature-dependent degree of polymerization and compare them to the UV melting curves. In the first model, we include a dimerization reaction for S-Nle-BTA monomers and homopolymerization43−45 of a-BTA but no coassembly. In this case, a fraction of 50% S-Nle-BTA, and thus a dilution of the a-BTA of about 50%, is needed to obtain a 13 °C drop of Te (Figure S5). This is significantly more than the 25% S-Nle-BTA needed to observe experimentally a 13 °C drop of Te. In the second model, we extend the first model with a reaction where S-Nle-BTA binds to the a-BTA aggregates and prohibits further polymerization; i.e., S-Nle-BTA acts as a chain-capper of a-BTA polymers. Interestingly, this chain-capper model (Figure S9) results in a constant Te rather than a decrease of Te as compared to the pure dilution. In a third model, we allow S-Nle-BTA to intercalate into the a-BTA aggregates. Also in this case, the Te increases as compared to the pure dilution (Figure S14), reminiscent of the case of complete copolymerization.41,42 Thus, all three copolymerization models predict a change in Te different than the one we experimentally observed, indicating that none of these models are likely scenarios.
Because the absence of an isodichroic point in the CD spectra (Figure 1A) points to the presence of additional species, we next considered the possibility of a competitive interaction in which a-BTAs are partially sequestered from the supramolecular polymers. In this way, the total available concentration of a-BTA to form polymers is decreased by competitive formation of other species. Based on DFT calculations and DOSY NMR at 5 mM concentration (Figures S36–S37), S-Nle-BTA is a likely candidate to sequester a-BTA from polymerizing by the formation of stable species such as the a-BTA/S-Nle-BTA dimer or/and the trimer a-BTA/S-Nle-BTA/a-BTA. DFT calculations show that the trimer a-BTA/S-Nle-BTA/a-BTA and the homodimer S-Nle-BTA/S-Nle-BTA have comparable stability. In fact, extension of the first model (homopolymers of a-BTA and dimers of S-Nle-BTA) into a competitor model with the possibility of forming a-BTA/S-Nle-BTA dimers already shows some decrease in Te (Figure S18). When the model in addition also includes a-BTA/S-Nle-BTA/a-BTA trimers of the same stability as that of the dimers, a sufficient decrease in Te is found. This observation indicates that sequestration of a-BTA by the formation of dimers and/or trimers is a plausible option to explain the observed experimental decrease in Te (Figure S22).
To corroborate the presence of dimers/trimers sequestering a-BTA, we subsequently studied the variation of the CD shape with temperature for the mixture comprising 10 mol % of S-Nle-BTA (Figure 2A). The shape of the CD cooling curves changes when decreasing the temperature from 80 to 10 °C, indicative of the appearance of different species when cooling the solution. Interestingly, at 80 °C (temperature well above the Te of a-BTA dominated stacks), the CD signal exhibits a weak double Cotton effect with two maxima at 225 and 260 nm (red trace Figure 2A), which is similar to the shape obtained with 50 mol % of S-Nle-BTA sergeant added at room temperature (Figure 1A, cyan trace). At 20 °C, the CD spectrum shows a maximum at 220 nm and a shoulder at ∼245 nm (blue trace, Figure 2A) indicative of a-BTA dominated stacks. These results indicate that, at elevated temperature, stable chiral species (heterodimers/trimers containing both S-Nle-BTA and a-BTA) are formed; then upon cooling, S-Nle-BTA interacts with pre-existing P- and M-type helical columnar aggregates and biases their helicity to give rise to a large CD signal.
Figure 2.

CD spectra at the ratio of S-Nle-BTA/a-BTA 1/9 (A) and 3/1 (B) at ctot = 50 μM in MCH at different temperatures.
Further analysis of the UV and CD data shows that above 55 °C the variations of UV and CD curves differ significantly (gray area in Figure 1D). Therefore, we conclude that the aggregates grow before their helicity is biased by the sergeant. This observation suggests that the sergeant S-Nle-BTA is not part of the nucleus of the a-BTA dominated aggregates and that S-Nle-BTA prefers, at least at elevated temperatures, to form a dimer/trimer rather than to copolymerize with a-BTA.
