

Summary
Recent studies in Type 1 Diabetes (T1D) support an emerging model of disease pathogenesis that involves intrinsic β‐cell fragility combined with defects in both innate and adaptive immune cell regulation. This combination of defects induces systematic changes leading to organ‐level atrophy and dysfunction of both the endocrine and exocrine portions of the pancreas, ultimately culminating in insulin deficiency and β‐cell destruction. In this review, we discuss the animal model data and human tissue studies that have informed our current understanding of the cross‐talk that occurs between β‐cells, the resident stroma, and immune cells that potentiate T1D. Specifically, we will review the cellular and molecular signatures emerging from studies on tissues derived from organ procurement programs, focusing on in situ defects occurring within the T1D islet microenvironment, many of which are not yet detectable by standard peripheral blood biomarkers. In addition to improved access to organ donor tissues, various methodological advances, including immune receptor repertoire sequencing and single‐cell molecular profiling, are poised to improve our understanding of antigen‐specific autoimmunity during disease development. Collectively, the knowledge gains from these studies at the islet–immune interface are enhancing our understanding of T1D heterogeneity, likely to be an essential component for instructing future efforts to develop targeted interventions to restore immune tolerance and preserve β‐cell mass and function.
Keywords: autoimmunity, diabetes, human, inflammation, islet
Introduction
Type 1 Diabetes (T1D) is classically thought to arise from dysregulated immune responses that culminate in the destruction of insulin producing β‐cells in the pancreatic islets 1. While there is general consensus for an organ‐specific autoimmune process, observations from clinical trials and studies on human organ donor tissues include not only stark differences among patients in rate of progression, age of onset, insulitis composition and frequency, and C‐peptide production at disease presentation but in addition, a surprising level of heterogeneity related to β‐cell death and dysfunction in subjects with long‐standing disease 2, 3. Beyond this, emerging data suggest that multiple pathways and cell subsets may be subject to dysregulation at the level of intra‐islet/paracrine interactions as well as between islets, the surrounding acinar pancreas tissue, and immune infiltrating cells. There is an outstanding need within the field to better understand the complicated interplay between genetic risk factors and potential environmental influences that control localized immune cell activation as well as residual β‐cell function and mass during T1D pathogenesis 4. While animal models have helped elucidate key pathways involved in T1D, tissue biobanks containing samples from cross‐sectional and/or organ donor sources now offer the opportunity to interrogate cell interactions at the islet:immune interface and when coupled with novel experimental platforms, provide a comprehensive picture of the cellular and molecular mechanisms contributing to human T1D.
Interrogating the islet: immune interactions in mouse models of T1D
Our understanding of the molecular mechanisms underlying T1D pathogenesis has been advanced significantly by leveraging animal models. In particular, the non‐obese diabetic (NOD) mouse 5, as well as humanized 6 and transgenic 7, 8, 9 models, have informed on central autoantigens [i.e. preproinsulin, glutamic acid decarboxylase 65 (GAD65), islet antigen‐2 (IA‐2), islet specific glucose‐6‐phosphatase catalytic subunit related protein (IGRP), zinc transporter 8 (ZnT8), and chromogranin A] targeted by autoreactive T cells 10, 11, 12, 13, 14, 15, 16 (including posttranslationally modified autoantigens) 12, 17. They have also provided insights into potential mechanisms behind the loss of tolerance 17, 18, and allowed for in vivo assessment of phenotype and function of human cells 19, 20. Recent studies have illuminated interspecies differences in islet cell morphology and function that may contribute to the lack of success in translating therapeutics from mouse models to human patients 21, 22, 23, 24. Indeed, therapies targeting effector T cells for depletion 25 and those inhibiting T cell co‐stimulation 26 successfully prevented immune cell infiltration of the pancreatic islets and symptomatic diabetes in NOD mice hundreds 27 of times and reversed in a handful of studies 28, 29, 30. In contrast, clinical trials of anti‐CD3 31, 32, anti‐thymocyte globulin (ATG) 33, 34, 35, abatacept (CTLA4‐Ig) 36, and alefacept (LFA‐3/IgG1) 37 have, at best, provided only temporary preservation of baseline C‐peptide production in subgroups of T1D patients while anti‐CD3 was recently reported to delay T1D onset in at‐risk individuals 38. Though an in‐depth analysis of the contributions of animal models is beyond the scope of this review, Table 1 summarizes a selection of key findings relevant to human disease made possible by in vivo models.
Table 1.
A selection of NOD mouse models facilitating studies on islet–immune interactions in T1D pathogenesis.
| In vivo T1D models | Critical findings | Advantages | Shortcomings | References |
|---|---|---|---|---|
| NOD mouse | CD4+ and CD8+ T cells, as well as B cells, are integral to disease development | Parallels the polygenic nature of T1D and importance of MHC | Differences in disease kinetics, inflammatory infiltrate composition, islet cell architecture, pancreas morphology, and number of PLN | 5, 6, 10, 11, 12, 14, 15, 16, 17, 22, 75, 120 |
| Islet antigen specific T cells recognize Ins B:9–23, Ins1 B5–14 and Ins1/2 A2–10 | ||||
| ChgA and IAPP are autoantigens and function to initiate disease and promote Th1 responses | Serves as a valuable pre‐clinical model for therapeutics | Differences in endocrine cell distribution, β‐cell regenerative capacity, and immune cell receptor expression and signaling | ||
| Antigen specific T cells recognize post‐translationally modified epitopes which may alter binding to MHC molecules | Capacity for transgenics, adoptive transfers and lineage tracing, and intervention testing in a spontaneous model system | |||
| NOD‐scid Il2rγ −/− (NSG) mouse | Adoptively transferred CD8+ T cells from T1D donors stimulated with pooled islet antigen peptides [IGRP, IAPP, Ins, and IA‐2] traffic to the islets and produce IFN‐γ | Lack endogenous T, B and NK cells | Increased β‐cell proliferative capacity as compared to humans | 6, 19 |
| Can be engrafted with human PBMCs | Alterations in homing, chemokine and cytokine use, and secondary lymphatics in xenogeneic system | |||
| NOD‐SCID BLT HLA‐DQ8 transgenic mouse | Demonstrated the in vivo pathogenicity of human HLA‐DQ8 restricted InsB:9–23 specific CD4+ T cells in exacerbating insulitis and β‐cell death | Possess a complete human lymphoid and myeloid immune cell repertoire | GVHD and wasting syndrome | 6, 20 |
| T cells are educated autologously and are HLA restricted | ||||
| HLA‐A2.1 transgenic NOD mouse | Accelerated disease compared to nontransgenic NOD mouse | Possession of human HLA molecules allow for testing a variety of agents, including adoptive cell therapy, and ASI on human cells in vivo | Often reduced disease incidence as compared to NOD | 7, 8, 9, 13 |
| Islet infiltrating T cells directly lyse HLA‐A2.1+ β‐cells | ||||
| NOD.β2mnull.HHD mouse | CD8+ islet infiltrating T cells from HLA‐A2.1 transgenic mice target an IGRP epitope cross‐reactive to human IGRP (IGRP228–236) | |||
| NOD.mβ2mnull.hβ2m.HLA‐A11 transgenic mouse | HLA‐A11 restricted CD8+ islet infiltrating T cells in HLA‐A11 transgenic mice recognize IGRP and Ins C‐peptide and are present prior to disease onset | |||
| Foxp3‐GFP‐Cre × R26‐YFPNOD transgenic mouse model | GFP–YFP+Foxp3– ex‐Treg which lost Foxp3 were identifiable and shown to have a pro‐inflammatory phenotype | Facilitates genetic lineage tracing | Potential for off‐target Cre recombination | 18 |
| Can identify plasticity in cell lineages and sort out these plastic populations to conduct functional studies | ||||
| Trafficking and localization can be visualized in situ |
ASI = Antigen specific immunotherapy; Ins = Insulin; ChgA = Chromogranin A; IAPP = Islet Amyloid Polypeptide; IGRP = Islet‐specific glucose‐6‐phosphatase catalytic subunit‐related protein; IA‐2 = Islet antigen‐2; BLT = Bone marrow, Liver, Thymus; NK = Natural Killer; PBMCs = Peripheral blood mononuclear cells; GVHD = Graft Versus Host Disease; Tregs = regulatory T cells.
