

Summary
Psoriasis is a common, inflammatory immune‐mediated skin disease mainly presenting with plaques whose pathogenesis is based on the central role of the interleukin (IL)‐23/IL‐17 axis. However, the mechanisms acting in papular lesions of early‐phase psoriasis are not fully understood. The aim of this study was to assess the involvement of autoinflammation, a state of sterile inflammation mainly driven by IL‐1 over‐production that has been recently hypothesized to act in the early phase of disease. Lesional skin of 10 patients with recent onset, untreated psoriasis has been investigated for expression of IL‐1β, IL‐17, IL‐23 and other cytokines involved in the disease in comparison with normal skin of 10 healthy controls using a protein array method. Immunohistochemical phenotyping of inflammatory infiltrate and co‐localization experiments with immunofluorescence confocal microscopy were conducted. IL‐1β was significantly more expressed in psoriasis than in normal skin (P < 0·0001). The chemokine IL‐8 was also over‐expressed in psoriasis (P = 0·03) while IL‐12, IL‐17, IL‐23, tumour necrosis factor‐α and interferon‐γ were only slightly more expressed in psoriasis than in normal skin, without reaching statistical significance. The inflammatory infiltrate consisted mainly of neutrophils with a relevant number of macrophages and dendritic cells and only scattered, predominantly T helper 1 lymphocytes. IL‐1β co‐localized mainly with CD66b, a neutrophil marker, suggesting that neutrophils were the major source of this cytokine. IL‐1β over‐expression in combination with low expression of cytokines that are predominant in late‐phase plaque psoriasis may support the role of autoinflammation in early‐phase disease, possibly paving the way to randomized trials with IL‐1 antagonists.
Keywords: autoinflammatory disease, cytokines, neutrophils, skin
Lesional skin of early‐phase psoriasis is hallmarked by interleukin IL‐1β over‐expression and low expression of both Th1‐and Th17‐related cytokines confirmed by immunohistochemical results. The major source of IL‐1β is represented by neutrophils as highlighted by confocal analysis. Over‐production of IL‐1β supports the role of autoinflammation in early‐phase psoriasis and pave the way to design‐controlled trials with IL‐1 antagonists.

Introduction
Psoriasis is a common, inflammatory immune‐mediated disease mainly affecting the skin and joints, that is genetically determined, influenced by epigenetic mechanisms and triggered by endogenous and exogenous factors 1, 2. Clinical phenotypes vary from stable plaque‐type psoriasis to early disease, manifesting mainly as papules or highly active flares presenting with pustular or papulopustular lesions 3.
The pathogenesis of the disease has now been deeply revised, switching from the classic view focused on the T helper type 1 (Th1) profile to the pathophysiological model based on the central role of the interleukin (IL)‐23/IL‐17 axis 4, 5, 6.
Pathophysiological studies are mainly conducted in stable plaque‐type psoriasis that is hallmarked by a predominant Th1 paradigm, while papular/papulopustular lesions of early‐phase psoriasis or highly inflammatory disease flares have only rarely been investigated 7. Indeed, although psoriasis prevalence is high (approximately 2% in Europe and North America) 8, approximately 70–80% of patients have mild psoriasis at onset that is controlled using topical therapies alone by dermatologists in private practice and general practitioners 9, making enrolling patients difficult for pathophysiological assessments in early phases of the disease.
Recently, some authors have pointed out the possible involvement of autoinflammation in the early phases of psoriasis 7, 10. Autoinflammation is a state of sterile inflammation that is driven mainly by IL‐1 over‐production and generally caused by mutations of genes regulating the innate immune response 11.
In order to characterize the cytokine profile of early‐phase psoriasis, we enrolled 10 patients with recent‐onset, untreated disease, assessing the lesional skin expression of IL‐1β, IL‐17, IL‐23 and the other cytokines mainly involved in the psoriasis pathogenesis by means of a protein array method. We found that IL‐1β is over‐expressed in lesional skin of early psoriasis and, using immunofluorescence confocal microscopy, demonstrated that innate immunity cells such as neutrophils are the main source of this cytokine, supporting the role of autoinflammation and making it conceivable to design trials with IL‐1 blockers in the early phases of the disease.
