

Summary
Tumor necrosis factor (TNF) receptor‐associated periodic syndrome (TRAPS) is an autoinflammatory disease that is caused by heterozygous mutations in the TNFRSF1A gene. Although more than 150 TNFRSF1A mutations have been reported to be associated with TRAPS phenotypes only a few, such as p.Thr79Met (T79M) and cysteine mutations, have been functionally analyzed. We identified two TRAPS patients in one family harboring a novel p.Gly87Val (G87V) mutation in addition to a p.Thr90Ile (T90I) mutation in TNFRSF1A. In this study, we examined the functional features of this novel G87V mutation. In‐vitro analyses using mutant TNF receptor 1 (TNF‐R1)‐over‐expressing cells demonstrated that this mutation alters the expression and function of TNF‐R1 similar to that with the previously identified pathogenic T79M mutation. Specifically, cell surface expression of the mutant TNF‐R1 in transfected cells was inhibited with both G87V and T79M mutations, whereas the T90I mutation did not affect this. Moreover, peripheral blood mononuclear cells (PBMCs) from TRAPS patients harboring the G87V and T90I mutations showed increased mitochondrial reactive oxygen species (ROS). Furthermore, the effect of various Toll‐like receptor (TLR) ligands on inflammatory responses was explored, revealing that PBMCs from TRAPS patients are hyper‐responsive to TLR‐2 and TLR‐4 ligands and that interleukin (IL)‐8 and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) are likely to be involved in the pathogenesis of TRAPS. These findings suggest that the newly identified G87V mutation is one of the causative mutations of TRAPS. Our findings based on unique TRAPS‐associated mutations provide novel insight for clearer understanding of inflammatory responses, which would be basic findings of developing a new therapeutic and prophylactic approach to TRAPS.
Keywords: autoinflammatory disease, TNF‐R1, TNF receptor‐associated periodic syndrome, TNFRSF1A
We found a novel G87V mutation in TNFRSF1A gene in two patients with TNF receptor‐associated periodic syndrome (TRAPS) in a family. Functional analyses of the G87V mutation have revealed that cell surface expression of the mutant TNF‐R1 was decreased, which is similar to that with the previously identified pathogenic T79M mutation. The peripheral blood mononuclear cells from TRAPS patients harboring the G87V mutation showed increased mitochondrial reactive oxygen species and enhanced production of IL‐8 and GM‐CSF in responses to TLR ligands.

Introduction
Tumor necrosis factor (TNF) receptor‐associated periodic syndrome (TRAPS; OMIM: 142680) is the second most common inherited autosomal dominant autoinflammatory disease 1, 2. It is clinically characterized by recurrent fevers, myalgia, skin rashes and abdominal pain 3, 4. Disease onset generally occurs in early childhood, but can also occur in adults, and the associated inflammatory attacks can last several weeks 3, 4. TRAPS is caused by mutations in the TNF TNFRSF1A gene located on chromosome 12p13, which encodes the 55‐kDa receptor for TNF receptor 1 (TNF‐R1, also referred to as CD120a) 5.
TNF‐R1 consists of an extracellular domain involved in ligand binding, a transmembrane domain, and an intracellular region 1, 5, 6. The binding of TNF to TNF‐R1 induces the recruitment of several intracellular adaptor proteins leading to downstream signaling events that activate inflammatory and apoptotic processes 1, 6, 7. Almost all known TRAPS mutations are point mutations that result in single amino acid substitutions in the extracellular domain of TNF‐R1 3, 4, 6. At present, more than 150 different TNFRSF1A mutations have been reported based on the Infevers database (https://infevers.umai-montpellier.fr/web/index.php) 8.
TRAPS mutations are distinguished, high‐ and low‐penetrance mutations 3, 9. The former type includes T79M and cysteine mutations such as C59R, C62Y, C81F and C117R 5, whereas the latter type includes T90I and R121Q mutations 10. The former types of mutations are associated with severe clinical phenotypes 4. Further, such high‐penetrance mutations affect the structure of the TNF‐R1 protein and interfere with its proper trafficking 1. As a result, mutant TNF‐R1 accumulates excessively in the endoplasmic reticulum (ER) 11, with increased ER stress subsequently causing inflammatory responses 11. In addition, decreased cell surface expression of mutant TNF‐R1 has been revealed in vitro in over‐expression cell models 3, 11, 12, 13, which reflects the aberrant intracellular accumulation of mutant TNF‐R1 protein. The latter type, low penetrance mutations, is associated with a milder form of TRAPS 14. However, the pathogenic mechanisms associated with low penetrance mutations are obscure. For example, dysregulated trafficking of mutant TNF‐R1 is not obvious in such mutations, as shown by the normal cell surface expression of mutant TNF‐R1 3, 11, 12. Although the pathogenic mechanisms underlying some high‐penetrance TRAPS mutations have been proposed 1, most TRAPS mutations have not been functionally analyzed and associated mechanisms have not been fully elucidated.
We recently identified a novel p.Gly87Val (G87V) mutation in the TNFRSF1A gene harbored by two TRAPS patients in one family. These individuals also carried a T90I mutation. In the present study, we examined the functional features of this novel G87V mutation, together with T90I, T79M and R121Q mutations. We found that based on TNR‐F1 over‐expression experiments using human embryonic kidney (HEK) 293 cells, the G87V mutation, but not the T90I mutation, behaves similarly to the previously characterized pathogenic T79M mutation, suggesting a possible causative role for the G87V mutation in TRAPS. Furthermore, peripheral blood mononuclear cells (PBMCs) from the TRAPS patients showed increased mitochondrial reactive oxygen species (ROS) and enhanced inflammatory responses to Toll‐like receptor (TLR) ligands.
Materials and methods
Patients
Patient A was a 65‐year‐old Japanese woman who suffered from various inflammatory episodes including periodic fever lasting for approximately 3 weeks, arthralgia/arthritis, myalgia, rash and scleritis since the age of 25. Further, a marked acute phase response was noted, as indicated by elevated C‐reactive protein. There were no findings suggestive of infection upon bacteriological examination. At the age of 30 years, her clinical symptoms regressed after the initiation of prednisolone, which was associated with improvements in laboratory data; however, febrile attacks and polyarthritis still occurred with low‐dose steroid therapy. The attacks recurred three to four times per year. Until the age of 60, she suffered from persistent arthritis and myalgia in addition to inflammatory attacks, despite continuing low‐dose prednisolone. The administration of etanercept has subsequently improved her symptoms.
Patient B was a 34‐year‐old Japanese woman who presented with periodic episodes of severe abdominal pain with fever since the age of 25. The attacks recurred three to four times per year. For every attack, her symptoms continued for 2–3 weeks. She was referred to our hospital for a consultation, and autoinflammatory disease was highly suspected based on her clinical history. Colchicine treatment did not ameliorate her symptoms. However, the administration of low‐dose prednisolone dramatically improved her inflammatory episodes.
Detection of TNFRSF1A mutations
A genetic test was performed as described previously 15. Genomic DNA was extracted from whole blood using a QIAamp DNA blood kit (QIAGEN, Venlo, the Netherlands). Two‐step polymerase chain reaction (PCR) was performed using the obtained DNA as a template. For the first PCR amplification step, all coding exons from the 12 genes (MEFV, NLRP3, TNFRSF1A, MVK, NOD2, IL1RN, NLRP12, PSTPIP1, PSMB8, NLRC4, PLCG2 and HO‐1) were amplified simultaneously by multiplex PCR in one tube. Secondary PCR amplification was performed by PCR with each primer for the 12 genes using the first PCR product as a template. The purified PCR products were then applied to a next‐generation sequencing system MiSeq platform using the MiSeq Reagent Kit version 3 600 cycles (Illumina, San Diego, CA, USA), according to the manufacturer’s instructions. The obtained data were analyzed as reported 15. The genetic test was approved by the Ethics Committee of Kyoto University (Kyoto, Japan) and the Kazusa DNA Research Institute (Chiba, Japan) and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all subjects.
Investigation of TNFRSF1A gene
As both G87V and T90I mutations were identified in exon 3 of TNFRSF1A, we performed subcloning to determine if these two mutations were, or were not, located on the same allele, as described previously 16, with some modifications. We amplified exon 3 of TNFRSF1A with the proofreading PCR enzyme KOD‐Plus polymerase (Toyobo, Tokyo, Japan) and subcloned it into the pCR2.1‐TOPO vector (Invitrogen, Waltham, MA, USA) after the addition of dA with an LA Taq polymerase (Takara Bio, Shiga, Japan). Subcloned amplicons were selected at random and retrieved by PCR with LA Taq polymerase. The PCR products were treated with ExoSAP‐IT (Thermo Fisher Scientific, Waltham, MA, USA) and proteinase K before direct sequencing. Sequencing was performed at the Kazusa DNA Research Institute using a BigDye Terminator kit (version 3.1) and an ABI 3730 DNA sequencer (Thermo Fisher Scientific).
