

Summary
Mounting evidence implicates hybrid insulin peptides (HIPs) as important autoantigens in the development of type 1 diabetes (T1D). These fusion peptides formed between insulin and other pancreatic beta cell‐derived peptides contain non‐genomically encoded amino acid sequences, making them plausible targets for autoreactive T cells in T1D. HIPs are detectable by mass spectrometry in human and murine islets and are targeted by diabetes‐inducing T cells in non‐obese diabetic mice as well as by T cells isolated from the residual pancreatic islets of human organ donors with T1D. The discovery of HIPs comes with numerous new challenges, as well as opportunities to study the pathogenesis of T1D. Here we review the original discovery of HIPs and describe recent studies investigating the role of HIP‐reactive T cells in the development of diabetes. We also discuss potential mechanisms that may be responsible for the generation of HIPs in beta cells and describe challenges that need to be addressed in the field of mass spectrometry to enable the discovery of new HIPs. The identification of these potentially disease‐driving antigens in T1D is of key interest to the field as it may provide new tools to predict, prevent and potentially reverse the disease.
Keywords: antigens, autoimmunity, diabetes, epitopes, peptides
Hybrid Insulin Peptides (HIPs), a newly‐discovered class of autoantigens in Type 1 Diabetes (T1D), are targeted by diabetes‐triggering CD4 T cells in mice and by CD4 T cells isolated from residual pancreatic islets of organ donors with T1D. Furthermore, mass spectrometric analyses have verified the presence of HIPs in human and murine islets. This article describes the discovery of HIPs and HIP‐reactive T cells and discusses potential mechanisms that could lead to the formation of HIPs in beta cells.
Introduction
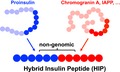
Post‐translational protein modifications alter the chemical makeup of proteins and have been implicated in the loss of self‐tolerance in various autoimmune diseases 1, 2, 3, 4, 5, 6, 7. In this context, the recent discovery of hybrid insulin peptides 8 (HIPs) as post‐translationally modified CD4 T cell epitopes in type 1 diabetes (T1D) has gained significant interest. HIPs form through a covalent cross‐linking reaction between the C‐terminal carboxylic acid group of proinsulin fragments and the N‐terminal amine group of other peptides (see Fig. 1). This leads to the formation of new linear peptides containing non‐genomically encoded amino acid sequences that can be presented to autoreactive T cells in context of a major histocompatibility complex (MHC). The presentation of such post‐translationally spliced peptides may provide a plausible explanation for the break of self‐tolerance in T1D. More than a decade ago, it was shown that the proteasome generates spliced peptides by joining non‐contiguous peptide fragments via reverse proteolysis, generating neoantigens that are presented by MHC class I and recognized by tumour‐infiltrating CD8 T cells 9, 10, 11. The discovery of spliced peptides and HIPs illustrates the previously unrecognized complexity of the proteome and the ever‐expanding boundaries of antigen diversity 12. In this article, we will review the growing number of manuscripts currently available that describe HIPs and their role as autoantigens and discuss the possible mechanisms that may lead to the formation of hybrid peptides in vivo.
Figure 1.
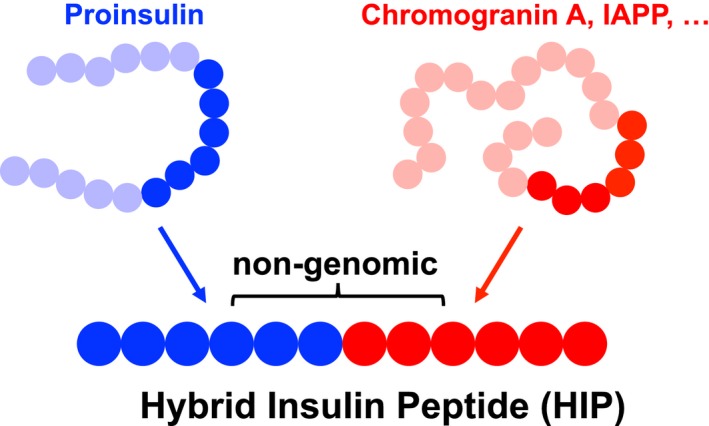
Hybrid peptide definition. Hybrid peptides form if peptides derived from two separate parent proteins are linked through a peptide bond. The resulting peptide contains a non‐genomically encoded amino acid sequence.