Patients and methods
Skin biopsies were obtained from 10 patients with early‐phase psoriasis (eight men and two women; mean age = 55·2 years, range = 33–74 years).
On clinical examination, all the patients had psoriasis manifesting as papular lesions on different sites of the body, which had been present for no more than 1 month; plaque‐type lesions were not visible. The mean values of psoriasis area and severity index (PASI), body surface area (BSA) and physician global assessment (PGA) were 9·6 (range = 6–14), 7% (range = 5–10%) and 2 (range = 1–3), respectively.
None of the patients had ever undergone any systemic treatment for psoriasis and the above lesions had not been treated with neither topical corticosteroids or other local agents for psoriasis.
Skin biopsies were taken from patients’ psoriasis lesional skin and divided into two specimens: the first was processed for routine histopathological examination, immunohistochemistry and immunofluorescence confocal microscopy studies, while the second was immediately frozen and stored in liquid nitrogen for protein array assessment. Histopathological findings were combined with clinical assessments for confirming the diagnosis of psoriasis and are not described in detail in the Results section.
Normal skin tissue specimens taken from 10 patients who underwent excision of benign skin tumours served as controls for the protein array studies.
Immunohistochemistry was used for phenotyping the psoriasis inflammatory infiltrate and for confirming the most significant findings of cytokine expression obtained on protein array. Co‐localization experiments were conducted using confocal microscopy, with the aim of identifying the cell populations potentially involved in releasing the main cytokines found over‐expressed on cytokine array.
The protocol was approved by Ethics Committee of IRCCS Ca’ Granda Ospedale Maggiore Policlinico of Milan and all subjects gave their informed consent before participating in the study.
Protein array
Each tissue sample was weighed and diced into very small pieces using a clean razor‐blade. Frozen tissue was sliced very thinly and thawed in radioimmunoprecipitation assay (RIPA) buffer (sc‐24948; Sigma‐Aldrich, St Louis, MO, USA) containing protease‐ and phosphatase‐inhibitors using 3 ml of ice‐cold RIPA buffer per gram of tissue. Samples were incubated on ice for 30 min, transferred to microcentrifuge tubes and centrifuged at 10 000 g for 10 min at 4°C. The supernatant was collected and the sample was centrifuged again. The new supernatant fluid was added to the previous one, this mixture representing the total cell lysate. In order to standardize the cell lysate of each tissue sample, we measured the total proteins in each sample using a microBCA kit (Thermo Scientific, Waltham, MA, USA). For each sample, we loaded a volume containing 100 µg of proteins in a glass‐slide format of cytokine antibody array (RayBio®, Norcross, GA, USA). The volume to be loaded was calculated by the following formula: volume (expressed in µl) = 100 µg/protein concentration (expressed in µg/µl). Each glass‐slide array contained 14 subarrays and was suitable for 14 samples. Each subarray allowed the evaluation of cytokine expression levels in a sample. Normalization of data at the end of the experiment provided semiquantitative results. The subarray was composed by specific antibodies against target molecules coated on the glass slide. After hybridization of the tissue lysate, each antibody bound its target molecule and unbound proteins were washed out. The slide was then incubated with biotin‐conjugated antibodies against the same target cytokines, washed and then incubated with cyanine (Cy)3‐conjugated streptavidin, creating a biotin–streptavidin‐Cy3 complex detectable using a microarray laser scanner. Using data extraction software, we could transform fluorescent signals into numerical data and, after normalization, we obtained an expression value of signal intensity for each molecule in each sample.
The molecules tested were: IL‐1β, IL‐8, IL‐12, IL‐17, IL‐23, tumour necrosis factor (TNF)‐α and interferon gamma (IFN)‐γ.
Immunoistochemistry
In order to define which cells were the most representative in psoriasis inflammatory infiltrate, formalin‐fixed paraffin‐embedded (FFPE) tissue of each psoriasis lesion biopsy was sectioned.