In‐silico analysis using online prediction tools
The possible pathogenicity of the G87V and T90I TNFRSF1A mutations was analyzed using in‐silico analysis tools. The following tools were used for this 17, 18, 19, 20: SIFT (Sorting Intolerant From Tolerant; https://sift.bii.a-star.edu.sg/), PolyPhen‐2 (Polymorphism Phenotyping version 2; http://genetics.bwh.harvard.edu/pph2/), PROVEAN (Protein Variation Effect Analyzer; http://provean.jcvi.org/index.php) and PANTHER (Protein ANalysis THrough Evolutionary Relationships; http://www.pantherdb.org/).
SIFT is a protein sequence homology‐based tool, which can predict the influence of amino acid substitutions 18. Scores range from 0 to 1, with a cut‐off of 0·05. Amino acid substitutions with a score less than 0·05 are predicted to be deleterious, whereas those with a score greater than or equal to 0·05 are predicted to be tolerated. PROVEAN measures the damaging effects of variations in protein sequences 17. A score equal to or below the threshold of −2·5 indicates a deleterious non‐synonymous single nucleotide polymorphism (nsSNP). PANTHER estimates the likelihood that a particular nsSNP will result in a functional alteration of the protein 19. A substitution score = 0 is considered functionally neutral, whereas negative values predict deleterious substitutions. A score cut‐off of −3 corresponds to a 50% probability that an nsSNP will be deleterious to the protein. PolyPhen‐2 is a sequence‐ and structure‐based method that determines the structural and functional consequences of nsSNPs 20. Scores range from 0 to 1. The classifications of the nsSNPs include ‘possibly damaging’ and ‘probably damaging’ (>0·5) or ‘benign’ (<0·5).
Wild‐type (WT) and mutant TNF‐R1 expression plasmid vectors
The cDNA fragment of human WT TNF‐R1 was obtained from its expression plasmid, as reported previously 3. This fragment was introduced into the pCMV6‐AcGFP vector (OriGene, Rockville, MD, USA), and the resulting protein was designated WT TNF‐R1‐green fluorescent protein (GFP). Expression plasmids for mutant TNF‐R1 were constructed by modifying the WT TNF‐R1‐GFP plasmid. Specifically, cDNA fragments encoding mutant human TNF‐R1, replacing either Thr79 with Met, Arg121 with Gln, Gly87 with Val, Thr90 with Ile or both Gly87 with Val and Thr90 with Ile, were introduced into the pCMV6‐AcGFP vector. The resulting proteins were designated TNF‐R1‐GFP T79M, R121Q, G87V, T90I and G87V/T90I, respectively. To generate TNF‐R1‐expression plasmids without GFP, the cDNA fragments of WT and mutant TNF‐R1 containing a stop codon were generated by PCR using each TNF‐R1‐GFP expression pCMV6‐AcGFP plasmid. The resulting cDNA fragments were then introduced into the pcDNA3.1 vector (Invitrogen). The DNA sequences of all constructs were confirmed by sequencing using the dideoxynucleotide chain termination method with a DNA Sequencing System model 3100 (Applied Biosystems, Foster City, CA, USA).
Flow cytometric analysis of TNF‐R1 expression
HEK293 cells (ATCC, Manassas, VA, USA) were cultured in Dulbecco’s modified essential medium supplemented with 10% heat‐inactivated fetal bovine serum (FBS) and penicillin/streptomycin on 10‐cm dishes at 37°C in a 5% CO2 humidified atmosphere. HEK293 cells were replated onto 10‐cm dishes at a density of 4·7 × 106 cells per dish and transfected with 15 μg of a WT or mutated TNF‐R1‐GFP expression plasmid using lipofectamine 3000 (Thermo Fisher Scientific), according to the manufacturer’s instructions. After transfection, the cells were cultured for 24–48 h in antibiotic‐free medium and then applied to the following flow cytometric analysis.
For cell‐surface TNF‐R1 staining, transfected cells were incubated with allophycocyanin (APC)‐conjugated mouse monoclonal anti‐human TNF‐R1 (FAB225A) (R&D Systems, Minneapolis, MN, USA) or isotype control antibody (130‐098‐846) (Miltenyi Biotec, Bergisch Gladbach, Germany) in staining buffer (phosphate‐buffered saline containing 2% FBS and 0·09% NaN3) on ice for 20 min in the dark. Intracellular staining was performed as reported previously 21. Briefly, cells were fixed in fixation buffer (BD Biosciences, Franklin Lakes, NJ, USA), permeabilized with Perm Buffer III (BD Biosciences), and then stained with APC‐conjugated anti‐TNF‐R1 and isotype control antibodies. After washing twice with staining buffer, cells were suspended in the staining buffer. The cell‐surface and intracellular expression of TNF‐R1 were analyzed with a flow cytometer FACSCanto II (BD Biosciences). GFP‐positive cells were gated and APC intensities were analyzed. For all measurements, a minimum of 3 × 104 events was evaluated. All data were analyzed using FlowJo software (BD Biosciences).
Real‐time quantitative polymerase chain reaction (qPCR) analysis
The qPCR analysis was performed as described previously 22, 23. Total RNA was extracted from HEK293 cells using RNAiso Plus (Takara Bio) and solubilized in RNase‐free water. The cDNA was synthesized using a PrimeScript RT reagent Kit with gDNA Eraser (Perfect Real Time; Takara Bio). The qPCR reactions were performed using SYBR Green PCR master mix (Takara Bio) with the StepOnePlus system (Thermo Fisher Scientific). TNFRSF1A mRNA expression levels were calculated by the ΔΔCt method normalizing to levels of the neomycin‐resistance gene to adjust for the influence of transfection efficiency in the samples. The values were normalized to a baseline control; namely, empty vector‐transfected cells. All qPCR reactions yielded products with single‐peak dissociation curves. Primers used in this study were as follows: TNFRSF1A (forward: 5′‐cacaagccacagagcctaga‐3′, reverse: 5′‐gaattccttccagcgcaac‐3′), HPRT (forward: 5′‐tgaccttgatttattttgcatacc‐3′, reverse: 5′‐cgagcaagacgttcagtcct‐3′) and neomycin‐resistance gene (forward: 5′‐ggccgcttttctggattcat‐3′, reverse: 5′‐agcaatatcacgggtagcca‐3′).
Western blot analysis
TNF‐R1 protein expression levels in HEK293 cells were determined by Western blotting, as described previously 24. HEK293 cells were transfected with the indicated plasmid vectors. One day after transfection, GFP‐positive cells were collected using a fluorescence activated cell sorter (FACS)Aria Ⅲ (BD Biosciences). Collected cells were soaked in the RIPA lysis buffer (Sigma‐Aldrich, St Louis, MO, USA) containing a protease inhibitor cocktail (P8340; Sigma‐Aldrich). After centrifugation (at 17 000 g for 15 min at 4°C), supernatants were collected and protein concentrations were determined using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific). Protein samples were resolved by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. After blocking with 5% skimmed milk in Tris‐buffered saline with 0·1% Tween‐20, the membranes were incubated with the indicated primary antibodies, followed by incubation with the appropriate horseradish peroxidase (HRP)‐conjugated, species‐specific secondary antibodies. Bands were detected using SuperSignal West Dura or Femto chemiluminescent substrate (Thermo Fisher Scientific) and visualized using an ImageQuant LAS‐4000 (GE Healthcare, Little Chalfont, UK). Actin was used as a loading control to normalize the amount of protein. The antibodies used in this study were as follows: anti‐TurboGFP antibody (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan) and anti‐actin antibody (A2066; Sigma‐Aldrich).
Nuclear factor kappa alpha (NF‐κB) transcriptional activity
The plasmids employed for the analysis of NF‐κB transcriptional activity were obtained as follows. The pNF‐κB‐MetLuc2 reporter plasmid with NF‐κB‐binding elements upstream of the gene encoding the secreted luciferase reporter was purchased from Takara Bio. The hybrid promoter sequence of the human elongation factor 1 promotor and R‐U5′ segment of the human T cell leukemia virus‐1 of the mammalian expression vector pSelect‐blasti (Invitrogen) was amplified by PCR and introduced into SEAP‐basic (Takara Bio). The resulting plasmid, designated pSEAP‐EFRU, constitutively expressed secreted alkaline phosphatase (SEAP) at high levels in various types of cell lines. The DNA sequences of pSEAP‐EFRU were confirmed by the dideoxynucleotide chain‐termination method with a DNA Sequencing System model 3100.
HEK293 cells were plated at 1 × 104 cells/well on 96‐well plates. The cells were then transfected with 50 ng of WT or mutant TNF‐R1 expression pcDNA3.1 plasmids with 10 ng of pNF‐κB‐MetLuc2 and pSEAP‐EFRU using PolyFect transfection reagent (Qiagen). After 24 h, culture supernatant was collected and luciferase activity was detected using the Ready‐to‐Glow Dual Secreted Reporter Assay (Takara Bio), according to the manufacturer’s protocol. Relative luciferase activity was determined by dividing the luciferase activity in each sample by SEAP activity to normalize values to transfection efficiency. The relative value of NF‐κB activity in cells expressing WT TNF‐R1 was depicted as 100%.