Discovery of HIPs utilizing CD4 T cell clones
A key reagent used in the discovery of HIPs was the CD4 T cell clone BDC‐2·5, which triggers diabetes in the non‐obese diabetic (NOD) mouse model of T1D 13, 14, 15. To identify the native beta cell antigen recognized by BDC‐2·5, a proteomic strategy was utilized that involved mass spectrometric analyses of antigenic beta cell tumour extracts 16 obtained from NOD‐rat insulin promoter T antigen (RIP‐Tag) mice 17. The tumour extracts were enriched for beta cell granules and subsequently fractionated by chromatography. The presence of antigen in fractions was then verified using the BDC‐2·5 clone. Proteins present in the fractions were then identified through mass spectrometric analyses. Previous studies in which synthetic peptide mimotopes were screened for antigenicity revealed that the amino acid motif WxRM on the C‐terminal side of the peptide was requisite for stimulation of BDC‐2·5 17, 18, 19. Of the proteins identified in peak antigenic fractions, only chromogranin A contained this motif. However, the native chromogranin A sequence (EDKRWSRMD) that contains the motif did not elicit a response by BDC‐2·5. Instead, the chromogranin A peptide WE14, a naturally occurring peptide cleavage product which starts with the WSRM sequence on its N‐terminus, was stimulatory, but only at supraphysiological concentrations. Mass spectrometric analyses verified the presence of WE14 in chromatographic fractions of beta cell tumour lysates 8. However, detectable ion intensities of WE14 in these fractions, which indicate relative peptide abundances of this peptide, did not correlate with the BDC‐2·5 antigen distribution profile over these fractions. In contrast, the mass spectral distribution of various C‐peptide fragments over these fractions matched the BDC‐2·5 antigen distribution profile 8. While this suggested that insulin C‐peptide may be a ligand for BDC‐2·5, this peptide was not recognized by the T cell clone.
These observations led to the hypothesis that a fragment of C‐peptide in combination with the N‐terminal region of WE14 may be a natural ligand for BDC‐2·5. To determine if such fusion peptides could be recognized by BDC‐2·5, a peptide cross‐linking strategy was utilized to synthesize a panel of HIPs containing insulin fragments linked via their C‐terminus to N‐terminal sequences of WE14 and other naturally occurring peptides. Upon screening this peptide library, one HIP‐sequence (2·5HIP) was not only recognized by BDC‐2·5, but also by two additional WE14‐reactive T cell clones (BDC‐10·1 and BDC‐9·46). This peptide, which was even more antigenic than established peptide mimotopes, was recognized by the three T cell clones at nanomolar concentrations. To strengthen the HIP hypothesis, the peptide library was screened with additional NOD CD4+ T cell clones. BDC‐6·9 and BDC‐9·3 20, 21, 22, two additional diabetogenic CD4 T cell clones, responded to a HIP sequence (6·9HIP) formed between an insulin C‐peptide fragment on the N‐terminal side linked to a naturally occurring peptide of islet amyloid polypeptide (IAPP). Additional evidence supporting chromogranin A and IAPP as a source of the antigenic ligands for BDC‐2·5 and BDC‐6·9 included data demonstrating that pancreatic islets of chromogranin A or IAPP‐deficient mice could not be used to elicit a response by BDC‐2·5 or BDC‐6·9, respectively 17, 22, 23. Furthermore, chromogranin A‐deficient NOD mice were protected from diabetes 23.
Identification of HIP‐reactive T cells in residual islets of organ donors with T1D
The existence of HIP‐reactive T cells that trigger diabetes in NOD mice led to a search for HIP‐reactive T cells in human subjects with type 1 diabetes. In a collaborative effort involving three laboratories, CD4 T cell lines and clones obtained from the residual pancreatic islets of organ donors with T1D were screened against a panel of 16 putative HIPs 8, 24. Two T cell clones, isolated from an organ donor, responded to the same HIP formed by the fusion of a proinsulin C‐peptide fragment to IAPP2. In addition, a T cell line isolated from a T1D organ donor responded to a HIP formed by the fusion of a proinsulin C‐peptide fragment linked to the sequence of a natural cleavage product of neuropeptide Y. In a follow‐up study by Babon et al. 24, additional HIP‐reactive T cells were identified. A T cell line grown from the residual islets of a T1D donor recognized a HIP formed between the previously mentioned C‐peptide fragment and the insulin A‐chain. Another T cell line isolated from a different organ donor recognized a HIP formed between the same C‐peptide fragment and a natural cleavage product of IAPP (IAPP1). Another T cell line isolated from this donor responded to a HIP formed between the same C‐peptide fragment and IAPP2. This sequence was also recognized by the two T cell clones described above 8.