After deparaffining and rehydrating, each tissue section was immersed in a retrieval buffer and boiled three times for 5 min in a pressure cooker, then washed with TRIS‐buffered saline (TBS) and incubated with the specific monoclonal antibody at room temperature for 45 min. Secondary antibodies used were biotinylated goat anti‐mouse and anti‐rabbit immunoglobulins (Dako REAL™, code K5005; DakoCytomation, Glostrup, Denmark) incubated at room temperature for 30 min. After incubation with the secondary antibody and another washing with TBS, pH 7·6, the sections were incubated with streptavidin conjugated with alkaline phosphatase (Dako REAL™, code K5005; DakoCytomation) at room temperature for 30 min. We used specific monoclonal antibodies to CD14 (EPR36; Abcam, Cambridge UK), CD163 (10D6; Leica Biosystems Newcastle Ltd, Newcastle upon Tyne, UK), CD11c (5D11; Leica Biosystems Newcastle Ltd), CD123 (9F5; BD Pharmingen, Franklin Lakes, NJ, USA); CD3 (polyclonal rabbit; DakoCytomation); CD4 (4B12, DakoCytomation); T‐bet (polyclonal rabbit; SantaCruz Biotechnology, Santa Cruz, CA, USA); IL‐1β (rabbit polyclonal; Abcam); IL‐17 (41802; R&D Systems, Minneapolis, MN, USA), TNF‐α (52B83; Monosan, Uden, the Netherlands) and IFN‐γ (IFNG/466; Abcam).
A red chromogen solution was prepared as indicated by the Dako REAL™ datasheet and used as an enzyme substrate, followed by counterstaining with Mayer’s haematoxylin. After air‐drying, each section was coverslipped using the VectaMount™ mounting medium (Vector Laboratories, Burlingame, CA, USA). A negative control was performed using a pool of mouse immunoglobulins (IgG1, IgG2a, IgG2b and IgM) as primary antibodies (negative control; Dako Cytomation).
Immunofluorescence confocal laser microscopy
After deparaffining and antigen retrieval, paraffin sections were treated briefly with 0·1 M glycine in phosphate‐buffered saline (PBS) pH7·4 followed by a buffer with 0·3% Triton X‐100 and incubated overnight at 4°C with the primary antibodies, namely IL‐1β (rabbit polyclonal; Abcam), CD163 (10D6 Leica Biosystems Newcastle Ltd), CD68 (PGM1; DakoCytomation), CD66b (G10F5; US Biologica, Swampscott, MA, USA) and CD1a, Mab010; DakoCytomation). The samples were washed and incubated for 1 h with appropriate conjugated secondary antibodies (Alexa Fluor donkey anti‐mouse 488 and Alexa Fluor donkey anti‐rabbit 555; Invitrogen/Thermo Fisher Scientific). The nuclei were counterstained with Toto‐3. Slides were mounted on glass slides with 95% glycerol in PBS.
Epifluorescence scanning images were acquired using a motorized Olympus BX63 fluorescence microscope equipped with the X‐cite 120 fluorescence illumination system (EXFO, Quebec, Canada), DP80 camera and software cellSens (Shinjuku Monolith, Tokyo, Japan). Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI).
Confocal microscopy was carried out using a Zeiss LSM 710 confocal microscope (GmbH 07745; Jena, Germany) equipped with a 458‐, 488‐, 514‐nm multiline argon laser, 561‐nm diode pumped solid state laser and a 633‐nm HeNe laser.
Statistics
Because the signal intensity data were positively skewed, they were log‐transformed before analysis.
The results are reported as anti‐log values of means with standard deviation (s.d.). Student’s t‐test for unpaired values was used to assess the statistical significance of differences between psoriasis and normal skin in the protein array studies. The significance level was set at P < 0·05.
Results
Cytokine expression
IL‐1β was significantly more expressed in psoriasis lesional skin (21·86 ± 1·74) than in normal skin (11·75 ± 1·78; P < 0·0001) (Fig. 1). The chemokine IL‐8 was also over‐expressed in psoriasis (21·56 ± 4·23 versus 17·25 ± 3·76; P = 0·03).
Figure 1.