Human PBMC culture and cytokine assays
Whole blood was obtained from TRAPS patients and healthy individuals. We enrolled 12 healthy individuals into this study: six men and six women, ranging in age from 28 to 74 years (mean = 40 years). PBMCs were isolated from whole blood by density gradient separation using Lymphoprep (Axis‐Shield, Oslo, Norway), as reported previously 25. PBMCs were incubated at 2 × 106 cells/ml on 96‐well plates in RPMI‐1640 medium containing 10% heat‐inactivated FBS and penicillin/streptomycin. These cells were then stimulated with TLR ligands or cytokines as follows: lipopolysaccharides (LPS) from Escherichia coli O111:B4 (0·01, 0·1 and 1·0 ng/ml, TLR‐4 ligand; Sigma‐Aldrich, St Louis, MO, USA), Pam3CSK4 (10 μg/ml, TLR‐1/‐2 ligand, a synthetic triacylated lipopeptide), HKLM (heat‐killed Listeria monocytogenes, 3 × 108 cells/ml, TLR‐2 ligand), FLA‐ST standard (flagellin from Salmonella typhimurium, 10 ng/ml, TLR‐5 ligand), FSL1 (1 ng/ml, TLR‐2/‐6 ligand, a synthetic lipopeptide derived from Mycoplasma salivarium; InvivoGen, San Diego, CA, USA), recombinant human interleukin (IL)‐1β (1 ng/ml) and recombinant human TNF (100 ng/ml; Peprotech, Rocky Hill, NJ, USA). After a 10‐h stimulation, the supernatant was collected, centrifuged to remove cells and debris, and stored at −80°C for cytokine analysis. Cytokine and chemokine profiles in the supernatant (1 : 3 dilution) were determined by the Milliplex human cytokine/chemokine magnetic bead premixed 29‐plex kit (Merck Millipore, Billerica, MA, USA), as reported previously 26. The measured cytokines and chemokines were as follows: epidermal growth factor (EGF), eotaxin, granulocyte colony‐stimulating factor (G‐CSF), granulocyte macrophage colony‐stimulating factor (GM‐CSF), interferon (IFN)‐α2, IFN‐γ, interleukin (IL)‐10, IL‐12p40, IL‐12p70, IL‐13, IL‐15, IL‐17A, IL‐1RA, IL‐1α, IL‐1β, IL‐2, IL‐3, IL‐4, IL‐5, IL‐6, IL‐7, IL‐8, IP‐10, monocyte chemoattractant protein (MCP)‐1, macrophage inflammatory protein (MIP)‐1α, MIP‐1β, TNF, TNF‐β and vascular endothelial growth factor (VEGF)‐A.
Experiments using human blood samples were approved by the Institutional Research Ethics Committee (no. 2829) at Kawasaki Medical School (Kurashiki, Japan). Written informed consent was obtained from patients and healthy individuals.
Mitochondrial superoxide measurements in PBMCs
Mitochondrial superoxide levels in PBMCs were measured by MitoSOX Red‐based flow cytometric measurements as reported previously 27, with some modifications. PBMCs (5 × 105 cells) were incubated in 1·5‐ml microtubes with MitoSOX Red superoxide indicator (Thermo Fisher) for 10 min. The cells were then analyzed on a flow cytometer FACSCanto II (BD Biosciences). Monocyte and lymphocyte subsets were identified based on size and granularity. Fluorescence intensity was measured at an excitation wavelength of 510 nm and an emission wavelength of 580 nm. For this analysis, a minimum of 1 × 105 events was evaluated. All data were analyzed using FlowJo software (BD Biosciences). Seven independent experiments were performed, and mean fluorescence intensity (MFI) values from healthy controls were averaged for each experiment. The MFI of each sample was then divided by the average value from the healthy controls for each experiment.
Statistical analysis
All values are provided as means ± standard deviation (s.d.). Statistical analyses were performed by the two‐tailed unpaired Student’s t‐test to compare two groups and one‐way analysis of variance (anova) (Tukey’s post‐hoc test) to compare three or more groups using GraphPad Prism version 5 (GraphPad Software, San Diego, CA, USA). P‐values less than 0·05 were considered statistically significant.
Results
Identification of G87V and T90I mutations in TNFRSF1A of TRAPS patients
As both patients from one family (Fig. 1a) enlisted for this study had been suffering from inflammatory episodes with periodic fever (Table 1), we strongly suspected the presence of a hereditary autoinflammatory disease. After obtaining written informed consent, we conducted genetic examinations to identify mutations in 12 autoinflammatory disease‐associated genes (MEFV, NLRP3, TNFRSF1A, MVK, NOD2, IL1RN, NLRP12, PSTPIP1, PSMB8, NLRC4, PLCG2 and HO‐1). This genetic test revealed that both patients had c.260 G>T (G87V: p.Gly87Val) heterozygous and c.269 C>T (T90I: p.Thr90Ile) heterozygous mutations in exon 3 of TNFRSF1A (Fig. 1b,c). Following subcloning, an examination revealed that both the G87V and T90I mutations were located on the same allele of the TNFRSF1A gene. Genetic testing also revealed that the patients harbored a MEFV gene mutation (p.Pro115Arg hetero), which is known as a non‐disease‐causing mutation 28. No other mutation was detected in the other tested genes. To investigate the inheritance of TNFRSF1A G87V and T90I mutations in the family, we conducted further genetic tests on other family members. Interestingly, two other family members of six individuals tested harbored the T90I heterozygous mutation without the G87V mutation (Fig. 1a). The two individuals with the T90I mutation alone did not present with periodic inflammatory episodes. Only the two patients with both G87V and T90I mutations exhibited such symptoms.
Figure 1.
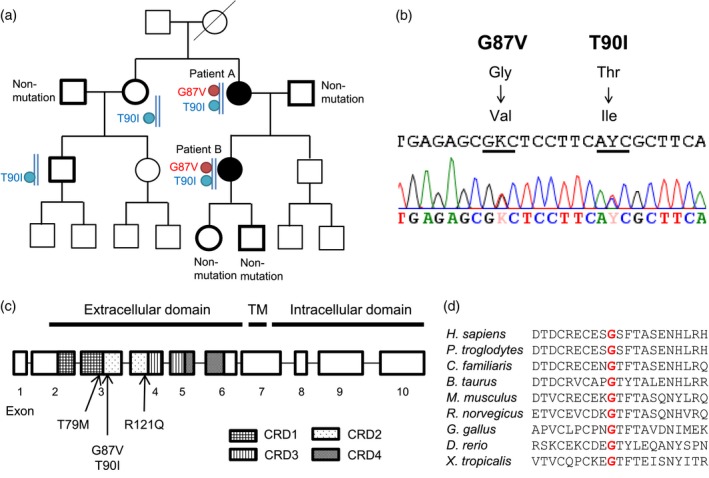
Gene mutations in TNFRSF1A. (a) Pedigree of the family of patients with tumor necrosis factor (TNF) receptor‐associated periodic syndrome (TRAPS). Patients (patients A and B) had both G87V and T90I mutations in the same allele of the TNFRSF1A gene. Patients with periodic inflammatory episodes are indicated as black symbols. Subcloning examination revealed that both G87V and T90I mutations are located on the same allele. The family members subjected to genetic tests are marked in bold. Types of mutations are indicated next to each symbol. (b) DNA sequence electropherograms of TNFRSF1A. Two locations of a heterozygous single‐base mutation, specifically c.260 G>T (G87V: p.Gly87Val) and c.269 C>T (T90I: p.Thr90Ile), in exon 3 were detected. Subcloning examination revealed that both the G87V and T90I mutations are located on the same allele of the TNFRSF1A gene. (c) Gene structure of TNFRSF1A and position of TRAPS mutations. TM = transmembrane domain; CRD = cysteine‐rich domain. T79M, G87V and T90I mutations are located in exon 3. The R121Q mutation is located in exon 4. (d) Comparison of peptide sequences among species. Red color indicates residue 87.
Table 1.
Clinical characteristics of tumor necrosis factor (TNF) receptor‐associated periodic syndrome (TRAPS) patients
| Patient A | Patient B | |
|---|---|---|
| Ethnicity/sex | Japanese/female | Japanese/female |
| Age at onset | 25 years | 24 years |
| Age at diagnosis | 65 years | 34 years |
| Clinical manifestations | Recurrence | Recurrence |
| Fever | Fever | |
| Scleritis | Arthritis | |
| Persistence | Myalgia | |
| Arthritis | Stomach pain | |
| Myalgia | Back pain | |
| Duration of attacks | 14–21 days | 14–21 days |
| WBC (/μl)* | 10 420 | 7360 |
| Neut (%)* | 91·0 | 73·5 |
| Lymph (%)* | 5·0 | 19·0 |
| CRP (mg/l)* | 180 | 48 |
| HGB (g/dl)* | 10·0 | 13·1 |
| HCT (%)* | 32·7 | 39·9 |
Representative results of blood examination, which were obtained during inflammatory episodes from the patients.
WBC = white blood cells; Neut = neutrophils; Lymph = lymphocytes; CRP = C‐reactive protein; HGB = hemoglobin; HCT = hematocrit.