Lessons from human T1D pathology
Insulitis has long been considered the histopathological hallmark of T1D 39, yet differences in pancreatic mass and volume have recently been observed in T1D subjects as well as autoantibody negative (AAb–) first‐degree relatives and autoantibody positive (AAb+) subjects 40, 41. These findings support the possibility that T1D may involve early atrophy or growth defects affecting the entire pancreas prior to clinical diagnosis, yet the disease symptoms almost exclusively reflect defects in β‐cell function and/or mass. However, there still remains a paucity of data surrounding starting pancreas mass and volume for these subjects. Hence, this section focuses primarily on immune‐islet interactions in T1D pathogenesis but also highlights recently reported alterations within the acinar pancreas from studies of primary human tissues obtained from living and organ donor cohorts 42, 43, 44, 45 (Table 2).
Table 2.
Tissue procurement programs for the study of T1D pathogenesis in human pancreata 121.
| Tissue Procurement Programs and Biobanks | Cohort information | Initiatives | Reference |
|---|---|---|---|
| Network for Pancreatic Organ Donors (nPOD) | Transplant quality pancreas and lymphoid tissues collected from deceased organ donors at T1D onset to established disease, along with AAb+ donors and non‐diabetic corresponding controls. Samples are processed freshly at the University of Florida, banked, and distributed to hundreds of approved investigators around the world. | Investigate pancreas architecture and extracellular matrix in health and T1D pathogenesis | 42, 47 |
| Define autoreactive T and B cell repertoire and antigen specificities with an interest in therapeutic intervention | |||
| Investigate the role of viruses in T1D | |||
| Attain comprehensive ‘omics data | |||
| Examine pancreata from organ donors who received a pancreas transplant | |||
| Exeter Archival Diabetes Biobank (EADB) | Pancreata collected at autopsy from predominantly young patients with recent‐onset T1D | Identification of age‐dependent differential insulitic profiles | 43, 50, 71 |
| Investigate enteroviral infection as a possible trigger for T1D | |||
| Diabetes Virus Detection Study (DiVID) | Pancreatic biopsies from living patients with T1D | Investigate potential viral triggers for T1D | 44 |
| Human Pancreas Analysis Program (HPAP) | Transplant quality pancreas and lymphoid tissues collected from deceased organ donors at T1D onset to established disease, along with AAb+ donors and non‐diabetic corresponding controls. Samples are processed and subjected to comprehensive analyses at Vanderbilt University and University of Pennsylvania. Metadata are made available to investigators via a publicly accessible database. | Utilize novel technologies to profile immune and endocrine cells over the natural history of disease. | 45 |
| Human Atlas of the Neonatal Development and Early Life‐Pancreas (HANDEL‐P) | Transplant quality pancreas collected from control organ donors without diabetes ranging from prenatal to 10 years of age | Understand the early changes, whether structural, immunological, β‐cell‐centric or otherwise, that occur in the normal human pancreas, unaffected by diabetes | 122 |
AAb+ = Autoantibody positive; T1D = type 1 diabetes.
Immunological determinants of human T1D
Within the pancreatic islets of T1D organ donors, CD8+ T cells comprise a majority of the infiltrating immune cells, with the remainder consisting primarily of B cells, CD4+ T cells, and macrophages 46. Yet, the severity of CD3+ infiltrate and residual β‐cell mass varies appreciably across T1D organ donors according to disease duration (Fig. 1a‐d) 47, 48. B cell infiltrate has also been shown to differ according to donor age at T1D onset with younger‐onset donors (<13 years of age) 49 exhibiting a “hyper‐immune” CD20Hi phenotype and fewer residual insulin containing islets (ICI), while older‐onset donors displayed fewer intra‐islet CD45+ immune cells and a CD20Lo profile within ICI (Fig. 1e‐f) 50. β‐cells expressing both insulin and the replication marker, Ki67, have been observed within insulitic islets of adult non‐diabetic donors with multiple AAb, potentially revealing simultaneous T cell mediated β‐cell destruction and attempts to restore endogenous β‐cell mass prior to T1D onset (Fig. 1g‐h) 51, 52. Stratifying patients by islet immune signatures may indicate different modes of pathogenesis or potentially, disease endotypes 53 that could be targeted with precision therapies.
Figure 1.
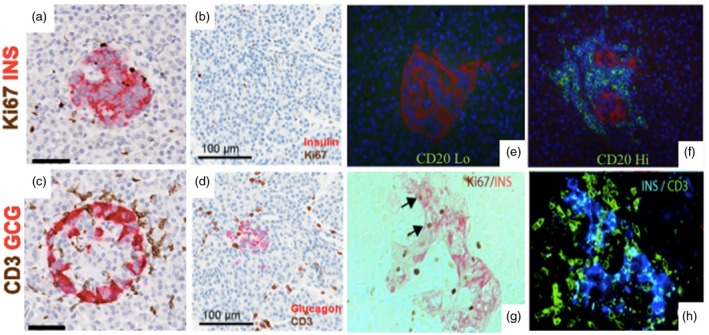
Heterogeneous islet morphology and immune cell infiltration across disease duration and according to age of T1D onset. Representative islets were imaged from serial paraffin sections stained for Ki67 plus insulin (INS) and CD3 plus glucagon (GCG) for a 13‐year‐old donor at T1D onset (nPOD 6228; a & c) and a 6 year old with 3 years T1D duration (nPOD 69; b & d) 47, 48. CD3+ infiltrate is present in both donors (c, d), though insulin containing islets are only observed in the first donor (a, b). Scale bars: a and c, 50μm; b and d, 100μm 47, 48. Pancreas samples from donors with recent‐onset T1D stained for CD20 (green) and glucagon (red), and nuclei (DAPI) exhibit differences in infiltrate composition, which can separate subjects based on hyper‐immune CD20Hi (nPOD 6052; e) and pauci‐immune CD20Lo profiles (nPOD 6070; f) 50. Histology of a 46 year old donor with ≥3 islet AAb shows both Ins+Ki67– β‐cells and Ins+Ki67+ cells replicating β‐cells (g, arrows) within islets that contain CD3+ T cell infiltrate (h) 51. Figures have been reprinted with permission from the American Diabetes Association 47, 48, 50, 51.
A second reproducible histological feature of T1D involves β‐cell hyperexpression of HLA Class I, which is observed most commonly within residual ICI and accompanied by elevated expression of the transcription factor STAT1 54. Whether this phenotype is the consequence or driver of lymphocyte infiltration and IFN‐γ production 55 within the islet is a subject of debate, but in either case, it is likely that HLA hyperexpression may facilitate surveillance by innate immune cells and β‐cell destruction by antigen‐specific CD8+ T cells. In a seminal report by Coppetiers et al., in situ HLA‐tetramer staining revealed CD8+ T cells specific for islet antigens – insulin, IGRP and IA‐2 – within insulitic lesions, though the persistence of β‐cell mass and number of islet antigen‐reactive T cell specificities varied across donors in accordance with disease duration 56. Similarly, CD4+ T cells isolated from the islets of organ donors with recent‐onset T1D exhibited reactivity to multiple GAD65 epitopes, as well as proinsulin76–90 and IA‐2545–562, measured by antigen‐stimulated IFNγ secretion 57. Moreover, islet‐infiltrating CD4+ T cell clones, when reconstructed to express T cell receptors (TCRs) as transductants, have been identified to respond to proinsulin, C‐peptide amino acids 19–35 (C:19–35), and insulin B:9–23 48. These data support a critical role for T cells in mediating islet antigen specific pathology.
In addition to examining autoantigen specificities, extensive adaptive immune cell repertoire analyses from peripheral blood and organ donor tissues of subjects with T1D have revealed disease‐related repertoire changes. We previously performed TCRβ chain and B cell immunoglobulin heavy chain (IgH) immunosequencing on lymphocytes isolated from the pancreatic draining lymph node (pLN), peripheral blood, spleen, and in one case, islet infiltrate from T1D and control nPOD donors. Overall, we observed limited receptor sharing in sorted CD4+ T cell subsets across tissues from a given donor, while CD8+ T cells demonstrated increased clonality, with one donor possessing shared complementarity determining region 3‐β (CDR3β) sequences across the islets, pLN, and peripheral blood mononuclear cells (PBMCs) 58. TCR sequencing of single sorted islet‐resident T cells has corroborated this oligoclonality of CD8+ T cells 48. TCR sequencing of islet‐infiltrating CD4+ T cells from a single donor revealed a bias in TRBV gene usage, with 30% of clones expressing TRBV5–1*01, and three clones expressing identical TCRs using TRBV5–1*01 recognized overlapping C‐peptide epitopes 59. Reactivity to these same epitopes was also found in PBMCs from living T1D patients 59. Identification of overlapping TCR sequences and reactivities within peripheral blood and the target organ suggests profiling of circulating autoreactive T cells could have disease predictive value or serve as a tool to assess efficacy of therapeutics aimed at reducing the autoreactive T cell pool.