Discovery of HIPs utilizing mass spectrometry
To establish the role of HIPs as naturally‐occurring T cell ligands, their presence in beta cell extracts had to be verified. Utilizing mass spectrometric analyses on islet cell extracts of both NOD mice 8, 22, 25 and non‐autoimmune BALB/c mice, the presence of both peptides, 2.5HIP and 6·9HIP, was confirmed. More recently, it was also verified that HIPs are present in primary human islets from non‐diabetic organ donors 25. In addition, Gonzales‐Duque et al. reported on the identification of several putative hybrid peptides presented by human leucocyte antigen (HLA) class I on the surface of a human beta cell line 26.
Efforts to identify HIPs and the closely related class of spliced peptides can be mutually beneficial. Generation of spliced peptides by the proteasome and presentation of these peptides by MHC class I molecules has now been studied for more than a decade 27, but reports on efforts to broadly identify spliced peptides have only recently started to emerge. Because the HIPs being studied in the context of T1D are composed of fragments of peptides enclosed in beta cell secretory granules, they may be formed by a different mechanism (possible mechanisms of HIP formation will be discussed later in this review). However, despite formation by potentially different mechanisms and differences in the donor proteins involved, both HIPs and proteasome‐spliced peptides involve the fusion of non‐contiguous amino acid sequences. Thus, the confident identification of HIPs and spliced peptides by mass spectrometry poses a unique set of challenges. The standard approach to assigning sequence interpretations for peptide tandem mass spectrometry (MS/MS) spectra is to compare each observed spectrum to the spectra predicted for peptides represented in a protein sequence database 28. A key challenge in identifying HIPs and spliced peptides by mass spectrometry is that these sequences are not represented in conventional databases. Custom databases including all possible HIPs/spliced peptides would be prohibitively large from a computational standpoint and would probably produce a large number of false identifications in standard searches.
In a recent study 29, Liepe et al. sought to estimate the frequency of spliced peptides in the HLA class I immunopeptidome of human cell lines (a lymphoblastoid line, a lymphoid line and primary human fibroblasts). In this study, the size of the protein database was greatly reduced by only considering possible cis‐spliced peptide sequences: those sequences that arise from splicing of two regions from the same protein molecule by removal of an intervening sequence. Trans‐spliced peptides, containing peptides from two separate parent proteins, were not considered. Because the biological samples being analysed were HLA class I‐eluted peptides, sequences were limited to 9–12 amino acids in length, the normal length for peptides bound to HLA class I. To further limit the database, only spliced peptides arising from joining of two sequences separated by a maximum of 25 intervening amino acid residues were included. Finally, only those peptides with an accurate mass that could be matched to a peptide in the sample were included in the final database.
The goal of the work by Liepe et al. was to assess the contribution of cis‐spliced peptides to the total HLA class I immunopeptidome. Others have taken a more focused approach aimed at identifying a subset of spliced peptides. Gonzales‐Duque et al., for example, rationally limited database size in order to identify HLA class I‐eluted peptides from a human beta cell line that may be CD8 T cell epitopes in T1D 26. In this study, both cis‐spliced and trans‐spliced sequences were considered. Database size was first limited by considering only a small list of proteins that are known or putative beta cell antigens. Then, based on previously reported findings 30, only those peptide sequences predicted to be mechanistically preferred N‐ and C‐terminal precursors for proteasome‐mediated peptide splicing were combined in silico to generate a spliced peptide database. In an analysis by Wiles et al. 25, primary mouse and human islets were analysed by mass spectrometry to identify HIPs. This study was focused on establishing rigorous validation guidelines, thus a limited HIP database design was used in which only insulin C‐peptide fragments were considered as N‐terminal precursors and a limited number of potential protein cleavage products were considered as C‐terminal precursors. In this study, a limited number of HIPs could be identified with high confidence in mouse and human islets.
As an alternative to searching large databases, other groups have used de‐novo sequencing‐based 31, 32, 33 approaches to identify fragmentation spectra corresponding to spliced peptides. Mylonas et al. performed de‐novo sequencing of MS/MS spectra to identify the sequences of peptides eluted from MHC class I molecules isolated from various human tissues and cell lines (melanoma tissues, B cell lines and fibroblast cells) 34. Sequences identified de novo were matched to genomically‐encoded protein sequences in databases. Sequences that could not be matched were then examined using an in‐house alignment tool to determine if they could be explained by a cis‐splicing event. Candidate spliced peptide sequences were added to a custom database, and data were then searched against this database using a standard search algorithm. Faridi et al. used a similar workflow to identify both cis‐ and trans‐spliced peptides 35.