Based on these results, we considered the following possibilities: (1) the G87V mutation is a newly identified disease‐causing mutation, (2) the T90I mutation is a causative mutation, as it has been reported to be a low‐penetrance mutation associated with TRAPS 3 and (3) the co‐existence of both G87V and T90I mutations would exert a disease‐causing effect. To clarify the causative role of G87V and T90I mutations in TRAPS, we performed in‐vitro experiments using mutant TNF‐R1‐expressing cells and compared the results to those of other known TRAPS‐linked mutations.
The G87V mutation is predicted to be highly damaging
The extracellular domain of TNF‐R1 consists of four cysteine‐rich domains characterized by structurally important intramolecular disulfide bonds 1, 6, 7. Both G87V and T90I mutations exist in exon 3, in which G87V and T90I are located in the cysteine‐rich domain (Fig. 1c). Both glycine at residue 87 and threonine at residue 90 of mature human TNF‐R1 are highly conserved among species, as shown in Fig1d. To predict the disease‐causing effect of G87V and T90I mutations, we performed in‐silico analysis using four prediction tools (SIFT, PROVEAN, PANTHER and Polyphen‐2), as shown in Table 2. All prediction tools used predicted that the G87V mutation would be a highly damaging amino acid substitution. Although the T90I mutation was also predicted to be probably damaging or deleterious in three of the analysis tools, SIFT predicted that it would be a tolerated mutation.
Table 2.
In‐silico functional analysis of TNFRSF1A mutations based on four different analysis tools
| AA change | SIFT | PROVEAN | PANTHER | PolyPhen2 | ||||
|---|---|---|---|---|---|---|---|---|
| G87V | Deleterious | 0·01 | Deleterious | −8.92 | Deleterious | −5.03 | Probably damaging | 1 |
| T90I | Tolerated | 0·19 | Deleterious | −4.647 | Deleterious | −4.19 | Probably damaging | 1 |
| T79M | Deleterious | 0·04 | Deleterious | −5.719 | Deleterious | −4.34 | Probably damaging | 1 |
| R121Q | Tolerated | 0·4 | Deleterious | −4.528 | Deleterious | −3.03 | Probably damaging | 1 |
AA = amino acid; SIFT = Sorting Intolerant From Tolerant; PROVEAN = (Protein Variation Effect Analyzer; PANTHER = Protein ANalysis THrough Evolutionary Relationships; PolyPhen‐2 = Polymorphism Phenotyping version 2.
The G87V mutation decreases the cell surface expression of TNF‐R1
TNF‐R1 is transported via the biosynthetic pathway through the ER to the Golgi storage pool 6, 29. In the presence of proinflammatory stimuli, TNF‐R1 is transported to the cell surface, whereas a small fraction of this protein is trafficked to the cell surface without stimuli 6, 29. With TRAPS, mutant TNF‐R1 is suggested to be retained in the ER 1, 29. Previous studies reported that the T79M‐mutant TNF‐R1 is not expressed on the cell surface 3, 11, 13, reflecting its intracellular retention, whereas the T90I‐ or R121Q‐mutant TNF‐R1 is expressed on the cell surface, similar to that observed with WT TNF‐R1 3, 11, 12.
To evaluate the effect of G87V and T90I mutations on TNF‐R1 trafficking, in‐vitro experiments were performed using HEK293 cells expressing WT and mutant TNF‐R1. First, we assessed TNF‐R1 trafficking by analyzing the cell surface and intracellular expression of TNF‐R1 by flow cytometry. It was confirmed that the T79M mutant was not expressed on the cell surface (Fig. 2), as reported previously 3, 11. Similar to this finding, the G87V mutant or G87/T90 double mutant was not expressed on the cell surface (Fig. 2). In contrast, the T90I and R121Q mutants were expressed on the cell surface, similar to WT TNF‐R1 (Fig. 2). These findings indicated that the presence of the G87V variant (G87V or G87V/T90I) is associated with decreased cell surface expression of mutant TNF‐R1.
Figure 2.
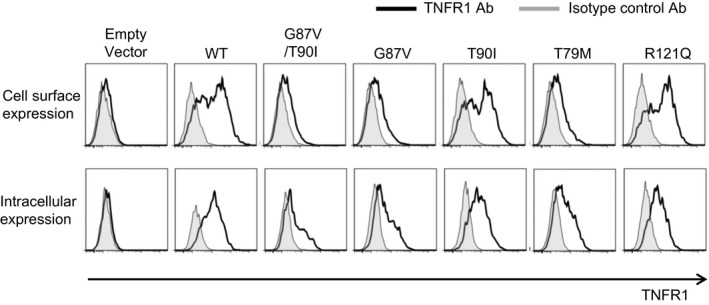
The cell surface expression of tumor necrosis factor (TNF) receptor type I (TNF‐R1) is impaired with the G87V TNFRSF1A mutation. HEK293 cells were transiently transfected with the indicated wild‐type (WT) or mutant TNF‐R1‐green fluorescent protein (GFP) fusion constructs. The cells were stained with the fluorochrome‐labeled anti‐TNF‐R1 antibody. Cell surface or intracellular expression of TNF‐R1 was determined by flow cytometry. The cells were gated based on the GFP‐positive cells, and then histograms representing TNF‐R1 expression were generated. T79M (c.236 C>T, p.Thr79Met) and R121Q (c.362 G>A, p.Arg121Gln) were used as controls. T79M and R121Q mutations are known to be related to severe and mild phenotypes of TRAPS, respectively. Data are representative of three independent experiments.
To exclude the possibilities that TNFRSF1A mutations regulate the expression levels of mRNA or protein, we examined mRNA and protein levels. We found that TNFRSF1A mRNA expression levels normalized to levels of neomycin‐resistance genes were comparable among WT and mutant TNF‐R1‐expressing cells (Supporting information, Fig. 1). To evaluate the expression levels of TNF‐R1 protein, we examined GFP expression levels in the gated cells as shown in Fig. 2. As TNF‐R1 was synthesized as a GFP‐fusion TNF‐R1 protein in this experiment, the levels of GFP reflected protein levels of TNF‐R1. This analysis revealed that GFP expression levels were comparable among WT and mutant TNF‐R1‐expressing cells (Supporting information, Fig. 2a). We further conducted Western blot analysis to determine TNF‐R1 protein levels in whole‐cell lysates. We found that TNF‐R1 protein levels, detected as GFP‐fusion TNF‐R1 proteins, were also comparable among WT and mutant TNF‐R1‐expressing cells (Supporting information, Fig. 2b). These findings indicate that the TNFRSF1A mutations tested in this study do not affect the expression levels of mRNA or protein.
The G87V mutation is associated with altered NF‐κB responses
To evaluate inflammatory responses mediated by TNF‐R1, we examined NF‐κB transcriptional activity in WT and mutant TNF‐R1‐expressing cells. TRAPS mutations such as T79M and C62G have been reported to paradoxically suppress NF‐κB activation in TNF‐R1 over‐expression experiments using transfected cells 11.
HEK293 cells were transfected with WT and mutant TNF‐R1 expression plasmids togther with the NF‐κB reporter plasmid. The over‐expression of WT TNF‐R1 was found to elevate NF‐κB transcriptional activity without the TNF ligand (Fig. 3). We also found that the T79M mutation significantly suppressed spontaneous NF‐κB activity (Fig. 3), in agreement with a previous report 11. Similar to the effect of the T79M mutation, both G87V and G87V/T90I double mutations significantly reduced spontaneous NF‐κB transcriptional activity by approximately 60% (Fig. 3). In contrast, the T90I mutation exerted a milder suppressive effect on NF‐κB transcriptional activity compared to that of the G87V and G87V/T90I double mutations (Fig. 3).
Figure 3.
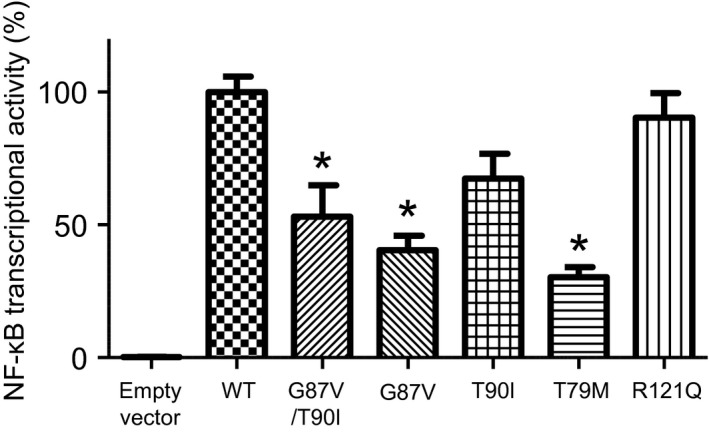
Nuclear factor‐kappa B (NF‐κB) promotor activity in HEK293 cells transfected with TNFRSF1A variants. HEK293 cells were transfected with wild‐type (WT) or mutant tumor necrosis factor receptor 1 (TNF‐R1)‐expression constructs together with an NF‐κB luciferase reporter vector and a secreted alkaline phosphatase (SEAP) expression vector, as a transfection efficiency control. After incubation for 24 h, the activity of each reporter enzyme was measured to calculate the relative NF‐κB transcriptional activity in each sample. The value in cells expressing WT TNF‐R1 is shown as 100%. Data represent the average of three independent experiments. Values are presented as means ± standard deviation (s.d.). *P < 0·05 between WT and indicated mutant TNF‐R1 constructs.