Altogether, these results point to a copolymerization of a fraction of S-Nle-BTA with a-BTA next to the competitive formation of S-Nle-BTA dimers and short aggregates (dimers/trimers) that sequester part of the a-BTA monomers away from polymerization. The question that remains is whether S-Nle-BTA copolymerizes with a-BTA aggregates by intercalation or by chain-capping. To investigate this question and gain insights into the distribution of the different species, the composition of each of the aggregates, and the polymer lengths, we extended both the chain-capper model and the intercalation model with the competitive formation of a-BTA/S-Nle-BTA dimers and a-BTA/S-Nle-BTA/a-BTA trimers. In both models, next to the parameters for the dimer, trimer, and a-BTA homopolymer formations, two interdependent parameters describe the process: (i) the energy difference between forming a new a-BTA/S-Nle-BTA bond in P-type aggregates as compared to a new a-BTA/a-BTA bond (ΔHABpen) and (ii) the additional mismatch penalty for forming such an a-BTA/S-Nle-BTA bond in an M-type aggregate of the nonpreferred helicity instead of in a P-type aggregate (ΔHMpen). For the intercalation model, ΔHABpen was determined to be about 7 kJ mol–1 and the mismatch penalty ΔHMpen to be at least 2 kJ mol–1 to obtain a good agreement with the experimental CD melting curves (Figures 1C and 3A and section 3.8 in the Supporting Information (SI)). These thermodynamic parameters indicate that the mixing of S-Nle-BTA into P- or M-type aggregates is less favorable than the addition of a-BTA and that only a low quantity of S-Nle-BTA monomers (about 1%) is incorporated into the a-BTA stacks (Figure S25). Most likely, the steric hindrance of the branched ester side chains in combination with the unfavorable orientation of the ester moieties hampers the formation of the macrodipole moment which is critical for the coassembly leading to long stacks.31,34,46 Thus, only limited amounts of S-Nle-BTA monomers can be incorporated into a-BTA aggregates, and upon further addition of S-Nle-BTA monomers, the formation of discrete dimers/trimers species is favored. In addition, this model reveals the distribution of different species present at each S-Nle-BTA fraction as a function of temperature (Figure 3B–F). At a low fraction of S-Nle-BTA (<10%), the cooperative formation of a-BTA aggregates dominates over the formation of dimers and trimers, with a predominance of P-type a-BTA aggregates over the M-type. The largest quantity of P-type polymers is obtained with 10% S-Nle-BTA added, which is in agreement with the experimental observations. Moreover, with a higher fraction of S-Nle-BTA (>25%), the formation of a-BTA/S-Nle-BTA dimers and mainly a-BTA/S-Nle-BTA/a-BTA trimers compete with P aggregates. From 50% of S-Nle-BTA added, the trimer species prevail over the a-BTA aggregates resulting in a change in the shape of the CD signal. These data also predict that, at 75% of S-Nle-BTA added, the ratio of species formed does not vary over temperature. This result was experimentally confirmed by measuring the CD spectra of this mixture at variable temperature. Indeed, a constant shape of the CD spectra was obtained between 20 and 80 °C (Figure 2B).
Figure 3.

(A) CD cooling curves calculated with the intercalation model containing the competitive formation of dimers/trimers at different ratios of S-Nle-BTA/a-BTA. (B–F) Calculated concentrations of BTAs in the various species types (P helical aggregate, M helical aggregate, dimers and trimer) and concentration of free monomers as a function of the temperature for several fractions of sergeants S-Nle-BTA 4% (B), 10% (C), 25% (D), 50% (E), and 75% (F) at ctot = 50 μM (in the legend, A is a-BTA and Nle is S-Nle-BTA).
Surprisingly, the chain-capper model with ΔHABpen null and a mismatch penalty ΔHMpen of 5 kJ/mol resulted in very similar predicted CD cooling curves and speciation plots (Figures S31–S32). As a result, it is not possible to differentiate between intercalation of S-Nle-BTA within a-BTA polymers and chain-capping of a-BTA polymers with S-Nle-BTA by comparing the calculated CD curves. Therefore, the question remains whether S-Nle-BTA copolymerizes with a-BTA aggregates by intercalation or by chain-capping next to the competitive sequestration of a-BTA monomers by S-Nle-BTA. However, according to the model predictions, the two scenarios can be differentiated by comparing the lengths of the polymers obtained. The chain-capper model predicts a prompt and strong decrease in mean polymer length for small fractions of chain-capper, whereas the intercalation model predicts a weaker decrease in length that moreover only starts above 25% S-Nle-BTA (Figure 4A, sections 3.8 and 3.9 in the SI). Thus, to differentiate between these two hypotheses, the polymer length of a-BTA was analyzed by static light scattering (SLS) experiments as a function of the percentage S-Nle-BTA added.