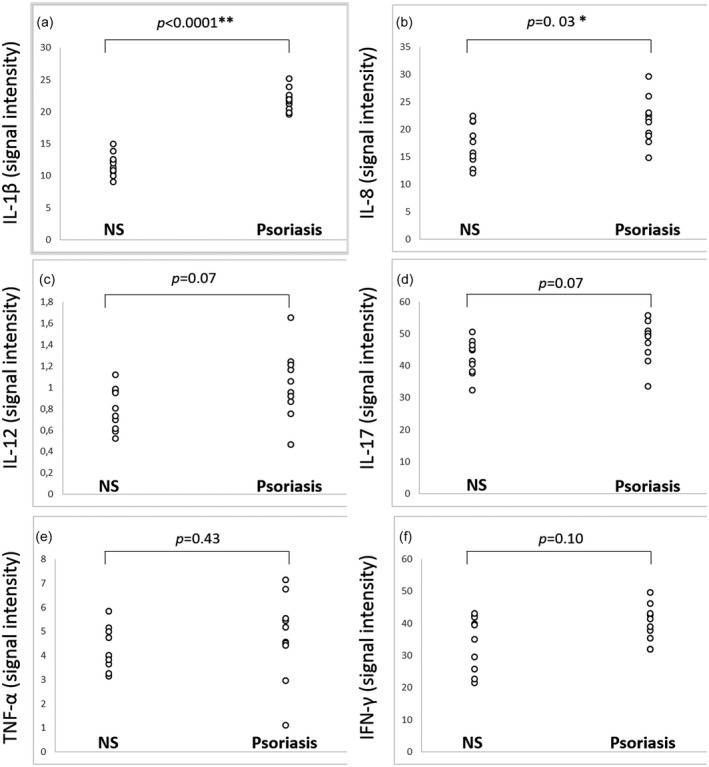
(a,b) Interleukin (IL)‐1β and IL‐8 in homogenate samples of psoriasis lesional skin of 10 patients, showing a statistically significant higher expression than in normal skin (NS) of 10 controls. (c–f) Expression levels of IL‐12, IL‐17, tumour necrosis factor (TNF)‐α and interferon (IFN) ‐γ are higher in psoriasis than in NS, but without reaching statistical significance. Signal intensity values for each cytokine were obtained transforming fluorescent signals into numerical data by means of a data extraction software.
IL‐12, IL‐17, IL‐23, TNF‐α and IFN‐γ were slightly more expressed in psoriasis (1·03 ± 0·32, 47·62 ± 6·45, 17·36 ± 2·54, 4·77 ± 1·75 and 39·86 ± 5·84, respectively) than in normal skin (0·80 ± 0·20, 42·51 ± 5·48, 14·95 ± 3·98, 4·27 ± 0·88 and 34·15 ± 8·54, respectively), without reaching statistical significance (P = 0·07, P = 0·07, P = 0·12, P = 0·43 and P = 0·10, respectively).
Immunohistochemical results
Phenotyping the psoriasis inflammatory infiltrate revealed abundant macrophages expressing CD11b, CD14 (Fig. 2a) and CD163 and dendritic cells expressing CD11c in the upper dermis; CD11c+ cells were predominantly present in the papillary dermis (Fig. 2b). Plasmacytoid dendritic cells were also well represented in superficial dermis, where they showed a perivascular distribution and tendency to form clusters, as detected by intense staining with the anti‐CD123 monoclonal antibody. (Fig. 2c) Only a few CD3+/CD4+ T lymphocytes (< 5% of the inflammatory infiltrate) were seen in the upper dermis; the majority of these cells displayed a Th1 profile, as demonstrated by the positivity of T‐bet, the master regulator of Th1 cells. An intense upper dermal staining with the anti‐IL‐1β monoclonal antibody was found; scanty intra‐epidermal IL‐1β+ cells were also evident (Fig. 2d).
Figure 2.
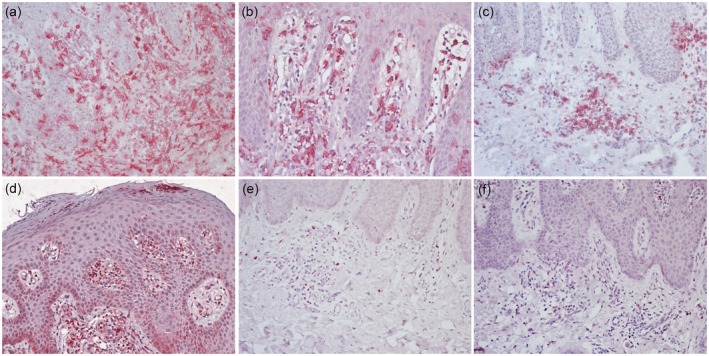
Immunohistochemical analysis of the inflammatory infiltrate and cytokine expression assessment in early‐phase psoriasis: (a) numerous macrophages expressing CD14 are evident in the superficial and papillary dermis. (b) CD11c+ dendritic cells are visible both in the dermis and epidermis. (c) Plasmacytoid dendritic cells, stained with anti‐CD123 monoclonal antibody, are abundantly represented in the dermis, particularly around vessels and at the top of dermal papillae. (d) Strong dermal expression of interleukin (IL)‐1β. (e) Scattered cells are positive with the anti‐IL‐17 monoclonal antibody. (f) Low dermal expression of interferon (IFN)‐γ.