Collectively, both G87V and G87V/T90I double mutations behaved similarly to the pathogenic T79M mutation in terms of cell surface expression and NF‐κB induction, whereas the T90I in G87V mutants did not result in any further reduction in NF‐κB activity induced by the over‐expression of mutant TNF‐R1. The presence of the G87V mutation alone is enough to evoke a characteristic change in TNF‐R1, and the co‐existence of T90I is not indispensable for this characteristic change. These findings indicate that the G87V mutation is a causative mutation associated with TRAPS.
Mitochondrial ROS production is increased in PBMCs from the TRAPS patients
Increased mitochondrial ROS levels in PBMCs from TRAPS patients have been reported 27, 30. This is considered to result from excessive ER stress, caused by the intracellular retention of incorrectly folded TNF‐R1 proteins 11, 27. We thus ascertained mitochondrial ROS in PBMCs from our TRAPS patients harboring the double mutation (patients A and B in Fig. 1a). We performed seven independent experiments using 16 PMBC samples from 12 healthy individuals and eight PBMC samples from the two TRAPS patients. We found that mitochondrial ROS levels were increased in both monocytes (Fig. 4a,b) and lymphocytes (Fig. 4c,d) from TRAPS patients compared to those in cells from healthy individuals. This increase in ROS production caused by the G87V/T90I mutation is consistent with the previous finding regarding pathogenic TRAPS mutations 27, 30.
Figure 4.
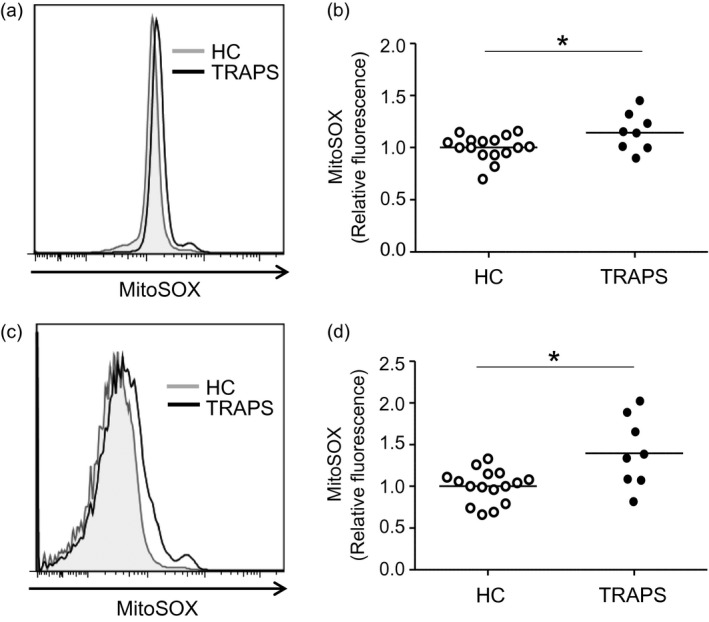
Mitochondrial reactive oxygen species (ROS) production in peripheral blood mononuclear cells (PBMCs) of tumor necrosis factor (TNF) receptor‐associated periodic syndrome (TRAPS) patients. PBMCs were isolated from healthy controls (HC; 16 PMBC samples from 12 individuals) and TRAPS patients (eight PBMC samples from two individuals) by density gradient separation. PBMCs were incubated with MitoSOX Red indicator for 10 min, and then ROS levels were determined by flow cytometry. Cells were gated based on monocytes (a,b) and lymphocytes (c,d) according to forward‐ and side‐scatter characteristics. Representative results of mitochondrial ROS production are shown in histograms (a,c). Data were normalized to the average values of healthy controls in each experiment. *P < 0·05.
The G87V mutation increases cytokine production in response to TLR ligands
PBMCs from TRAPS patients are reported to be hyper‐responsive to inflammatory stimuli such as LPS and TNF, which subsequently promotes the production of inflammatory cytokines such as IL‐6, IL‐1 and TNF 27, 31. To evaluate susceptibility to various inflammatory stimuli, PBMCs from our TRAPS patients (patients A and B in Fig. 1a) and healthy controls were treated with TLR ligands and inflammatory cytokines (TNF and IL‐1β). To determine the effect of previously untested stimuli, we also tested TLR‐2, ‐5 and ‐6 ligands in addition to a TLR‐4 ligand (LPS). In a series of experiments, four PBMC samples from the two TRAPS patients (PBMCs were collected twice from each patient) and four PBMC samples from four individual healthy controls were used. To assess the inflammatory responses of these PBMCs, we measured the levels of 29 cytokines and chemokines in the culture supernatant using multiplex assay systems.
We found that PBMCs from the TRAPS patients tended to exhibit increased IL‐6 production in response to low‐concentration (0·1 ng/ml) LPS treatment (Fig. 5), in accordance with previous findings 27, 31. Of note, FSL‐1 (TLR‐2/‐6 ligand) stimulation enhanced IL‐8 production in the TRAPS PBMCs compared to that in control PBMCs (Fig. 5). Additionally, we found increased production of GM‐CSF in response to LPS in the TRAPS PBMCs (Fig. 5). However, increased secretion of IL‐1α, IL‐1β, and TNF in the TRAPS PBMCs was not observed (Fig. 5). In addition, the levels of other cytokines/chemokines, including IL‐1RA, MIP‐1α, IL‐10, MCP‐1, G‐CSF, MIP‐1β, EGF, eotaxin, IL‐7, IP‐10, IFN‐α2 and VEGF, were comparable between the TRAPS and healthy control PBMCs (Supporting information, Fig. 3). The following cytokines were below the measurable limits: IFN‐γ, IL‐12p40, IL‐12p70, IL‐13, IL‐15, IL‐17A, IL‐2, IL‐3, IL‐4, IL‐5 and TNF‐β. Collectively, this exploratory analysis suggested increased inflammatory responses against TLR ligands.
Figure 5.
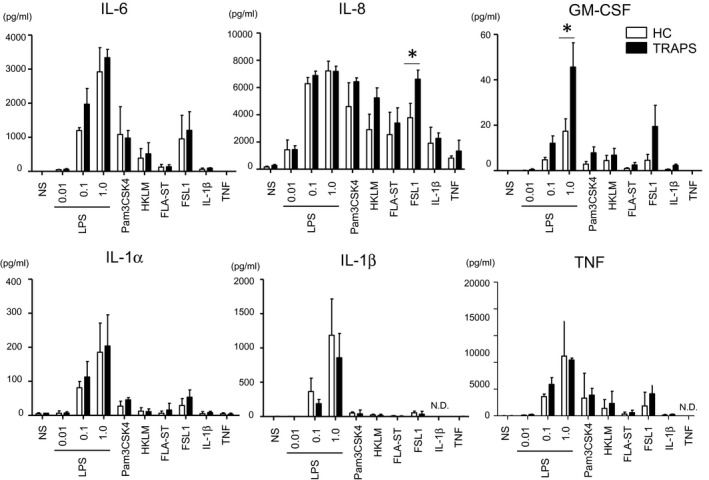
Expression of multiple cytokines in the culture supernatant of peripheral blood mononuclear cells (PBMCs) from healthy controls and the patients with tumor necrosis factor (TNF) receptor‐associated periodic syndrome (TRAPS). PBMCs from healthy controls (HC; n = 4, collected samples = 4) and the TRAPS patients (n = 2, collected samples = 4) were incubated for 10 h with lipopolysaccharide (LPS) (0·01, 0·1, 1·0 ng/ml), Pam3CSK (10 μg/ml), HKLM (3 × 108 cells/ml), FLA‐ST (10 ng/ml), FSL1 (1 ng/ml), interleukin (IL)‐1β (1 ng/ml) and TNF (100 ng/ml). The expression levels of indicated cytokines and chemokines were determined using a multiplex assay system. Values are presented at the means ± standard deviation (s.d.). *P < 0·05; n.d. = not determined; n.s. = no stimulation; LPS = lipopolysaccharide; HKLM, heat‐killed Listeria monocytogenes; FLA‐ST, flagellin from Salmonella typhimurium; IL, interleukin; GM‐CSF, granulocyte–macrophage colony‐stimulating factor.
Discussion
In this study we identified two TRAPS patients in one family, and genetic tests revealed that both had G87V and T90I mutations in the same allele of the TNFRSF1A gene, whereas two other family members harbored only the T90I mutation. The G87V mutation was detected only in patients with inflammatory episodes, whereas individuals with only the T90I mutation did not develop a periodic fever. The association between the presence of the G87V mutation and inflammatory episodes led us to speculate that G87V has a causative role in TRAPS. To ascertain the effect of this mutation we conducted in‐vitro experiments, and the results suggested that this mutation is a novel pathogenic mutation associated with TRAPS.