Intrinsic markers of β‐cell stress and dysfunction in T1D
Due to the significant influence of genetics on T1D pathogenesis, studies investigating the expression of candidate genes in the islets and their functional implications are of great interest. Furthermore, the finding that disease‐associated T1D loci often map to noncoding regions, specifically regulatory elements 60, suggests a role for these loci in regulating gene expression. There is evidence to suggest T1D candidate genes not only impact immune cell repertoire and activity but also, may function at the level of the islet to regulate β‐cell apoptosis and immune interactions 61 (Fig. 2), though there is a currently a paucity of mechanistic data. Recent studies combining transcriptomic, epigenomic, and proteomic data to assess the influence of T1D risk loci on β‐cell dysfunction have shown isolated human islets treated with IFN‐γ and IL‐1β in culture to exhibit differential transcriptomic and epigenetic remodeling. This was mediated by inflammatory response transcription factor recruitment, DNA looping and chromatin acetylation, cumulatively leading to upregulation of genes and proteins involved in the inflammatory response and apoptosis 62, implicating these as mechanisms likely underpinning β‐cell dysfunction. In particular, knockdown of T1D candidate gene GLIS3 in human islets has been shown to sensitize β‐cells to IFN‐γ and IL‐1β induced death 63, found to be mediated by a reduction in alternative splicing factor SRp55 which normally negatively regulates apoptosis and endoplasmic reticulum (ER) stress 64. Thus, crosstalk between T1D candidate genes and splicing regulators may result in increased β‐cell susceptibility to immune‐mediated as well as endogenous stress related dysfunction.
Figure 2.
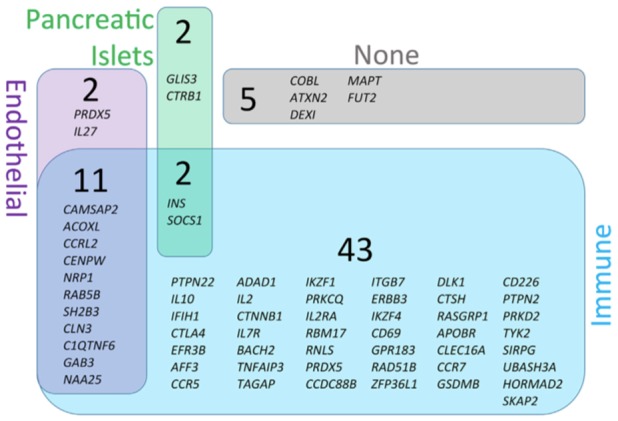
T1D candidate genes are highly expressed in endothelial, islet, and immune tissues. T1D risk loci were collected from ImmunoBase (http://www.immunobase.org), tissue expression was identified per tissue (genevisible.com/search), and a venn diagram was created based on relative abundance of expression in immune (blue), endothelial (purple), and pancreatic islet tissue (green) 61. Figure has been reprinted with permission from Frontiers in Endocrinology 61.
Recent attention has been directed toward the role of β‐cell stress in T1D pathology, due to findings of increased levels of ER stress markers such as C/EBP homologous protein (CHOP) glucose‐regulated protein 78 (GRP78, also known as IgH binding protein (BiP)) in the islets of T1D subjects 65. These function as part of the Unfolded Protein Response (UPR) to promote cell death and aid in protein folding, respectively 66. Aberrant protein processing, in the form of misfolded or alternatively spliced isoforms, may result in the generation and presentation of neoantigens 67. Indeed, T cells reactive to post‐translationally modified proteins such as a citrullinated form of GRP78292–305 and islet amyloid polypeptide (IAPP)65–84, as well as defective ribosomal insulin gene products (DRiPs) and peptides formed from the fusion of insulin C‐peptide fragments with chromogranin A or IAPP (termed hybrid insulin peptides, HIPs) have been isolated from the islets of T1D donors 57, 68. Thus, β‐cells may play a role in their own destruction through stress‐induced protein processing defects, which may serve to induce a critical break in tolerance or to exacerbate existing pathology by expanding the β‐cell antigenic repertoire.
The search for viral triggers of T1D
Extensive efforts to identify precise environmental triggers that might initiate or potentiate the autoimmune process have proven challenging thus far, but examination of donor pancreata have revealed viral response signatures which could offer clues into early T1D etiology. Biopsies of pancreas from living donors with recent‐onset T1D, aged 24–35 years, through DiViD contained some indications of viral capsid protein VP1 immunostaining, HLA Class I hyperexpression, and enterovirus RNA within the islets 69, possibly the result of a β‐cell tropism mediated by expression of a specific isoform of the Coxsackie and Adenovirus Receptor 70. Furthermore, islets infected with coxsackievirus B (CVB) in‐vitro demonstrated reduced insulin mRNA expression and insulin secretion 71, suggesting viral infection could impair β‐cell function. Additional studies have shown CVB infection to induce the production of type III interferons, the magnitude of this response being associated with the single nucleotide polymorphism (SNP) rs1990760 within the T1D candidate gene, IFIH1, suggesting a role for this T1D risk locus in influencing viral clearance 72. These results mirror those from The Environmental Determinants of Diabetes in the Young (TEDDY) study, where susceptibility to enteroviral infection was shown to correlate with HLA genotype, with HLA‐DQ2/DQ8 individuals, who have the highest risk of developing T1D, presenting with more enteroviral infections 73. Hence, in situ and clinical data together suggest a model in which T1D susceptibility loci may predispose to viral infection that could, in turn, promote immune dysregulation. However, it is also possible that the anti‐viral immune signature could be impacted by a viral infection that does not immediately localize to the islet, or alternatively, host viral sensing machinery is detecting a build‐up of endogenous mRNA and triggering a form of “sterile” inflammation and viral sensing pathways 74. Thus, caution must be used when interpreting studies of an anti‐viral signature in the islet microenvironment.
Immune dysfunction within the pLN, migration, and trafficking
The critical role of pLN in initiating antigen‐specific T cell responses, as demonstrated by disease protection upon early (3 weeks) excision in the NOD 75, suggests defective signaling or activation within the pLN could contribute to islet‐directed immune dysfunction in human T1D. A recent study observed the pLN to be disorganized, containing fewer secondary follicles and follicular dendritic cell (fDC) networks in donors with recent‐onset T1D (≤1.5 years duration) compared to non‐diabetic controls and long‐duration (4–19 years) T1D donors 76. Both B and TFH cells have been implicated in T1D, with increased islet infiltrating CD20+ B cells being associated with rapid progression to T1D 49 and with TFH frequency and IL‐21 production being increased in the peripheral blood of T1D subjects 77, collectively highlighting the need to determine the source(s) of pLN germinal center dysfunction in T1D.
In order for diabetogenic immune cells to infiltrate the pancreas and exert their effector functions, they must bypass inhibitory immune checkpoints (e.g. PD‐L1:PD‐1) and migrate down chemokine gradients. Thus, an area of great therapeutic interest involves the development of agents to modulate these processes. The PD1:PD‐L1 interaction has been therapeutically targeted in cancer to enhance CD8+ T cell‐mediated tumor destruction, though the use of these immune checkpoint inhibitors sometimes results in the manifestation of immune‐related adverse events, including T1D 78. Increased PD‐L1 expression was observed in isolated islets and human β‐cell lines exposed to IFNα and IFNγ treatment in culture via JAK‐STAT dependent signaling 54. Importantly, increased PD‐L1 expression was also observed in the ICI of T1D organ donors suggesting that PD‐L1 upregulation might protect residual β‐cells in the context of the proinflammatory milieu 79. The interferon inducible chemokine CXCL10 was also expressed in ICI of recent‐onset T1D subjects, along with its receptor CXCR3 on infiltrating lymphocytes 80, 81, cumulatively providing a potential mechanism wherein local interferon production might drive lymphocyte infiltration via chemotactic cues, resulting in the selective destruction of β‐cells that fail to upregulate PD‐L1.