Figure 4.
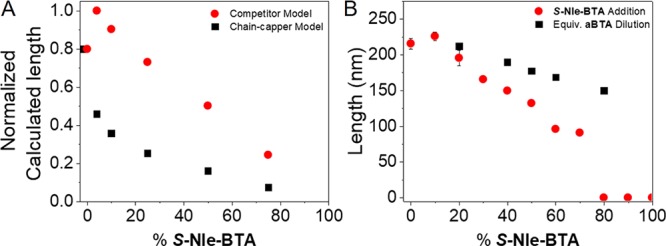
(A) Normalized calculated mean polymer length (number of monomers per stack) as a function of the S-Nle-BTA content predicted with the competitor model (red trace) and the chain-capper model (black trace). (B) Measured weight-average length of the supramolecular polymer as a function of the S-Nle-BTA content (red trace) compared with the corresponding dilution (black trace) determined via SLS at ctot = 0.5 mM in MCH at 20 °C.
Supramolecular Copolymerization Studied by Static Scattering Techniques
Solutions of various ratios of a-BTA/S-Nle-BTA at 0.5 mM in MCH were investigated by small-angle X-ray scattering (SAXS) and SLS.47 The weight-average length of the supramolecular copolymers as a function of the mixture’s composition was obtained by fitting the scattering curves to a cylinder model with a fixed radius of 6 nm (radius obtained from SAXS measurements Figure S38, Figure S39). Based on these fits, the interaction of S-Nle-BTA with a-BTA results in a decrease of the supramolecular fiber length (Figure 4B) but this reduction becomes only significant above 25% S-Nle-BTA added. Based on this weak decrease in polymer length and in accordance with the model predictions (Figure 4A, Figures S28–S30 and S33–S34), we conclude that S-Nle-BTA does not act as a chain-capper (which would result in an immediate decrease of the length) but rather intercalates into a-BTA dominated polymers.
To confirm the nature of the assemblies formed and whether S-Nle-BTA intercalates in a-BTA dominated polymers by the formation of hydrogen bonds via the amide or the ester, we studied FT-IR spectroscopy of 2 mM MCH solutions (Figure S40). The a-BTA aggregates and S-Nle-BTA dimers display different spectroscopic signatures characteristic of bonded and free amide, respectively.31,32 Upon addition of 10% S-Nle-BTA to a-BTA, the C=O stretch I exhibits two maxima at 1750 cm–1 (free ester C=O) and at 1735 cm–1 (bonded ester C=O) of similar intensity, which indicates the coexistence of free ester and bonded ester. This observation indicates the intercalation of S-Nle-BTA into a-BTA with formation of intermolecular hydrogen bonds via the amide in the a-BTA polymers, leaving the ester free.
Discussion
Based on the model prediction supported by spectroscopy and light scattering experiments, the entire copolymerization process can be summarized (Scheme 2). At high temperature, monomers are molecularly dissolved and a fraction forms chiral short aggregates, in the form of dimers S-Nle-BTA/S-Nle-BTA, a-BTA/S-Nle-BTA and trimer a-BTA/S-Nle-BTA/a-BTA, which reduce the pool of a-BTA monomers available for polymerization, thus resulting in considerably lower Te’s of polymer growth. Upon cooling, a-BTAs nucleate and grow into racemic cooperative 1D stacks as both P- and M-type, self-sorted from the chiral species. At lower temperature, small amounts of chiral S-Nle-BTAs interact with the a-BTA aggregates, resulting in more P-type aggregates than M-type aggregates and yielding an increase of the CD signal. However, the incorporation of S-Nle-BTA into the a-BTA dominated polymers competes with the formation of the short aggregates, so that only a small percentage (about 1%) of S-Nle-BTAs copolymerizes into a-BTA stacks resulting in a moderate CD signal (Scheme 2, left side). Most likely, the steric hindrance and the unfavorable orientation of the ester dipolar units hamper further incorporation of S-Nle-BTA, leading to the formation of discrete short species. Upon further addition of S-Nle-BTA, the sequestration of a-BTA by the competitive formation of dimers and trimers results in a decrease of the P- and M-type polymer length (Scheme 2, right side). With a large ratio of S-Nle-BTA, mostly S-Nle-BTA dimers assemble and contribute with high CD intensity.
Scheme 2. Schematic Representation of the Supramolecular Copolymers Formed by a-BTA and S-Nle-BTA at Room Temperature as a Function of the Composition.