Many molecular mechanisms have been implicated in the pathogenesis of TRAPS, including autophagy dysregulation 12, 32 and microRNA‐mediated regulation 33. One of the most widely accepted disease mechanisms is impaired TNF‐R1 trafficking 1, 29. Changes in secondary structure result in impaired TNF‐R1 trafficking, leading to the intracellular retention of mutant TNF‐R1 in the ER. Consequently, excessive ER stress causes an associated unfolded protein response, which is followed by inflammatory responses that are characterized by the spontaneous activation of NF‐κB and mitogen‐activated protein kinases 31. Such mechanisms might account for the pathogenesis of TRAPS associated with high penetrance mutations such as T79M and cysteine mutations. However, the mechanisms underlying the effects of low penetrance mutations on this disease are poorly understood.
The present study demonstrates that G87V‐mutant TNF‐R1‐expressing cells behave similarly to those expressing a previously identified pathogenic T79M mutation. The G87V mutation diminished cell surface TNF‐R1 expression and decreased spontaneous NF‐κB activation, which was in accordance with previous studies on the T79M mutation 3, 11. Such findings were not observed for T90I‐ or R121Q‐mutant TNF‐R1‐expressing cells. These results indicate that G87V and G87V/T90I mutations, but not T90I alone, cause similar conformational changes to those induced by the T79M mutation. As G87V is sufficient to induce this effect, this novel variant in TNFRSF1A is suggested to be a mutation that is responsible for TRAPS pathogenesis.
It should be noted that we cannot exclude the possibility that T90I is associated with the inflammatory phenotypes of TRAPS patients. As the T90I mutation is a low‐penetrance mutation associated with TRAPS 3, 34, it is possible that only some of the carriers develop TRAPS phenotypes. Moreover, the co‐existence of G87V and T90I mutations might have a synergistic effect on the induction of TRAPS. In the family described in this report, the G87V missense mutation was speculated to arise de novo in patient A, which was passed on to patient B, although we have not performed a genetic test on the parents of patient A. The identification of a TRAPS patient harboring the G87V mutation alone in TNFRSF1A would directly prove the causative role of this variant in human TRAPS.
In this study, we have shown that expression levels of mutant TNF‐R1 protein are comparable to that of WT TNF‐R1 protein in whole‐cell lysates (Supporting information, Fig. 2). Conversely, mutant TNF‐R1 expression appeared to be decreased in G87V/T90I‐, G87V‐ and T79M‐TNF‐R1‐expressing cells based on intracellular staining (Fig. 2). We speculated that this decrease resulted from diminished antibody binding due to the aberrant aggregation of mutant proteins, as reported previously 11. Alternatively, conformational changes caused by these mutations might have affected the binding capacity of the antibody, as impaired binding capacities of antibodies to mutant TNF‐R1 have been reported previously 35.
Previous reports have shown that, in PBMCs from TRAPS patients, even minor triggers that stimulate the innate immune system can induce the production of inflammatory cytokines such as IL‐1, IL‐6 and TNF 12, 27, 31. However, the exact mediators of inflammatory episodes in TRAPS patients are largely unknown. LPS has been shown to be a stimulator in various experimental settings, and increased responsiveness to LPS has been shown based on in‐vitro experiments and in‐vivo murine TRAPS models 27, 31. In TRAPS mutation knock‐in mouse models, mutant mice are susceptible to low‐dose LPS, which results in increased mortality compared to that in WT mice 31. In agreement with the previous findings, our study demonstrates that low‐dose LPS tends to enhance IL‐6 production in TRAPS PMBC cultures. Despite supporting evidence that LPS can trigger inflammation, including that of this study, there is no obvious clinical association between periodic episodes and bacterial inflammation. Thus, the involvement of other triggering factors must be investigated. To explore this, in the present study, we tested other TLR ligands (TLR‐2, 5‐ and ‐6 ligands) in addition to a TLR‐4 ligand (LPS). We found that PBMCs from TRAPS patients might be hyper‐responsive to TLR‐2/‐6 ligands, as shown by the increased production of IL‐8 in response to a TLR‐2/‐6 ligand (FSL1, a synthetic lipopeptide), compared to that in PBMCs from healthy individuals. Considering these findings, TRAPS mutations are likely to lower the threshold of inflammatory responses to various TLR ligands, including pathogen‐associated molecular patterns and damage‐associated molecular patterns. Indeed, a recent study has reported that PBMCs from TRAPS patients are susceptible to a TLR‐9 ligand 36. Apart from TLR ligands, heparin has been suggested to be a natural stimulant of inflammation 37, which might imply the possible involvement of the transmembrane protein 184A, a receptor for heparin 38. Moreover, female hormones are suggested to modulate the severity of inflammation 39. To develop a new therapeutic and prophylactic approach to periodic inflammatory episodes, further research will be required to determine if inflammatory episodes are caused by specific stimuli and to identify such triggers.
In terms of inflammatory mediators, the present study suggests the possible involvement of IL‐8 and GM‐CSF, as shown by increased secretion from PBMCs with the G87V mutation in response to TLR ligands. The increased expression of IL‐8 and GM‐CSF is also in accordance with previous reports 40, 41, 42. IL‐8 and GM‐CSF probably play roles in promoting inflammation by recruiting inflammatory cells and activating the generation of myeloid cells. Previous studies also suggested the involvement of IL‐1β, IL‐6 and TNF in the etiology of TRAPS 12, 31. The involvement of IL‐1β and TNF is supported by the therapeutic effect of canakinumab (anti‐IL‐1β antibody) 43, 44 and etanercept (soluble TNF receptor p75) 45. In our study, IL‐6 and TNF secretion from PBMCs with the G87V mutation tended to be increased in response to lower concentrations (0·1 ng/ml) of LPS compared to those in cells from healthy controls. However, increased secretion of IL‐1 was not observed in our experimental settings. Differences in experimental settings between ours and previous studies might have affected the results of cytokine production; further, some unknown genetic factors other than TNFRSF1A might have modulated the severity of such inflammatory responses. It should be noted that this is an exploratory analysis, because sample sizes and types of mutations were limited in our PBMC experiments. Further analyses are required to evaluate the detailed mechanisms underlying these augmented inflammatory responses by increasing the sample size.
Our study also suggests the possible involvement of lymphocytes in the pathogenesis of TRAPS. In cultures of TRAPS PBMCs harboring the G87V mutation, mitochondrial ROS production was increased in lymphocytes and in monocytes. Although most previous studies focused on the effect of TRAPS mutations in monocytes 27, 30, 46, such variations might also affect the functions of lymphocytes. This notion is supported by a previous study showing the dysregulation of T cell subsets in TRAPS patients 47. Although dysfunctions in autoimmune systems are not attributed to the development of TRAPS, the dysregulation of adaptive immune cell components might contribute to the autoinflammatory features of TRAPS, in association with activated innate immune responses. As TNF‐R1 is ubiquitously expressed in various cells, analyses of the effects of TRAPS mutations in various cell types including lymphocytes would provide a clearer understanding of the pathogenesis of TRAPS.
In conclusion, the newly identified G87V mutation in TNFRSF1A is considered a causative mutation of TRAPS, as it behaves similarly to the previously identified pathogenic T79M mutation. The possible involvement of TLR‐2/‐6 ligand and cytokines (IL‐8 and GM‐CSF) is also suggested to be a pathophysiological mechanism underlying TRAPS. Our findings, obtained from patients harboring unique mutations, provide novel insight to understand more clearly the inflammatory responses associated with TRAPS, which represent basic findings that might lead to the development of new therapeutic and prophylactic approaches for TRAPS.
Author contributions
S. T., H. M., Y. M. and T. M. designed the study. S. T., H. M., M. I., H. H., Y. H. and T. M. conducted the study and collected data. S. T., H. M., M. I., H. H., Y. H. and T. M. analyzed the data. S. T., H. M., M. I., A. N., H. H., K. I., Y. M. and T. M. interpreted the data. S. T., Y. M. and T. M/ wrote the original draft. All authors contributed to the review and editing of the manuscript and its approval. S. T., Y. M. and T. M. take responsibility for the integrity of the data analysis.
Disclosures
S. T., A. N., H. H., Y. M. and T. M. have received scholarship donations from Chugai Pharmaceutical. R. N. has received scholarship donations from Novartis. The funders had no role in the design of the study, in the collection, analyses or interpretation of data, in the writing of the manuscript or in the decision to publish the results.
Supporting information
Fig. S1. TNFRSF1A mRNA expression levels in WT or mutant TNFR1‐expressing HEK293 cells. (a) An agarose gel electrophoretic image of PCR products amplified from the TNFRSF1A gene. RNA samples were collected from WT TNFR1‐expressing cells. The RNA samples were treated with DNase to eliminate contaminating plasmid DNA and then used to synthesize cDNA. To confirm the absence of plasmid DNA, the cDNA and indicated RNA samples were applied to PCR, and the yielded PCR products were electrophoresed. The image indicates that contaminating plasmid DNA was properly removed by DNase treatment. RT, reverse transcription. (b,c) Quantitative‐PCR analysis of TNFRSF1A (b) and neomycin‐resistance gene (c) expression. HEK293 cells were transfected with WT or mutant TNFR1‐expression vectors. After a 24‐h culture, RNA samples were collected. Gene expression levels relative to HPRT were calculated and normalized to expression levels in HEK293 cells transfected with a WT TNFR1 vector. HPRT, hypoxanthine phosphoribosyltransferase. (d) TNFRSF1A gene expression relative to levels of the neomycin‐resistance gene. Gene expression levels were determined and normalized to expression in HEK293 cells transfected with a WT TNFR1 vector. Values are presented at means ± SD.