The contributions of pancreas architecture to T1D pathology
As noted above, T1D is clinically identified following β‐cell dysfunction/destruction sufficient to induce hyperglycemia, but the entire pancreas appears to be compromised in donors with recent‐onset and pre‐T1D. Deficits in exocrine pancreas mass and function have been identified in subjects with and at‐risk for T1D. Indeed, reduced secretion of pancreatic enzymes, such as trypsinogen 82, amylase, and lipase 83, as well as a smaller pancreas organ as measured by weight or volume 40, 41, are potentially reflective of exocrine damage accompanying T1D pathology. Increased complement deposition was also observed on the vascular endothelium and extracellular matrix (ECM) surrounding vessels and ducts within the exocrine pancreas tissue in T1D versus control donors 84, indicating localized inflammation. Moreover, studies examining immune infiltration of the exocrine pancreas have found CD8+ T cells, CD4+ T cells and CD11c+ dendritic cells (DCs) to be present at higher numbers in T1D versus control donors, with CD11c+ cells being increased in both AAb+ donors (prior to disease onset) and short‐duration T1D donors 85. These findings raise important questions surrounding mechanisms underlying loss of exocrine pancreas mass and function as well as the potential for interactions between islet and exocrine tissues via the insulo‐acinar portal system wherein exocrine pancreas defects may contribute to endocrine cell death (or vice versa).
Immune cell infiltration around and within islets is thought to be, in part, facilitated by defects in ECM, namely in basement membrane integrity, which permit leukocyte penetration 86. The ECM component hyaluronan (HA) has been shown in vitro to promote lymphocyte adhesion and migration 87, proliferation 88, and antigen presentation 89, suggesting low molecular weight HA could drive immune activation and infiltration in T1D. Indeed, accumulation of HA has been observed in islets of young recent‐onset (0–8 weeks duration) T1D donors and was associated with CD45+ infiltrate 90. Inhibiting HA synthesis impairs antigen presentation in vitro, and in vivo treatment promotes tolerance in a mouse model of autoimmune encephalitis 91, suggesting that targeting components of ECM may be beneficial in T1D.
Islets require a high degree of vascularity for optimal β‐ and α‐cell function in sensing and maintaining glucose homeostasis 92, a function that is directed in part by branches of the sympathetic nervous system63. However, high vascularity may also contribute toward immune infiltration in T1D. Canzano et al. showed reduced islet vessel diameter in T1D subjects as compared to controls, but T1D subjects with residual ICI more closely resembled control subjects, making it difficult to discern whether changes in microvasculature precede or follow loss of functional β‐cell mass 93. At the same time, T1D islets exhibit α‐cell dysfunction, which clinically results in hyperglycemia complicated by hyperglucagonemia or hypoglycemia with failed counterregulation. Though the exact mechanisms remain unknown, it has been speculated that α‐cell failure may reflect a loss of islet cell paracrine signaling 94 or a response to the local inflammatory milieu. Indeed, RNA‐sequencing of human T1D islet α‐cells has revealed decreased expression of genes involved in calcium signaling and electrical activity, with partial resolution when transplanted into a non‐autoimmune environment 95. Thus, loss of islet vasculature may contribute toward α‐ and β‐cell dysfunction by impeding communication between and across islets, serving to exacerbate inflammation‐induced dysfunction.
Intra‐islet vascular defects could potentially point to underlying sympathetic nervous system dysfunction as sympathetic neurons are thought to control vascular tone and thus, determine responsiveness to both glucose and neurotransmitters important for β‐cell function 96. Reduced sympathetic nerve density observed in the islets of recent‐onset and long‐duration T1D versus control donors supports this notion 97. Moreover, β‐cells have been shown to express the inhibitory neurotransmitter gamma‐aminobutyric acid (GABA) produced by GAD, of which the 65kD isoform is a potent autoantigen in T1D 98. Interestingly, positive allosteric modulators of GABA promote β‐cell proliferation in vitro 99, indicating a potential therapeutic role for this molecule in replenishing lost β‐cell mass. GABA receptors are also expressed on immune cells 100, and GABA was recently demonstrated to function in an immunoregulatory capacity, suppressing T cell proliferation in vitro as well as TH1 and TH2 cytokine production by PBMCs isolated from patients with T1D 101. GABA has also been suggested to influence α‐ to β‐cell conversion 102, though this area remains controversial and requires additional study 103. Thus, GABA could potentially serve in a regenerative context to promote β‐cell expansion or to bolster the efficacy of islet cell transplantation. The aforementioned findings illuminate the complex T1D pathology involving dysfunction throughout the pancreas organ and across multiple physiological systems, emphasizing the need for more comprehensive approaches to study and treat the underlying mechanisms of T1D pathogenesis.
Multiplexed analyses of human pancreas tissue
High‐throughput single cell transcriptomic profiling of immune and islet cells 104 provides a means to assess the contributions of candidate genes/gene expression toward T1D‐associated phenotypic and functional defects at greater depth and with less starting material. Thus, information gleaned from valuable donor tissue is maximized, and it is possible to identify rare cell subsets that might be masked with bulk sequencing efforts. Using this technology to interrogate the β‐cell transcriptome has revealed population heterogeneity in the form of differential expression of genes involved in β‐cell function (i.e. UCN3) and ER stress inducible genes (e.g. HERPUD1, DDIT3, and HSP5, the latter of which encodes GRP78/BiP) 105. This suggests the existence of β‐cell populations that are predisposed to stress and might preferentially be destroyed in an autoimmune environment. Additionally, the Assay for Transposase Accessible Chromatin with sequencing (ATAC‐seq, Table 3), previously used to examine cell‐type specific regulatory elements in endocrine cells 106, has recently been scaled down to single cell resolution and optimized for use in frozen tissue 107. Hence, studies investigating complex DNA regulatory networks from banked tissue samples are expected to uncover defects in epigenetic regulation which function to promote T1D at the level of the islet–immune interface.
Table 3.
Emerging technologies for studies of T1D pathology.
| Emerging Technologies/Platforms | Applications | Reference |
|---|---|---|
| Single cell transcriptomics/ proteomics | Assess expression of candidate genes and their protein products for paired cell genotype/phenotype data, as well as adaptive immune cell repertoire analysis | 104 |
| Single cell ATAC‐sequencing | Investigate the contribution of dysregulated gene networks, particularly in candidate loci, to immune or β‐cell function | 106, 107 |
| CyTOF | Following labeling with metal tagged antibodies, deep phenotyping of single cell suspensions can be performed | 110 |
| IMC | By applying metal tagged antibodies to fixed tissue sections, deep phenotyping of cells can be performed in situ | 111, 112 |
| Laser capture microdissection | Identify differentially expressed genes and proteins to develop disease‐predictive biomarkers or targeted therapeutics | 108 |
| nanoPOTS | Identify novel post‐translational modifications within a single islet and the proteomic basis for islet heterogeneity | 109 |
| CODEX | Examine disease‐related tissue and cell subset reorganization, as well as perform multiplexed deep phenotyping | 113 |
| Tissue clearing (e.g. X‐CLARITY, PARS) | Visualization of intact morphology, vasculature, innervation, and extracellular matrix | 114 |
| RNA single‐molecule FISH | Identify the dynamics of candidate gene and checkpoint molecule expression in all subsets present in the tissue microenvironment, examine how they influence cell phenotype and function | 115 |
| Microphysiological Systems | Introduce targeted therapeutics or innate and adaptive immune cells to assess their contribution to disease development/treatment | 116 |
| Investigate the underlying cause of the islet anti‐viral immune signature | ||
| Examine the role of hybrid insulin peptides (HIPs), defective ribosomal products (DRiPs) and neoantigens in β‐cell stress or destruction | ||
| Tissue Slice | Profile live immune and endocrine cells across the pancreas in the native tissue environment | 24 |
| Assess the role of chemokines and adhesion molecules | ||
| Determine which cells are directly pathogenic vs bystander or tissue resident cells |
nanoPOTS = Nanodroplet processing in one pot for trace samples; PARS = Perfusion‐assisted agent release in situ; ATAC = Assay for Transposase Accessible Chromatin; CyTOF = Cytometry time of flight; IMC = Imaging mass cytometry.