Only a few cells expressing IL‐17 (Fig. 2e), TNF‐α and IFN‐γ (Fig. 2f) were revealed in the superficial dermis.
Confocal microscopy findings
In order to identify the cells that were IL‐1β producers in early‐phase psoriasis, double immunofluorescence staining was performed on FFPE tissues and analysed by means of confocal laser microscopy.
Immunofluorescence stainings were performed for IL‐1β in combination with CD66b, CD1a and a mixture of CD68/CD163 to highlight neutrophils, dendritic cells and macrophages, respectively.
The major number of double‐stained cells were seen combining IL‐1β and CD66b and were present both in the dermis and epidermis, supporting that neutrophils were the main source of this cytokine in early‐phase psoriasis lesional skin (Fig. 3). Nevertheless, some cells showing positivity only for IL‐1β and not for CD66b were visible in the upper dermal inflammatory infiltrate and at the level of both subcorneal and basal epidermis, suggesting that cell populations other than neutrophils were possibly IL‐1β‐producers.
Figure 3.
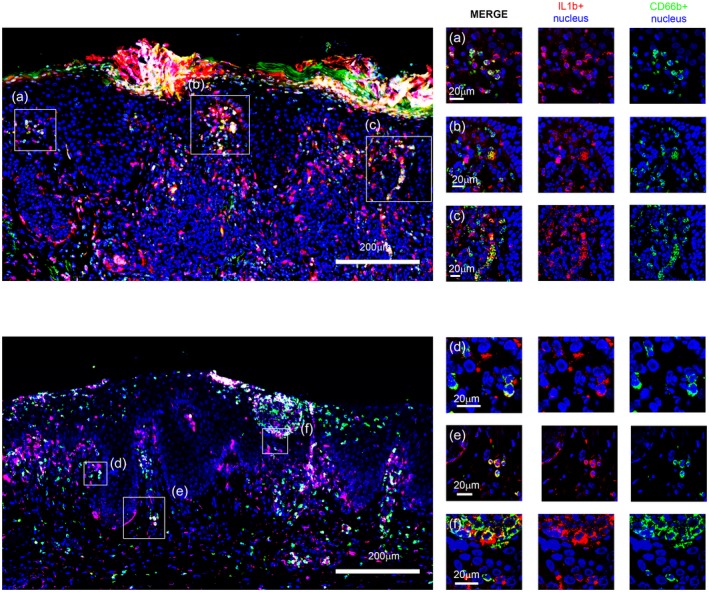
Immunofluorescence staining (left panel) and confocal analysis (right panel) for interleukin (IL)‐1β (red) and CD66b (green) in formalin‐fixed, paraffin‐embedded tissues of psoriasis lesional skin. Nuclei were stained with Toto‐3 (blue). Representative images at small magnification showing neutrophils both in the dermis and in the epidermis (left panels). CD66b+ neutrophils expressing IL‐1β are uniformly distributed in the skin (yellow). (a–f). Squared areas are visible at higher magnification in confocal laser scanning micrographs (right panels).
Assessing the nature of these cell populations, we found that several epidermal and dermal cells were dendrocytes, as they were expressing both IL‐1β and CD1a (Fig. 4), while scattered dermal cells were macrophages on the basis of merging of IL‐1β and CD68/CD163 mixture (Fig. 5).
Figure 4.