For clarity, only the predominant species are represented.
With this detailed knowledge of the system in hand, we reanalyzed the first gelation study presented. The self-supporting properties of the a-BTA gel at 100 mM is lost by the presence of only 10 mol % of S-Nle-BTA (Figure S1), although such a small amount of S-Nle-BTA does not affect the molecular weight of the polymeric aggregate at 0.5 mM (Figure 4B). The decrease of polymer length is more pronounced at high concentration than at low concentration. Based on the modeling, the trend in polymer length goes in two regimes, and the transition between these two regimes depends on the concentration. Upon increasing the S-Nle-BTA/a-BTA ratio, the polymer length first increases and then decreases. First, at a low ratio of S-Nle-BTA content, S-Nle-BTA intercalates into a-BTA stacks. As a result, the helicity is biased in favor of P-type helical stacks, so that the concentration of monomers forming P stacks increases, and the length of these P-type helical stacks increases. Then, at a higher ratio of S-Nle-BTA, the a-BTA length decreases due to the decrease of a-BTA concentration, the destabilization of the a-BTA dominated stacks, and the formation of competitive short species. The transition between these two regimes is concentration dependent because the composition of the system varies with concentration. The concentrations of dimers and trimers scale much slower with the free monomer concentration than the concentration of polymer does. In other words, at low concentration, the homodimers, heterodimers, and trimers can sequester relatively more a-BTA than at high concentration. As a result, the quantity of S-Nle-BTA that intercalates into a-BTA stacks increases with concentration and the destabilization of the stacks is reached at lower content of S-Nle-BTA. Then, the transition between the two regimes of polymer length shifts to lower content of S-Nle-BTA at higher concentration. In particular, with 10% S-Nle-BTA content, at high concentration (the 100 mM gelation study) the length decreases, whereas at lower concentration (the 0.5 mM SLS study) the length slightly increases.
The model developed was then used to gain insights into the effect of a chain-capper or a competitor on the length of the polymers. The chain-capper interacts with the chain-end and thus inhibits further growth, while the competitor preferentially stabilizes the monomers and therefore pushes the thermodynamic equilibrium to depolymerization. The challenge is to understand the subtle differences between mixing the monomers with a strong competitor and mixing the monomers with a poor chain-capper. The polymer lengths were calculated as a function of the energy difference (ΔHA/Bdiff) between adding a chain-capper instead of an additional monomer to a polymer (Figure 5, red trace) and the energy difference between sequestrating the monomer into competitive species instead of polymerizing (Figure 5, green trace). At positive values of ΔHA/Bdiff, the polymerization of the monomers is more favorable than the chain-capping or competitive interaction. Therefore, the addition of a chain-capper or a competitor does not significantly affect the length of the fibers. Whereas at negative values of ΔHA/Bdiff, the chain-capping or competitive interaction is more favorable than the polymerization of the monomers. Consequently, the length of the polymers decreases with the chain-capper yielding shorter polymers than the competitor. At the ΔHA/Bdiff close to null, small variations in the interactions between the components lead to large changes in the polymer lengths. As a result, this plot also shows that a strong competitor (ΔHA/Bdiff < 0 kJ mol–1) yields polymer of similar lengths as a poor chain-capper (ΔHA/Bdiff > 0 kJ mol–1). This analysis confirms the issues encountered to design and characterize a chain-capper for cooperative supramolecular polymers and rationalizes the subtle differences between chain-capping and competitive aggregation pathways on polymer lengths.
Figure 5.

Calculated weight-average mean length of the cooperative P-polymers over the energy difference ΔHA/Bdiff with the addition of 10% (A) and 25% (B) of a chain-capper (red trace) and a competitor forming dimers and trimers (green trace) at 293 K, ctot = 50 μM.
Conclusions
In conclusion, the analysis of experimental data with a newly developed theoretical model allows us to unveil the details of a two-component supramolecular polymerization. The thermodynamic parameters, the distribution of species, the composition, and the lengths of polymers were calculated at different feed ratios of the two monomers. Moreover, the topical issue of chain-capper in cooperative supramolecular polymers was discussed and the model was used to predict the impact of the addition of a chain-capper and a competitor on the length of supramolecular polymers.