A novel algorithm for identifying spliced peptides was recently reported by Rolfs et al. 36. This algorithm, called Neo‐Fusion, employed a unique way of searching traditional databases, thus eliminating the need for the generation and searching of large custom spliced peptide databases. The first stage of the Neo‐Fusion algorithm disregards the mass of the observed peptide. Instead, from a standard protein database, an N‐terminal ion database and a C‐terminal ion database are generated (e.g. a b‐ion and a y‐ion database, respectively, for data generated using collision‐induced dissociation). Ions in the observed spectrum are compared to predicted ions in these databases to identify N‐ and C‐terminal parent peptides. The two parent peptides sequences and every possible truncation of each are then spliced in silico to identify hypothetical splice products with the appropriate mass. Candidates that pass this stage are used to generate a new database, which is then used in a traditional database search.
After potential spliced peptides have been identified in a sample, validation of these matches is critical. As pointed out in a recent review 37, validation in peptidomics studies, where identifications are often based on confidence in an interpretation of a single spectrum, is more critical than in traditional proteomics studies where identification of a protein is based on the detection of several spectra corresponding to different peptides of the same protein. Furthermore, because hybrid peptides often differ from genomically encoded sequences by only a few amino acid residues, a single spectrum can in some cases be matched with similar confidence to both a spliced and a non‐spliced sequence 25, 34, 36. Reliable, broadly implementable protocols for identifying spliced and hybrid peptides using tandem mass spectrometry data must be established. This need is highlighted by the highly disparate results reported in two of the works discussed here: Liepe et al. estimated that cis‐spliced peptides constitute more than 30% of the peptide sequences displayed on the cell surface by MHC class I molecules 29, while Mylonas et al. estimated that this value should be 2–6% 34. In the recent report by Wiles et al. on the identification of HIPs in mouse and human islets 25, several strict criteria for validating mass spectrometry‐based HIP identifications were proposed, allowing a high degree of confidence in the identification of HIPs. More work in this area is needed in order to enable the confident identification of HIPs and spliced peptides by mass spectrometry.
HIP‐reactive T cells as disease biomarkers
While HIP‐reactive T cells have been identified in residual islets of T1D organ donors, a compelling question is whether they can serve as biomarkers of the disease process. To study this question, Baker et al. used MHC class II tetramer reagents specific for 2·5HIP and 6·9HIP‐reactive T cells to analyse whole pancreas and isolated islets of NOD mice for the presence of HIP tetramer‐positive (tet+) CD4 T cells 38. The authors observed significant increases in the percentages of 2·5HIP and 6·9HIP tet+ cells in the pancreata of NOD mice at 10 weeks of age. In contrast, this significant increase could not be observed at 10 weeks when using tetramer reagents that stain T cells specific for two insulin B:9–23 mimotope sequences. Upon the direct analysis of isolated islets obtained from 6‐ to 20‐week‐old prediabetic NOD mice, it was determined that the abundance of 2·5HIP tet+ T cells averaged approximately 4·5% of the total infiltrating CD4 T cells, exceeding the abundance of 6·9HIP tet+ cells, which averaged approximately 1%. Furthermore, 2·5HIP‐specific T cells in the pancreatic lymph nodes acquired an antigen‐experienced phenotype (CD44high and CD62Llow). Both 2·5HIP and 6·9HIP tet+ CD4 T cells were detectable in the peripheral blood of NOD mice with increasing frequency as the mice aged. These data imply that 2·5HIP and 6·9HIP tet+ cells may serve as indicators for the disease process.
In a study by Ito et al., a NOD mouse with a mutated MHC class II‐associated invariant chain peptide (CLIP) segment was generated and used to study the infiltration of islets by HIP‐specific T cells 39. These mice had a significantly decreased incidence of diabetes, and the authors determined that islet infiltration was diminished in this model. In particular, the percentage of islet‐infiltrating CD4 T cells that were 6·9HIP tet+ was significantly reduced in 8‐, 9‐ and 12‐week‐old mice, and the percentage of 2·5HIP tet+ cells was reduced at 8 but not at 9 or 12 weeks of age. In contrast, the percentage of insulin B:12–20 and B:13–21 tet+ cells was not significantly different at these time‐points. The reduction of HIP tet+ cells was attributed to a decreased presentation of HIPs by antigen‐presenting cells as a result of the CLIP mutation. These observations further substantiate the disease‐driving role of HIP‐reactive CD4 T cells in the development of diabetes in NOD mice and their potential role as biomarkers for disease.