Single cell assays such as mass cytometry (CyTOF), along with other new methods designed to profile the proteome at high resolution and with minimal cell input 108, 109, can offer insights into cellular heterogeneity but cannot discern cell‐cell interactions or orientation within the native tissue environment (Table 3). In contrast, Imaging Mass Cytometry (IMC), which facilitates visualization of tissue sections labeled with antibodies conjugated to rare metal isotopes using high‐resolution laser ablation and time‐of‐flight mass spectrometry, allows for high‐parameter in situ data acquisition without the compensation issues plaguing traditional immunofluorescence analyses 110, 111 (Table 3). Simultaneous detection of 36 parameters from a single tissue section by IMC coupled with analysis of pseudotime dynamics indicated that β‐cells lose identifying markers such as insulin, proinsulin, IAPP, and protein tyrosine phosphatase receptor type N (PTPRN) prior to their destruction 111, potentially indicating a direct role for β‐cells in inciting their own destruction by autoreactive T cells. Additionally, intra‐ and peri‐islet T cell numbers were negatively correlated with disease duration and residual β‐cell mass, with new‐onset T1D donors possessing the highest degree of infiltration, regardless of the presence of β‐cells (defined as islet cells expressing both Pancreatic And Duodenal Homeobox 1 (PDX1) and NKX6‐1) 111. Similarly, Wang et al. observed increased Granzyme B+ macrophages and CD8+ T cells in pancreas tissues from T1D donors, with β‐cell containing islets (defined by expression of insulin, NKX6.1 and PDX1) enriched in CD8+ T cells and CD20+ B cells 112, likely identifying islets that are actively being targeted by autoreactive immune cells. Defects in peri‐islet collagen and decreased intra‐islet vascularization were also observed in human donors with T1D as compared to controls 112. Cumulatively, these studies suggest that morphological, genetic, and proteomic dysfunction work in concert to influence aberrant immune responses, β‐cell dysfunction or loss of β‐cell identity, and β‐cell destruction in T1D (Fig. 3).
Figure 3.
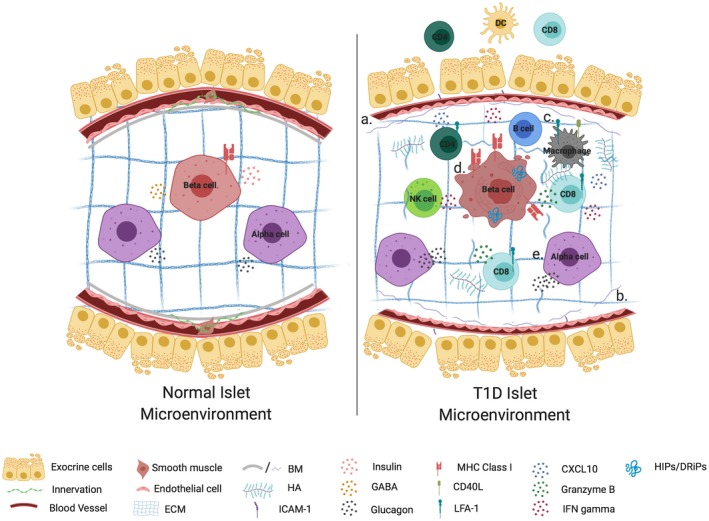
Islet–immune interactions and their impact on β‐cell function in health and T1D. An islet in a normal microenvironment is surrounded by intact extracellular matrix and exocrine tissue, while provided sufficient vascularization and innervation to promote glucose homeostasis and immunoregulation through insulin and glucagon secretion and release of GABA (left). In T1D (a) reduced vessel diameter and innervation impair islet‐cell function, while morphological dysfunction in the form of (b) loss of basement membrane (BM) and extracellular matrix (ECM) integrity as well as hyaluronan (HA) accumulation permit (c) the infiltration and activation of lymphocytes in both the exocrine and intra‐islet tissue. These immune cells make use of chemokine gradients and promote a diabetogenic milieu through the release of pro‐inflammatory mediators. Cumulatively these defects contribute toward β‐cell dysfunction in the form of (d) MHC class I hyperexpression, loss of insulin and GABA expression. They also may serve to magnify endogenous β‐cell stress presented as protein processing defects, which in turn function as neoantigens to exacerbate immune activation, all of which culminate in β‐cell death. In addition to β‐cell defects, α‐cell dysfunction (e) contributes to impaired glucose homeostasis in T1D through hyperglucagonemia or failed counterregulatory responses. Figure created with Biorender.
A novel method developed by Nolan and colleagues, co‐detection by indexing (CODEX), enables multiplexed in situ deep phenotyping through the use of polymerase‐driven iterative antibody visualization, with up to 66 antigens able to be codetected on a standard 3–4 color microscope by iterative hybridization and staining 113 (Table 3). A comprehensive image is then constructed from the combination of images, allowing associations to be drawn between cell phenotype and spatial arrangement in the tissue microenvironment. Our lab and others are in the process of applying this technique to banked donor tissues spanning the natural history of disease. We expect this approach to further aid in deciphering the timeline of changes in morphology, immune and endocrine cell phenotypes, the nature of cell‐cell interactions within the islets and throughout the pancreas, and the profile of β‐cells that are more susceptible to autoimmune attack. Additionally, tissue clearing methods such as CLARITY, which allow for whole organ imaging without morphological or cellular perturbations 114 (Table 3), have enabled visualization of age and disease related changes in pancreas architecture, vascularization, and innervation across the pre‐ and post‐natal periods as well as in pediatric T1D tissues 23. Cleared tissues offer compatibility with new methods of single mRNA transcript visualization 115, allowing measurement of localization of transcripts within the whole organ. Collectively, the ongoing work leveraging these aforementioned advancements will provide a more comprehensive view of T1D pathology at the islet:immune interface related phenotypic, transcriptomic, and morphological changes as well as the molecular and cellular events underpinning disease development.
Ex vivo platforms for studying islet–immune interactions in human T1D
Novel platforms for ex vivo studies of live human islets have been developed to mimic the human islet microenvironment and model islet–immune cell interactions (Table 3). One such technology is the recently developed islet‐on‐a‐chip microphysiological system (MPS), which allows laminar flow of immunotherapeutic agents or specific immune cells over human pancreatic islets with immunofluorescent imaging in real time, providing a way to monitor β‐cell function and phenotype in response to any number of putatively diabetogenic or protective stimuli 116. Indeed, in a recent study, Lenguito et al. were able to maintain viable islets and reliably detect insulin secretion in response to glucose and KCl stimulation 116, illustrating the capacity to further leverage this MPS to interrogate the etiological role of host‐pathogen interactions; specific antigen targets, lymphocyte activation state, as well as function of autoreactive T and B cells at the islet–immune interface within an isogenic system; and the potential for regulatory T cells (Tregs) to inhibit β‐cell destruction. Importantly, we expect that such information might facilitate the development and ex vivo functional validation of immune and β‐cell directed therapeutics.
Islet isolation will introduce confounding effects, such as induced stress or bias for healthy islets, and cannot recapitulate the islet‐acinar tissue interactions that occur in vivo. Thus, a promising tissue slice platform was developed by the Speier lab 24 facilitating studies that link endocrine and exocrine secretory function with morphological features of intact pancreatic tissues maintained under physiological conditions using a perfusion system. Responses to well characterized stimuli for the secretion of hormones [e.g. insulin (glucose, KCl) and glucagon (KCl, arginine)] and enzymes [amylase, lipase and trypsinogen (CCK‐8, carbachol)] 117 can be assessed in real time under steady state conditions, and the experimental system can likely be modulated with the addition of inflammatory cytokines or other soluble factors to mimic or mollify the T1D microenvironment. The potential drawbacks of this platform include the confounding influence of pancreatic enzymes, as well as lack of innervation and blood flow limiting tissue thickness to limit hypoxia‐induced stress. Comparison of human and mouse pancreas slices illustrated differences in endocrine and epithelial cell contacts, with human slices showing increased β‐cell‐α‐cell contacts 21, 118, illustrating the importance of paracrine signaling in optimal human pancreas function. Furthermore, reduced β‐cell connectivity in human slices as compared to mouse slices illustrates interspecies differences in islet cell‐cell communication 21, a factor that potentially contributes to poor translation of T1D therapies from mouse to humans. Multifluorescent 3‐D imaging of human pancreas slices has also provided in‐depth structural analysis of the individual islet vasculature, the surrounding duct cells and ECM 119. These high‐resolution analyses may serve to identify subtle changes occurring within the microenvironment in AAb+ pre‐T1D donors prior to diagnosis. Co‐registration of endocrine and exocrine functional profiles with precise anatomical location and visualization of immune cell infiltration will expand our understanding of mechanistic relationships between islet, acinar and immune defects in T1D as well as our ability to test novel therapeutics directed at one or more of these components.