This work provides an alternative strategy to control the length of cooperative supramolecular polymers. Contrary to the studies on living supramolecular polymerization with the use of initiator,13−16 the length of the polymers is here controlled by a competitive pathway which traps the monomers into inactive species, i.e. species that do not participate in the polymerization process. In this context, new insights are given for the rational design of supramolecular monomers for control over the polymers length as well as contribute to a general understanding of two-component cooperative supramolecular polymerization. Hereby, a step forward is made for the construction of multicomponent supramolecular systems and adaptive materials.48 The work presented will also help to elucidate mixtures of amides and esters.28,49−51
Acknowledgments
This work was financially supported by The Netherlands Organization for Scientific Research (NWO-TOP PUNT Grant 10018944 and NWO-VENI Grant 722.017.003) and the Dutch Ministry of Education, Culture and Science (Gravity Program 024.001.035). G.V. thanks Lafayette de Windt and Mathijs Mabesoone for fruitful discussions. We thank Dr. Patrick Brocorens for kindly providing the coordinates of the MM/MD calculations of the S-Nle-BTA dimers. The ICMS Animation Studio is acknowledged for providing the artwork.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b09443.
Experimental procedures, characterization data and Figures S1–S40 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Flory P. J.Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, 1953. [Google Scholar]
- Odian G. G.Principle of Polymerization, 3d ed.; Wiley: 1991. [Google Scholar]
- Rudin A.The Elements of Polymer Science and Engineering, 3rd ed.; Elsevier: 2012. [Google Scholar]
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A. P. H. J.; Sijbesma R. P.; Meijer E. W. Supramolecular Polymerization. Chem. Rev. 2009, 109, 5687–5754. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Sijbesma R. P.; Beijer F. H.; Brunsveld L.; Folmer B. J. B.; Hirschberg J. H. K. K.; Lange R. F. M.; Lowe J. K. L.; Meijer E. W. Reversible Polymers Formed from Self-Complementary Monomers Using Quadruple Hydrogen Bonding. Science 1997, 278, 1601–1604. 10.1126/science.278.5343.1601. [DOI] [PubMed] [Google Scholar]
- Michelsen U.; Hunter C. A. Self-Assembled Porphyrin Polymers. Angew. Chem., Int. Ed. 2000, 39, 764.. [DOI] [PubMed] [Google Scholar]
- Castellano R. K.; Nuckolls C.; Rebek J. Jr. Reversibly-Formed Polymeric Capsules. Polym. News 2000, 25, 44–52. [Google Scholar]
- Folmer B. J. B.; Cavini E.; Sijbesma R. P.; Meijer E. W. Photo-Induced Depolymerization of Reversible Supramolecular Polymers. Chem. Commun. 1998, 1847–1848. 10.1039/a804797i. [DOI] [Google Scholar]
- Terech P.; Coutin A. Structure of a Transient Network Made up of Entangled Monomolecular Organometallic Wires in Organic Liquids. Effects of an Endcapping Molecule. Langmuir 1999, 15, 5513–5525. 10.1021/la990184z. [DOI] [Google Scholar]
- Yagai S.; Iwashima T.; Karatsu T.; Kitamura A. Synthesis and Noncovalent Polymerization of Self-Complementary Hydrogen-Bonding Supramolecular Synthons: N,N′-Disubstituted 4,6-diamino-pyrimidin-2(1H)-ones. Chem. Commun. 2004, 1114–1115. 10.1039/B401132E. [DOI] [PubMed] [Google Scholar]