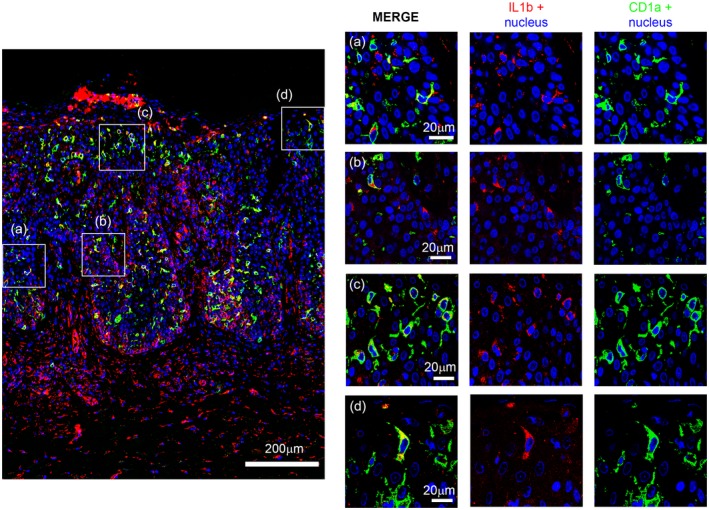
Scanning micrograph of immunofluorescent staining with CD1a (green) and interleukin (IL)‐1β (red) shows a large number of double positive cells (yellow). In the right panel confocal laser analysis shows in detail some double‐labelled cells.
Figure 5.
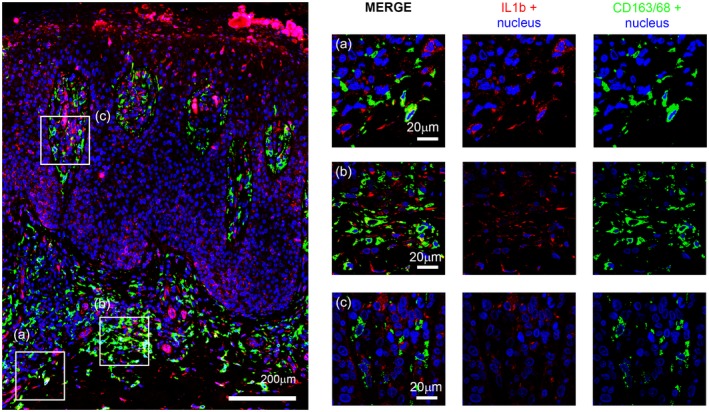
CD163/68 (green) and interleukin (IL)‐1β (red) double‐label immunofluorescence staining. Confocal analysis zoomed view in the right panel. High number of CD163‐positive cells, few of which express IL‐1β, are seen in the dermis.
Discussion
The most important finding of the present study was the over‐expression of IL‐1β in the lesional skin of patients with early‐phase psoriasis, as detected by means of cytokine array. Interestingly, neutrophils proved to be the major source of IL‐1β, as supported by the co‐localization experiments conducted on confocal laser microscopy using CD66b, a classic marker of neutrophils.
Histopathological and immunohistochemical examinations have revealed a high number of neutrophils in the upper dermal inflammatory infiltrate, with a significant presence of these cells also in the context of the epidermis, particularly beneath the corneum layer.
IL‐1β is a pivotal cytokine in the innate immune response, and it is well known that its over‐production plays a crucial role in the autoinflammation, which is a state of sterile inflammation not mediated by circulating autoantibodies and autoreactive T cells 7, 11. Our study may support the hypothesis that the clinical development of early lesions in psoriasis would be linked to periodic waves of autoinflammation, represented by a burst of neutrophils and cytokines related to the IL‐1 family 7. Some studies have demonstrated that other members of the IL‐family, including IL‐1α and IL‐36, may cross‐talk with IL‐1β in triggering and orchestrating the autoinflammatory process in psoriasis 12, 13, most notably in its pustular variants 14, 15, as in neutrophilic dermatoses that have been recently included in the spectrum of autoinflammatory diseases 16, 17, 18.
Indeed, the concept of autoinflammation can be extended beyond the group of monogenic periodic fever syndromes 19 and pyogenic arthritis, acne and pyoderma gangrenosum (PAPA) syndrome 20, which are the prototypical autoinflammatory diseases, as it has been suggested that different skin disorders, notably psoriasis, show a mixed pattern of inflammation with components of both adaptive and innate immunity. This view is also supported by the fact that innate immune cells such as neutrophils, macrophages and mast cells have been reported to be abundantly represented in the early‐phase inflammatory infiltrate of psoriasis 7.