Mechanistic considerations of hybrid peptide formation
Hybrid peptides form through a covalent cross‐linking reaction between a C‐terminal carboxylic acid group of one peptide and the N‐terminal amine group of another peptide (see Fig. 1). Under physiological conditions, the reaction that leads to the formation of such a covalent bond does not take place spontaneously. Instead, the carboxylic acid group needs to be chemically activated prior to the formation of a new peptide bond. There are several putative mechanisms that could allow such a reaction to take place in vivo (Fig. 2).
Figure 2.
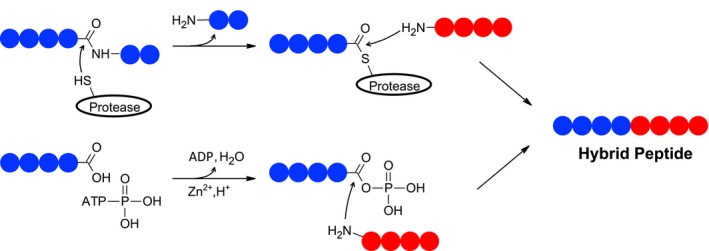
Potential mechanisms of hybrid peptide formation. A prerequisite for the formation of a hybrid peptide is the chemical activation of a peptide's C‐terminal carboxylic acid group prior to the formation of a new peptide bond. Such activation can occur through the formation of an intermediate between a peptide's C‐terminal carboxylic acid group and (a) a protease (thioester and ester) or (b) a phosphate‐group (mixed phosphoanhydrite).
One mechanism of HIP formation may occur through reversed proteolysis (Fig. 2a), as previously implicated in the generation of spliced peptides by the proteasome 9, 10, 27. During a reversed proteolytic reaction, a peptide bond is cleaved by a protease. In the case of non‐metalloproteases or non‐aspartyl proteases, this reaction leads to the formation of an activated intermediate between the functional group of the protease and the carboxylic acid group of the targeted peptide bond. The activated intermediate can react with the N‐terminal amine of another peptide leading to the formation of a hybrid peptide. The beta cell secretory granules, which contain several enzymes with proteolytic activity, are a possible location for HIP formation. Proteases inside these granules include dipeptidyl peptidase 2 40, carboxypeptidases E 40, 41 and N, cathepsins B, D, F and L 40, prohormone convertases I/III and II 42 and tripeptidyl‐peptidase 1 40. While the specificities of some of these proteases are poorly characterized, some, such as the prohormone convertases and carboxypeptidase E, which play important roles in processing of proinsulin, are known to target specific amino acid motifs. For example, prohormone convertase is an endopeptidase that cleaves proteins at the C‐terminal side of basic residue pairs, and carboxypeptidase E subsequently removes the exposed basic residues 43. Because of their specificities, it is unlikely that these proteases would be capable of generating the HIP sequences that have been characterized thus far. Because carboxypeptidase N is a metalloprotease 44, it does not form a reactive intermediate with the N‐terminal portion of a peptide substrate and would therefore not generate a spliced sequence by the same mechanism described for proteasome‐mediated splicing. However, it could potentially generate a spliced sequence by a similar mechanism. In addition to beta cell granules, crinophagic bodies 45 or lysosomes could also be a location of HIP formation. Unanue et al. identified several putative HIPs by mass spectrometry in beta cell extracts that were enriched for crinophagic bodies through differential centrifugation 45. A factor that may contribute to hybrid peptide formation in beta cell granules or crinophagic bodies is the high local peptide concentrations which can drive the peptide splicing reaction through molecular crowding 46. The identification of a proteolytic enzyme that participates in the formation of HIPs may provide a therapeutic target for T1D. Ablation or inhibition of such an enzyme would interrupt the HIP formation process, thereby depleting beta cells of potential disease driving T cell epitopes.
Another plausible mechanism of HIP formation may involve a non‐enzymatic reaction utilizing adenosine triphosphate (ATP) as a reagent 47. Secretory vesicles of beta cells and other tissues contain high concentrations of ATP 48 which, in the presence of various divalent metal ions, has been shown to activate carboxylic acid groups through the formation of a mixed phosphoanhydride intermediate 47 (Fig. 2b). This chemical activation of carboxylic acid groups may, in the presence of other peptides, lead to the formation of hybrid peptides (Fig. 2b). This non‐enzymatic reaction utilizes divalent metal ions, such as Be2+, Ni2+, Co2+, Mn2+ and Zn2+, as catalysts. The reaction's yield peaks at pH 5.2 47, which is close to the pH reported in beta cell granules 49, 50. The beta cell granules, which contain high concentrations of ATP and Zn2+, as well as a slightly acidic environment, could therefore, in the presence of high peptide concentrations, provide an ideal environment of HIP formation through this mechanism. Interestingly, the divalent metal ions that are known to catalyze this reaction have been associated with other medical conditions, including berylliosis as well as cobalt, nickel and manganese allergies.