- Ciferri A.Supramolecular polymers; CRC Press, 2005. [Google Scholar]
- Yang L.; Tan X.; Wang Z.; Zhang X. Supramolecular Polymers: Historical Development, Preparation, Characterization, and Functions. Chem. Rev. 2015, 115 (15), 7196–7239. 10.1021/cr500633b. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Stepanenko V.; Sugiyasu K.; Takeuchi M.; Wurthner F. Mechanism of Self-Assembly Process and Seeded Supramolecular Polymerization of Perylene Bisimide Organogelator. J. Am. Chem. Soc. 2015, 137, 3300–3307. 10.1021/ja511952c. [DOI] [PubMed] [Google Scholar]
- Kang J.; Miyajima D.; Mori T.; Inoue Y.; Itoh Y.; Aida T. A Rational Strategy for the Realization of Chain-Growth Supramolecular Polymerization. Science 2015, 347, 646–651. 10.1126/science.aaa4249. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Sugiyasu K.; Manna S.; Samitsu S.; Takeuchi M. Living Supramolecular Polymerization Realized Through a Biomimetic Approach. Nat. Chem. 2014, 6, 188–195. 10.1038/nchem.1849. [DOI] [PubMed] [Google Scholar]
- Endo M.; Fukui T.; Jung S. H.; Yagai S.; Takeuchi M.; Sugiyasu K. Photoregulated Living Supramolecular Polymerization Established by Combining Energy Landscapes of Photoisomerization and Nucleation-Elongation Processes. J. Am. Chem. Soc. 2016, 138, 14347–14353. 10.1021/jacs.6b08145. [DOI] [PubMed] [Google Scholar]
- Lortie F.; Boileau S.; Bouteiller L.; Chassenieux C.; Lauprêtre F. Chain Stopper-Assisted Characterization of Supramolecular Polymers. Macromolecules 2005, 38, 5283–5287. 10.1021/ma050168a. [DOI] [Google Scholar]
- Knoben W.; Besseling N. A. M.; Cohen Stuart M. A. Rheology of a Reversible Supramolecular Polymer Studied by Comparison of the Effects of Temperature and Chain Stoppers. J. Chem. Phys. 2007, 126, 024907 10.1063/1.2409296. [DOI] [PubMed] [Google Scholar]
- Pal D. S.; Kar H.; Ghosh S. Phototriggered Supramolecular Polymerization. Chem. - Eur. J. 2016, 22, 16872–16877. 10.1002/chem.201603691. [DOI] [PubMed] [Google Scholar]
- Smulders M. M. J.; Nieuwenhuizen M. M. L.; Grossman M.; Filot I. A. W.; Lee C. C.; de Greef T. F. A.; Schenning A. P. H. J.; Palmans A. R. A.; Meijer E. W. Cooperative Two-Component Self-Assembly of Mono- and Ditopic Monomers. Macromolecules 2011, 44, 6581–6587. 10.1021/ma201255q. [DOI] [Google Scholar]
- Karunakaran S. C.; Cafferty B. J.; Pelaez-Fernandez M.; Neselu K.; Schmidt-Krey I.; Fernandez-Niieves A.; Schuster G. B.; Hud N. V. Exquisite Regulation of Supramolecular Equilibrium Polymers in Water: Chain Stoppers Control Length, Polydispersity and Viscoelasticity. Polym. Chem. 2018, 9, 5268–5277. 10.1039/C8PY01146J. [DOI] [Google Scholar]
- Sanguramath R. A.; Nealey P. F.; Shenhar R. Quasi-Block Copolymers Based on a General Polymeric Chain Stopper. Chem. - Eur. J. 2016, 22, 10203–10210. 10.1002/chem.201600478. [DOI] [PubMed] [Google Scholar]
- Korevaar P. A.; Schaefer C.; de Greef T. F. A.; Meijer E. W. Controlling Chemical Self-Assembly by Solvent-Dependent Dynamics. J. Am. Chem. Soc. 2012, 134, 13482–13491. 10.1021/ja305512g. [DOI] [PubMed] [Google Scholar]
- Venkata Rao K.; Miyajima D.; Nihonyanagi A.; Aida T. Thermally Bisignate Supramolecular Polymerization. Nat. Chem. 2017, 9, 1133–1139. 10.1038/nchem.2812. [DOI] [PubMed] [Google Scholar]