It is well known that mutations in the genes encoding components of inflammasomes, which are molecular platforms responsible for processing IL‐1β via the caspase 1‐dependent cleavage of its inactive precursor, lead to dysregulation of the IL‐1 inflammatory pathway and the resulting monogenic autoinflammatory diseases 21.
Recently, gain‐of‐function mutations in caspase recruitment domain family member 14 (CARD14), an important mediator of nuclear factor kappa‐B (NF‐κB), have been reported to be involved in familial cases of psoriasis 22.
Thus, initially, psoriasis seems to reproduce the pathogenic model of both classic autoinflammatory diseases, such as PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum and acne), and neutrophilic dermatoses, such as non‐syndromic pyoderma gangrenosum, which are due to mutations of genes regulating the innate immunity and share with psoriasis the same cascade of cytokine release and cell activation 11, 17, 18. In fact, high levels of proinflammatory cytokines such as IL‐1 and the classic chemokine IL‐8, the latter found also over‐expressed in our study, have been revealed in lesional skin even of patients with non‐syndromic pyoderma gangrenosum 17, 18. Also, the link between psoriasis and different autoinflammatory conditions is given from the clinical ground, as the occurrence of a severe erythrodermic and pustular presentation of psoriasis is present in syndromes such as deficiency of IL‐1 receptor antagonist (DIRA) and deficiency of IL‐36 receptor antagonist (DITRA) 14, 15.
Another important result of our study was the low expression of cytokines such as TNF‐α, IFN‐γ, IL‐12, IL‐23 and IL‐17, as detected using both cytokine array and immunohistochemistry. The role of IL‐17 in the pathophysiology of psoriasis is well recognized, and is also supported by a number of clinical trials successfully using antibodies targeting IL‐17. The low expression of IL‐17 seen in our study is in line with the recent concept of ‘bimodal immune activation’ 7, hypothesizing that the first autoinflammatory flare of psoriasis is followed by accumulation in the lesional skin of Th17 cells that produce mast cells, co‐operating with neutrophils, 23, 24 and γδT cells 25, proinflammatory cytokines such as IL‐17 and IL‐22 cross‐talking with the IL‐12/IL‐23 axis 26. According to this model 7, the late phase of the disease, clinically manifesting as plaque psoriasis, is characterized by the predominance of Th1 cells in the inflammatory infiltrate which, in contrast, are scanty in the early phase, and by over‐expression of Th1‐related cytokines, particularly TNF‐α and IFN‐γ. In our study, the immunohistochemical characterization of the early‐phase psoriasis inflammatory infiltrate showed only a low number of T lymphocytes, the majority of which displaying a Th1 profile. In contrast, innate immunity cells other than neutrophils, such as plasmacytoid dendritic cells and macrophages, were both well represented in the inflammatory infiltrate, possibly co‐operating with neutrophils in creating the IL‐1 cytokine milieu hallmarking the early stage of psoriasis. These findings on the phenotyping of the infiltrate fit well with the bimodal immune activation model of psoriasis.
The main limitation of the present study is the limited number of the assessed samples, which can, however, be counterbalanced by the difficulty in collecting patients with recent‐onset untreated psoriasis.
The IL‐1‐driven autoinflammatory state of early‐phase psoriasis could pave the way to controlled trials with IL‐1 antagonists, potentially blocking the following Th1/Th17‐mediated inflammatory cascade.
Disclosures
None to declare.
Author contributions
D. F., L. V., S. T. and A. V. M. designed the study. D. F. and A. V. M. wrote the paper and A. V. M. revised the overall intellectual content of the manuscript. D. F., L. V. and B. V. performed all the experiments. S. T., A. C., E. B. and A. V. M. performed the diagnosis and followed the patients. E. B. and B. E. L. revised all slides. All authors revised and approved the final version of the paper.