Proteasome‐generated spliced peptides are presented by MHC class I and recognized by CD8 T cells and have been implicated in anti‐tumour immunity 27. In contrast, HIPs have thus far only been studied as MHC class II‐restricted antigens for CD4 T cells. Such presentation could be mediated by antigen‐presenting cells, such as dendritic cells in the islet or pancreatic lymph nodes that have taken up beta cell material, potentially in the form of dead beta cells or secreted HIPs. However, it has also been demonstrated that mouse 51, 52 – and, more recently, human 53 – beta cells can express MHC class II. Thus, it is possible that HIPs are directly presented on MHC class II molecules to CD4 T cells by beta cells themselves. The possibility that HIPs are presented on MHC class I molecules at the surface of beta cells and could be recognized by pathogenic CD8 T cells also merits consideration, particularly as hyperexpression of MHC class I by beta cells is a hallmark of T1D in humans 54.
Conclusions
The identification of hybrid insulin peptides as target epitopes for diabetogenic CD4 T cells involved several decades of work. BDC‐2·5 was among the first HIP‐reactive T cell clones isolated from diabetic NOD mice and was the key reagent making this discovery possible. Advances in mass spectrometry have also contributed to the discovery of HIPs. Ongoing efforts involve the identification of new HIPs in human islets, characterization of HIP‐reactive T cells as biomarkers for disease, understanding of the mechanism of HIP formation and the development of antigen‐specific tolerance induction strategies utilizing HIPs. Efforts also need to be made to determine if hybrid peptides form in other tissues, as the results of such work could provide new insights into various autoimmune diseases.
Author contributions
Both authors listed have made a substantial, direct and intellectual contribution to the work and approved it for publication.
Disclosures
The authors declare no conflicts of interest.
Acknowledgements
T. D.: ADA Pathway to Stop Diabetes (1‐15‐ACE‐14) NIDDK R01 (1R01DK119529‐01); T. A. W.: JDRF Postdoctoral Fellowship (3‐PDF‐2019‐746‐A‐N).
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Historical and new insights into pathogenesis of type 1 diabetes. Clinical and Experimental Immunology 2019, 198: 292–293.
Birth and coming of age of islet autoantibodies. Clinical and Experimental Immunology 2019, 198: 294–305.
Immune cell trafficking to the islets during type 1 diabetes. Clinical and Experimental Immunology 2019, 198: 314–325.
Islet‐immune interactions in type 1 diabetes: the nexus of beta cell destruction. Clinical and Experimental Immunology 2019, 198: 326–340.
References
- 1. Valesini G, Gerardi MC, Iannuccelli C, Pacucci VA, Pendolino M, Shoenfeld Y. Citrullination and autoimmunity. Autoimmun Rev 2015; 14:490–7. [DOI] [PubMed] [Google Scholar]
- 2. McGinty JW, Marre ML, Bajzik V, Piganelli JD, James EA. T cell epitopes and post‐translationally modified epitopes in type 1 diabetes. Curr Diab Rep 2015; 15:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nguyen H, James EA. Immune recognition of citrullinated epitopes. Immunology 2016; 149:131–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harbige J, Eichmann M, Peakman M. New insights into non‐conventional epitopes as T cell targets: the missing link for breaking immune tolerance in autoimmune disease? J Autoimmun 2017; 84:12–20. [DOI] [PubMed] [Google Scholar]
- 5. Sollid LM. The roles of MHC class II genes and post‐translational modification in celiac disease. Immunogenetics 2017; 69:605–16. [DOI] [PubMed] [Google Scholar]
- 6. James EA, Pietropaolo M, Mamula MJ. Immune recognition of beta‐cells: neoepitopes as key players in the loss of tolerance. Diabetes 2018; 67:1035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mannering SI, Di Carluccio AR, Elso CM. Neoepitopes: a new take on beta cell autoimmunity in type 1 diabetes. Diabetologia 2019; 62:351–6. [DOI] [PubMed] [Google Scholar]
- 8. Delong T, Wiles TA, Baker RL et al Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016; 351:711–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanada K, Yewdell JW, Yang JC. Immune recognition of a human renal cancer antigen through post‐translational protein splicing. Nature 2004; 427:252–6. [DOI] [PubMed] [Google Scholar]