Implications for natural history studies and interventions
The aforementioned knowledge gains surrounding the islet:immune interface have collectively advanced our understanding of T1D pathogenesis in humans. Animal model studies continue to inform on mechanisms underlying β‐cell destruction, namely migration and activation of autoantigen‐specific T cells, which are essential to T1D pathology. However, numerous effective immunomodulatory therapies in mice have failed to meet their primary clinical endpoints in human trials, suggesting a need for additional and complementary models to recapitulate the human disease. With the development of ex vivo experimental platforms (e.g. islet‐on‐a‐chip MPS and pancreas slices), we are poised to investigate the complex interplay between the various pancreas cell types at play (e.g. islet, immune, neural, vascular, and exocrine) from fresh human donor tissues. Indeed, studies of human pancreata have shown T1D pathogenesis to extend far beyond the β‐cell with defects affecting multiple cell types across the entire pancreas, though we have yet to identify the precise disease initiating event(s). Hence, multiparameter analyses coupled with advanced bioinformatics and statistical methods will be essential to linking complex datasets to determine the underlying mechanisms of β‐cell destruction, to identify disease predictive biomarkers of pathogenic events occurring within the endocrine or exocrine pancreas, and to develop targeted approaches to alleviate autoimmunity and restore β‐cell mass. Evidence is building for multifactorial defects within the β‐cells, islet niche, acinar pancreas, and immune compartment, and a combinatorial strategy targeting multiple arms of T1D pathogenesis will likely be necessary to effectively treat the disease.
Author contributions
LP wrote the manuscript, AP reviewed/edited the manuscript and contributed to discussion, TMB conceived of the article and wrote the manuscript.
Disclosures
The authors declare that no conflicts of interest exist pertaining to this manuscript.
Acknowledgements
The authors would like to thank the Brusko and Atkinson laboratories for their continued support and guidance when preparing this manuscript. These studies were supported by grants from the NIH (P01 AI042288, R01 DK106191 and HIRN UC4 DK104194) as well as a grant from The Leona M. and Harry B. Helmsley Charitable Trust.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Historical and new insights into pathogenesis of type 1 diabetes. Clinical and Experimental Immunology 2019, 198: 292–293.
Birth and coming of age of islet autoantibodies. Clinical and Experimental Immunology 2019, 198: 294–305.
HIPs and HIP‐reactive T cells. Clinical and Experimental Immunology 2019, 198: 306–313.
Immune cell trafficking to the islets during type 1 diabetes. Clinical and Experimental Immunology 2019, 198: 314–325.
References
- 1. Atkinson M. Type 1 Diabetes. Lancet 2013; 383:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lohr M, Kloppel G. Residual insulin positivity and pancreatic atrophy in relation to duration of chronic type 1 (insulin‐dependent) diabetes mellitus and microangiopathy. Diabetologia 1987; 30:757–62. [DOI] [PubMed] [Google Scholar]
- 3. Oram RA, Jones AG, Besser RE et al The majority of patients with long‐duration type 1 diabetes are insulin microsecretors and have functioning beta cells. Diabetologia 2014; 57:187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Achenbach P, Bonifacio E, Koczwara K, Ziegler A‐G. Natural History of Type 1. Diabetes 2005; 54:S25–S31. [DOI] [PubMed] [Google Scholar]
- 5. Pearson JA, Wong FS, Wen L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun 2016; 66:76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brehm MA, Powers AC, Shultz LD, Greiner DL. Advancing animal models of human type 1 diabetes by engraftment of functional human tissues in immunodeficient mice. Cold Spring Harb Perspect Med 2012; 2:a007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pow Sang L, Surls J, Mendoza M, Casares S, Brumeanu T. HLA‐DR*0401 expression in the NOD mice prevents the development of autoimmune diabetes by multiple alterations in the T‐cell compartment. Cell Immunol 2015; 298:54–65. [DOI] [PubMed] [Google Scholar]
- 8. Marron MP, Graser RT, Chapman HD, Serreze DV. Functional evidence for the mediation of diabetogenic T cell responses by HLA‐A2.1 MHC class I molecules through transgenic expression in NOD mice . Proc Natl Acad Sci U S A 2002; 99:13753‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takaki T, Marron MP, Mathews CE et al HLA‐A*0201‐restricted T cells from humanized NOD mice recognize autoantigens of potential clinical relevance to type 1 diabetes. J Immunol 2006; 176:3257–65. [DOI] [PubMed] [Google Scholar]
- 10. Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD‐scid/scid mice. Relative contributions of CD4+ and CD8+ T‐cells from diabetic versus prediabetic NOD.NON‐Thy‐1a donors. Diabetes 1993; 42:44–55. [DOI] [PubMed] [Google Scholar]
- 11. Jarchum I, Baker JC, Yamada T et al In vivo cytotoxicity of insulin‐specific CD8+ T‐cells in HLA‐A*0201 transgenic NOD mice. Diabetes 2007; 56:2551–60. [DOI] [PubMed] [Google Scholar]
- 12. Crawford F, Stadinski B, Jin N et al Specificity and detection of insulin‐reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A 2011; 108:16729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Antal Z, Baker JC, Smith C et al Beyond HLA‐A*0201: new HLA‐transgenic nonobese diabetic mouse models of type 1 diabetes identify the insulin C‐peptide as a rich source of CD8+ T cell epitopes. J Immunol 2012; 188:5766–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stadinski BD, Delong T, Reisdorph N et al Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol 2010; 11:225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baker RL, Delong T, Barbour G, Bradley B, Nakayama M, Haskins K. Cutting edge: CD4 T cells reactive to an islet amyloid polypeptide peptide accumulate in the pancreas and contribute to disease pathogenesis in nonobese diabetic mice. J Immunol 2013; 191:3990–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baker RL, Bradley B, Wiles TA et al Cutting Edge: Nonobese Diabetic Mice Deficient in Chromogranin A Are Protected from Autoimmune Diabetes. J Immunol 2016; 196:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A 2010; 107:10978–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou X, Bailey‐Bucktrout SL, Jeker LT et al Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo . Nat Immunol 2009; 10:1000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Whitfield‐Larry F, Young EF, Talmage G et al HLA‐A2‐matched peripheral blood mononuclear cells from type 1 diabetic patients, but not nondiabetic donors, transfer insulitis to NOD‐scid/gammac(null)/HLA‐A2 transgenic mice concurrent with the expansion of islet‐specific CD8+ T cells. Diabetes 2011; 60:1726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan S, Li Y, Xia J et al Type 1 diabetes induction in humanized mice. Proc Natl Acad Sci U S A 2017; 114:10954–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cohrs CM, Chen C, Jahn SR et al Vessel Network Architecture of Adult Human Islets Promotes Distinct Cell‐Cell Interactions In Situ and Is Altered After Transplantation. Endocrinology 2017; 158:1373–85. [DOI] [PubMed] [Google Scholar]
- 22. Brissova M, Fowler MJ, Nicholson WE et al Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 2005; 53:1087–97. [DOI] [PubMed] [Google Scholar]
- 23. Hsueh B, Burns VM, Pauerstein P et al Pathways to clinical CLARITY: volumetric analysis of irregular, soft, and heterogeneous tissues in development and disease. Sci Rep 2017; 7:5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marciniak A, Cohrs CM, Tsata V et al Using pancreas tissue slices for in situ studies of islet of Langerhans and acinar cell biology. Nat Protoc 2014; 9:2809–22. [DOI] [PubMed] [Google Scholar]
- 25. Chatenoud L, Thervet E, Primo J, Bach JF. Anti‐CD3 antibody induces long‐term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A 1994; 91:123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lenschow DJ, Ho SC, Sattar H et al Differential effects of anti‐B7‐1 and anti‐B7‐2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med 1995; 181:1145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kachapati K, Adams D, Bednar K, Ridgway WM. The non‐obese diabetic (NOD) mouse as a model of human type 1 diabetes. Methods Mol Biol 2012; 933:3–16. [DOI] [PubMed] [Google Scholar]
- 28. Ablamunits V, Henegariu O, Hansen JB et al Synergistic reversal of type 1 diabetes in NOD mice with anti‐CD3 and interleukin‐1 blockade: evidence of improved immune regulation. Diabetes 2012; 61:145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grinberg‐Bleyer Y, Baeyens A, You S et al IL‐2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 2010; 207:1871–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takiishi T, Cook DP, Korf H et al Reversal of Diabetes in NOD Mice by Clinical‐Grade Proinsulin and IL‐10‐Secreting Lactococcus lactis in Combination With Low‐Dose Anti‐CD3 Depends on the Induction of Foxp3‐Positive T Cells. Diabetes 2017; 66:448–59. [DOI] [PubMed] [Google Scholar]