Fig. S2. TNFR1 protein expression levels in WT or mutant TNFR1‐expressing HEK293 cells. (a,b) HEK293 cells were transiently transfected with WT or mutant TNFR1‐GFP expression vectors. TNFR1 protein expression levels in the cells were evaluated by detecting green fluorescent protein (GFP), since TNFR1 was produced as a GFP‐fusion protein. (a) Flow cytometry for GFP expression. After culturing HEK293 cells transfected with WT or mutant TNFR1‐GFP expression vectors for 24 h, GFP‐positive cells were analyzed by flow cytometry. The cells were gated based on GFP‐positive cells, and then histograms representing GFP expression levels were generated. (b) Immunoblot analysis of GFP. HEK293 cells were transfected with WT or mutant TNFR1‐expression vectors. After a 24‐h culture, GFP‐positive cells were collected using a FACSAria Ⅲ cell sorter, and protein samples were collected. GFP‐fusion TNFR1 protein levels were determined using an anti‐TurboGFP antibody.
Fig. S3. Expression of multiple cytokines in the culture supernatant of peripheral blood mononuclear cells (PBMCs) from healthy controls and patients with TNF receptor‐associated periodic syndrome (TRAPS). PBMCs from healthy controls (HC; n = 4, collected samples = 4) and TRAPS patients (n = 2, collected samples = 4) were incubated for 10 h with LPS (0.01, 0.1, 1.0 ng/ml), Pam3CSK (10 μg/ml), HKLM (3 × 108 cells/ml), FLA‐ST (10 ng/ml), FSL1 (1 ng/ml), IL‐1β (1 ng/ml), and TNF (100 ng/ml). The expression levels of indicated cytokines and chemokines were determined using a multiplex assay system. Values are presented at the means ± SD. *P < 0.05. N.D. = not determined. NS, no stimulation; LPS, lipopolysaccharide; HKLM, heat‐killed Listeria monocytogenes; FLA‐ST, flagellin from Salmonella typhimurium; IL, interleukin; IL‐1RA, interleukin‐1 receptor antagonist; MIP‐1α, macrophage inflammatory protein‐1 α; MCP‐1, monocyte chemotactic protein‐1; G‐CSF, granulocyte colony stimulating factor; MIP‐1β, macrophage inflammatory protein‐1β; TNF, tumor necrosis factor; EGF, epidermal growth factor; IP‐10, interferon‐inducible protein 10; IFNα2, interferon α2; VEGF, vascular endothelial growth factor.
Acknowledgments
We would like to thank Dr S. Inokuchi (Department of Rheumatology, Kyusyu University) for kindly providing TNF‐R1 expression plasmids and the staff at the Department of Pediatrics, Kyoto University Graduate School of Medicine for performing genetic tests and providing critical suggestions. We are grateful to Dr T. Otsuki, Dr S. Lee (Department of Hygiene, Kawasaki Medical School), Dr K. Kawahara, Dr S. Fujita, Dr T. Akagi, Y. Mino, H. Nakashima, N. Takemasa, M. Yoshimoto, N. Obara (Department of Rheumatology, Kawasaki Medical School), Y. Tsukada and S. Tarumoto (undergraduate students, Kawasaki Medical School) for their technical assistance. We are also indebted to the staff at the Central Research Institute of Kawasaki Medical School. Additionally, we thank Editage for English language editing. This work was supported by grants from The Kawasaki Foundation for Medical Science and Medical Welfare (S. T.), Japan Rheumatism Foundation (S. T.), JSPS KAKENHI (19K08923 to H. H.) and Kawasaki Medical School (30B‐053 to H. H.).
References
- 1. Jarosz‐Griffiths HH, Holbrook J, Lara‐Reyna S, McDermott MF. TNF receptor signalling in autoinflammatory diseases. Int Immunol 2019. 10.1093/intimm/dxz024. [DOI] [PubMed] [Google Scholar]
- 2. Ozen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next‐of‐kin. Nat Rev Rheumatol 2014; 10:135–47. [DOI] [PubMed] [Google Scholar]
- 3. Ueda N, Ida H, Washio M et al . Clinical and genetic features of patients with TNFRSF1A variants in Japan: findings of a nationwide survey. Arthritis Rheumatol 2016; 68:2760–71. [DOI] [PubMed] [Google Scholar]
- 4. Lachmann HJ, Papa R, Gerhold K et al . The phenotype of TNF receptor‐associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the Eurofever/EUROTRAPS international registry. Ann Rheum Dis 2014; 73:2160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McDermott MF, Aksentijevich I, Galon J et al . Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97:133–44. [DOI] [PubMed] [Google Scholar]
- 6. Lobito AA, Gabriel TL, Medema JP, Kimberley FC. Disease causing mutations in the TNF and TNFR superfamilies: Focus on molecular mechanisms driving disease. Trends Mol Med 2011; 17:494–505. [DOI] [PubMed] [Google Scholar]
- 7. Shi JH, Sun SC. Tumor necrosis factor receptor‐associated factor regulation of nuclear factor kappaB and mitogen‐activated protein kinase pathways. Front Immunol 2018; 9:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Milhavet F, Cuisset L, Hoffman HM et al . The Infevers Autoinflammatory Mutation Online Registry: update with new genes and functions. Hum Mutat 2008; 29:803–8. [DOI] [PubMed] [Google Scholar]
- 9. Hull KM, Drewe E, Aksentijevich I et al . The TNF receptor‐associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine 2002; 81:349–68. [DOI] [PubMed] [Google Scholar]
- 10. Cantarini L, Rigante D, Merlini G et al . The expanding spectrum of low‐penetrance TNFRSF1A gene variants in adults presenting with recurrent inflammatory attacks: clinical manifestations and long‐term follow‐up. Semin Arthritis Rheum 2014; 43:818–23. [DOI] [PubMed] [Google Scholar]
- 11. Lobito AA, Kimberley FC, Muppidi JR et al . Abnormal disulfide‐linked oligomerization results in ER retention and altered signaling by TNFR1 mutants in TNFR1‐associated periodic fever syndrome (TRAPS). Blood 2006; 108:1320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bachetti T, Chiesa S, Castagnola P et al Autophagy contributes to inflammation in patients with TNFR‐associated periodic syndrome (TRAPS). Ann Rheum Dis 2013; 72:1044–52. [DOI] [PubMed] [Google Scholar]
- 13. Todd I, Radford PM, Draper‐Morgan KA et al . Mutant forms of tumour necrosis factor receptor I that occur in TNF‐receptor‐associated periodic syndrome retain signalling functions but show abnormal behaviour. Immunology 2004; 113:65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pelagatti MA, Meini A, Caorsi R et al . Long‐term clinical profile of children with the low‐penetrance R92Q mutation of the TNFRSF1A gene. Arthritis Rheum 2011; 63:1141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakayama M, Oda H, Nakagawa K et al Accurate clinical genetic testing for autoinflammatory diseases using the next‐generation sequencing platform MiSeq. Biochem Biophys Rep 2017; 9:146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanaka N, Izawa K, Saito MK et al . High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum 2011; 63:3625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLOS ONE 2012; 7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31:3812–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas PD, Campbell MJ, Kejariwal A et al . PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 2003; 13:2129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adzhubei IA, Schmidt S, Peshkin L et al . A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iseki M, Omori‐Miyake M, Xu W et al . Thymic stromal lymphopoietin (TSLP)‐induced polyclonal B‐cell activation and autoimmunity are mediated by CD4+ T cells and IL‐4. Int Immunol 2012; 24:183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukai T, Ishida S, Ishikawa R et al . SH3BP2 cherubism mutation potentiates TNF‐alpha‐induced osteoclastogenesis via NFATc1 and TNF‐alpha‐mediated inflammatory bone loss. J Bone Mineral Res 2014; 29:2618–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mukai T, Gallant R, Ishida S et al . Loss of SH3 domain‐binding protein 2 function suppresses bone destruction in tumor necrosis factor‐driven and collagen‐induced arthritis in mice. Arthritis Rheum 2015; 67:656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nagasu A, Mukai T, Iseki M et al . Sh3bp2 gain‐of‐function mutation ameliorates lupus phenotypes in B6.MRL‐Fas(lpr) mice. Cells 2019; 8:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujita S, Mukai T, Mito T et al . Pharmacological inhibition of tankyrase induces bone loss in mice by increasing osteoclastogenesis. Bone 2018; 106:156–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katsumata R, Ishii M, Lee S et al . Cytokine profile and immunoglobulin E‐mediated serological food hypersensitivity in patients with irritable bowel syndrome with diarrhea. J Neurogastroenterol Motility 2018; 24:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bulua AC, Simon A, Maddipati R et al . Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1‐associated periodic syndrome (TRAPS). J Exp Med 2011; 208:519–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Migita K, Izumi Y, Jiuchi Y et al . Familial Mediterranean fever is no longer a rare disease in Japan. Arthritis Res Therapy 2016; 18:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Turner MD, Chaudhry A, Nedjai B. Tumour necrosis factor receptor trafficking dysfunction opens the TRAPS door to pro‐inflammatory cytokine secretion. Biosci Rep 2012; 32:105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dickie LJ, Aziz AM, Savic S et al . Involvement of X‐box binding protein 1 and reactive oxygen species pathways in the pathogenesis of tumour necrosis factor receptor‐associated periodic syndrome. Ann Rheum Dis 2012; 71:2035–43. [DOI] [PubMed] [Google Scholar]