- Anfinsen C. B. Principles that Govern the Folding of Protein Chains. Science 1973, 181 (4096), 223–230. 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Helmich F.; Lee C. C.; Nieuwenhuizen M. M. L.; Gielen J. C.; Christianen P. C. M.; Larsen A.; Fytas G.; Leclere P. E. L. G.; Schenning A. P. H. J.; Meijer E. W. Dilution-Induced Self-Assembly of Porphyrin Aggregates: A Consequence of Coupled Equilibria. Angew. Chem., Int. Ed. 2010, 49, 3939–3942. 10.1002/anie.201000162. [DOI] [PubMed] [Google Scholar]
- Cantekin S.; de Greef T. F. A.; Palmans A. R. A. Benzene-1,3,5-tricarboxamide: a Versatile Ordering Moiety for Supramolecular Chemistry. Chem. Soc. Rev. 2012, 41, 6125–6137. 10.1039/c2cs35156k. [DOI] [PubMed] [Google Scholar]
- Veld M. A. J.; Haveman D.; Palmans A. R. A.; Meijer E. W. Sterically Demanding Benzene-1,3,5-tricarboxamides: Tuning the Mechanisms of Supramolecular Polymerization and Chiral Amplification. Soft Matter 2011, 7, 524–531. 10.1039/C0SM00516A. [DOI] [Google Scholar]
- Smulders M. M. J.; Schenning A. P. H. J.; Meijer E. W. Insight into the Mechanisms of Cooperative Self-Assembly: The “Sergeants-and-Soldiers” Principle of Chiral and Achiral C3-Symmetrical Discotic Triamides. J. Am. Chem. Soc. 2008, 130, 606–611. 10.1021/ja075987k. [DOI] [PubMed] [Google Scholar]
- Kulkarni C.; Meijer E. W.; Palmans A. R. A. Cooperativity Scale: A Structure–Mechanism Correlation in the Self-Assembly of Benzene-1,3,5-tricarboxamides. Acc. Chem. Res. 2017, 50, 1928–1936. 10.1021/acs.accounts.7b00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmarchelier A.; Alvarenga B. G.; Caumes X.; Dubreucq L.; Troufflard C.; Tessier M.; Vanthuyne N.; Idé J.; Maistriaux T.; Beljonne D.; Brocorens P.; Lazzaroni R.; Raynal M.; Bouteiller L. Tuning the Nature and Stability of Self-Assemblies Formed by Ester Benzene 1,3,5-tricarboxamides: the Crucial Role Played by the Substituents. Soft Matter 2016, 12, 7824–7838. 10.1039/C6SM01601D. [DOI] [PubMed] [Google Scholar]
- Desmarchelier A.; Raynal M.; Brocorens P.; Vanthuyne N.; Bouteiller L. Revisiting The Assembly of Amino Ester-Based Benzene-1,3,5-tricarboxamides: Chiral Rods in Solution. Chem. Commun. 2015, 51, 7397–7400. 10.1039/C5CC01513H. [DOI] [PubMed] [Google Scholar]
- Hanabusa K.; Kawakami A.; Kimura M.; Shirai H. Remarkable Viscoelasticity of Organic Solvents Containing Trialkyl-1,3,5-benzenetricarboxamides and Their Intermolecular Hydrogen Bonding. Chem. Lett. 1997, 26, 191–192. 10.1246/cl.1997.191. [DOI] [Google Scholar]
- de Loos M.; van Esch J. H.; Kellogg R. M.; Feringa B. L. C-3-symmetric, Amino Acid Based Organogelators and Thickeners: A Systematic Study of Structure-Property Relations. Tetrahedron 2007, 63, 7285–7301. 10.1016/j.tet.2007.02.066. [DOI] [Google Scholar]
- Palmans A. R. A.; Vekemans J. A. J. M.; Hikmet R. A.; Fischer H.; Meijer E. W. Lyotropic Liquid-Crystalline Behavior in Disc-Shaped Compounds Incorporating the 3,3′-Di(acylamino)-2,2′-bipyridine Unit. Adv. Mater. 1998, 10, 873–876. . [DOI] [Google Scholar]
- Sakamoto A.; Ogata D.; Shikata T.; Urakawa O.; Hanabusa K. Large Macro-Dipoles Generated in a Supramolecular Polymer of N,N′,N″-Tris(3,7-dimethyloctyl)benzene-1,3,5-tricarboxamide in n-Decane. Polymer 2006, 47, 956–960. 10.1016/j.polymer.2005.12.019. [DOI] [Google Scholar]
- The concept of “Sergeant and Soldiers” experiment is here extended to two monomers with different side chains but a similar core.