- 10. Vigneron N, Stroobant V, Chapiro J et al An antigenic peptide produced by peptide splicing in the proteasome. Science 2004; 304:587–90. [DOI] [PubMed] [Google Scholar]
- 11. Liepe J, Ovaa H, Mishto M. Why do proteases mess up with antigen presentation by re‐shuffling antigen sequences? Curr Opin Immunol 2018; 52:81–6. [DOI] [PubMed] [Google Scholar]
- 12. Mannering SI, So M, Elso CM, Kay TWH. Shuffling peptides to create T‐cell epitopes: does the immune system play cards? Immunol Cell Biol 2018; 96:34–40. [DOI] [PubMed] [Google Scholar]
- 13. Haskins K, Portas M, Bradley B, Wegmann D, Lafferty K. T‐lymphocyte clone specific for pancreatic islet antigen. Diabetes 1988; 37:1444–8. [DOI] [PubMed] [Google Scholar]
- 14. Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic islet‐specific T‐cell clones from nonobese diabetic mice. Proc Natl Acad Sci USA 1989; 86:8000–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haskins K. Pathogenic T‐cell clones in autoimmune diabetes: more lessons from the NOD mouse. Adv Immunol 2005; 87:123–62. [DOI] [PubMed] [Google Scholar]
- 16. Bergman B, Haskins K. Islet‐specific T‐cell clones from the NOD mouse respond to beta‐granule antigen. Diabetes 1994; 43:197–203. [DOI] [PubMed] [Google Scholar]
- 17. Stadinski BD, Delong T, Reisdorph N et al Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol 2010; 11:225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoshida K, Martin T, Yamamoto K et al Evidence for shared recognition of a peptide ligand by a diverse panel of non‐obese diabetic mice‐derived, islet‐specific, diabetogenic T cell clones. Int Immunol 2002; 14:1439–47. [DOI] [PubMed] [Google Scholar]
- 19. Judkowski V, Pinilla C, Schroder K, Tucker L, Sarvetnick N, Wilson DB. Identification of MHC class II‐restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol 2001; 166:908–17. [DOI] [PubMed] [Google Scholar]
- 20. Bradley BJ, Wang YY, Lafferty KJ, Haskins K. In vivo activity of an islet‐reactive T‐cell clone. J Autoimmun 1990; 3:449–56. [DOI] [PubMed] [Google Scholar]
- 21. Peterson JD, Haskins K. Transfer of diabetes in the NOD‐SCID mouse by CD4 T‐cell clones. Differential requirement for CD8 T‐cells. Diabetes 1996; 45:328–36. [DOI] [PubMed] [Google Scholar]
- 22. Wiles TA, Delong T, Baker RL, et al An insulin‐IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non‐obese diabetic mouse. J Autoimmun 2017; 78:11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baker RL, Bradley B, Wiles TA et al Cutting edge: nonobese diabetic mice deficient in chromogranin A are protected from autoimmune diabetes. J Immunol 2016; 196:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Babon JA, DeNicola ME, Blodgett DM et al Analysis of self‐antigen specificity of islet‐infiltrating T cells from human donors with type 1 diabetes. Nat Med 2016; 22:1482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aaron Wiles T, Powell R, Michel CR et al Identification of hybrid insulin peptides (HIPs) in mouse and human islets by mass spectrometry. J Proteome Res 2019; 18:814–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gonzalez‐Duque S, Azoury ME, Colli ML et al Conventional and neo‐antigenic peptides presented by beta cells are targeted by circulating naive CD8+ T cells in type 1 diabetic and healthy donors. Cell Metab 2018; 28:1–15. [DOI] [PubMed] [Google Scholar]
- 27. Vigneron N, Ferrari V, Stroobant V, Abi Habib J, Van den Eynde BJ. Peptide splicing by the proteasome. J Biol Chem 2017; 292:21170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kertesz‐Farkas A, Reiz B, Myers MP, Pongor S. Database searching in mass spectrometry based proteomics. Curr Bioinform 2012; 7:221–30. [Google Scholar]
- 29. Liepe J, Marino F, Sidney J et al A large fraction of HLA class I ligands are proteasome‐generated spliced peptides. Science 2016; 354:354–8. [DOI] [PubMed] [Google Scholar]
- 30. Berkers CR, de Jong A, Schuurman KG et al Definition of proteasomal peptide splicing rules for high‐efficiency spliced peptide presentation by MHC class I molecules. J Immunol 2015; 195:4085–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hughes C, Ma B, Lajoie GA. De novo sequencing methods in proteomics. Methods Mol Biol 2010; 604:105–21. [DOI] [PubMed] [Google Scholar]