- 31. Hagopian W, Ferry RJ Jr, Sherry N et al Teplizumab preserves C‐peptide in recent‐onset type 1 diabetes: two‐year results from the randomized, placebo‐controlled Protege trial. Diabetes 2013; 62:3901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herold KC, Gitelman SE, Ehlers MR et al Teplizumab (anti‐CD3 mAb) treatment preserves C‐peptide responses in patients with new‐onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013; 62:3766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haller MJ, Gitelman SE, Gottlieb PA et al Antithymocyte Globulin Plus G‐CSF Combination Therapy Leads to Sustained Immunomodulatory and Metabolic Effects in a Subset of Responders With Established Type 1 Diabetes. Diabetes 2016; 65:3765–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haller MJ, Schatz DA, Skyler JS et al Diabetes TrialNet ATGGSG. Low‐Dose Anti‐Thymocyte Globulin (ATG) Preserves beta‐Cell Function and Improves HbA1c in New‐Onset Type 1 Diabetes. Diabetes Care 2018; 41:1917–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haller MJ, Long SA, Blanchfield JL et al Low‐Dose Anti‐Thymocyte Globulin Preserves C‐Peptide, Reduces HbA1c, and Increases Regulatory to Conventional T‐Cell Ratios in New‐Onset Type 1 Diabetes: Two‐Year Clinical Trial Data. Diabetes 2019; 68:1267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Orban T, Bundy B, Becker DJ et al Diabetes TrialNet Abatacept Study G. Costimulation modulation with abatacept in patients with recent‐onset type 1 diabetes: follow‐up 1 year after cessation of treatment. Diabetes Care 2014; 37:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rigby MR, Harris KM, Pinckney A et al Alefacept provides sustained clinical and immunological effects in new‐onset type 1 diabetes patients. J Clin Invest 2015; 125:3285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Herold KC, Bundy BN, Long SA et al An Anti‐CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 1965; 14:619–33. [DOI] [PubMed] [Google Scholar]
- 40. Campbell‐Thompson ML, Kaddis JS, Wasserfall C et al The influence of type 1 diabetes on pancreatic weight. Diabetologia 2016; 59:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Williams AJ, Thrower SL, Sequeiros IM et al Pancreatic volume is reduced in adult patients with recently diagnosed type 1 diabetes. J Clin Endocrinol Metab 2012; 97:E2109–113. [DOI] [PubMed] [Google Scholar]
- 42. Campbell‐Thompson M, Wasserfall C, Kaddis J et al Network for Pancreatic Organ Donors with Diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev 2012; 28:608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Exeter Archival Diabetes Biobank . 2015. Available at: http://foulis.vub.ac.be (accessed June 6, 2019).
- 44. Krogvold L, Edwin B, Buanes T et al Detection of a Low‐Grade Enteroviral Infection in the Islets of Langerhans of Living Patients Newly Diagnosed With Type 1 Diabetes. Diabetes 2015; 64:1682–7. [DOI] [PubMed] [Google Scholar]
- 45. Human Pancreas Analysis Program . 2018. Available at: https://hpap.pmacs.upenn.edu (accessed June 6, 2019).
- 46. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009; 155:173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Campbell‐Thompson M, Fu A, Kaddis JS et al Insulitis and beta‐Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 2016; 65:719–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Michels AW, Landry LG, McDaniel KA et al Islet‐Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes 2017; 66:722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leete P, Willcox A, Krogvold L et al Differential Insulitic Profiles Determine the Extent of beta‐Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes 2016; 65:1362–9. [DOI] [PubMed] [Google Scholar]
- 50. Arif S, Leete P, Nguyen V et al Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 2014; 63:3835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. In't Veld P, Lievens D, De Grijse J et al Screening for insulitis in adult autoantibody‐positive organ donors. Diabetes 2007; 56:2400–4. [DOI] [PubMed] [Google Scholar]
- 52. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Evidence of increased islet cell proliferation in patients with recent‐onset type 1 diabetes. Diabetologia 2010; 53:2020–8. [DOI] [PubMed] [Google Scholar]
- 53. Battaglia M, Anderson MS, Buckner JH et al Understanding and preventing type 1 diabetes through the unique working model of TrialNet. Diabetologia 2017; 60:2139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Richardson SJ, Rodriguez‐Calvo T, Gerling IC et al Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 2016; 59:2448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moore F, Naamane N, Colli ML et al STAT1 is a master regulator of pancreatic {beta}‐cell apoptosis and islet inflammation. J Biol Chem 2011; 286:929–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Coppieters KT, Dotta F, Amirian N et al Demonstration of islet‐autoreactive CD8 T cells in insulitic lesions from recent onset and long‐term type 1 diabetes patients. J Exp Med 2012; 209:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Babon JA, DeNicola ME, Blodgett DM et al Analysis of self‐antigen specificity of islet‐infiltrating T cells from human donors with type 1 diabetes. Nat Med 2016; 22:1482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seay HR, Yusko E, Rothweiler SJ et al Tissue distribution and clonal diversity of the T and B cell repertoire in type 1 diabetes. JCI Insight 2016; 1:e88242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pathiraja V, Kuehlich JP, Campbell PD et al Proinsulin‐specific, HLA‐DQ8, and HLA‐DQ8‐transdimer‐restricted CD4+ T cells infiltrate islets in type 1 diabetes. Diabetes 2015; 64:172–82. [DOI] [PubMed] [Google Scholar]
- 60. Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res 2012; 22:1748–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wallet MA, Santostefano KE, Terada N, Brusko TM. Isogenic Cellular Systems Model the Impact of Genetic Risk Variants in the Pathogenesis of Type 1 Diabetes. Front Endocrinol (Lausanne) 2017; 8:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ramos‐Rodríguez M, Raurell‐Villa H, Colli ML et al The impact of pro‐inflammatory cytokines on the β‐cell regulatory landscape provides new insights into the genetics of type 1 diabetes . BioRxiv 2019; 560193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nogueira TC, Paula FM, Villate O et al GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3‐only protein Bim. PLoS Genet 2013; 9:e1003532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Juan‐Mateu J, Alvelos MI, Turatsinze JV et al SRp55 Regulates a Splicing Network That Controls Human Pancreatic beta‐Cell Function and Survival. Diabetes 2018; 67:423–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Marhfour I, Lopez XM, Lefkaditis D et al Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012; 55:2417–20. [DOI] [PubMed] [Google Scholar]
- 66. Brozzi F, Eizirik DL. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Ups J Med Sci 2016; 121:133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 2013; 56:234–41. [DOI] [PubMed] [Google Scholar]
- 68. Kracht MJ, van Lummel M, Nikolic T et al Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med 2017; 23:501–7. [DOI] [PubMed] [Google Scholar]
- 69. Richardson SJ, Leete P, Bone AJ, Foulis AK, Morgan NG. Expression of the enteroviral capsid protein VP1 in the islet cells of patients with type 1 diabetes is associated with induction of protein kinase R and downregulation of Mcl‐1. Diabetologia 2013; 56:185–93. [DOI] [PubMed] [Google Scholar]
- 70. Ifie E, Russell MA, Dhayal S et al Unexpected subcellular distribution of a specific isoform of the Coxsackie and adenovirus receptor, CAR‐SIV, in human pancreatic beta cells. Diabetologia 2018; 61:2344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hodik M, Skog O, Lukinius A et al Enterovirus infection of human islets of Langerhans affects beta‐cell function resulting in disintegrated islets, decreased glucose stimulated insulin secretion and loss of Golgi structure. BMJ Open Diabetes Res Care 2016; 4:e000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Domsgen E, Lind K, Kong L et al An IFIH1 gene polymorphism associated with risk for autoimmunity regulates canonical antiviral defence pathways in Coxsackievirus infected human pancreatic islets. Sci Rep 2016; 6:39378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sioofy‐Khojine AB, Oikarinen S, Honkanen H, Huhtala H, Lehtonen JP, Briese T, Hyoty H, Group TS . Molecular epidemiology of enteroviruses in young children at increased risk of type 1 diabetes. PLoS ONE 2018; 13:e0201959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. West AP. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017; 391:54–63. [DOI] [PubMed] [Google Scholar]