References
- 1. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509. [DOI] [PubMed] [Google Scholar]
- 2. Deng Y, Chang C, Lu Q. The inflammatory response in psoriasis: a comprehensive review. Clin Rev Allergy Immunol 2016; 50:377–89. [DOI] [PubMed] [Google Scholar]
- 3. Griffiths CE, Christophers E, Barker JN et al A classification of psoriasis vulgaris according to phenotype. Br J Dermatol 2007; 156:258–62. [DOI] [PubMed] [Google Scholar]
- 4. Krueger JG, Fretzin S, Suárez‐Fariñas M et al IL‐17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol 2012; 130:145–154.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lowes MA, Russell CB, Martin DA, Towne JE, Krueger JG. The IL‐23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol 2013; 34:174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martin DA, Towne JE, Kricorian G et al The emerging role of IL‐17 in the pathogenesis of psoriasis: preclinical and clinical findings. J Invest Dermatol 2013; 133:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Christophers E, Metzler G, Röcken M. Bimodal immune activation in psoriasis. Br J Dermatol 2014; 170:59–65. [DOI] [PubMed] [Google Scholar]
- 8. Christophers E. Psoriasis – epidemiology and clinical spectrum. Clin Exp Dermatol 2001; 26:314–20. [DOI] [PubMed] [Google Scholar]
- 9. Boehncke WH, Schön MP. Psoriasis. Lancet 2015; 386:983–94. [DOI] [PubMed] [Google Scholar]
- 10. Hedrich CM. Shaping the spectrum – from autoinflammation to autoimmunity. Clin Immunol 2016; 165:21–8. [DOI] [PubMed] [Google Scholar]
- 11. Kastner DL, Aksentijevich I, Goldbach‐Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell 2010; 140:784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Milora KA, Fu H, Dubaz O, Jensen LE. Unprocessed interleukin‐36α regulates psoriasis‐like skin inflammation in cooperation with interleukin‐1. J Invest Dermatol 2015; 135:2992–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnston A, Xing X, Wolterink L et al IL‐1 and IL‐36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol 2017; 140:109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aksentijevich I, Masters SL, Ferguson PJ et al An autoinflammatory disease with deficiency of the interleukin‐1‐receptor antagonist. N Engl J Med 2009; 360:2426–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marrakchi S, Guigue P, Renshaw BR et al Interleukin‐36‐receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med 2011; 365:620–8. [DOI] [PubMed] [Google Scholar]
- 16. Lukens JR, Kanneganti TD. SHP‐1 and IL‐1α conspire to provoke neutrophilic dermatoses. Rare Dis 2014; 2:e27742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marzano AV, Fanoni D, Antiga E et al Expression of cytokines, chemokines and other effector molecules in two prototypic autoinflammatory skin diseases, pyoderma gangrenosum and Sweet’s syndrome. Clin Exp Immunol 2014; 178:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marzano AV, Damiani G, Ceccherini I, Berti E, Gattorno M, Cugno M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol 2017; 176:1588–98. [DOI] [PubMed] [Google Scholar]
- 19. Gattorno M, Hofer M, Federici S et al Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis 2019;78:1025–1032. [DOI] [PubMed] [Google Scholar]
- 20. Marzano AV, Borghi A, Meroni PL, Cugno M. Pyoderma gangrenosum and its syndromic forms: evidence for a link with autoinflammation. Br J Dermatol 2016; 175:882–91. [DOI] [PubMed] [Google Scholar]
- 21. de Jesus AA, Canna SW, Liu Y, Goldbach‐Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol 2015; 33:823–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jordan CT, Cao L, Roberson ED et al PSORS2 is due to mutations in CARD14. Am J Hum Genet 2012; 90:784–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keijsers RRMC, Hendriks AGM, van Erp PEJ et al In vivo induction of cutaneous inflammation results in the accumulation of extracellular trap‐forming neutrophils expressing RORγt and IL‐17. J Invest Dermatol 2014; 134:1276–84. [DOI] [PubMed] [Google Scholar]
- 24. Lin AM, Rubin CJ, Khandpur R et al Mast cells and neutrophils release IL‐17 through extracellular trap formation in psoriasis. J Immunol 2011; 187:490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laggner U, Di Meglio P, Perera GK et al Identification of a novel proinflammatory human skin‐homing Vγ9Vδ2 T cell subset with a potential role in psoriasis. J Immunol 2011; 187:2783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chiricozzi A, Romanelli P, Volpe E, Borsellino G, Romanelli M. Scanning the immunopathogenesis of psoriasis. Int J Mol Sci 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]