- 32. Medzihradszky KF, Chalkley RJ. Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrom Rev 2015; 34:43–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tran NH, Zhang X, Xin L, Shan B, Li M. De novo peptide sequencing by deep learning. Proc Natl Acad Sci USA 2017; 114:8247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mylonas R, Beer I, Iseli C et al Estimating the contribution of proteasomal spliced peptides to the HLA‐I ligandome. Mol Cell Proteomics 2018; 17:2346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Faridi P, Li C, Ramarathinam SH et al A subset of HLA‐I peptides are not genomically templated: evidence for cis‐ and trans‐spliced peptide ligands. Sci Immunol 2018; 3:eaar3947. [DOI] [PubMed] [Google Scholar]
- 36. Rolfs Z, Solntsev SK, Shortreed MR, Frey BL, Smith LM. Global identification of post‐translationally spliced peptides with neo‐fusion. J Proteome Res 2018. 10.1021/acs.jproteome.8b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Faridi P, Purcell AW, Croft NP. In immunopeptidomics we need a sniper instead of a shotgun. Proteomics 2018; 18:e1700464. [DOI] [PubMed] [Google Scholar]
- 38. Baker RL, Jamison BL, Wiles TA et al CD4 T cells reactive to hybrid insulin peptides are indicators of disease activity in the NOD mouse. Diabetes 2018; 67:1836–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ito Y, Ashenberg O, Pyrdol J et al Rapid CLIP dissociation from MHC II promotes an unusual antigen presentation pathway in autoimmunity. J Exp Med 2018; 215:2617–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brunner Y, Coute Y, Iezzi M et al Proteomics analysis of insulin secretory granules. Mol Cell Proteomics 2007; 6:1007–17. [DOI] [PubMed] [Google Scholar]
- 41. Rindler MJ. Carboxypeptidase E, a peripheral membrane protein implicated in the targeting of hormones to secretory granules, co‐aggregates with granule content proteins at acidic pH. J Biol Chem 1998; 273:31180–5. [DOI] [PubMed] [Google Scholar]
- 42. Itoh Y, Tanaka S, Takekoshi S, Itoh J, Osamura RY. Prohormone convertases (PC1/3 and PC2) in rat and human pancreas and islet cell tumors: subcellular immunohistochemical analysis. Pathol Int 1996; 46:726–37. [DOI] [PubMed] [Google Scholar]
- 43. Davidson HW. (Pro)Insulin processing: a historical perspective. Cell Biochem Biophys 2004; 40(Suppl 3):143–58. [DOI] [PubMed] [Google Scholar]
- 44. Rawlings ND, Barrett AJ. Evolutionary families of metallopeptidases. Methods Enzymol 1995; 248:183–228. [DOI] [PubMed] [Google Scholar]
- 45. Wan X, Zinselmeyer BH, Zakharov PN et al Pancreatic islets communicate with lymphoid tissues via exocytosis of insulin peptides. Nature 2018; 560:107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mishto M, Goede A, Taube KT et al Driving forces of proteasome‐catalyzed peptide splicing in yeast and humans. Mol Cell Proteomics 2012; 11:1008–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lowenstein JM, Schatz MN. The nonenzymatic activation of acetate by adenosine triphosphate‐bivalent metal chelates. J Biol Chem 1961; 236:305–7. [PubMed] [Google Scholar]
- 48. Estevez‐Herrera J, Dominguez N, Pardo MR et al The crucial component of secretory vesicles. Proc Natl Acad Sci USA 2016; 113:E4098–E4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hutton JC. The internal pH and membrane potential of the insulin‐secretory granule. Biochem J 1982; 204:171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suckale J, Solimena M. The insulin secretory granule as a signaling hub. Trends Endocrinol Metab 2010; 21:599–609. [DOI] [PubMed] [Google Scholar]
- 51. Walter U, Toepfer T, Dittmar KE et al Pancreatic NOD beta cells express MHC class II protein and the frequency of I‐A(g7) mRNA‐expressing beta cells strongly increases during progression to autoimmune diabetes. Diabetologia 2003; 46:1106–14. [DOI] [PubMed] [Google Scholar]
- 52. Zhao Y, Scott NA, Quah HS et al Mouse pancreatic beta cells express MHC class II and stimulate CD4(+) T cells to proliferate. Eur J Immunol 2015; 45:2494–503. [DOI] [PubMed] [Google Scholar]
- 53. Russell MA, Redick SD, Blodgett DM et al HLA class II antigen processing and presentation pathway components demonstrated by transcriptome and protein analyses of islet beta‐cells from donors with Type 1 diabetes. Diabetes 2019; 68:988–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Richardson SJ, Rodriguez‐Calvo T, Gerling IC et al Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 2016; 59:2448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]