- 75. Gagnerault MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J Exp Med 2002; 196:369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Willcox A, Richardson SJ, Walker LSK, Kent SC, Morgan NG, Gillespie KM. Germinal centre frequency is decreased in pancreatic lymph nodes from individuals with recent‐onset type 1 diabetes. Diabetologia 2017; 60:1294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ferreira RC, Simons HZ, Thompson WS et al IL‐21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia 2015; 58:781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Stamatouli AM, Quandt Z, Perdigoto AL et al Collateral Damage: Insulin‐Dependent Diabetes Induced With Checkpoint Inhibitors. Diabetes 2018; 67:1471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Colli ML, Hill JLE, Marroqui L et al PDL1 is expressed in the islets of people with type 1 diabetes and is up‐regulated by interferons‐α and‐γ via IRF1 induction . EBioMedicine 2018; 36:367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Uno S, Imagawa A, Saisho K et al Expression of chemokines, CXC chemokine ligand 10 (CXCL10) and CXCR80 in the inflamed islets of patients with recent‐onset autoimmune type 1 diabetes. Endocr J 2010; 57:991–6. [DOI] [PubMed] [Google Scholar]
- 81. Roep BO, Kleijwegt FS, van Halteren AG et al Islet inflammation and CXCL10 in recent‐onset type 1 diabetes. Clin Exp Immunol 2010; 159:338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li X, Campbell‐Thompson M, Wasserfall CH et al Serum Trypsinogen Levels in Type 1 Diabetes. Diabetes Care 2017; 40:577–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Madole MB, Iyer CM, Madivalar MT. Wadde SK, Howale DS. Evaluation of Biochemical Markers Serum Amylase and Serum Lipase for the Assessment of Pancreatic Exocrine Function in Diabetes Mellitus. J Clin Diagn Res 2016; 10:BC01–BC4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rowe P, Wasserfall C, Croker B et al Increased complement activation in human type 1 diabetes pancreata. Diabetes Care 2013; 36:3815–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Campbell‐Thompson M, Rodriguez‐Calvo T, Battaglia M. Abnormalities of the Exocrine Pancreas in Type 1 Diabetes. Curr Diab Rep 2015; 15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dolensek J, Rupnik MS, Stozer A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015; 7:e1024405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ariel A, Lider O, Brill A et al Induction of interactions between CD44 and hyaluronic acid by a short exposure of human T cells to diverse pro‐inflammatory mediators. Immunology 2000; 100:345–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mahaffey CL, Mummert ME. Hyaluronan synthesis is required for IL‐2‐mediated T cell proliferation. J Immunol 2007; 179:8191–9. [DOI] [PubMed] [Google Scholar]
- 89. Bollyky PL, Evanko SP, Wu RP et al Th1 cytokines promote T‐cell binding to antigen‐presenting cells via enhanced hyaluronan production and accumulation at the immune synapse. Cell Mol Immunol 2010; 7:211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bogdani M, Johnson PY, Potter‐Perigo S et al Hyaluronan and hyaluronan‐binding proteins accumulate in both human type 1 diabetic islets and lymphoid tissues and associate with inflammatory cells in insulitis. Diabetes 2014; 63:2727–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nagy N, Kuipers HF, Frymoyer AR et al 4‐methylumbelliferone treatment and hyaluronan inhibition as a therapeutic strategy in inflammation, autoimmunity, and cancer. Front Immunol 2015; 6:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Reinert RB, Brissova M, Shostak A et al Vascular endothelial growth factor‐a and islet vascularization are necessary in developing, but not adult, pancreatic islets. Diabetes 2013; 62:4154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Canzano JS, Nasif LH, Butterworth EA, Fu DA, Atkinson MA, Campbell‐Thompson M. Islet Microvasculature Alterations With Loss of Beta‐cells in Patients With Type 1 Diabetes. J Histochem Cytochem 2018; 22155418778546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Caicedo A. Paracrine and autocrine interactions in the human islet: more than meets the eye. Semin Cell Dev Biol 2013; 24:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Brissova M, Haliyur R, Saunders D et al α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep 2018; 22:2667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rodriguez‐Diaz R, Abdulreda MH, Formoso AL et al Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab 2011; 14:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Mundinger TO, Mei Q, Foulis AK, Fligner CL, Hull RL, Taborsky GJ Jr. Human Type 1 Diabetes Is Characterized by an Early, Marked, Sustained, and Islet‐Selective Loss of Sympathetic Nerves. Diabetes 2016; 65:2322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Braun M, Ramracheya R, Bengtsson M et al γ‐aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta‐cells. Diabetes 2010; 59:1694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tian J, Dang H, Middleton B, Kaufman DL. Clinically applicable GABA receptor positive allosteric modulators promote ss‐cell replication. Sci Rep 2017; 7:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Barragan A, Weidner JM, Jin Z, Korpi ER, Birnir B. GABAergic signalling in the immune system. Acta Physiol (Oxf) 2015; 213:819–27. [DOI] [PubMed] [Google Scholar]
- 101. Bhandage AK, Jin Z, Korol SV et al GABA Regulates Release of Inflammatory Cytokines From Peripheral Blood Mononuclear Cells and CD4(+) T Cells and Is Immunosuppressive in Type 1 Diabetes. EBioMedicine 2018; 30:283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ben‐Othman N, Vieira A, Courtney M et al Long‐Term GABA Administration Induces Alpha Cell‐Mediated Beta‐like Cell Neogenesis. Cell 2017; 168:73–85.e11. [DOI] [PubMed] [Google Scholar]
- 103. Ackermann AM, Moss NG, Kaestner KH. GABA and Artesunate Do Not Induce Pancreatic α‐to‐β Cell Transdifferentiation in vivo . Cell Metab 2018; 28:787–92.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Picelli S. Single‐cell RNA‐sequencing: The future of genome biology is now. RNA Biol 2017; 14:637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Baron M, Veres A, Wolock SL et al A Single‐Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter‐ and Intra‐cell Population Structure. Cell Syst 2016; 3:346–60.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ackermann AM, Wang Z, Schug J, Naji A, Kaestner KH. Integration of ATAC‐seq and RNA‐seq identifies human alpha cell and beta cell signature genes. Mol Metab 2016; 5:233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Preissl S, Fang R, Huang H et al Single‐nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell‐type‐specific transcriptional regulation. Nat Neurosci 2018; 21:432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nyalwidhe JO, Grzesik WJ, Burch TC et al Comparative quantitative proteomic analysis of disease stratified laser captured microdissected human islets identifies proteins and pathways potentially related to type 1 diabetes. PLoS ONE 2017; 12:e0183908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zhu Y, Piehowski PD, Zhao R et al Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat Commun 2018; 9:882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Giesen C, Wang HA, Schapiro D et al Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods 2014; 11:417–22. [DOI] [PubMed] [Google Scholar]
- 111. Damond N, Engler S, Zanotelli VRT et al A Map of Human Type 1 Diabetes Progression by Imaging Mass Cytometry. Cell Metab 2019; 29:755–68.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wang YJ, Traum D, Schug J, Gao L, Liu C, Atkinson MA, Powers AC, Feldman MD, Naji A, Chang KM, Kaestner KH. Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab 2019; 29:769–83.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Goltsev Y, Samusik N, Kennedy‐Darling J et al Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018; 174:968–81.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Yang B, Treweek JB, Kulkarni RP et al Single‐cell phenotyping within transparent intact tissue through whole‐body clearing. Cell 2014; 158:945–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Shah S, Lubeck E, Schwarzkopf M et al Single‐molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing. Development 2016; 143:2862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lenguito G, Chaimov D, Weitz JR et al Resealable, optically accessible, PDMS‐free fluidic platform for ex vivo interrogation of pancreatic islets. Lab Chip 2017; 17:772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Liang T, Dolai S, Xie L et al Ex vivo human pancreatic slice preparations offer a valuable model for studying pancreatic exocrine biology. J Biol Chem 2017; 292:5957–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bosco D, Armanet M, Morel P et al Unique arrangement of alpha‐ and beta‐cells in human islets of Langerhans. Diabetes 2010; 59:1202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Fowler JL, Lee SS, Wesner ZC, Olehnik SK, Kron SJ, Hara M. Three‐Dimensional Analysis of the Human Pancreas. Endocrinology 2018; 159:1393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Steiner DJ, Kim A, Miller K, Hara M. Pancreatic islet plasticity: interspecies comparison of islet architecture and composition. Islets 2010; 2:135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kaestner KH, Powers AC, Naji A, Atkinson MA. NIH Initiative to Improve Understanding of the Pancreas, Islet, and Autoimmunity in Type 1 Diabetes: The Human Pancreas Analysis Program (HPAP). Diabetes 2019; 68:1394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Helmsley Charitable Trust . Available at: https://helmsleytrust.org (accessed June 6, 2019).