- 31. Simon A, Park H, Maddipati R et al . Concerted action of wild‐type and mutant TNF receptors enhances inflammation in TNF receptor 1‐associated periodic fever syndrome. Proc Natl Acad Sci USA 2010; 107:9801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hua Y, Shen M, McDonald C, Yao Q. Autophagy dysfunction in autoinflammatory diseases. J Autoimmun 2018; 88:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harrison SR, Scambler T, Oubussad L et al . Inositol‐requiring enzyme 1‐mediated downregulation of MicroRNA (miR)‐146a and miR‐155 in primary dermal fibroblasts across three TNFRSF1A mutations results in hyperresponsiveness to lipopolysaccharide. Front Immunol 2018; 9:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ida H, Kawasaki E, Miyashita T et al . A novel mutation (T61I) in the gene encoding tumour necrosis factor receptor superfamily 1A (TNFRSF1A) in a Japanese patient with tumour necrosis factor receptor‐associated periodic syndrome (TRAPS) associated with systemic lupus erythematosus. Rheumatology (Oxf) 2004; 43:1292–9. [DOI] [PubMed] [Google Scholar]
- 35. Todd I, Radford PM, Daffa N, Bainbridge SE, Powell RJ, Tighe PJ. Mutant tumor necrosis factor receptor associated with tumor necrosis factor receptor‐associated periodic syndrome is altered antigenically and is retained within patients' leukocytes. Arthritis Rheum 2007; 56:2765–73. [DOI] [PubMed] [Google Scholar]
- 36. Negm OH, Singh S, Abduljabbar W et al . Patients with TNF receptor associated periodic syndrome (TRAPS) are hypersensitive to Toll‐like receptor 9 stimulation. Clin Exp Immunol 2019. 10.1111/cei.13306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ohmori S, Hino R, Nakamura M, Tokura Y. Heparin serves as a natural stimulant of the inflammasome and exacerbates the symptoms of tumor necrosis factor receptor‐associated periodic syndrome (TRAPS). J Dermatol Sci 2012; 66:82–4. [DOI] [PubMed] [Google Scholar]
- 38. Pugh RJ, Slee JB, Farwell SL et al . Transmembrane protein 184A Is a receptor required for vascular smooth muscle cell responses to heparin. J Biol Chem 2016; 291:5326–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kriegel MA, Huffmeier U, Scherb E et al . Tumor necrosis factor receptor‐associated periodic syndrome characterized by a mutation affecting the cleavage site of the receptor: implications for pathogenesis. Arthritis Rheum 2003; 48:2386–8. [DOI] [PubMed] [Google Scholar]
- 40. Rebelo SL, Amel‐Kashipaz MR, Radford PM et al Novel markers of inflammation identified in tumor necrosis factor receptor‐associated periodic syndrome (TRAPS) by transcriptomic analysis of effects of TRAPS‐associated tumor necrosis factor receptor type I mutations in an endothelial cell line. Arthritis Rheum 2009; 60:269–80. [DOI] [PubMed] [Google Scholar]
- 41. Nedjai B, Hitman GA, Quillinan N et al . Proinflammatory action of the antiinflammatory drug infliximab in tumor necrosis factor receptor‐associated periodic syndrome. Arthritis Rheum 2009; 60:619–25. [DOI] [PubMed] [Google Scholar]
- 42. Greco E, Aita A, Galozzi P et al . The novel S59P mutation in the TNFRSF1A gene identified in an adult onset TNF receptor associated periodic syndrome (TRAPS) constitutively activates NF‐kappaB pathway. Arthritis Res Ther 2015; 17:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gattorno M, Obici L, Cattalini M et al . Canakinumab treatment for patients with active recurrent or chronic TNF receptor‐associated periodic syndrome (TRAPS): an open‐label, phase II study. Ann Rheum Dis 2017; 76:173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Benedetti F, Gattorno M, Anton J et al . Canakinumab for the Treatment of autoinflammatory recurrent fever syndromes. N Engl J Med 2018; 378:1908–19. [DOI] [PubMed] [Google Scholar]
- 45. Kusuhara K, Hoshina T, Saito M et al . Successful treatment of a patient with tumor necrosis factor receptor‐associated periodic syndrome using a half‐dose of etanercept. Pediatr Int 2012; 54:552–5. [DOI] [PubMed] [Google Scholar]
- 46. Todd I, Radford PM, Ziegler‐Heitbrock L, Ghaemmaghami AM, Powell RJ, Tighe PJ. Elevated CD16 expression by monocytes from patients with tumor necrosis factor receptor‐associated periodic syndrome. Arthritis Rheum 2007; 56:4182–8. [DOI] [PubMed] [Google Scholar]
- 47. Pucino V, Lucherini OM, Perna F et al . Differential impact of high and low penetrance TNFRSF1A gene mutations on conventional and regulatory CD4+ T cell functions in TNFR1‐associated periodic syndrome. J Leukoc Biol 2016; 99:761–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. TNFRSF1A mRNA expression levels in WT or mutant TNFR1‐expressing HEK293 cells. (a) An agarose gel electrophoretic image of PCR products amplified from the TNFRSF1A gene. RNA samples were collected from WT TNFR1‐expressing cells. The RNA samples were treated with DNase to eliminate contaminating plasmid DNA and then used to synthesize cDNA. To confirm the absence of plasmid DNA, the cDNA and indicated RNA samples were applied to PCR, and the yielded PCR products were electrophoresed. The image indicates that contaminating plasmid DNA was properly removed by DNase treatment. RT, reverse transcription. (b,c) Quantitative‐PCR analysis of TNFRSF1A (b) and neomycin‐resistance gene (c) expression. HEK293 cells were transfected with WT or mutant TNFR1‐expression vectors. After a 24‐h culture, RNA samples were collected. Gene expression levels relative to HPRT were calculated and normalized to expression levels in HEK293 cells transfected with a WT TNFR1 vector. HPRT, hypoxanthine phosphoribosyltransferase. (d) TNFRSF1A gene expression relative to levels of the neomycin‐resistance gene. Gene expression levels were determined and normalized to expression in HEK293 cells transfected with a WT TNFR1 vector. Values are presented at means ± SD.
Fig. S2. TNFR1 protein expression levels in WT or mutant TNFR1‐expressing HEK293 cells. (a,b) HEK293 cells were transiently transfected with WT or mutant TNFR1‐GFP expression vectors. TNFR1 protein expression levels in the cells were evaluated by detecting green fluorescent protein (GFP), since TNFR1 was produced as a GFP‐fusion protein. (a) Flow cytometry for GFP expression. After culturing HEK293 cells transfected with WT or mutant TNFR1‐GFP expression vectors for 24 h, GFP‐positive cells were analyzed by flow cytometry. The cells were gated based on GFP‐positive cells, and then histograms representing GFP expression levels were generated. (b) Immunoblot analysis of GFP. HEK293 cells were transfected with WT or mutant TNFR1‐expression vectors. After a 24‐h culture, GFP‐positive cells were collected using a FACSAria Ⅲ cell sorter, and protein samples were collected. GFP‐fusion TNFR1 protein levels were determined using an anti‐TurboGFP antibody.
Fig. S3. Expression of multiple cytokines in the culture supernatant of peripheral blood mononuclear cells (PBMCs) from healthy controls and patients with TNF receptor‐associated periodic syndrome (TRAPS). PBMCs from healthy controls (HC; n = 4, collected samples = 4) and TRAPS patients (n = 2, collected samples = 4) were incubated for 10 h with LPS (0.01, 0.1, 1.0 ng/ml), Pam3CSK (10 μg/ml), HKLM (3 × 108 cells/ml), FLA‐ST (10 ng/ml), FSL1 (1 ng/ml), IL‐1β (1 ng/ml), and TNF (100 ng/ml). The expression levels of indicated cytokines and chemokines were determined using a multiplex assay system. Values are presented at the means ± SD. *P < 0.05. N.D. = not determined. NS, no stimulation; LPS, lipopolysaccharide; HKLM, heat‐killed Listeria monocytogenes; FLA‐ST, flagellin from Salmonella typhimurium; IL, interleukin; IL‐1RA, interleukin‐1 receptor antagonist; MIP‐1α, macrophage inflammatory protein‐1 α; MCP‐1, monocyte chemotactic protein‐1; G‐CSF, granulocyte colony stimulating factor; MIP‐1β, macrophage inflammatory protein‐1β; TNF, tumor necrosis factor; EGF, epidermal growth factor; IP‐10, interferon‐inducible protein 10; IFNα2, interferon α2; VEGF, vascular endothelial growth factor.