- Green M. M.; Reidy M. P.; Johnson R. D.; Darling G.; O’Leary D. J.; Willson G. Macromolecular Stereochemistry: the Out-of-Proportion Influence of Optically Active Comonomers on the Conformational Characteristics of Polyisocyanates. The Sergeants and Soldiers Experiment. J. Am. Chem. Soc. 1989, 111, 6452–6454. 10.1021/ja00198a084. [DOI] [Google Scholar]
- Palmans A. R. A.; Vekemans J. A. J. M.; Havinga E. E.; Meijer E. W. Sergeants-and-Soldiers Principle in Chiral Columnar Stacks of Disc-Shaped Molecules with C3 Symmetry. Angew. Chem., Int. Ed. Engl. 1997, 36, 2648–265. 10.1002/anie.199726481. [DOI] [Google Scholar]
- Prins L. J.; Timmerman P.; Reinhoudt D. N. Amplification of Chirality: The “Sergeants and Soldiers” Principle Applied to Dynamic Hydrogen-Bonded Assemblies. J. Am. Chem. Soc. 2001, 123, 10153–10163. 10.1021/ja010610e. [DOI] [PubMed] [Google Scholar]
- Das A.; Vantomme G.; Markvoort A. J.; ten Eikelder H. M. M.; Garcia-Iglesias M.; Palmans A. R. A.; Meijer E. W. Supramolecular Copolymers: Structure and Composition Revealed by Theoretical Modeling. J. Am. Chem. Soc. 2017, 139, 7036–7044. 10.1021/jacs.7b02835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Eikelder H. M. M.; Adelizzi B.; Palmans A. R. A.; Markvoort A. J. Equilibrium Model for Supramolecular Copolymerizations. J. Phys. Chem. B 2019, 123 (30), 6627–6642. 10.1021/acs.jpcb.9b04373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Eikelder H. M. M.; Markvoort A. J.; de Greef T. F. A.; Hilbers P. A. J. An Equilibrium Model for Chiral Amplification in Supramolecular Polymers. J. Phys. Chem. B 2012, 116, 5291–5301. 10.1021/jp300622m. [DOI] [PubMed] [Google Scholar]
- Markvoort A. J.; ten Eikelder H. M. M.; Hilbers P. A. J.; de Greef T. F. A.; Meijer E. W. Theoretical Models of Nonlinear Effects in Two-Component Cooperative Supramolecular Copolymerizations. Nat. Commun. 2011, 2, 509. 10.1038/ncomms1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korevaar P. A.; Grenier C.; Markvoort A. J.; Schenning A. P. H. J.; de Greef T. F. A.; Meijer E. W. Model-Driven Optimization of Multicomponent Self-Assembly Processes. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17205–17210. 10.1073/pnas.1310092110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filot I. A. W.; Palmans A. R. A.; Hilbers P. A. J.; van Santen R. A.; Pidko E. A.; de Greef T. F. A. Understanding Cooperativity in Hydrogen-Bond-Induced Supramolecular Polymerization: A Density Functional Theory Study. J. Phys. Chem. B 2010, 114 (43), 13667–13674. 10.1021/jp1072928. [DOI] [PubMed] [Google Scholar]
- Knoben W.; Besseling N. A. M.; Cohen Stuart M. A. Chain Stoppers in Reversible Supramolecular Polymer Solutions Studied by Static and Dynamic Light Scattering and Osmometry. Macromolecules 2006, 39, 2643–2653. 10.1021/ma0518914. [DOI] [Google Scholar]
- Vantomme G.; Meijer E. W. The Construction of Supramolecular Systems. Science 2019, 363 (6434), 1396–1397. (49) Desmarchelier, A.; Caumes, X.; Raynal, M.; Vidal-Ferran, A.; van Leeuwen, P.W.N.M.; Bouteiller, L. Correlation between the selectivity and the structure of an asymmetric catalyst built on a chirally amplified supramolecular helical scaffold. J. Am. Chem. Soc. 2016, 138, 4908-4916. (50) Zimbron, J.M.; Caumes, X.; Li, Y.; Thomas, C.M.; Raynal, M.; Bouteiller, L. Real-time control of the enantioselectivity of a supramolecular catalyst allows selecting the configuration of consecutively formed stereogenic centers. Angew. Chem. Int. Ed. 2017, 56, 14016-14019. (51) Li, Y.; Caumes, X.; Raynal, M.; Bouteiller, L. Modulation of catalyst enantioselectivity through reversible assembly of supramolecular helices. Chem. Commun. 2019, 55, 2162-2165. 10.1126/science.aav4677. [DOI] [PubMed] [Google Scholar]
- Desmarchelier A.; Caumes X.; Raynal M.; Vidal-Ferran A.; van Leeuwen P. W. N. M.; Bouteiller L. Correlation between the selectivity and the structure of an asymmetric catalyst built on a chirally amplified supramolecular helical scaffold. J. Am. Chem. Soc. 2016, 138, 4906–4916. 10.1021/jacs.6b01306. [DOI] [PubMed] [Google Scholar]
- Zimbron J. M.; Caumes X.; Li Y.; Thomas C. M.; Raynal M.; Bouteiller L. Real-time control of the enantioselectivity of a supramolecular catalyst allows selecting the configuration of consecutively formed stereogenic centers. Angew. Chem., Int. Ed. 2017, 56, 14016–14019. 10.1002/anie.201706757. [DOI] [PubMed] [Google Scholar]
- Li Y.; Caumes X.; Raynal M.; Bouteiller L. Modulation of catalyst enantioselectivity through reversible assembly of supramolecular helices. Chem. Commun. 2019, 55, 2162–2165. 10.1039/C8CC09819K. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.